

Abstract
The use of synergistic antibiotic combinations has emerged as a viable approach to contain the rapid spread of antibiotic‐resistant pathogens. Here we report the discovery of a new strongly synergistic pair – microcin J25 and sulfamonomethoxine. The former is a lasso peptide that inhibits the function of RNA polymerase and the latter is a sulfonamide antibacterial agent that disrupts the folate pathway. Key to our discovery was a screening strategy that focuses on an antibiotic (microcin J25) that targets a hub (transcription) in the densely interconnected network of cellular pathways. The rationale was that disrupting such a hub likely weakens the entire network, generating weak links that potentiate the growth inhibitory effect of other antibiotics. We found that MccJ25 potentiates five other antibiotics as well. These results showcase the merit of taking a more targeted approach in the search and study of synergistic antibiotic pairs.
Keywords: antibiotics, metabolic network, microcin J25 (MccJ25), resistance development, sulfonamide, synergy
The lasso peptide microcin J25 and sulfamonomethoxine each disrupt a hub, transcription and folate biosynthesis, respectively, in the intricately interconnected cellular network. They act in synergy to inhibit bacterial growth and suppress resistance development.

Introduction
The average human life expectancy in developed countries has increased by more than 30 years since the 1950s, and, to a large extent, this gain can be attributed to the wide availability of effective antibiotics. [1] Nevertheless, drug‐resistant microbial pathogens have been spreading at an alarming speed in recent years. They threaten to make once life‐saving antibiotics obsolete and chip away the achievements of modern healthcare accumulated over the past several decades. Combination therapy, especially the use of antibiotics that act in synergy, has emerged as a viable remedy to combat drug‐resistant microbial pathogens. [2] As one antibiotic enhances the potency of the other in a synergistic pair, such a regimen generally requires the use of either antibiotics at lower doses and reduces the risk of adverse side effects. A strong synergistic pair of antibiotics also suppresses resistance development, [3] and in some cases, reinvigorates the use of otherwise defunct antibiotics. [4] Despite these benefits, few synergistic antibiotic combinations are known and even fewer are used in clinics.
Antibiotic synergism generally falls into two broad categories. It may arise from one antibiotic in a pair increasing target access for the other, which we term access enabling mode. For example, penicillin weakens the cell envelope to increase the permeability of streptomycin, so that more streptomycin molecules make their way across the cell membrane to bind to the ribosomal 30S subunit, manifesting more potent bacterial growth inhibition. [5] In contrast, many synergistic combinations operate in a mechanism cooperation mode and consist of antibiotics targeting nearby biosynthetic steps within an essential pathway, resulting in an apparent growth inhibitory effect that is more potent than the expected sum of individual antibiotics (Figure 1a). The fosfomycin‐carbapenem and sulfonamide‐trimethoprim combinations both fall into this category. In the former pair, fosfomycin and carbapenem inhibit the UDP−N‐acetylglucosamine enolpyruvyl transferase (MurA) and the dd‐transpeptidase of the peptidoglycan biosynthesis pathway, respectively. [6] In the latter pair, sulfonamide and trimethoprim inhibit the production of dihydropteroate (DHP) and tetrahydrofolate (THF), respectively, both of which are critical biosynthetic intermediates in the folate pathway. [7] We believe that, instead of seeing each pathway as a linear sequence of events, it is more appropriate to view the numerous cellular pathways as a network (Figure 1b). While a generalizable theory for antibiotic synergy remains elusive, such a viewpoint suggests that scouting around an extensively interconnected pathway is a logical strategy if one were to search for new synergistic pairs.
Figure 1.

a, Synergistic antibiotic combinations may operate in a mechanism cooperation mode and consist of antibiotics targeting nearby biosynthetic steps within an essential pathway. b, We view the numerous metabolic pathways in a cell as a densely interconnected network. Disrupting a hub (blue‐filled circle) will weaken itself and many other pathways (open circles, dotted edges), resulting in an overall debilitated network that is much more sensitive toward other disruptions. In this study, we used the lasso peptide MccJ25, an antibiotic known to bind and inhibit the RNAP complex, to disrupt transcription (the hub) and investigate whether it potentiates the activity of other antibiotics.
