

Abstract
Background
While the advent of cystic fibrosis transmembrane conductance regulator (CFTR) modulator use has improved daily life and long‐term prognosis of CF for many with approved CFTR mutations, approximately 10% of people with CF (pwCF) have only symptomatic treatments available.
Methods
Between June 10 and July 1, 2021, Emily's Entourage distributed a 38‐question anonymous survey targeted at pwCF not benefitting from approved modulators via social media and email to pwCF and CF advocacy groups in and outside the United States regarding health status, impact of CF, unmet needs, and clinical research interest.
Results
There were 431 survey respondents representing pwCF on five continents. The majority of pwCF had moderate lung disease (50.3%). Ineligibility based on CFTR mutation (64.1%) was the most frequently reported reason pwCF were not on modulators. PwCF reported the most impacted aspects of life were mental (66.7%) and physical (40.7%) health. Financial concerns and feelings of isolation were commonly reported. Witnessing improvements for peers with access to modulators was both uplifting and disheartening. The majority of pwCF would be interested in participating in future clinical research (77.6%), although some living outside of the United States cited lack of opportunity to participate in clinical trials as a barrier.
Conclusions
PwCF who are ineligible, intolerant, or lack access to modulators have a high burden of disease impacting their physical and mental health. Although most are happy for those who are benefiting from modulators, they are eager for the opportunity to experience similar improvements for themselves, and willing to participate in clinical trials of new therapies.
Keywords: 10%, CFTR modulator, cystic fibrosis, nonsense mutation, orphan disease
1. INTRODUCTION
Cystic fibrosis (CF), a genetic disorder that can occur in people of any race or ethnicity, is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Clinical manifestations resulting from CFTR protein dysfunction frequently include impaired mucociliary clearance and chronic lung infections, chronic sinusitis, and pancreatic insufficiency leading to malabsorption, amongst others.
In the last decade, treatment options for people with CF (pwCF) have changed dramatically. 1 , 2 While treatments in the preceding decades were directed at the signs and symptoms of CF, there are now four approved oral therapies that treat the basic defect in CF. 3 , 4 , 5 , 6 , 7 This class of drugs, CFTR modulators, improve CFTR protein transport and function, and in so doing dramatically improve lung function, weight, and quality of life. 8 Although the first approved CFTR modulator therapy was only available to treat approximately 4% of pwCF; currently, approximately 90% of the population of pwCF has a mutation that is likely to be responsive to CFTR modulators. 9
While the advent of CFTR modulator use has improved daily life and the long‐term prognosis of CF for many with approved CFTR mutations, approximately 10% of pwCF still only have symptomatic treatments available. 9 Because of their rare mutations, Black, Indigenous, and People of Color (BIPOC) are overrepresented in this group of pwCF. 10 Additionally, there are pwCF who do not tolerate CFTR modulators because of side effects. 11 Finally, while all four drugs have approval in the United States with broad in vitro based application, approval varies widely by nation, with some nations having no access to CFTR modulators. 11 Although registries can be utilized to describe clinical characteristics of those not eligible for modulators in one region or country, registries do not capture the burden of care individuals in that group or their family members live with on a day‐to‐day basis nor their perceptions about participating in clinical trials. 9 , 12 , 13 We sought to understand the demographics, clinical characteristics, care perceptions, treatment needs, mental health impacts of lack of access, and clinical research participation interest of the approximately 10% of pwCF who do not have access or are unable to tolerate CFTR modulator therapy.
