

Abstract
Acute lymphoblastic leukemia (ALL) is a malignancy that can be subdivided into distinct entities based on clinical, immunophenotypic and genomic features, including mutations, structural variants (SVs), and copy number alterations (CNA). Chromosome banding analysis (CBA) and Fluorescent In‐Situ Hybridization (FISH) together with Multiple Ligation‐dependent Probe Amplification (MLPA), array and PCR‐based methods form the backbone of routine diagnostics. This approach is labor‐intensive, time‐consuming and costly. New molecular technologies now exist that can detect SVs and CNAs in one test. Here we apply one such technology, optical genome mapping (OGM), to the diagnostic work‐up of 41 ALL cases. Compared to our standard testing pathway, OGM identified all recurrent CNAs and SVs as well as additional recurrent SVs and the resulting fusion genes. Based on the genomic profile obtained by OGM, 32 patients could be assigned to one of the major cytogenetic risk groups compared to 23 with the standard approach. The latter identified 24/34 recurrent chromosomal abnormalities, while OGM identified 33/34, misinterpreting only 1 case with low hypodiploidy. The results of MLPA were concordant in 100% of cases. Overall, there was excellent concordance between the results. OGM increased the detection rate and cytogenetic resolution, and abrogated the need for cascade testing, resulting in reduced turnaround times. OGM also provided opportunities for better patient stratification and accurate treatment options. However, for comprehensive cytogenomic testing, OGM still needs to be complemented with CBA or SNP‐array to detect ploidy changes and with BCR::ABL1 FISH to assign patients as soon as possible to targeted therapy.
1. INTRODUCTION
Acute lymphoblastic leukemia (ALL) is a hematopoietic malignancy defined by the accumulation of lymphoid progenitor cells in the blood and bone marrow. It can be divided into two broad groups depending on cell lineage with the majority of cases (85%) belonging to the B‐ and only 15% to the T‐cell lineage. 1 , 2 ALL represents the most common childhood cancer and approximately 60% of cases occur in individuals under 20 years of age with the peak incidence at 1–4 years. 3 , 4
ALL is a heterogeneous disease and a combination of clinical, immunophenotypic and genomic features are used to define distinct entities that are biologically homogeneous and clinically relevant. 1 , 3 Cytogenetic analysis has been routinely performed at diagnosis for more than thirty years and the identification of recurrent structural and numerical chromosome abnormalities provided the first prognostic indicators for risk stratification. This is reflected in the World Health Organization (WHO) classification of B‐cell lymphoblastic leukemia/lymphoma (B‐ALL/‐LBL) which defines sub‐groups based on the presence of primary genomic abnormalities. 5 In T‐cell ALL (T‐ALL) WHO mentions recurrent cytogenetic aberrations but does not yet propose distinct subgroups.
However, the low resolution of chromosomes in leukemia together with the presence of cryptic abnormalities and the poor proliferation of neoplastic cells in culture means that accurate identification of rearrangements and imbalances by chromosome banding analysis (CBA) only is sometimes limited. 6 , 7 Some of these limitations are overcome by the introduction of routine Fluorescent In‐Situ Hybridization (FISH) analysis and this combined approach is the mainstay of routine diagnostics. Whilst this strategy allows the identification of the most frequent or prognostically important subgroups, it will not detect rare or new variants resulting in a paucity of genetic information for some patients.
To address this, RNA and DNA sequencing and array‐based technologies have been applied in the last decade. This has resulted in the identification of novel cryptic chromosome abnormalities and pheno‐copies of existing ALL sub‐groups allowing further genetic subtypes of B‐ALL to be defined. In particular, this includes Ph‐like ALL that can benefit from Tyrosine Kinase Inhibitor (TKI) therapy. 8 , 9 , 10 Several focal gene deletions were also identified, some of which have prognostic significance. 11 , 12 , 13 Consequently, in recent years diagnostic genetic testing of ALL has become more extensive requiring a combination of CBA, FISH, arrays or Multiple Ligation‐dependent Probe Amplification (MLPA), and PCR‐based methods. 6 , 14
The current diagnostic strategy is labor‐intensive, time‐consuming, and costly. In addition, due to cost, cascade testing is often performed which can lead to increased turnaround times (TATs) that do not respond to the needs of clinical trials that require rapid results for stratification into different treatment arms. It is therefore interesting for laboratories to explore new technologies that can simplify and enhance current testing pathways.
An interesting new technology is optical genome mapping (OGM), which uses ultra‐long linear DNA molecules that are enzymatically labeled at specific sequence motifs. OGM allows detection of genome‐wide numerical (> 500 bp) and structural aberrations, including balanced rearrangements, in one assay with a TAT of one week. This technology has already been applied to different cohorts of patients with hematological neoplasms with promising results. 15 , 16 , 17 , 18
Here, we examine the application of OGM for the genetic characterization of newly diagnosed ALL patients to determine whether this approach can eventually replace all, or part of, the current testing strategy. It is expected that OGM will not only lead to an improved detection rate of chromosome abnormalities but could also facilitate the identification of novel aberrations and contribute to the better‐individualized treatment of patients.
2. METHODS
2.1. Sample selection
A series of 41 ALL patients (29 B‐ALL and 12 T‐ALL) at diagnosis were included. Thirty‐eight cases were retrospective and included 21 cases (16 B‐ and 5 T‐ALL) with previously identified recurrent abnormalities representative of most genetic subtypes and 16 cases with a failed or normal karyotype (chromosome banding analysis or CBA). The latter included 8 T‐ALLs positively selected for OGM analysis due to the lack of karyotype information. The 38 retrospective cases were used as a training set to validate the bioinformatics pathway. Three prospective cases (14, 15, and 16) were first analyzed using OGM (blind) and the results were subsequently compared to those of the standard testing pathway.
The study was conducted in compliance with the principles of the Declaration of Helsinki, Good Clinical Practice, and all applicable regulatory requirements. Approval by the Ethical Committee of the University Hospitals Leuven (Belgium) was obtained.
2.2. Conventional testing
All 41 samples were analyzed by the conventional pathway (Table S1) including CBA, FISH, MLPA (P335‐B1 ALL‐IKZF1, MRC‐Holland), RT‐PCR and IG/TR monoclonality. All analyses were performed according to standard techniques or manufacturer's instructions. A minimum of 10 metaphases were analyzed for cases with an abnormal karyotype and 20 for cases with a normal karyotype. 6 Cases with less than 20 normal metaphases were considered failures. For FISH, 200 interphase nuclei were examined on cultured cells and metaphase FISH was also performed when possible to confirm or elucidate gene rearrangements. 6 Cascade interphase FISH analysis was performed for both B‐ and T‐ALL. Additional FISH and molecular experiments were undertaken in some cases to clarify the karyotype.
2.3. Optical genome mapping
Peripheral blood (PB) and bone marrow (BM) from newly diagnosed ALL patients were used. For ethylenediaminetetraacetic acid (EDTA) samples, 650 μL was stored directly at −80°C without any further processing steps or additives. For heparin samples, 10% 0.5 M EDTA was added before freezing at −80°C. For retrospective cases, analysis was performed on previously‐stored diagnostic material, some of which were cell pellets prepared with dimethyl sulfoxide (DMSO) and fetal calf serum (FCS). All samples were stored at −80°C within 6 days of collection.
Prior to genomic DNA (gDNA) isolation, samples frozen at −80°C were thawed in a 37°C water bath. Cell pellets stored with DMSO were resuspended in PBS with 10% FCS, centrifuged at 400 g for 15 min and, after removal of the supernatant, were resuspended in PBS (final volume between 0.5 and 3 mL, depending on the pellet size). Subsequently, ultra‐high molecular weight (UHMW) DNA was extracted from ~1.5 × 106 white blood cells according to the manufacturer's instructions (Bionano Genomics, San Diego USA). Briefly, after counting, white blood cells were pelleted (2200 g for 2 min) and treated with proteinase K and lysis and binding buffer to release gDNA. After proteinase K inactivation gDNA was bound to a paramagnetic nanobind disk. After washing, UHMW DNA (typically sized from 50 kb to ≥1 Mb) was eluted in an appropriate buffer and left to homogenize at room temperature overnight (up to 48 h).