Transcription is a hub where numerous biological pathways intersect. For example, transcription and replication both require physical access to the DNA template, and conflicts between these two processes are inevitable as the replisome moves approximately ten‐fold faster than the transcription machinery. [8] The transcription machinery must also communicate with the ribosome(s) as an mRNA molecule is translated while it is being transcribed in a prokaryotic cell. [9] Transcription‐translation coupling has manifested in the electron micrograph that has been a staple in biochemistry textbooks, wherein a DNA strand is associated with multiple mRNA transcripts, each of which is occupied by multiple ribosomes. [10] Our hypothesis was that disrupting transcription, where numerous biological pathways intersect, may generate weak links elsewhere in the network to potentiate the activity of other antibiotics. Focusing on such a “privileged” pathway may be an effective strategy in searching for new antibiotic synergy. Furthermore, RNA polymerases (RNAP) are known to be highly conserved among bacteria and yet distinct from their human counterparts. [11] The bacterial RNAP complex is therefore both an appropriate target for developing new antimicrobial agents [12] and an ideal starting point to search for new synergistic antibiotic combinations. Herein, we report the discovery and subsequent mechanistic studies of the microcin J25 (MccJ25)‐sulfamonomethoxine (SMM) combination as a new strongly synergistic pair of antibiotics. MccJ25 was also found to potentiate five other antibiotics to lesser extents.
Results and Discussion
To investigate whether the disruption of a hub in the network of cellular pathways, i. e., transcription, will potentiate the activity of other antibiotics, we chose to conduct a systematic screen around the known RNAP inhibitor MccJ25. MccJ25 is a natural product that belongs to the ribosomally synthesized and post‐translationally modified peptide (RiPP) superfamily and exhibits potent antibacterial activity against Enterobacteria, including Escherichia coli and Salmonella Newport, by targeting the RNA polymerase (RNAP). It was regarded as a promising candidate for treating bacterial infections since its discovery, [13] displaying a minimum inhibitory concentration (MIC) of 0.0625 to 0.5 μg/mL against various pathogenic strains. [14] It is not only stable under gastrointestinal tract conditions but also showed no detectable toxicity. [15] As a lasso peptide, MccJ25 has also drawn considerable interest from chemists for its unique structure, wherein the tail is threaded through and mechanically locked inside the noose. [16] Resistance conferring mutations and structural studies indicated that MccJ25 interacts with the β’ subunit (RpoC) of the RNAP complex. [17] It blocks the secondary channel through which NTP substrates enter the active site, thereby disrupting transcription in bacteria, and shows a partial competitive mechanism of inhibition with respect to NTP. [18]
The bacterial RNAP complex consists of two α subunits and one of each of the β, β′, and ω subunits. [19] Transcription entails a series of events that begins with promoter recognition and is followed by unwinding of the DNA template at the transcription start site. [20] After abortive transcription, the RNAP leaves the promoter region and elongation ensues, forming a dynamic DNA/RNAP/mRNA ternary complex that ratchets along the DNA template. [21] Transcription ends at specific terminator sequences that weakens the interaction between DNA and the ternary complex, resulting in the dissociation of the ternary complex and the disintegration of the RNAP complex itself. [22] Disrupting any of these carefully choreographed steps will throw transcription into disarray and negatively impact bacterial growth. However, rifampicin is the only antibiotic currently used in the clinics that targets transcription. [12] We wanted to close this knowledge gap and decided to conduct a systematic screen in search of new synergistic antibiotic combinations, wherein one or both antibiotics function by interfering with transcription.