2. MATERIAL AND METHODS
2.1. Survey development
The 38‐question survey (displayed in its entirety in Figure S1) questions were written or adapted by Emily's Entourage (EE) staff, with input and feedback from various stakeholders, including CF Foundation Community Voice, the Cystic Fibrosis Research Institute, and a family member of a person with CF. The page preceding the survey entry stated that the survey was directed toward those not benefiting from modulators and intended for individuals with CF and their family members or legal guardians. Potential respondents were not told that they would be excluded for any reason. Question 1, asked the survey respondent to describe her/his/their relationship to CF, and questions 2 and 3 asked about caregiver experience. The remaining survey questions are based on information or experiences of the individual with CF; for such questions, respondents with CF were asked to respond to the question for her/him/themselves. All other respondents were asked to respond to the questions based on the experience of their family member with CF (Figure S1). Survey questions included multiple‐choice, Likert‐type scales, and free‐text responses. Before distribution, the survey was tested by a group of members of the CF community composed of patients, caregivers, and advocates. The survey authors reviewed feedback and incorporated suggestions where appropriate.
Between June 10 and July 1, 2021, the survey was distributed via social media (Twitter, Facebook, Instagram, and LinkedIn) and email to CF patients, families, and advocacy organizations in the United States (US), Canada, United Kingdom (UK), Australia, and Israel, who forwarded the survey to any potential respondents. Additionally, EE sent the survey to its entire CF Nonsense Mutation Patient Registry, a global registry of people with one or two copies of a nonsense mutation of CF. The survey was officially closed on September 14, 2021.
2.2. Statistics
Survey answers were summarized and reported as n and/or % respondents answering each question.
2.3. Free text survey responses
Responses to the open‐ended questions in section six of the survey were independently reviewed for themes by Liza Kramer and Jennifer L. Taylor‐Cousar. Theme‐representative quotations were agreed upon and included in the body of the manuscript and associated figures.
2.4. Ethics statement
Acknowledgment of the anonymous nature of the survey, planned use and publication of the data, and consent for such use of the data preceded the survey. Before beginning the survey, respondents were informed that they could leave the survey at any point (please see Figure S1). Respondents were not required to answer all questions before proceeding to subsequent questions. Emily's Entourage collected anonymous survey data which would be considered exempt from Institutional Review Board review.
3. RESULTS
3.1. Relationship to CF of survey respondents
A total of 431 people responded to the survey including 187 (43.4%) parents of pwCF, 182 (42.1%) adults with CF, 34 (7.9%) other family members of pwCF, 16 (3.7%) pwCF under the age of 18 years, 7 (1.6%) who reported none of the above relationships and 5 (1.2%) spouses of pwCF.
3.2. Demographics of people with CF
A higher percentage of women (n = 174/282, 61.7%) than men (n = 99/282, 35.1%) responded. Most respondents self‐reported their race and ethnicity as White (n = 233/282, 82.6%) and not Hispanic or Latinx (n = 32/282, 83.7%), respectively (Table 1). Respondents in the US (including Puerto Rico and the Virgin Islands) comprised 69.9% (n = 257/375) of the group, with the other 30.1% (n = 113/375) of respondents living outside the US (representing five continents). The majority (n = 199/375, 53.1%) of pwCF were between 18 and 44 years of age (Table 1). When asked about insurance coverage, private insurance coverage was most common (n = 205/375, 54.7%) followed by Medicaid or state insurance programs (n = 75/375, 20%) and Medicare (n = 49/375,13.1%); 3.5% (n = 13/375) of pwCF had no insurance, and 23.5% (n = 88/375) responded that they were “outside the U.S.” (Table 1).
Table 1.