Seven hundred and fifty nanograms of DNA were labeled using a sequence‐specific DLE‐1 Direct Labeling Enzyme that attaches a green fluorophore to a specific 6 bp sequence, present around 15 times per 100 kb in the human genome. Subsequently, the labeled UHMW DNA was loaded onto a Saphyr chip and scanned on the Saphyr instrument (Bionano Genomics, San Diego USA). Up to 6 samples were analyzed in parallel. Single double‐stranded DNA molecules pass through the nanochannels where the fluorescent tags are read as a barcode.
For each sample, we aimed to generate 1300 Gb of data to obtain, on average, an effective genome coverage of about 300x with a theoretical mean variant allele frequency (VAF) sensitivity of 5% (equivalent to aberrations present in heterozygous state in 10% of the cells). Sample preparation took up to 4 days, while the instrument run took another 1–2 days. Quality and run parameters were assessed according to the manufacturer's instructions and included: the total DNA collected ≥150 kb, the map rate (the % of Bionano molecules that align to the reference), the N50 (parameter to qualify molecule length) (≥ 20 kb), the N50 (≥ 150 kb), the average label density (in labels/100 kb), the positive and negative label variance (respectively indicating the percentage of the labels absent in the reference and the percentage of reference labels absent in the molecules) and the effective coverage of the reference.
2.4. De novo assembly and structural variant calling
Samples were analyzed with 2 pipelines: the De Novo Assembly Pipeline, highly sensitive in detecting structural variants (SV) in a diploid genome; and the Rare Variant Pipeline, specifically designed to detect SVs at low allele frequency. A copy number aberrations (CNA) pipeline is embedded in both pipelines. In accordance with guidelines, 6 , 19 filter settings were set to detect all CNA ≥ 5 Mb; of these only CNA detected with a high confidence score (= 1) were directly reported. SVs sized between 500 bp and 5 Mb were only reported when encompassing clinically relevant loci associated with ALL, listed in Table S2, or if they were associated with an unbalanced structural rearrangement.
Applied filter settings and software versions are available in the Supplementary Methods (Data S1). The number of SVs retained with each filter step is provided in Table S3 and illustrated for case 19 in Figure S1.
2.5. Interpretation of OGM results
The validity of the filtering algorithms was assessed and optimized by comparing the aberrations identified in the 38 retrospective cases by the standard techniques to OGM. The same filtering was then applied to 3 prospective cases tested blindly.
2.6. Comparison of results
The OGM results were considered concordant with existing standard pathway results if the same abnormalities were detected by both approaches. Where karyotypes included marker chromosomes or rearranged chromosomes containing material of unknown origin (e.g., der, add) the results were considered concordant if the overall aberrations identified by OGM were consistent with the abnormalities described. Cases with normal karyotypes were also considered concordant if the same abnormalities were identified by the standard FISH panel (Table S1) and OGM. Variations in assigned breakpoints within the same chromosome arm were not considered discordant.
Results were considered discordant when the abnormalities identified by one of the approaches were inconsistent. Cases with normal karyotype and FISH results and an abnormal profile by OGM were also considered discordant.
Additional testing was undertaken to resolve any discordances including karyotype review, supplementary FISH or molecular tests and RNA‐Seq. More details can be found in the Supplementary Methods (Data S1).
3. RESULTS
3.1. Technical characteristics of the OGM analysis
We first evaluated the technical performance of the OGM analysis. A cohort of 41 ALL patients at diagnosis was analyzed. Of these, 29 were B‐ALL cases (12 adult and 17 pediatric) and 12 were T‐ALL (3 adult and 9 pediatric).
OGM analysis resulted in an average label density of 14/100 kb, a map rate of 79% and average effective genome coverage of 312x with a theoretical mean VAF sensitivity of 5%. OGM quality parameters for all cases are available in Table S4.
3.2. Incidence of clonal abnormalities detected by the different techniques
Identification of structural and numerical abnormalities is pivotal for correct risk stratification in ALL. We, therefore, assessed how OGM compares to the standard testing pathway in its ability to detect different clonal aberrations. Figures 1 and S2 show an example of the applied validation strategy (case 15 and 3).
FIGURE 1.
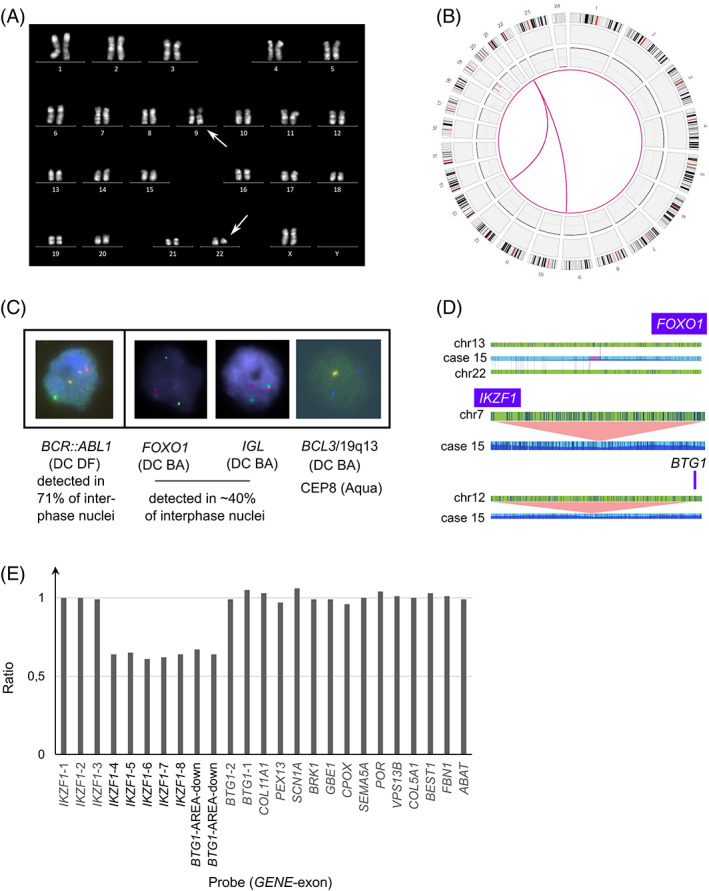
Example of the workflow used to validate optical genome mapping as a diagnostic tool in acute lymphoblastic leukemia (case 15). (A) Chromosome banding analysis (R‐banding) identified a typical t(9;22)(q34;q11) BCR::ABL1. Arrows indicate aberrant chromosome 9 and 22. (B) OGM results represented as a circos plot revealing 4 large chromosomal aberrations (>5 Mb) including a t(9;22)(q34;q11), t(13;22)(q14;q11) and 2 deletions affecting the long and short arm of chromosome 19. The SV track was not shown in the plot for clarity as it also includes unreported SVs. (C) Standard FISH analysis confirmed the presence of a BCR::ABL1 rearrangement in 71% of interphase nuclei. Extra FISH experiments were performed to confirm the additional OGM findings. FISH identified rearrangements of both FOXO1 and IGL in ~40% of interphase nuclei, confirming the presence of the t(13;22)(q14;q11). In addition, FISH confirmed the 19q deletion detected by OGM, illustrated by the loss of one red/green (‘yellow’) BCL3/19q13 signal whilst two blue signals were observed for the control probe encompassing centromere 8. The following probes were used: LSI BCR (SG)/ABL (SO) (DC DF) [9q34/22q11, Vysis]; LSI FOXO1 (DC BA) [13q14, Vysis]; XL IGL (DC BA) [22q11, Metasystems] and BCL3 (DC BA) [19q13, Empire Genomics] together with CEP8 (SA) [Vysis]. (D) Detailed analysis of the clinically relevant structural variants and copy number aberrations (as provided by the genome browser view from the Bionano Access Software) revealed involvement of the FOXO1 gene in the t(13;22)(q14;q11) and 2 submicroscopic deletions encompassing exon 4–8 of IKZF1 and the downstream region of BTG1. (E) Graph illustrating the loss of IKZF1 exon 4–8 and the downstream area of BTG1 as shown by MLPA analysis. The final ratio for the IKZF1 probes (exons 1–8) and BTG1 probes (area: downstream, exons 1–2) is given compared to the reference probes. MLPA was performed according to manufacturer's specifications (SALSA P335‐C1, MRC‐Holland); data analysis was performed with Coffalyzer. Normal range: 0,7‐1,3. Abbreviations: OGM optical genome mapping, SV structural variant, FISH fluorescent in‐situ hybridization, DC DF dual color dual fusion probe, SG spectrum Green, SO spectrum Orange, DC BA dual color break apart probe, SA single color spectrum Aqua, MLPA multiple ligation‐dependent probe amplification, CEP centromeric probe [Color figure can be viewed at wileyonlinelibrary.com]
For the 29 B‐ALL cases, clonal abnormalities were identified by CBA in 23 cases (79%). The standard FISH panel (Table S1) identified clonal abnormalities in 21/29 cases (72%), including 4 patients with a normal karyotype. The overall clonal detection rate by CBA combined with standard FISH in B‐ALL cases was 93%.