We organized known antibiotics into five broad categories, i. e., those that disrupt DNA replication, transcription, translation, folate biosynthesis, and cell envelope integrity. As antibiotics in each category act in different ways still, at least one from each sub‐category was selected to compile a 19‐member collection of antibiotics that covers all common mechanisms of action (MOA) (Table 1). In light of its RNAP targeting MOA and other positive attributes described above, we designed our synergy screens around MccJ25. Notably, resistant mutants of MccJ25 showed no cross‐resistance to rifampicin as the two antibiotics effect transcription inhibition in distinct ways.[ 17a , 17b , 23 ] MccJ25 was prepared according to literature procedures [24] and paired with each of the 19 members in a checkerboard assay (Figure 2), which is used routinely to assess the impact on potency when two antibiotics are administered in combination. We set up our checkerboard assays against E. coli MG1655 by mixing a pair of two‐fold dilution series starting at the respective minimum inhibitory concentrations (MIC) of each antibiotic. The effect of combining two antibiotics was quantitated by the minimum fractional inhibitory concentration index (FICIm, see Experimental Section). [25] Most antibiotics we tested were indifferent to the presence of MccJ25 (0.5<FICIm≤4.0, shown in shades of gray, Figure 2) and none was antagonistic (FICIm>4.0). Five antibiotics in our screen showed weak synergy (cerulenin, chloramphenicol, clarithromycin, ofloxacin, tobramycin) when used in combination with MccJ25 (FICIm=0.5, shown in shades of blue, Figure 2f, 2j, 2l, 2m, and 2q). These antibiotics act on a wide range of cellular pathways. Cerulenin treatment reduces the availability of membrane building blocks as it inhibits lipid biosynthesis (fatty acids and steroids), which likely results in a weakened membrane and an increase in MccJ25 uptake. Ofloxacin targets DNA topoisomerases, and the rest of the antibiotics – chloramphenicol, clarithromycin, and tobramycin – all inhibit protein synthesis by binding to the ribosome at various positions. The identification of these synergistic pairs is in line with our hypothesis that disrupting transcription, a hub of complex cellular processes, creates weak links in numerous other pathways in the network to potentiate the activity of other antibiotics.
Table 1.
Antibiotics used in this study. MICs against E. coli MG1655 were determined by the broth dilution method.
|
Drug |
Target Cellular Process |
Mechanism |
MIC (μg/mL) |
|---|---|---|---|
|
Ampicillin |
Cell wall |
Blocks DD‐transpeptidase |
16 |
|
Ceftazidime |
128 |
||
|
Cycloserine D |
Inhibits alanine racemase and d‐alanine:d‐alanine ligase |
64 |
|
|
Fosfomycin |
Inactivates UDP−N‐acetylglucosamine‐3‐enolpyruvyltransferase |
4 |
|
|
Polymyxin B |
Membrane |
Binds to lipopolysaccharides |
0.25 |
|
Cerulenin |
Binds to ß‐keto‐acyl ACP synthase |
32 |
|
|
Fusidic Acid |
Protein synthesis |
Prevents the turnover of EF‐G |
256 |
|
Puromycin |
Causes premature chain termination |
128 |
|
|
Minocycline |
Binds to 30S; interferes with binding of tRNA to ribosome |
0.5‐2 |
|
|
Amikacin |
Binds to 30S; causes mRNA codon to be misread |
2‐4 |
|
|
Kanamycin |
4 |
||
|
Tobramycin |
4 |
||
|
Clarithromycin |
Binds to 50S; blocks the polypeptide exit tunnel |
32 |
|
|
Chloramphenicol |
Binds to 50S; inhibits formation of peptide bond |
4 |
|
|
Trimethoprim |
Folate pathway |
Inhibits dihydrofolate reductase |
0.063 |
|
Sulfamonomethoxine |
Inhibits dihydropteroate synthase |
64–128 |
|
|
Ofloxacin |
DNA |
Inhibits type II and IV topoisomerase |
0.031 |
|
Novobiocin |
Inhibits DNA gyros GyrB subunit |
64 |
|
|
Rifampicin |
RNA synthesis |
Inhibits RNA polymerase |
8 |
Figure 2.

a–s, Nineteen antibiotics were tested in combination with MccJ25 against E. coli MG1655 in checkerboard assays. Representative results from at least three independent experiments (n=3) for each combination are shown. Relative bacterial growth in each well based on OD600 was shown in colored shades; raw data can be found in the Supporting Information (Figure S1). Indifferent (0.5<FICIm≤4.0), weakly synergistic (FICIm=0.5), and strongly synergistic (FICIm<0.5) combinations are shown in shades of gray, blue, and red, respectively. t, All assays were set up by combining two‐fold serial dilutions of MccJ25 and an antibiotic from the 19‐member collection (Table 1); MccJ25 is plotted on the horizontal axis and the other antibiotic on the vertical axis. The minimum fractional inhibitory concentration index (FICIm) for each combination is shown on the upper right corner of each graph.