Demographics of people with CF described in the survey
| Demographic characteristic | N (%)a |
|---|---|
| Age | Total n = 375 |
| <10 years | 80 (21.3) |
| 10–17 years | 61 (16.3) |
| 18–24 years | 59 (15.7) |
| 25–34 years | 85 (22.7) |
| 35–44 years | 55 (14.7) |
| ≥45 years | 35 (9.3) |
| Gender identity | Total n = 282 |
| Gender queer/nonbinary | 3 (1.1) |
| Manb | 99 (35.1) |
| Trans manb | 0 |
| Trans womanb | 0 |
| Womanb | 174 (61.7) |
| Prefer not to answer | 3 (1.1) |
| Other | 3 (1.1) |
| Race | Total n = 282 |
| American Indian/Alaska Native | 7 (2.5) |
| Black/African American | 7 (2.5) |
| East Asian | 2 (0.7) |
| Middle Eastern/North African | 4 (1.5) |
| Multiracial | 11 (3.9) |
| Native Hawaiian/Other Pacific Islander | 1 (0.4) |
| South Asian | 12 (4.3) |
| White/Caucasian | 233 (82.6) |
| Other | 20 (7.1) |
| Ethnicity | Total n = 282 |
| Hispanic or Latinx | 32 (11.3) |
| Not Hispanic or Latinx | 236 (83.7) |
| Prefer not to answer | 14 (5.0) |
| Country of residence | Total n = 375 |
| Outside the US | 113 (30.1) |
| US | 257 (69.9) |
| Insurance coveragec | Total n = 375 |
| Medicaid/state programs | 75 (20) |
| Medicare | 49 (13.1) |
| Military health care | 8 (2.1) |
| No insurance | 13 (3.5) |
| Outside the US | 88 (23.5) |
| Private | 205 (54.7) |
Based on participant choice and question composition, n for respondents for questions varied.
Survey (Figure S1) listed female, male, trans woman/female, and trans man/male, but the terminology is corrected here for accuracy.
Answers not mutually exclusive.
3.3. Clinical characteristics
Approximately 26% (n = 88/334) of pwCF had mild lung disease (baseline ppFEV1 ≥ 90%), 50.3% (n = 168/334) had moderate disease (percent predicted forced expiratory volume in 1 s [ppFEV1] 40%–89%) and 11.1% (n = 37/334) had severe disease (ppFEV1 < 40%; Table 2). The most commonly reported complications included pulmonary exacerbations (n = 178/334, 53.3%), sinus disease (n = 142/334, 42.5%), and CF‐related diabetes (29.9%; Table 2). Although the majority of pwCF have moderate to severe disease with multiple complications, more than half rated their physical (n = 180/334, 53.9%) and mental (n = 181/334, 54.2%) health as good to excellent (data not shown).
Table 2.
Clinical characteristics of people with CF
| Clinical characteristic | N (%), Total n = 334 |
|---|---|
| Reported baseline ppFEV1 | |
| ≥90% | 88 (26.4) |
| 70%–89% | 101 (30.2) |
| 40%–69% | 67 (20.1) |
| <40% | 37 (11.1) |
| Unsure/prefer not to say | 41 (12.3) |
| Presence of complications of CF | |
| CF‐related arthritis | 29 (8.7) |
| CF‐related diabetes | 100 (29.9) |
| CF‐related liver disease | 47 (14.1) |
| History of lung transplant | 24 (7.2) |
| Pulmonary exacerbations | 178 (53.3) |
| Sinus disease | 142 (42.5) |
| Most common chronic infections | |
| Aspergillus | 38 (11.4) |
| B. cepacia | 17 (5.1) |
| MDR‐PA | 26 (7.8) |
| MRSA | 51 (15.3) |
| MSSA | 122 (36.5) |
| No growth | 40 (12.0) |
| NTM | 30 (9.0) |
| P. aeruginosa | 152 (45.5) |
| Don't know | 24 (7.2) |
| Other | 16 (4.5) |
Abbreviations: B. cepacia, Burkholderia cepacia; CF, cystic fibrosis; MDRA‐PA, multidrug‐resistant Pseudomonas aeruginosa; MRSA, methicillin‐resistant Staphylococcus aureus; MSSA, methicillin‐sensitive Staphylococcus aureus; NTM, nontuberculous mycobacteria; P. aeruginosa, Pseudomonas aeruginosa.
More than 90% of pwCF had at least one class I mutation (Figure 1A) and were ineligible for a modulator based on CFTR mutations (n = 214/334, 64.1%; Figure 1B). The percentage of pwCF who self‐identified as a BIPOC individual who was ineligible for modulators (n = 29/40, 72.5%) was higher than those who self‐identified as White (n = 152/233, 65.2%). Lung transplantation, CF liver disease, intolerable side effects, age ineligibility, and lack of access were reported as reasons that some people were not on modulators. Twenty‐four pwCF responded to the survey who were eligible and on modulators, and n = 6 pwCF were not eligible for modulators, but using a modulator off‐label.