For the 12 T‐ALL cases, clonal abnormalities were detected by CBA in 4 cases (33%). Three cases had a normal karyotype, five had a failed karyotype. The standard FISH probes were applied to all cases and clonal abnormalities were detected in 7 cases, including 4 of the 8 cases with a normal or failed karyotype. FISH analysis was inconclusive (borderline result) in a further case (20) with a failed karyotype, the suspected abnormality was not confirmed by OGM. CBA combined with standard FISH identified at least one clonal abnormality in 8/12 T‐ALL cases (67%).
OGM on the other hand identified a clonal abnormality in all B‐ and T‐ALL cases (100%) providing an increased detection rate compared to the standard testing pathway in both B‐ and T‐ALL.
However, in some cases, there was no full concordance between the identified abnormalities. OGM failed to distinguish some subclonal aberrations, observed by CBA in 7 patients (case 7, 9, 13, 17, 22, 32, 40) and only identified the abnormalities present in 1 of the 2 independent clones detected in another (B‐ALL, case 8). The latter had an independent clone with t(X;4) detected by CBA and confirmed by metaphase FISH (using whole chromosome painting probes) that was not detected by OGM. FISH confirmed that the breakpoint on the X chromosome was located in the centromeric region. It is well established that in regions concentrated around centromere and telomere regions OGM molecule alignment can be unreliable.
In general, however, non‐detection by OGM was due to misinterpretation of CBA or to the abnormalities being present in a small subset of CBA metaphases (7–18%). Whilst OGM was unable to detect the subclonal aberrations present in these cases, it was able to detect aberrations present at this, or lower frequencies in others. It is therefore difficult to establish OGM sensitivity with respect to CBA results. By performing additional interphase FISH (data available in Supplementary Methods (Data S1) and Figure S3), we could estimate the true size of the clones and established that aberrations present in at least 15% of ALL cells can be reliably detected using OGM.
There were only two cases where we were unable to explain the discordant results. In case 36 (B‐ALL), CBA suspected a large structural rearrangement involving the short arm of chromosome 2 and the long arm of chromosome 14 in the majority of metaphases (90%) that was not confirmed by OGM. A review of the karyotype still yielded a high suspicion of a translocation, whilst OGM showed no evidence of chromosome 2 nor 14 involvement. It is possible that the breakpoints occur in repetitive regions not covered by OGM. Similarly, in case 22 (T‐ALL), OGM reported a DUX4::FRG2B rearrangement which could not be confirmed with FISH nor RNA seq. This false‐positive result was probably due to a similar labeling pattern in both regions.
The full results for all techniques are summarized in Table 1. Additional details are available in Table S5.
TABLE 1.
Standard genetic testing pathway results compared to Optical Genome Mapping in ALL patients
| WHO classification | Karyotype FISH/RT‐PCR | Recurrent deletions detected by MLPA a | OGM formulab | Recurrent deletions detected by OGM a | Recurrent SVs and ploidy detected by OGM | |
|---|---|---|---|---|---|---|
| 1 | B‐ALL with TCF3::PBX1 |
46,XX,der(19)t(1;19)(q23;p13)[5]/46,XX[5] PBX1 and TCF3 rearranged |
ND |
ogm[GRCh37] 1q23.3q44(164940579_248630818)x3,t(1;19)(q23;p13.3)(164775625;1617441),ins(1;19)(q23;p13.3p13.3)(164775625;1617441_1759114inv),19p13.3(pter_1617442)x1 |
None | TCF3::PBX1 |
| 2 | B‐ALL NOS |
46,XX[20] No abnormalities detected by FISH |
None |
ogm[GRCh37] t(12;22)(p13.31;q13.2)(6795009;41525645) |
EP300 ::ZNF384 | |
| 3 | B‐ALL with ETV6::RUNX1 |
46,XY,del(6)(q16q24),add(11)(q23),?add(12)(p?),add(12)(q23),add(21)(q21),inc[11]/46,XY[2] Metaphase FISH complex t(11;12;21) ETV6::RUNX1 rearrangement |
Exon 2–6 PAX5, exon 8 ETV6 |
ogm[GRCh37] 6q15q23.2(90570707_132994084)×1,9p13.2p13.2(36924868_37031738)×1,t(11;12;21)(q24.2;p13;q22.11)(124542267;12034841;36326359),12p13.2p12.1(12034841_2497337)×1,21q21.1q22.12(16755788_36235050)×3 |
Exon 2–6 PAX5, exon 6–8 ETV6 |
ETV6::RUNX1 |
| 4 | B‐ALL with ETV6::RUNX1 |
46,XX[20] ETV6::RUNX1 rearrangement |
Exon 1–8 PAX5 |
ogm[GRCh37] Xq24q28(118659748_155008232)×3,t(X;21)(q24;q22.3)(118596968;48117142),9p13.2(36853610_37382413)×1,t(12;21)(p13.2;q22.12)(12034841;36420383) |
Exon 1–8 PAX5 |
ETV6::RUNX1 |
| 6 | B‐ALL NOS |
46,XX,del(3)(p13p26),der(9)t(3;9)(p13;p13)[6]/46,XX[1] No abnormalities detected by FISH |
Exon 7–10 PAX5, biallelic CDKN2A/2B |
ogm[GRCh38] t(3;9)(p13;p13.2)(71063635;36957140),9p24.3p13.2(14566_36957140)×1, 9p21.3(21825731_22009703)×0 |
Exon 7–10 PAX5, biallelic CDKN2A/2B |
PAX5::FOXP1 |
| 7 | B‐ALL with hyperdiploidy |
56,XX,+X,+4,+6,+8,+10,+14,+14,+17,+21,+21[2]/57,sl,+11[5]/46,XX[4] FISH compatible with hyperdiploid clone |
Exon 8 ETV6 |
ogm[GRCh38] (X)×3,(4)×3,(6)×3,(8)×3,(10)×3,(11)×3,12p13.2(11870531_119814480)×1,(14)×4,(17)×3,(21)×4 | Exon 6–8 ETV6 | |
| 8 | B‐ALL, BCR::ABL1‐like |
46,XX,t(8:9)(p22;p24)[1]/46,sl,der(8;9)(q10;q10),inc[6]/46,X,t(X;4)(p11;q1?3)[4]/46,XX[11] PCM1 and JAK2 rearranged |
ND | ogm[GRCh38] t(8;9)(p22;p24.1)(18021361;5060274),t(8;9)(q12.2;p24.1)(60671418;5961314),9p24.1p22.3(7306311_16265816)×1,9p21.3p21.2(21951394_27261605)×0,25 | PCM1::JAK2 | |
| 9 | B‐ALL NOS |
46,XX,del(9)(q22q33),der(19)t(1;19)(q23;p13)[6]/46,sl,del(13)(q13q21)[3]/46,XX[1] No TCF3::PBX1 rearrangement detected by FISH |
None | ogm[GRCh37] 1q21.2q44(149529883_249237532)×3,t(1;19)(q21.1;q13.43)(144837767;59111086),9q21.11q22.33(70321158_101956617)×1,19q13.43(59128983_qter)×1 | None | |