One antibiotic pairing, the MccJ25‐SMM combination, showed strong synergy (FICIm=0.25, shown in shades of red, Figure 2o). SMM targets the dihydropteroate synthase and inhibits the production of 7,8‐dihydropteroate (DHP), which is the immediate precursor for dihydrofolate (DHF) biosynthesis in the folate pathway. Interestingly, the dihydrofolate reductase inhibitor trimethoprim is indifferent to the presence of MccJ25. These observations underscore the challenge to build a generalizable theoretical framework for the accurate prediction of antibiotic synergism.
Encouraged by the observed MccJ25‐SMM synergy, we next assessed its generality. Two other common sulfonamide antibacterial agents, sulfamethoxazole and sulfadiazine, were tested and both potentiated the action of MccJ25, albeit to a lesser extent (Figure S2, Supporting Information). We also tested two additional E. coli strains, BCRC 13B0198 and BCRC 13B0202, both of which are drug‐resistant clinical isolates. They are resistant to multiple commonly used antibiotics, including members of the β‐lactam, cephalosporin, fluoroquinolone, lincosamide, sulfonamide, and vancomycin families, and unfortunately, did not respond to the MccJ25‐SMM combination treatment.
While the newly discovered synergistic pair MccJ25‐SMM may not display the potency and generality necessary for clinical applications, their interaction represents a previously unrecognized mechanism cooperation to exert synergy and is mechanistically an intriguing question itself. We therefore decided to investigate the MccJ25‐SMM combination in greater detail using E. coli MG1655 as a model system. Synergistic antibiotic pairs often (but not always) expedite bacterial killing and suppress resistance development, and we tested whether our newly discovered pair has this feature. The time‐kill trace of MccJ25‐SMM did not show a steeper drop as compared to that of MccJ25 alone (Figure 3a), likely because SMM by itself is a bacteriostatic antibiotic. On the other hand, the presence of a very small amount of SMM was able to suppress the rise of resistant mutants as compared to MccJ25 alone. Specifically, SMM at 1/4× and 1/8× MIC resulted in a 5.9 and 2.4‐fold reduction in resistance development (Figure 3b), quantitated by the number of colonies formed after exposure to one or both antibiotics. Supplementing SMM at 1/2× MIC completely suppressed the emergence of resistant colonies. These results show that combining MccJ25 with SMM effectively reduces resistance development in E. coli.
Figure 3.

a, Time‐kill assays of SMM and MccJ25 alone and in combination were performed against E. coli MG1655 (n=3). b, Resistance development rate of E. coli MG1655 against MccJ25 alone and in combinations with different concentrations of SMM, represented as concentrations relative to their respective MICs (n=3). **p<0.01, ***p<0.001, **** p<0.0001. p‐values of pairwise comparisons were calculated by the Student's t‐test.
The folate pathway is connected to numerous metabolic pathways by providing various folate derivatives as a single‐carbon donor. Compounds of the folate family are involved in the biosynthesis of methionine, purines (AMP and GMP), and deoxythymidine monophosphate (dTMP) (Figure 4a). The interconversion of serine and glycine requires 5,10‐methylenetetrahydrofolate (5,10‐methylene‐THF) as a cofactor. Disrupting the folate pathway will therefore perturb the homeostasis of amino acids, nucleotides, and deoxynucleotides and negatively impact translation, transcription, and replication. The following experiments were performed to better understand the underlying mechanism of the newly discovered MccJ25‐SMM synergy. First, we found that including THF (100 μg/mL) in the growth medium led to an increase in the FICIm (0.63, Figure 4b), suggesting that the observed synergy is indeed the result of THF deprivation as opposed to other secondary effects. We then tested whether the presence of added nucleotide or amino acid(s), supplemented at twice their respective physiological concentrations in E. coli, influences the outcome of the MccJ25‐SMM checkerboard assay. We saw no apparent change when methionine and serine/glycine were supplemented; guanosine 5′‐triphosphate (GTP) supplementation showed no change either (Figure S3, Supporting Information); ATP supplementation was not tested as too many biological processes would be affected. Since dTMP is biosynthesized from dUMP using 5,10‐methylene‐THF as the single‐carbon donor, we investigated the possibility that the observed synergy originates from a reduced intracellular supply of dTMP. The combination of MccJ25 and 5‐fluorouracil (5FU), a thymidylate synthase inhibitor, showed strong synergism in the checkerboard assay (FICIm = 0.38 Figure 4c). Together these results point to a previously unrecognized synergy in the mechanism cooperation mode between the inhibition of transcription (MccJ25), replication (5FU), and folate biosynthesis (SMM).