Figure 1.
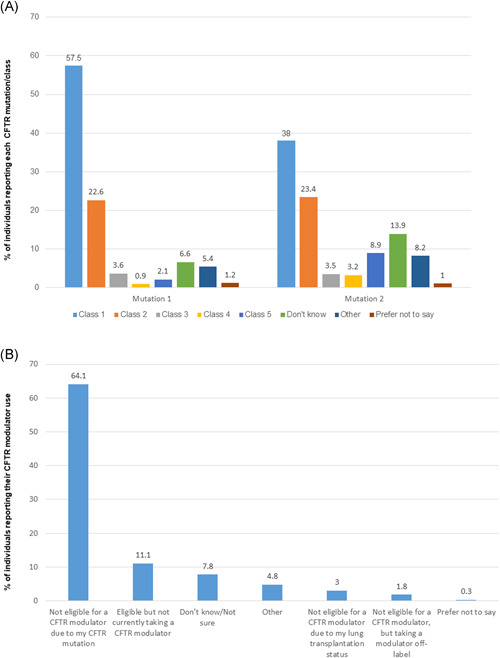
Respondents were asked to report CFTR mutation class and CFTR modulator eligibility. (A) Respondents self‐reported CFTR mutation class for each of their two mutations using this survey‐provided reference: Class 1: Protein production mutations—including nonsense mutations (i.e., G542X, W1282X, and R553X), some splice mutations and deletions; Class 2: Protein processing mutations—including F508del, N1303K, and I507del; Class 3: Gating mutations—including G551D and S549N; Class 4: Conduction mutations—including D1152H, R347P, and R117H; Class 5: Insufficient protein mutations—including some splice mutations (i.e., 3849+1‐ kb C‐‐>T); Other, please specify (free text; mutation 1 [n = 332], mutation 2 [n = 316]), *of the 44 respondents who reported their first or second mutations as “other,” based on data available in CFTR2, the majority of mutations were adjudicated as class I mutations 14 (Table S1) (B) self‐reported eligibility and use of CFTR modulators (n = 334) [Color figure can be viewed at wileyonlinelibrary.com]
3.4. Impact of disease and burden of therapy
The symptoms that were most commonly reported to occur on a regular basis were gastrointestinal issues (n = 216/334, 64.7%), excessive cough (n = 180/334, 53.9%), fatigue (n = 170/334, 50.9%), shortness of breath (n = 134/334, 40.1%), mental health issues (n = 116/327, 34.7%), chest tightness (n = 113/334, 33.8%), and chronic pain (n = 60/334,18.0%). Individuals reported use of both pulmonary and non‐pulmonary treatments to manage the signs and symptoms of CF and its complications (Table S2). Respiratory therapies used by more than 50% of pwCF (n = 323) included airway clearance, oral/inhaled bronchodilators, dornase alfa, hypertonic saline, and sinus rinses. Gastrointestinal therapies used by more than 50% of pwCF (n = 323) included fat‐soluble vitamins and pancreatic enzymes. While only 4.3% (n = 14/323) of individuals spend more than 4 h per day doing medical treatments, 76.5% (n = 247/323) spend between 1 and 4 h (Figure S2). On an annual basis (for a time period that included the COVID pandemic), more than half of individuals with CF spent one to four nights in the hospital for a CF‐related concern (n = 184/334, 55.1%), and 13.8% (n = 46/334) spent five or more nights in the hospital. More than 86.1% (n = 267/310) of those surveyed reported that CF has a moderate to significant impact on their or their family member's life, and 93.5% (n = 188/201) of caregivers reported that CF had a moderate to significant impact on daily life, with mental (n = 136/204, 66.7%) and physical (n = 83/204, 40.7%) health impacts being highly reported by caregivers. Respondents rated the four most burdensome aspects of CF to be treatment burden and time required for daily therapies (n = 218/310, 70.3%), avoiding germs (n = 178/310, 57.4%), feeling isolated or like a burden on others (n = 166/310, 53.6%) and disruption of hospitalizations to daily life and obligations (n = 140/310, 45.2%). Financial concerns such as fear of running out of treatments, ability to afford prescribed drugs, vitamins, and treatments, and obtaining and keeping insurance coverage were also frequently reported (Table S3).