| 10 | B‐ALL with ETV6::RUNX1 |
46,XX,‐8,der(12)t(8;12)(q13;p13),+21[7]/46,XX[3] ETV6::RUNX1 rearrangement |
Exon 1–6 PAX5, ETV6 |
ogm[GRCh38] 8p23.3p11.23(61805_38264830)×1,t(8;12)(p11.23;p12.1)(38274465;26217459),9p13.2(36955993_372945461),12p13.33p12.1(14568_26225920)×1,t(12;21)(p13.2;q22.12)(11881907;35043931),(21)×3 |
Exon 1–6 PAX5, ETV6 |
ETV6::RUNX1 |
| 12 | B‐ALL with low hypodiploidy |
46,XX[20] FISH compatible with low hypodiploid clone |
None |
ogm[GRCh38] (X)×3,(1)×3,(5)×3,(6)×3,(8)×3,(10)×3,(11)×3,(14)×3,(15)×3,(18)×3,(19)×3,(21)×3,(22)×3 After manual correction of the ploidy:36,XX,–2,–3,–4,–7,–9,–12,dup(12p12.1p11.21)(22140659_31024594),–13,–16,–17,–20 |
None | After manual correction of ploidy: low hypodiploid clone |
| 13 | B‐ALL NOS |
45,X,‐Y[4]/46,XY,i(9)(q10),inc[3]/46,XY[16] No abnormalities detected by FISH |
None | ogm[GRCh38] t(1;19)(q23.3;p13.3)(164684214;1617442) |
None |
TCF3::PBX1 |
| 14 | B‐ALL NOS |
48,XY,+X,del(1)(q25q44),add(5)(q32),?der(7),+21[6]/46,XY[4] Trisomy X and 21 |
Exon 5–7 IKZF1, CDKN2A/2B |
ogm[GRCh37] (X)×2,t(1;5)(q22;q32)(156445725;149451946),5q35.2q35.3(175705336_180899715)×1,7p12.2(50451066_50467354)×1,9p21.3(21897504_22003647)×1,9p21.3(21975985_22019207)×1,17q24.3q25.3(70828023_77521027)×1,19q13.31q13.33(45144628_51391878)×1,(21)×3 |
Exon 5–7 IKZF1, CDKN2A/2B |
MEF2D::CSF1R |
| 15 | B‐ALL with BCR::ABL1 |
46,XX,t(9;22)(q34;q11)[2]/46,XX[6] BCR::ABL1 rearrangement |
Exon 4–8 IKZF1, downstream BTG1 | ogm[GRCh37] 7p12.2(50389810_51209685)×1,t(9;22)(q34.12;q11.23)(133647045;23548004),12q21.33(91794497_92538906)×1,t(13;22)(q14.11;q11.22)(41019582;22364735),19p13.12p12(16140309_21286406)×1,19q13.31q13.41(45026283_51442852)×1 | Exon 4–8 IKZF1, downstream BTG1 |
BCR::ABL1 IGL::FOXO1 |
| 16 | B‐ALL with low hypodiploidy |
32,XX,‐2,‐3,‐4,‐6,‐7,‐8,‐11,‐12,‐13,‐14,‐15,‐16,‐17,‐19[2]/37‐46,XX,inc[3] No structural or numerical abnormalities detected by FISH |
None | ogm[GRCh37] (X)×3,(1)×3,(5)×3,(9)×3,(10)×3,(11)×3,(18)×3,(19)×3,(21)×3,(22)×3 | None |
Manual correction of ploidy impossible due to low blast count: hyperdiploid clone |
| 17 | B‐ALL with iAMP21 |
47,XX,+X,add(3)(p21),del(4)(q?),del(12)(p11p13),add(14)(p11),‐21,+mar,inc[2]/45,sl,‐X,del(7)(p11p21)[2],add(10)(p11)[2],add(10)(p12),‐del(12),+add(12)(p11),‐mar[cp4]/46,XX[6] RUNX1 amplification by FISH |
ETV6, biallelic exon 19–26 RB1 | ogm[GRCh37] inv(2)(q33.1q35)(202530525;218107974),t(3;8)(p26.3;q21.2)(61829;84698449),4q21.22q26(82562646_118148833)×1,t(4;10)(q35.1;p12.2)(183938118;24572275),8q21.2q24.3(84747232_146301511)×3,10p15.3p12.2(64453_24569782)×4,10p12.2p11.21(24572275_35188421)×3,12p13.33p12.1(62597_32082761)×1,13q14.2(48981801_49089572)×0,14q11.2q32.33(20412883_106295617)×2,93,(21)cth,21q22.1q22.2(32113440_41074917)×8,25 |
ETV6, biallelic exon 18–27 RB1 gene |
Amplification RUNX1 |
| 18 | B‐ALL with hyperdiploidy |
55,XX,+4,+6,+8,+10,+14,+17,+18,+21,+21[4]/46,XX[6] FISH compatible with hyperdiploid clone |
None | ogm[GRCh38] (4)×3,(6)×3,(8)×3,(10)×3,(14)×3,(17)×3,(18)×3,(21)×4 | None | High hyperdiploid clone |
| 27 | B‐ALL NOS |
46,XY[23] No abnormalities detected by FISH |
None | ogm[GRCh38] inv(9)(p24.1p13.2)(5081915;36992699) | None | PAX5::JAK2 |
| 29 | B‐ALL with ETV6::RUNX1 |
46,XX[20] ETV6::RUNX1 rearrangement |
Exon 2–6 PAX5, ETV6 | ogm[GRCh38] 9p13.2(36924871_37031741)×1,10p15.3p11.23(2148472_29605241)×3, t(10;12)(p11.23;p12.3)(29628820;15233701),12p13.33p12.3(14568_15233701)×1,t(12;21)(p13.2;q22.12)(11870531;35029693) |
Exon 2–6 PAX5, ETV6 |
ETV6::RUNX1 |
| 30 | B‐ALL with BCR::ABL1 |
46,XX,t(9;22)(q34;q11)[8]/46,sl,add(16)(p12)[2] BCR::ABL1 rearrangement |
Exon 4–7 IKZF1, CDKN2A/2B, PAX5, exon 19–26 RB1, exon 2 BTG1 + downstream |
ogm[GRCh38] t(1;16)(q21.1;p13.3)(143361930;14134),7p12.2(50324505_50399656)×1,t(9;22)(q34.12;q11.23)(130709559;23244051),9p21.3(21925732_22165462)×1,9p13.2(36759965_37121784)×1,12q21.33(91882647_92145130)×1,13q14.2(48402465_48510295)×1 |
Exon 3–7 IKZ1,CDKN2A/2B, PAX5,exon 18–27 RB1, exon 2 BTG1 + downstream |
BCR::ABL1 |
| 31 | B‐ALL NOS |
46,XX,idic(9)(p1?3)[7]/46,XX[3] No abnormalities detected by FISH |
CDKN2A/2B, PAX5 | ogm[GRCh38] 9p24.3p12(14566_39591818)×1,9p12q34.3(39788526_136344539)×3 | CDKN2A/2B, PAX5 | |
| 32 | B‐ALL with hyperdiploidy |
56,XX,+X,+4,+6,+8,+10,+17,+18,+21,+21,+mar[7]/55,sl,t(15;15)(q21;q24),‐17[2]/46,XX[2] FISH compatible with hyperdiploid clone, unbalanced rearrangement of IGH |
CDKN2A/2B, exon 1 ETV6 | ogm[GRCh38] (X)×3,(4)×3,(6)×3,(8)×3,9p21.3(21636919_23285480)×1,(10)×3, 12p13.2(11635931_11654206)×1,(14)×3,t(14;14)(q11.2;q32.33)(22422983;106294644),(17)×3,(18)×3,(21)×4 |
CDKN2A/2B, exon 1 ETV6 |
TRA::IGH |
| 34 | B‐ALL NOS |
91<4n>,XXYY,‐17,idic(17)(p11)[8]/92<4n>,XXYY[1]/46,XY[5] FISH compatible with tetraploid clone |
Not informative (low blast count) | ogm[GRCh38] ins(9;?)(p13.2;?)(9:37012419_37031741ins(?:?_?),17p13.3p11.2(1315080_16299764)×1,78 | TP53 |
PAX5::ZNF318 Tetraploidy not detected |
| 35 | B‐ALL with hyperdiploidy |
55,XY,+X,+4,+6,+8,+11,+14,add(17)(p13),+18,+21,+21,inc[7]/46,XY[7] FISH compatible with hyperdiploid clone |
CDKN2A/2B | ogm[GRCh38] (X)×2,(4)×3,(6)×3,(8)×3,9p21.3(20360830_23290064)×1,(11)×3,(14)×3,t(17;17)(p13.3;q21.2)(320531;42625238)17q21.2q25.3(42621068_83246392)×3,(18)×3,(21)×4 |
CDKN2A/2B |
High hyperdiploid clone |
| 36 | B‐ALL NOS |
46,XY,‐2,?t(9;17)(p13;q21),del(11)(q21q23),?der(14)t(2;14)(?p11;q32),+mar[8]/46,XY[1] Loss of entire KMT2A gene by FISH |
None | ogm[GRCh38] t(9;17)(p13.2;q22.12)(37205108;48932814),10p15.3p11.22(18514_31936573)×3,t(10;11)(p11.22;q22.1)(32029500;100293717),11q22.1q25(100233393_135069565)×1 | KMT2A | |