Figure 4.
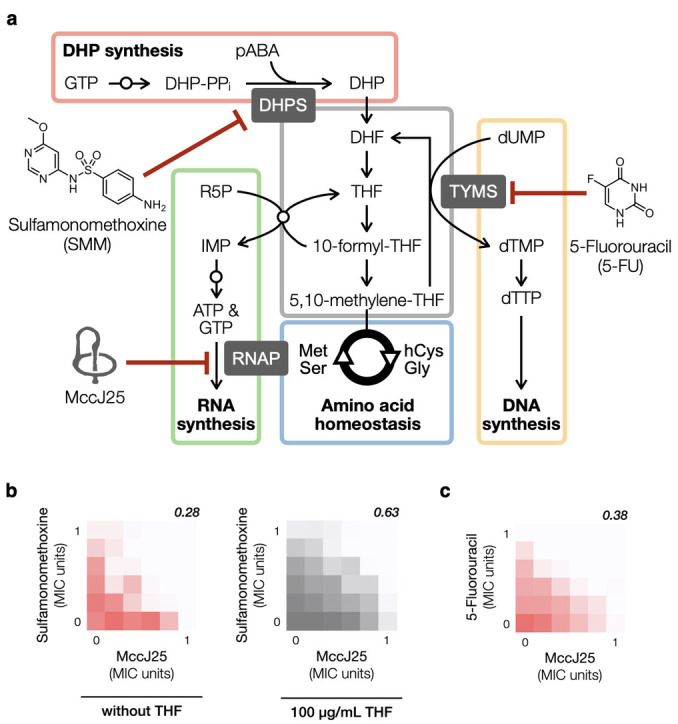
a, The folate pathway is connected to many other pathways critical to the survival of a cell, including DNA synthesis, RNA synthesis, amino acid homeostasis, etc. Small open circles overlaid on arrows denote the presence of multiple steps for the indicated metabolic reaction. b–c, Additional checkerboard assays were performed to investigate the underlying mechanism of MccJ25‐SMM synergy. Representative results from at least three independent experiments (n=3) for each combination are shown. b, MccJ25 and SMM showed no synergy in the presence of THF (100 μg/mL). c, MccJ25 and 5FU show strong synergy (FICIm=0.38).
Conclusion
We reported herein the discovery of MccJ25 and SMM as a new pair of antibiotics that act in synergy. The extent to which this synergistic pair was able to suppress resistance development was also described. MccJ25 has garnered extensive interest since its discovery in 1992. [13] It is a potent antibiotic that inhibits transcription via a novel mechanism, i. e., by obstructing the RNAP complex secondary channel through which NTP molecules access the transcription active site. Chemists are also captivated by the threaded lasso structure of MccJ25 and to date are still trying to delineate the biosynthetic details that result in such a unique structure. [26] Our observations add another dimension to an antibiotic that has already attracted the attention of scientists across many disciplines.
We carried out studies to obtain further mechanistic insights into the observed MccJ25‐SMM synergy. A transcription inhibitory MOA in E. coli has been clearly established for MccJ25, and sulfonamide antibacterial agents are well‐known inhibitors of the dihydropteroate synthase. A shortage of dihydropteroate, the precursor for DHF biosynthesis, disrupts the folate pathway and many other biosynthetic pathways with which it intersects. We believe that this is the underlying reason for our observed synergy between antibiotics that inhibit the synthesis of DNA (5FU), RNA (MccJ25), and metabolites of the folate family (SMM).