The aspects of life that pwCF felt were most impacted by CF were mental health (n = 140/310, 45.5%), plans for the future (n = 140/310, 45.2%) and ability to spend time with friends/participating in social activities (n = 123/310, 39.7%; Table S4). Other areas of life commonly reported to be impacted included school issues (like staying on top of school or handling medication and health issues at school (n = 73/310, 26.5%), financial stability (n = 79/310, 25.5%), staying healthy for dependents (n = 71/310, 22.9%) or participating in sports/hobbies/extracurricular activities (n = 71/310, 22.9%). Respondents reported that the four most concerning aspects of CF were lung function issues (n = 236/310, 76.1%), bacterial colonization (n = 170/310, 54.8%), GI and nutritional issues (38.7%, n = 120/310) and mental and emotional health (n = 119/310, 38.4%).
3.5. Potential clinical research participation
Out of 292 respondents who answered the question regarding whether they/their family member would be interested in participating in future clinical trials, 76.7% (n = 222) responded affirmatively. While some respondents were unsure about participation (n = 49/292, 16.8%), only 12 (n = 12/292, 4.1%) respondents said they/their family member were uninterested in participating. When asked why the individual/the family member had not or would not participate in a clinical trial, the most common reason was lack of qualification (n = 108/292, 37.0%), but concerns about possible negative side effects/health deterioration (n = 97/292, 33.2%), travel distance to study site (n = 79/292, 27.1%), not being asked/being unaware of clinical trials (n = 67/292, 23.0%), and scheduling conflicts/time commitment (n = 62/292, 21.2%) were other frequently listed barriers (Table S5). Respondents living outside of the US cited lack of opportunity to participate in clinical trials. Concerns about fertility/family planning, not meeting inclusion criteria, and potential compromise of insurance benefits were listed as “other” concerns. For those who did want to participate in trials, the most commonly reported reasons were the possibility of improving one's own health (n = 252/292, 86.3%,) and the desire to help find a cure (n = 221/292, 75.7%; Figure 2). Trial features deemed extremely or somewhat important by the majority of respondents were access to drug after the trial ends (n = 270/292, 92.5%) and addition of more clinical trial locations (n = 225/286, 78.7%; Figure S3). For a new therapy associated with potential health risks, respondents reported that the two most important considerations for deciding whether to participate in the trial were the likelihood of treatment benefits outweighing risks (53.8%, n = 157/292) and the reversibility of risks if they developed (n = 120/292, 41.1%). Careful risk monitoring of the study (n = 90/292, 30.8%) and the impact of treatment on a symptom that has profound life impact (n = 76/292, 26.2%) were also of import.
Figure 2.

Respondents were asked to select all reasons that applied for their main rationale for participation in a CF clinical trial. N = 292. CF, cystic fibrosis [Color figure can be viewed at wileyonlinelibrary.com]
3.6. Perceptions regarding access to CFTR modulators for some but not all people with CF and treatment options
When survey respondents were asked about how it feels to see others in the CF community benefit from modulators when they/their family member were unable to benefit, 238 entered free‐text comments. Most felt happy and excited for those who were benefitting, but other recurring themes included “bitter sweet,” “envious,” “sad,” “frustrating,” “disappointing,” “angry,” “worried,” and “left behind.” One respondent said, “We feel left behind. Isolated. Like our child is being left behind because they have rare mutations. We feel scared for the future without a cure or treatment. And desperate for something to pin our hopes on.” Twenty‐eight people responded to the question, “How does it feel to be eligible for a CFTR modulator while many in the CF community still lack eligibility based on their mutations or access?” Among those who were not eligible but taking a modulator off‐label, several respondents expressed fear of loss of access, and common themes among those eligible and on modulators were feeling “lucky” but also “guilty” and “sad” for those not eligible. One respondent stated, “It is great for one daughter, but didn't work for my other daughter. Looking forward to the next CFTR modulator that can help everyone.”