| 37 | B‐ALL with hyperdiploidy |
60,XY,‐5[5],+6,+8,+10,+11[5],+12,+14[5],+15,+16[5],+17,+18[3],+21,+21[5],+3‐7mar,inc[cp6]/46,XY[3] FISH compatible with hyperdiploid clone |
CDKN2A/2B, deletion of exon 2 ETV6 (only 2 copies) | ogm[GRCh38] (X)×2,(4)×3,(5)×3,t(4;5)(q34.3;q31.1)(179501025;136258772),t(5;5)(q33.1;q35.3)(152558057;187110343),t(5;10)(q33.2;q23.31)(153843504;90199407),t(5;10)(q35.1;q24.2)(171252456;97709101),9p21.3(21801680_22314347)×1,(6)×3,(8)×3,(10)×4,(12)×3,12p13.2(11699080_11790985)×1,(14)×3,(16)×3,(17)×3,(18)×3,(21)×4 |
CDKN2A/2B, exon 2 ETV6 |
High hyperdiploid clone |
| 38 | B‐ALL, BCR::ABL1‐like |
46,XX[20] IGH::CRLF2 rearrangement |
IKZF1, exon 1–2 ETV6, exon 2 + downstream BTG1 |
ogm[GRCh38] 3p21.31p12.2(44984632_81647686)×1,7p14.1p12.1(36363239_50695470)×1,12p13.2 (11635931_11818565)×1,12q21.33(91882647_92145130)×1,17p13.3p11.2(66653_19154549)×1,17q11.1q25.3(26692353_79373278)×3 | IKZF1, exon 1–2 ETV6, exon 2 + downstream BTG1 | Software version 1.6 or higher: t(X;14)(p22.33;q32) IGH::CRLF2 |
| 39 | B‐ALL NOS |
46,XX,+8,‐20[1]/46,sl,der(9)t(9;20)(p13;p11)[16]/46,XX[9] No abnormalities detected by FISH |
Biallelic CDKN2A/2B, exon 10 PAX5 |
ogm[GRCh38] (8)×3,9p24.3p13.2(14566_37056711)×1,(9)(p21.3)(21897505_22009703)×0,t(9;20)(p13.2; q11.21)(36866463;32456682),20q11.21q13.33(32456682_64333718)×1 | Biallelic CDKN2A/2B, exon 9–10 PAX5 | PAX5::ASLX1 |
| 40 | B‐ALL NOS |
47,XX,+21[9]/47,sl,add(3),add(6),‐12,add(14),+mar[1]/47,sl,add(3) (q13),del(4)(q13q24),add(6)(p11),‐12,add(14)(p11),+mar[2]/46,XX[8] Trisomy 21 |
None | ogm[GRCh38] (21)×3 | None | |
| 41 | B‐ALL with TCF3::PBX1 |
46,XX,del(6)(q16q25),der(19)t(1;19)(q23;p13)[10] TCF3::PBX1 rearrangement |
None |
ogm[GRCh38] 1q23.3q44(164679641_248943333)×3,t(1;19)(q23.3;p13.3)(164694931;1617442),6q14.1q22.1(77866354_115226336)×1,19p13.3(pter_1617442)×1 |
None |
TCF3::PBX1 |
| 5 | T‐ALL |
46,XY[15] No abnormalities detected by FISH |
ogm[GRCh38] t(1;14)(p33;q32)(47227936;106586104),14q32(106586104_106607463)×1 |
IG H::TAL1 |
||
| 11 | T‐ALL |
46,XY[11] TRB::HOXA10 |
ogm[GRCh37] inv(7)(p15.2q34)(27231194;142508528) |
TRB::HOXA10 | ||
| 19 | T‐ALL |
46,XY,t(11;14)(p13;q11)[3]/46,XY[12] FISH TRD::LMO2 rearrangement/RT‐PCR: STIL::TAL1 rearrangement |
ogm[GRCh38] 1p33(47227936_47325742)×1,t(11;14)(p13;q11.2)(33816961;22082444) |
STIL::TAL1 TRD::LMO2 |
||
| 20 | T‐ALL |
46,XY[1] Suspected rearrangement of TRA/D by FISH |
ogm[GRCh38] t(5;11)(q31.1;p11.2)(134127916;47361335) |
TCF7::SPI1 | ||
| 21 | T‐ALL |
46,XY[27] No abnormalities detected by FISH |
ogm[GRCh38] 9p21.3p21.2(21082754_26785046)×0,38,t(14;14)(q11.2;q13.3 )(22442014;36521900) | TRA::NKX2‐1 | ||
| 22 | T‐ALL |
46,XY,del(6)(q15q23),t(7;11)(q34;p13),add(16)(p12)[11]/46,sl,del(8)(q12q21)[2]/46,XY[2] TRB::LMO2 rearrangement by FISH |
ogm[GRCh38] t(4;10)(q35.2;q26.3)(190058514;133630087),6q11.1q21(62507864_110148938)×1,t(7;11)(q34;p13)(142810844;33848837) |
TRB::LMO2 t(4;10) DUX4::FRG2B False positive (not confirmed by FISH) |
||
| 23 | T‐ALL |
46,XY,?del(10)(q23q25)[6]/46,XY[17] No abnormalities detected by FISH/RT‐PCR STIL::TAL1 |
ogm[GRCh38] 1p33(47227936_47324493)×1 |
STIL::TAL1 |
||
| 24 | T‐ALL |
46,XY[15] TLX3 rearrangement by FISH |
ogm[GRCh38] t(5;14)(q35.1;q32.2)(171333079;98622030),13q12.2q21.31(27781122_63486427)×1 |
?BCL11B::TLX3 |
||
| 25 | T‐ALL |
46,XY,add(6)(q22),add(14)(q2?4)[16]/46,XY[1] No abnormalities detected by FISH |
ogm[GRCh38] t(6;6)(q24.2;q25.3)(144332634;157040528) or inv(6)(q24.2q25.3),t(6;14)(q24.2;q32.2)(144332634;99277645),t(6;14)(q25.3;q32.2)(156271735;99266630) |
BCL11B::? |
||
| 26 | T‐ALL |
46,XX[20] TRA/D rearrangement by FISH |
ogm[GRCh38] t(2;14)(q22.3;q11.2)(144622655;22496144) | TRA::ZEB2 | ||
| 28 | T‐ALL |
46,XY[16] TRB rearrangement by FISH |
ogm[GRCh38] t(7;7)(p15.2;q34)(27549839;142784716),7q34q36.3(142784716_159334984)×1,12p12.2p12.1(20367120_26146317)×1 | TRB::HIBADH | ||
| 42 | T‐ALL |
46,XY,add(4)(q21),add(7)(q34),idic(9)(p13),der(9;9)(p10;p10),inc[2]/46,XY[8] TRB rearrangement by FISH |
ogm[GRCh37] 4p16.3p15.1(12985_29399657)×1,inv(4)(p15.1p13)(29414444;43347003),t(7;10)(q34;q24.31)(142508527;102897948),inv(8)(p21.3p11.21)(19781043;41512623),9p24.3p13.1(14566_38792295)×1,9p21.3(21535833_24382513)×0,9q21.11q34.2(70321158_137096384)×3 | TRB::TLX1 | ||
Note: Bold refined breakpoints, resolving cytogenetic findings and identification of additional clinically relevant SVs.
Abbreviations: ALL, acute lymphoblastic leukemia, MLPA, multiple ligation dependent probe amplification, ND, not determined, OGM, optical genome mapping, SV, structural variant.
If no exons are mentioned, the entire gene is deleted.
OGM formula abnormalities adapted according to guidelines.
3.3. Ability of OGM to detect disease defining abnormalities
3.3.1. Concordance to detect recurrent SVs
Having evaluated first the general sensitivity of OGM to detect clonal aberrations, we next tested the capacity of OGM to identify specific lesions. We first focused on the detection of SVs typically present in B‐ and T‐ALL. Seventeen recurrent SVs (as defined by Iacobucci et al.) were identified in 26 ALL patients by the standard testing panel. 20 Of note, in one T‐ALL patient (case 19) 2 recurrent SVs were identified. All the observed SVs were also detected by OGM analysis (Table 2). Initially, the IGH::CRLF2 fusion was not detected by OGM in case 38 (B‐ALL) but, after manual inspection of the OGM profile, the translocation was visible but had not been called by the Bionano analytical pipeline (data not shown). The software has since been upgraded and this variant is called by the new version.