The rationale for our MccJ25‐centric assays in searching for new synergistic antibiotics was based on the expectation that disrupting the hub of an intricate and densely interconnected network, i. e., transcription, will create multiple weak links to potentiate the activity of other antibiotics. It is perhaps not surprising that the antibiotic emerging from our screen that synergizes strongly with MccJ25, SMM, exerts its function by disrupting yet another metabolic hub – the folate pathway. In fact, sulfonamide antibiotics are known to synergize with the dihydrofolate reductase inhibitor trimethoprim and this is one of the very few synergistic antibiotic combinations that is used in the clinic. Whereas SMM is also known to synergize with rifampicin, another antibiotic that inhibits transcription, our assays show a strictly additive effect when MccJ25 is used in combination with trimethoprim and rifampicin (Figure 2p and 2s).
By not viewing cellular pathways individually as linear sequences of events, but rather as an intricately interconnected network, we set up screens around an antibiotic (MccJ25) that disrupts a critical hub (transcription) in this network. In addition to identifying the strongly synergistic MccJ25‐SMM combination, we noticed that MccJ25 potentiates (albeit weakly) the growth inhibitory effect of five other antibiotics (shown in shades of blue, Figure 2), providing multiple entry points toward gaining a deeper insight into antibiotic interactions and exploring antibiotic synergistic actions further. Another strongly synergistic combination (MccJ25‐5FU) was discovered during the course of mechanistic studies (Figure 4c). Our screening strategy seemed to have pointed us toward an effective approach in finding new synergistic antibiotic pairs. Nevertheless, we are still far from a thorough understanding of antibiotic interactions and a lot more work is necessary for a full grasp of its complexity.
Experimental Section
MccJ25 production and purification. MccJ25 was produced and purified based on published protocols with minor modifications. Briefly, E. coli BL21(DE3) carrying pTUC202 was cultivated in M9 medium supplemented with 0.4 % (w/v) glucose and 2 mM MgSO4 at 37 °C for four days. Cells were removed by centrifugation. The supernatant was heated in boiling water for 10 min and extracted with 2 volumes of 1‐butanol. The organic layer was collected and dried in vacuo. The resulting residue was redissolved in a minimum amount of aqueous acetonitrile (5 %, v/v) and loaded onto a solid‐phase extraction cartridge (Sep‐Pak C18 Vac, Waters) that had been pre‐washed with acetonitrile and water, successively. Water and acetonitrile supplemented with 0.1 % (v/v) formic acid were used as the mobile phase, denoted as solvent A and B, respectively. MccJ25 typically elutes at 25 to 35 %B when the cartridge was washed with 5 % (v/v) stepwise increments of solvent B. Fractions that contained MccJ25 were pooled and subjected to a second round of cartridge purification to yield materials for various antibiosis assays. A DMSO stock solution (16 mg/mL) was prepared from lyophilized MccJ25 powder and stored as aliquots at −20 °C. Quality assessment was performed by reversed‐phase HPLC (Waters) with an analytical SHARPSIL‐U C18 column (250×4.6 mm ID, 100 Å, 5 μm) (Figure S4). The identity of MccJ25 was confirmed by mass spectrometry (microTOF‐QII, Bruker); HRMS (ESI‐TOF) calculated for C101H141N23O27 [M+2H]2+: 1054.0178, found: 1054.0184 (Figure S5).
MIC determination. Susceptibility of E. coli MG1655 to individual antibiotics was performed in 96‐well microtiter plates using the broth dilution method. First, LB medium was added to all wells (50 μL). Antibiotic solutions in LB broth (50 μL, 128 μg/mL) were added to the first wells of each row and diluted serially (1/2×) across the plate. The last two wells were reserved for positive (no drug) and negative (no bacteria) controls. Overnight cultures in LB medium grown from a single colony (200 rpm, 37 °C) were diluted 5,000‐fold and added to each well (50 μL). MIC values were determined by visual inspection after static incubation at 30 °C for 22 h. All assays were performed at least in triplicate.