The 249 respondents who answered the question regarding what they hoped for in a therapy while they are waiting for a cure ranged from improved lung function to reduced burden of care, reduced infection, and prolonged life expectancy. Respondents felt that finding a cure for CF (n = 252) would mean “a normal life,” “planning for the future,” and “freedom.” Finally, respondents were asked if there was anything else they would like to add about living with CF and treatment options. Out of the 90 respondents, representative comments are shown in Figure 3.
Figure 3.

Free text responses from respondents on living with CF and treatment options. CF, cystic fibrosis [Color figure can be viewed at wileyonlinelibrary.com]
3.7. Limitations
Although responses were received from individuals across five continents, because the social constructs of race and ethnicity are viewed differently around the world, and because the survey was distributed electronically and by social media and only in English, the responses may not be representative of the entire population of individuals with CF. Furthermore, while the majority of respondents answered most of the survey questions, there was a 100% response rate for only the first question, thus creating additional potential ascertainment bias. An additional limitation is that pwCF and their caregivers were solicited anonymously for information and there was no link to medical records for verification of clinical information. However, pwCF and their family members are often more knowledgeable about their medical history than those without chronic disease. Finally, while the survey design input was received from other patient organizations with experience conducting surveys, the survey is not a validated one. Potential bias exists regarding those who chose to respond to the survey.
4. DISCUSSION
Although the discovery of the CFTR gene in 1989 created hope for an imminent cure for the disease, it was not until 2012 when the first CF drug was approved that slowed disease progression by treating the basic defect. 3 , 15 , 16 , 17 In 2021, there are now four approved therapies in this drug class, two of which are highly effective and for which approximately 90% of the population of pwCF is eligible based on their CFTR mutations. 3 , 4 , 5 , 6 , 7 While these approvals have been life‐changing for those eligible who also have access and tolerate the therapies, there is a group of pwCF who have not had the opportunity to experience these changes. This survey captured the demographics, clinical characteristics, and perceptions of pwCF or their family members across a wide age range from five continents.
The majority of respondents had moderate to severe disease, and consequently, the therapeutic burden that they reported was required to maintain their health was substantial and expected. Reported recurrent symptoms and complications were consistent with those reported in CF registries for pwCF. 9 , 12 , 13 Importantly, respondents were clear that beyond the burden of disease, as reported by others, the psychosocial impacts of living with CF for pwCF and for their caregivers are profound. 18 Furthermore, the psychological impact of the burden and worry that having CF has on daily life has been compounded by the differential access to life‐changing therapies available to many but not all pwCF.