TABLE 2.
Recurrent chromosome abnormalities of clinical relevance detected by OGM versus current testing strategy
| Genetic subtype a | Recurrent chromosomal abnormality | Case ID | Number of detected variants | ||
|---|---|---|---|---|---|
| CBA/FISH/RT‐PCR | OGM | ||||
| B‐ALL | t(12;21) | t(12;21)(p13;q22) [ETV6::RUNX1] | 3;4;10;29 | 4 | 4 |
| t(1;19) | t(1;19)(q23;p13.3) [TCF3::PBX1] | 1;13;41 | 2 | 3 | |
| BCR::ABL1 | t(9;22)(q34;q11) [BCR::ABL1] | 15;30 | 2 | 2 | |
| BCR::ABL1‐like | t(8;9)(p22;p24) [PCM1::JAK2] | 8 | 1 | 1 | |
| inv(9)(p24p13) [PAX5::JAK2] | 27 | 0 | 1 | ||
| t(1;5)(q22;q32) [MEF2D::CSF1R] | 14 | 0 | 1 | ||
| CRLF2 rearrangements | t(X;14)(p22.33;q32) [IGH::CRLF2] | 38 | 1 | 1 | |
| ZNF384 rearrangements | t(12;22)(p13.31;q13.2) [EP300::ZNF384] | 2 | 0 | 1 | |
| PAX5 rearrangements | dic(9;20)(p13;q11) [PAX5::ASXL1] | 39 | 0 | 1 | |
| t(3;9)(p13;p13.2) [PAX5::FOXP1] | 6 | 0 | 1 | ||
| ins(9;?)(p13.2;?) [PAX5::ZNF318] | 34 | 0 | 1 | ||
| iAMP21 | iAMP21 | 17 | 1 | 1 | |
| T‐ALL | TAL1 deregulation | STIL::TAL1 | 19;23 | 2 | 2 |
| IGH::TAL1 | 5 | 0 | 1 | ||
| LMO2 deregulation | t(11;14)(p13;q11) [TRA::LMO2] | 19 | 1 | 1 | |
| t(7;11)(q34;p13) [TRB::LMO2] | 22 | 1 | 1 | ||
| TLX1 (HOX11) deregulation | t(7;10)(q34;q24) [TRB::TLX1] | 42 | 0 | 1 | |
| TLX3 (HOX11L2) deregulation | t(5;14)(q35;q32) [BCL11B::TLX3] | 24 | 1 | 1 | |
| HOXA10 deregulation | inv(7)(p15q34) [TRB::HOXA10] | 11 | 1 | 1 | |
| t(7;7)(p15.2;q34) [TRB::HIBADH] | 28 | 0 | 1 | ||
| Subtotal recurrent SVs | 17/27 | 27/27 | |||
| B‐ALL | Aneuploidy | High hyperdiploidy | 7;18;32;35;37 | 5 | 5 |
| Low hypodiploidy | 12;16 | 2 | 1 | ||
| Total | 24/34 | 33/34 | |||
adapted from Iacobucci I, Mullighan CG. Genetic Basis of Acute Lymphoblastic Leukemia. J Clin Oncol 2017;35(9):975–983.
In addition to the SVs detected by the standard testing pathway, OGM analysis identified a further 10 recurrent SVs (Table 2). All of these were confirmed by FISH and/or molecular techniques. Some cases presented with a normal karyotype (2, 5, 27, 28), while others had an abnormal karyotype. In the latter group, the additional SVs were either not observed in the karyotype (B‐ALL, case 34, PAX5 rearrangement) or were not recognized as a recurrent SV due to low‐resolution banding (6, 14, 39, 42). In one B‐ALL case (13) an isochromosome for the long arms of a chromosome 9 [i(9q)] was observed in poor quality metaphases whilst a t(1;19)TCF3::PBX1 was identified by OGM. After karyotype review, a t(1;19) and i(9q) were present in 3/72 metaphases. The FISH analysis also confirmed a t(1;19) in 35% of cells and a trisomy 9q in only 6.5% of cells. The t(1;19) was thus missed by cytogenetics due to poor quality chromosomes and the i(9q) was missed by OGM due to the small size of the subclone.
In the T‐ALL cases, FISH identified rearrangements of TRA/D, TRB, and TLX3 in 7 patients. Identification of the partner chromosome however required additional analysis and in 3 cases (24, 28, 42) the partner remained undetermined even after extensive FISH testing. OGM confirmed these rearrangements and in addition identified the partner gene in all cases.
3.3.2. Concordance to identify aneuploidy
In B‐ALL, aneuploidy represents an important risk factor with high hyperdiploidy (gain of ≥5 chromosomes) being linked to a favorable outcome, whilst low hypodiploid cases (31–39 chromosomes) have a very poor prognosis. 3 , 21 Hence, precise documentation of the chromosome count is mandatory at diagnosis.
OGM CNA pipeline correctly determined the chromosome copy number in 38/41 cases, including 5 high hyperdiploid samples (7, 18, 32, 35, 37). Discordances were seen in 3 cases where there was a gain or loss of an almost entire haploid/diploid set (changes in ploidy). In 2 cases with low hypodiploidy (12, 16), the chromosomal gains were correctly identified by OGM but the results were incorrectly interpreted as hyperdiploid (Table 2). The OGM software allows you to manually plot the zygosity states of all the SVs (using filter set B, described in Supplementary Methods (Data S1) section, on the “De Novo Assembly” data), chromosome per chromosome. Loss of heterozygosity (LOH) for certain chromosomes might be indicative of the loss of one copy of these chromosomes (or of copy‐neutral LOH). By correlating zygosity states with karyotype or FISH results, the baseline could be corrected for one of the two cases (case 12) resulting in the same low hypodiploid karyotype as detected with conventional cytogenetic tests. However, this manual approach was not successful for case 16 probably due to the lower blast count (~40%).
Importantly, in both low hypodiploid cases, there were discordances between the results of CBA, FISH, MLPA and OGM. One case had a normal karyotype, losses by FISH and gains by MLPA and OGM, and the other had an abnormal karyotype, normal FISH and MLPA and gains by OGM. This cohort also included a case with a tetraploid clone (34), detected by CBA and FISH. OGM correctly identified the abnormalities present but misinterpreted the ploidy as diploid. Overall, there was no one technique that was 100% successful at reliably identifying patients with ploidy alterations.
3.3.3. Conclusion
To summarize, there was an overall good concordance between the different approaches to detect the major cytogenetic risk groups (Table 2). OGM analysis identified 32 of the 33 patients that could be classified into the known recurrent cytogenetic subgroups compared to 23 by traditional genomics. Within these 33 patients, the standard approach identified 24/34 recurrent chromosomal abnormalities, while OGM identified 33/34, misinterpreting only one case with low hypodiploidy.
3.4. Accuracy of OGM to detect focal deletions
In B‐ALL several small focal submicroscopic deletions of prognostic significance have been identified that require molecular techniques, such as MLPA, for their identification. Based on the copy number alterations present, patients can then be classified into different risk stratification groups. 22
The ability of OGM to detect these small deletions was compared to the results of MLPA analysis and we assessed how this impacted risk assignment (Table S6).
MLPA was performed in 27/29 of B‐ALL patients. Seventeen had an abnormal and 10 cases a normal profile. For two patients we had insufficient material to permit MLPA analysis.
The MLPA results were concordant with OGM in 100% of the cases (27/27). Biallelic gene deletions, as well as downstream deletions of BTG1, were correctly identified by both techniques. In 4 cases OGM also detected gain of the PAR1 region due to gain of chromosome X but this has no known clinical impact. The smallest focal deletion picked up in the current cohort using OGM was a 17.7 kb deletion of exons 5–7 of IKZF1 (case 14). Risk assignment into good or poor UKALL CNA profiles was identical based on MLPA versus OGM results. 22
3.5. Refinement of abnormal karyotypes and resolution of abnormalities of unknown origin by OGM
As shown in Table 1, OGM enhanced the karyotype in cases where the chromosomal origin of some rearrangements was unknown (marker and derivative chromosomes). As well as confirming deletions, OGM identified the genomic imbalances and determined the underlying structural rearrangements enabling abnormalities to be redefined as balanced or unbalanced translocations. In case 17 (B‐ALL) for example, a very complex karyotype with monosomy 21 and the presence of a marker chromosome could be redefined by OGM as chromothripsis of chromosome 21 confirming also the iAMP21 previously documented by FISH.