Checkerboard assay. Serial dilutions of MccJ25 and a select antibiotic were prepared separately. They were then combined at equal volumes in a 96‐well microtiter plate to generate a 12×8 grid of two serially diluted antibiotics; the results were presented as a 6×6 grid starting at the respective MIC of the two antibiotics. Overnight E. coli MG1655 cultures in LB medium grown from a single colony (200 rpm, 37 °C) were diluted 5,000‐fold and added to each well. The plate was incubated statically at 30 °C for 22 h and bacterial growth was quantified by optical density recorded at 600 nm (OD600). All assays were done at least in triplicate. The type of interaction between two antibiotics was categorized based on the minimum Fractional Inhibitory Concentration Index (FICIm),
where MICA and MICB denote the individual MIC of each antibiotic, and A and B denote their respective MIC in the presence of the other antibiotic. Antibiotic pairs with FICIm<0.5 and FICIm=0.5 are categorized as strongly and weakly synergistic, respectively. Those with 0.5<FICIm≤4.0 were categorized as indifferent to the presence of each other, and FICI≥4.0 are antagonistic (not observed in our studies).
Time‐kill assay. An overnight culture was grown from a single E. coli MG1655 colony in Müller‐Hinton broth (MHB). The culture was used to start fresh MHB cultures at 106 CFU/mL supplemented with MccJ25 (1 μg/mL), SMM (0.512 mg/mL), or both antibiotics (MccJ25 at 0.5 μg/mL and SMM at 0.256 mg/mL). These cultures were incubated at 37 °C with agitation, serially diluted, and spotted on MHB agar plates after 0, 1, 2, 4, and 8 h. The plates were incubated at 37 °C until the formation of visible colonies. The number of colonies at each timepoint was counted and plotted into a time‐kill trace. This assay was performed in triplicate.
Resistance development assay. An overnight culture was grown from a single E. coli MG1655 colony in MHB. Overnight cultures were washed with fresh MHB, serially diluted, and spotted on MHB agar plates supplemented with 1 μg/mL MccJ25 and varying concentrations of SMM. The plates were incubated at 37 °C until the formation of visible colonies. The number of colonies at each timepoint was counted, and the number of CFU relative to that of the antibiotic‐free control was used as a proxy for overall resistance development.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by General Research Grant no. 109WFA0110175 and 110WFA0112053 of the Ministry of Science and Technology, Taiwan. We thank Professor Nai‐Chun Lin (National Taiwan University) for sharing E. coli MG1655 and Professor Mohamed A. Marahiel (Philipps‐Universität Marburg) for sharing pTUC202.
P.-H. Chen, L.-K. Sung, J. D. Hegemann, J. Chu, ChemMedChem 2022, 17, e202200075.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.Center for Disease Control and Prevention. National Center for Health Statistics | Life Expectancy. https://www.cdc.gov/nchs/fastats/life-expectancy.htm (accessed Nov. 3, 2021).