In order for a new therapy to be approved, it must go through rigorous evaluation in different phases of clinical trial design, culminating with large, multicenter randomized trials that include large numbers of participants in the treatment versus placebo arms of the study. 19 While it has been feasible in CF to conduct such trials up to this point, the number of people with this orphan disease (impacting less than 200,000 people in the US) who do not qualify for a CFTR modulator likely includes less than 8000 people worldwide and only approximately 3000 people in the US. Once inclusion and exclusion criteria are applied, and interest in trial participation considered, the pool of potential research participants is further diminished. 20 In this survey, the majority of people who are not on a modulator are interested in participating in clinical research not only because it will possibly benefit them, but also because it will enable the discovery of a cure for all with CF. However, respondents were also clear that careful trial monitoring of risks and access to therapy following the end of the trial would influence their desire to participate. As others have shown, one of the most common reasons potential respondents had not been in clinical trials previously was because they had not been asked to consider enrollment. 21 Also as others have shown, there are a disproportionate number of individuals who are BIPOC in the remaining group of pwCF not eligible for modulators. The historic and ongoing mistreatment in medicine and clinical research of many BIPOC individuals may make them wary of participation. Thus, additional training and culturally sensitive approaches to the presentation of trial opportunities will be necessary. 10 , 22 , 23
In summary, the lives of the many pwCF have been dramatically improved by the development and approval of CFTR modulators. However, for some pwCF, lack of eligibility based on mutations or age, lack of access based on approvals and cost, or drug intolerance prevents use of modulators. Many of those without modulators have significant physical, mental, and social impacts from their disease. Although most are happy for those who have benefited from modulators, the current therapeutic environment has left them feeling scared, forgotten, and neglected. The majority of those in this group are interested in participating in trials, both to benefit themselves and others. Partnership between CF basic scientists, clinical investigators, biopharmaceutical companies, advocacy organizations, regulators, and the community is essential to ensure that therapies are developed expeditiously and are accessible for all with CF.
CONFLICTS OF INTEREST
Emily Kramer‐Golinkoff is a founding member of Emily's Entourage. In the past 3 years, her financial and commercial interests were with Medidata Solutions, Inc and Translate Bio, in terms of speaking at meetings/travel. Her noncommercial interests included consulting for the University of Pennsylvania Health System and speaking at meetings/travel from Cystic Fibrosis Research Incorporated, Personalized Medicine Coalition, Academy Health, and BIO International. Amanda Camacho was employed by Emily's Entourage at the time of the design and conduct of the survey as well as at the time of the writing of the original version of the manuscript. Liza Kramer had commercial interests (past 3 years) in terms of speaking at meetings/travel for Translate Bio, Cystic Fibrosis Research Incorporated, and Personalized Medicine Coalition. Jennifer L. Taylor‐Cousar, in the past 3 years, has displayed her financial and commercial interests as faculty for an institution that is part of the CF TDN. She had been site PI on studies for Vertex, Bayer, Celtaxys, Eloxx, Nivalis, and Proteostasis; on advisory boards for Genentech, Gilead, AbbVie, Insmed, and Vertex; done consulting/provided clinical trial design advice for Vertex, Celtaxys, Proteostasis, Santhera, 4DMT, and Polarean; and served on the data monitoring committee for AbbVie. She is a professional member of the CFF Clinical Research Executive Committee, CF TDN Women's Health Research Working Group, and ATS Clinical Problems Programming and Scientific Grant Review Committees. On a noncommercial basis, she had received a grant from the CFF. She also has nonprofit relationships with the CFF Board of Trustees and Emily's Entourage Scientific Advisory Board.
AUTHOR CONTRIBUTIONS
Emily Kramer‐Golinkof designed and distributed the survey, contributed to the writing of the first draft, and edited all subsequent drafts; Amanda Camacho designed and distributed the survey, contributed to the writing of the first draft, and edited all subsequent drafts; Liza Kramer designed and distributed the survey, contributed to the writing of the first draft, and edited all subsequent drafts. Jennifer L. Taylor‐Cousar reviewed the data collected by Emily Kramer‐Golinkof, Amanda Camacho, and Liza Kramer, wrote the first draft of the manuscript, and edited all subsequent drafts.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
The authors would like to acknowledge the vital contributions of people with CF and their families that participated in the survey and contributed data for this study. The authors would also like to thank Siri Vaeth, MSW (Cystic Fibrosis Research, Inc.), Enid Aliaj, MA (Cystic Fibrosis Foundation), and Martin Moore and Cystic Fibrosis Foundation Community Voice for providing guidance in survey development, and those who helped with survey testing: Laura Bonnell; Michelle Shore, LPC and LLB; Rebecca Cosgriff, MA; Martin Moore; Erin Moore; Siri Vaeth, MSW; Enid Aliaj, MA and Lucy Allen, PhD. This study was supported by Emily's Entourage.