OGM analysis also allows a more precise assignment of chromosome breakpoints and, following analysis, breakpoints were adjusted in some cases. Although most were minor changes (with the results considered concordant) this led to the reclassification of the type of rearrangement in some cases. For instance, a translocation was reclassified as a dicentric chromosome in case 39 (B‐ALL): dic(9;20)(p13.2;q11.21)[PAX5::ASXL1], a recurrent abnormality known to be associated with intermediate risk. 11
Similarly, in case 14 refinement of the breakpoints led to the identification of a MEF2D::CSF1R translocation, while in case 27, a PAX5::JAK2 fusion gene was identified solely by OGM. Both cases could be redefined as BCR::ABL1 like B‐ALL instead of B‐ALL, NOS. Other SVs that were detected only by OGM, included an EP300::ZNF384 fusion gene (case 2) and other important translocations involving PAX5 and FOXO1 (cases 6, 15, 34). These translocations may influence B‐ALL outcome. 8 , 23 , 24 , 25
For 7 T‐ALL/LBL cases (5, 20, 21, 25, 26, 28, 42) OGM was the sole method to either detect additional SVs, including a TCF7::SPI1 fusion gene and BCL11B, TAL1 rearrangements or to identify TRA/TRB fusion partners. All variants were confirmed using FISH and/or RNA seq. The BCL11B rearrangement in case 25 designated by OGM was due to a t(6;14) only rarely described in T‐ALL. 26
In addition to identifying otherwise non‐detected abnormalities, the resolution of breakpoints by OGM also disproved putative rearrangements. In case 9 (B‐ALL), the t(1;19)(q23;p13)[TCF3::PBX1] suspected by CBA could not be confirmed by FISH analysis. Indeed, OGM revealed that no TCF3::PBX1 was present, and the breakpoint was reassigned to the long arm of chromosome 19.
4. DISCUSSION
In this study, we evaluated the opportunities of OGM in the diagnostic work‐up of ALL and compared OGM to our standard testing pathway. Overall, we experienced an excellent concordance between the results of the conventional approach and OGM. There were however some significant discrepancies, which highlight the limitations and advantages associated with each of the techniques used.
First, our study emphasizes that the poor resolution of chromosomes and the complexity of the karyotype sometimes means that the nature of certain abnormalities cannot be fully determined, and critical abnormalities may be overlooked. Non‐detected aberrations by CBA may also result from the poor proliferation of leukemic cells in culture and overgrowth by normal cells. Although the routine use of disease‐specific FISH panels increases the diagnostic yield by identifying both abnormalities in non‐proliferating cells and cryptic abnormalities, our cohort contained cases where no abnormalities were detectable by combined CBA/FISH. This highlights the limitation of FISH to detect only the aberrations included in the diagnostic panels.
OGM identified abnormalities in all cases with normal CBA and FISH, thus increasing the overall detection rate. Disease‐defining abnormalities were detected in 32 ALL patients compared to only 23 by our standard testing pathway. Interestingly, based on OGM analysis 3 patients could be reclassified, the first (case 13) as a B‐ALL with TCF3::PBX1 and the other 2 (case 14 and 27) as BCR::ABL1 like B‐ALL instead of B‐ALL, NOS.
At present, the WHO discriminates no subgroups among T‐ALL/‐LBL patients. Although currently not needed for prognostic purposes, the detection of SVs and CNAs by OGM might define personalized treatment options in the future.
Secondly, OGM allowed the correction of karyotypes and chromosomal breakpoints. OGM was not only able to identify balanced and non‐balanced rearrangements but, as it offers more precise mapping of breakpoints, it allowed improved identification of both known and novel/rare abnormalities of clinical significance. In T‐ALL, OGM abrogated the need for (elaborate) cascade FISH testing in cases with T‐cell receptor rearrangements to identify the fusion partner, resulting in an important reduction of the TAT.
However, OGM also has limitations. In some cases, it was unable to identify abnormalities present only in a subset of cells by CBA. While CBA provides information at the single‐cell level and can therefore inform on clonal architecture, OGM analysis provides an overall representation of abnormalities present at the population level. Although the resulting discordance can be explained by the sensitivity of molecular techniques to identify low‐level abnormalities, it also reflects the fact that CBA detects abnormalities present in proliferating cells while OGM demonstrates abnormalities present in non‐proliferating cells. The incidence of aberrations and estimation of clone sizes, determined by CBA, is further influenced by the selection of metaphases for analysis. OGM, unlike some other molecular‐based copy number technologies, does not include an amplification step and thus provides a more accurate estimation of aberration incidence. FISH analysis performed on interphase nuclei of cultured cells also provides a good estimation of clone size and demonstrated that OGM could reliably detect abnormalities present in at least 15% of cells.
Although in this cohort, the non‐detection of subclones was of no clinical relevance, there is a risk of missing clinically relevant SVs present only in a subset of the cells. Furthermore, for pathologies where karyotype complexity has prognostic significance, for example MDS, the emergence of a subclone with additional abnormalities could impact risk stratification and outcome. Nevertheless, the sensitivity of cytogenetics to detect small clones can also be questioned since only 20 metaphases are analyzed.
A second limitation of OGM is the non‐detection of chromosomal abnormalities due to the location of the breakpoints. OGM can detect all balanced rearrangements except those that occur within highly repetitive regions of the genome such as centromeres and the short arms of the acrocentric chromosomes. However, these loci generally contain no actionable genes concerning ALL and are not considered driver abnormalities. Still, an important consideration is that also iso−/isodicentric chromosomes could be misinterpreted, whilst Robertsonian translocations go completely undetected by OGM. This includes rob(15;21), an important predisposing factor to ALL with iAMP21. 27 , 28
Interestingly, the OGM software failed to detect a t(X;14), an abnormality associated with intermediate to poor prognosis, 11 , 29 , 30 , 31 , 32 , 33 , 34 although it was clearly visible during manual inspection. The non‐detection of abnormalities involving the PAR1 region has previously been reported. 16 , 18 Following this finding the OGM analytical software has been updated and the new version now calls this IGH::CRLF2 translocation. Nevertheless, the region is currently always manually inspected in our routine setting. Of note, in contrast to the studies by Lestringant et al., and Lühmann et al., OGM was able to detect a P2RY8::CRLF2 rearrangement in one case (confirmed by FISH) outwith this study. 16 , 18 Moreover, Lestringant et al. also reported cross‐contamination between samples, a problem we did not encounter. 16
Besides potentially missing important alterations, OGM sometimes also provides false‐positive results. We encountered a DUX4::FRG2B rearrangement that could not be confirmed by FISH nor by RNA sequencing. This error is probably due to a similar labeling pattern of both regions on chromosomes 4 and 10. DUX4 rearrangements are recurrent genetic alterations associated with favorable outcomes in B‐ALL. 8 To avoid prognostic inaccuracies confirmation of the rearrangement with a second technique is therefore strongly advised.
Importantly, this study has highlighted the problem of ploidy assessment in B‐ALL, a major drawback in a pathology that includes ploidy as a key risk stratification subgroup. This is a well‐known limitation of molecular‐based karyotype technologies that do not include any allele‐specific information. Low hypodiploid clones frequently co‐exist with a duplicated near triploid clone. As OGM presents an average copy number this may result in a copy number gain of 3, as was seen in cases 12 and 16. Manual zygosity plotting of the variants detected by OGM can help resolve ploidy problems but is less reliable in cases with low blast counts. We anticipate improvements of the LOH calling by the De Novo Assembly pipeline. For example, calling missing or additional labels due to single nucleotide polymorphisms in the 6‐mer recognition motif, as recently suggested by Neveling et al., will also improve baseline corrections. 17
Since OGM is currently unable to entirely replace CBA, we combine OGM with CBA in our diagnostic workup of ALL. We also continue to perform BCR::ABL1 FISH to avoid delays in TKI‐based therapy. This new approach is less labor‐intensive (avoiding cascade FISH panel testing and MLPA or array‐based techniques) and can be done at roughly half the cost of the combination of traditional methods (FISH, MLPA and PCR‐based methods). Since implementing OGM in the routine diagnostic setting, a further 59 ALL cases have been analyzed, and all were successfully resolved using OGM.