- 2. Worthington R. J., Melander C., Trends Biotechnol. 2013, 31, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu X., Xu L., Yuan G., Wang Y., Qu Y., Zhou M., Sci. Rep. 2018, 8, 7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Malik M., Li L., Zhao X., Kerns R. J., Berger J. M., Drlica K., J. Antimicrob. Chemother. 2014, 69, 3227–3235; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Cassir N., Rolain J. M., Brouqui P., Front. Microbiol. 2014, 5, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Plotz P. H., Davis B. D., Science 1962, 135, 1067–1068. [DOI] [PubMed] [Google Scholar]
- 6. Samonis G., Maraki S., Karageorgopoulos D. E., Vouloumanou E. K., Falagas M. E., Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 695–701. [DOI] [PubMed] [Google Scholar]
- 7. Bushby S. R., J. Infect. Dis. 1973, 128, 442–462. [DOI] [PubMed] [Google Scholar]
- 8. Hamperl S., Cimprich K. A., Cell 2016, 167, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Irastortza-Olaziregi M., Amster-Choder O., Front. Microbiol. 2020, 11, 624830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O. L. Miller Jr. , Hamkalo B. A., C. A. Thomas Jr. , Science 1970, 169, 392–395. [DOI] [PubMed] [Google Scholar]
- 11. Werner F., Grohmann D., Nat. Rev. Microbiol. 2011, 9, 85–98. [DOI] [PubMed] [Google Scholar]
- 12. Villain-Guillot P., Bastide L., Gualtieri M., Leonetti J. P., Drug Discov. Today 2007, 12, 200–208. [DOI] [PubMed] [Google Scholar]
- 13. Salomon R. A., Farias R. N., J. Bacteriol. 1992, 174, 7428–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Martin-Gomez H., Jorba M., Albericio F., Vinas M., Tulla-Puche J., Int. J. Mol. Sci. 2019, 20; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. de Cristobal R. E., Solbiati J. O., Zenoff A. M., Vincent P. A., Salomon R. A., Yuzenkova J., Severinov K., Farias R. N., J. Bacteriol. 2006, 188, 3324–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Naimi S., Zirah S., Hammami R., Fernandez B., Rebuffat S., Fliss I., Front. Microbiol. 2018, 9, 1764; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Yu H., et al., J. Agric. Food Chem. 2018, 66, 11301–11310. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Rosengren K. J., Blond A., Afonso C., Tabet J. C., Rebuffat S., Craik D. J., Biochemistry 2004, 43, 4696–4702; [DOI] [PubMed] [Google Scholar]
- 16b. Rosengren K. J., Clark R. J., Daly N. L., Goransson U., Jones A., Craik D. J., J. Am. Chem. Soc. 2003, 125, 12464–12474. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Yuzenkova J., et al., J. Biol. Chem. 2002, 277, 50867–50875; [DOI] [PubMed] [Google Scholar]
- 17b. Delgado M. A., Rintoul M. R., Farias R. N., Salomon R. A., J. Bacteriol. 2001, 183, 4543–4550; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Braffman N. R., Piscotta F. J., Hauver J., Campbell E. A., Link A. J., Darst S. A., Proc. Natl. Acad. Sci. USA 2019, 116, 1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mukhopadhyay J., Sineva E., Knight J., Levy R. M., Ebright R. H., Mol. Cell. 2004, 14, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Murakami K. S., Masuda S., Darst S. A., Science 2002, 296, 1280–1284; [DOI] [PubMed] [Google Scholar]
- 19b. Murakami K. S., Masuda S., Campbell E. A., Muzzin O., Darst S. A., Science 2002, 296, 1285–1290; [DOI] [PubMed] [Google Scholar]
- 19c. Vassylyev D. G., Sekine S., Laptenko O., Lee J., Vassylyeva M. N., Borukhov S., Yokoyama S., Nature 2002, 417, 712–719. [DOI] [PubMed] [Google Scholar]
- 20. Browning D. F., Busby S. J., Nat. Rev. Microbiol. 2004, 2, 57–65. [DOI] [PubMed] [Google Scholar]
- 21. Korzheva N., Mustaev A., Kozlov M., Malhotra A., Nikiforov V., Goldfarb A., Darst S. A., Science 2000, 289, 619–625. [DOI] [PubMed] [Google Scholar]
- 22. Ray-Soni A., Bellecourt M. J., Landick R., Annu. Rev. Biochem. 2016, 85, 319–347. [DOI] [PubMed] [Google Scholar]
- 23. Ho M. X., Hudson B. P., Das K., Arnold E., Ebright R. H., Curr. Opin. Struct. Biol. 2009, 19, 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Hegemann J. D., et al., J. Med. Chem. 2014, 57, 5829–5834; [DOI] [PubMed] [Google Scholar]
- 24b. Solbiati J. O., Ciaccio M., Farias R. N., Salomon R. A., J. Bacteriol. 1996, 178, 3661–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Odds F. C., J. Antimicrob. Chemother. 2003, 52, 1. [DOI] [PubMed] [Google Scholar]
- 26. Duquesne S., Destoumieux-Garzon D., Zirah S., Goulard C., Peduzzi J., Rebuffat S., Chem. Biol. 2007, 14, 793–803. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.