Kramer‐Golinkoff E, Camacho A, Kramer L, Taylor‐Cousar JL. A survey: Understanding the health and perspectives of people with CF not benefiting from CFTR modulators. Pediatric Pulmonology. 2022;57:1253‐1261. 10.1002/ppul.25859
Ms. Camacho was employed by Emily's Entourage at the time of the design and conduct of the survey as well as at the time of the writing of the manuscript.
REFERENCES
- 1. Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. 2018;17(2):153‐178. [DOI] [PubMed] [Google Scholar]
- 2. Bell SC, Mall MA, Gutierrez H, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med. 2020;8(1):65‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor‐ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(18):1783‐1784. [DOI] [PubMed] [Google Scholar]
- 5. Taylor‐Cousar JL, Munck A, McKone EF, et al. Tezacaftor‐ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377(21):2013‐2023. [DOI] [PubMed] [Google Scholar]
- 6. Middleton PG, Mall MA, Drevinek P, et al. Elexacaftor‐tezacaftor‐ivacaftor for cystic fibrosis with a single Phe508del Allele. N Engl J Med. 2019;381(19):1809‐1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double‐blind, randomised, phase 3 trial. Lancet. 2019;394(10212):1940‐1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor‐Cousar JL, Mall MA, Ramsey BW, et al. Clinical development of triple‐combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ Open Res. 2019;5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.2019 Annual Data Report. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient-Registry-Annual-Data-Report.pdf
- 10. McGarry ME, McColley SA. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr Pulmonol. 2021;121:800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barry PJ, Taylor‐Cousar JL. Triple combination cystic fibrosis transmembrane conductance regulator modulator therapy in the real world – opportunities and challenges. Curr Opin Pulm Med. 2021;27(6):554‐566. [DOI] [PubMed] [Google Scholar]
- 12.UK Cystic Fibrosis Registry Annual Data Report 2019. UK Trust; 2021. Accessed May 3, 2021. https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/2019%20Registry%20Annual%20Data%20report_Sep%202020.pdf
- 13.Australian Cystic Fibrosis Data Registry Annual Report. Monash University; 2019. Accessed October 21, 2019. https://www.cysticfibrosis.org.au/dataregistry
- 14.Clinical and Functional TRanslation of CFTR (CFTR2). Accessed October 21, 2021. http://cftr2.org
- 15. Rommens JM, Iannuzzi MC, Kerem BS, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245(4922):1059‐1065. [DOI] [PubMed] [Google Scholar]
- 16. Riordan JR, Rommens JM, Kerem BS, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066‐1073. [DOI] [PubMed] [Google Scholar]
- 17. Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073‐1080. [DOI] [PubMed] [Google Scholar]
- 18. Quittner AL, Goldbeck L, Abbott J, et al. Prevalence of depression and anxiety in patients with cystic fibrosis and parent caregivers: results of The International Depression Epidemiological Study across nine countries. Thorax. 2014;69(12):1090‐1097. [DOI] [PubMed] [Google Scholar]
- 19.Development & Approval Process|Drugs; 2019. Accessed October 21, 2021. https://www.fda.gov/drugs/development-approval-process-drugs
- 20. Mayer‐Hamblett N, van Koningsbruggen‐Rietschel S, Nichols DP, et al. Building global development strategies for cf therapeutics during a transitional cftr modulator era. J Cyst Fibros. 2020;19(5):677‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dobra R, Elborn JS, Madge S, et al. Guiding the rational design of patient‐centred drug trials in Cystic Fibrosis: a Delphi study. J Cyst Fibros. 2021;20:986‐993. [DOI] [PubMed] [Google Scholar]
- 22. Miller F, Miller P. Transgenerational trauma and trust restoration. AMA J Ethics. 2021;23(6):E480‐E486. [DOI] [PubMed] [Google Scholar]
- 23. Wright M, Wright T. Health disparity in CF: perspectives from a lived experience. Pediatr Pulmonol. 2021;57(Suppl 1):S13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.