In summary, OGM addresses some of the limitations associated with conventional cytogenomic testing, simplifying the workflow and avoiding the need for complementary FISH analysis to identify partner genes. This approach reduces the overall number of tests required as well as TATs and labor time. Results are comparable to CBA with improved diagnostic yield. The simplified flow allows detection of most major genomic risk markers in one test and means that the clinician receives one comprehensive integrated report rather than multiple individual reports. However, OGM still needs to be complemented with CBA to detect ploidy changes and the presence of subclones, and with IG/TR clonality assays for future disease monitoring.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
Supporting information
Figure S1. Illustration of the filter settings for structural variant calling in case 19.
Figure S2. Complex translocation involving chromosome 11, 12 and 21 resolved by OGM (case 3).
Figure S3. Clone size of low level chromosomal abnormalities.
Table S1. Standard testing pathway in B‐ and T‐ALL at diagnosis.
Table S2. Regions with known submicroscopic copy number changes in ALL.
Table S3. Number of structural variants retained with each filter step for 41 ALL cases.
Table S4. Optical genome mapping quality parameters of 41 ALL cases.
Table S5. The full results for all techniques including the standard genetic testing pathway and the optical genome mapping.
Table S6. Comparison of OGM and MLPA.
ACKNOWLEDGMENTS
The authors would like to thank all laboratory technicians that performed experiments in the context of the validation of the Bionano Saphyr Optical Genome Mapping device, with specific thanks to Chelsea Hayen and Geneviève Michils for technical and intellectual input.
Rack K, De Bie J, Ameye G, et al. Optimizing the diagnostic workflow for acute lymphoblastic leukemia by optical genome mapping. Am J Hematol. 2022;97(5):548-561. doi: 10.1002/ajh.26487
Katrina Rack and Jolien De Bie are co‐first authors. Lucienne Michaux and Barbara Dewaele are co‐senior authors.
Funding information This project was supported by an Impulse budget from the University Hospitals Leuven and funded by KU Leuven (C14/18/104, Jan Cools, Kim De Keersmaecker, Johan Maertens, Heidi Segers).
DATA AVAILABILITY STATEMENT
Original data and protocols are available after specific request.
REFERENCES
- 1. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541‐1552. [DOI] [PubMed] [Google Scholar]
- 2. Moorman AV, Chilton L, Wilkinson J, Ensor HM, Bown N, Proctor SJ. A population‐based cytogenetic study of adults with acute lymphoblastic leukemia. Blood. 2010;115(2):206‐214. [DOI] [PubMed] [Google Scholar]
- 3. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395(10230):1146‐1162. [DOI] [PubMed] [Google Scholar]
- 4. Cancer Research UK . https://www.cancerresearchuk.org/ (accessed November 27, 2021).
- 5. Borowitz MJ, Chan JKC, Downing JR, Le Beau MM, Arber DA. Precursor lymphoid neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised. 4th ed. IARC; 2017:200‐209. [Google Scholar]
- 6. Rack KA, van den Berg E, Haferlach C, et al. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia. 2019;33(8):1851‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weise A, Liehr T. Cytogenetics. In: Liehr T, ed. Cytogenomics. Academic Press; 2021:25‐34. [Google Scholar]
- 8. Li J, Dai Y, Wu L, et al. Emerging molecular subtypes and therapeutic targets in B‐cell precursor acute lymphoblastic leukemia. Front Med. 2021;15(3):347‐371. [DOI] [PubMed] [Google Scholar]
- 9. Brown LM, Lonsdale A, Zhu A, et al. The application of RNA sequencing for the diagnosis and genomic classification of pediatric acute lymphoblastic leukemia. Blood Adv. 2020;4(5):930‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reshmi SC, Harvey RC, Roberts KG, et al. Targetable kinase gene fusions in high‐risk B‐ALL: a study from the Children's oncology group. Blood. 2017;129(25):3352‐3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105(11):2524‐2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwab CJ, Chilton L, Morrison H, et al. Genes commonly deleted in childhood B‐cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica. 2013;98(7):1081‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B‐cell precursor acute lymphoblastic leukemia. Haematologica. 2016;101(4):407‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harrison CJ, Haas O, Harbott J, et al. Detection of prognostically relevant genetic abnormalities in childhood B‐cell precursor acute lymphoblastic leukaemia: recommendations from the biology and diagnosis Committee of the International Berlin‐Frankfürt‐Münster study group. Br J Haematol. 2010;151(2):132‐142. [DOI] [PubMed] [Google Scholar]
- 15. Dixon JR, Xu J, Dileep V, et al. Integrative detection and analysis of structural variation in cancer genomes. Nat Genet. 2018;50(10):1388‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lestringant V, Duployez N, Penther D, et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosomes Cancer. 2021;60(10):657‐667. [DOI] [PubMed] [Google Scholar]
- 17. Neveling K, Mantere T, Vermeulen S, et al. Next‐generation cytogenetics: comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am J Hum Genet. 2021;108(8):1423‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lühmann JL, Stelter M, Wolter M, et al. The clinical utility of optical genome mapping for the assessment of genomic aberrations in acute lymphoblastic leukemia. Cancers (Basel). 2021;13(17):4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoumans J, Suela J, Hastings R, et al. Guidelines for genomic array analysis in acquired haematological neoplastic disorders. Genes Chromosomes Cancer. 2016;55(5):480‐491. [DOI] [PubMed] [Google Scholar]
- 20. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35(9):975‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Safavi S, Paulsson K. Near‐haploid and low‐hypodiploid acute lymphoblastic leukemia: two distinct subtypes with consistently poor prognosis. Blood. 2017;129(4):420‐423. [DOI] [PubMed] [Google Scholar]
- 22. Moorman AV, Enshaei A, Schwab C, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124(9):1434‐1444. [DOI] [PubMed] [Google Scholar]
- 23. Hirabayashi S, Ohki K, Nakabayashi K, et al. ZNF384‐related fusion genes define a subgroup of childhood B‐cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica. 2017;102(1):118‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gu Z, Churchman ML, Roberts KG, et al. PAX5‐driven subtypes of B‐progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51(2):296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng Q, Jiang C, Liu H, et al. Down‐regulated FOXO1 in refractory/relapse childhood B‐cell acute lymphoblastic leukemia. Front Oncol. 2020;10:579673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Di Giacomo D, La Starza R, Gorello P, et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T‐lymphoid and myeloid immature acute leukemia. Blood. 2021;138(9):773‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Y, Schwab C, Ryan SL, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508(7494):98‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harrison CJ, Schwab C. Constitutional abnormalities of chromosome 21 predispose to iAMP21‐acute lymphoblastic leukaemia. Eur J Med Genet. 2016;59(3):162‐165. [DOI] [PubMed] [Google Scholar]
- 29. Buitenkamp TD, Pieters R, Gallimore NE, et al. Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leuk. 2012;26(10):2204‐2211. [DOI] [PubMed] [Google Scholar]
- 30. Dou H, Chen X, Huang Y, et al. Prognostic significance of P2RY8‐CRLF2 and CRLF2 overexpression may vary across risk subgroups of childhood B‐cell acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2017;56(2):135‐146. [DOI] [PubMed] [Google Scholar]
- 31. Jain N, Roberts KG, Jabbour E, et al. Ph‐like acute lymphoblastic leukemia: a high‐risk subtype in adults. Blood. 2017;129(5):572‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herold T, Schneider S, Metzeler KH, et al. Adults with Philadelphia chromosome–like acute lymphoblastic leukemia frequently have IGH‐CRLF2 and JAK2 mutations, persistence of minimal residual disease and poor prognosis. Haematologica. 2017;102(1):130‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harrison CJ, Johansson B. Acute lymphoblastic leukemia. In: Heim S, Mitelman F, eds. Cancer Cytogenetics: Chromosomal and Molecular Genetic Aberrations of Tumor Cells. Fourth ed. Wiley Blackwell; 2015:p204. [Google Scholar]
- 34. Chen IM, Harvey RC, Mullighan CG, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's oncology group study. Blood. 2012;119(15):3512‐3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Illustration of the filter settings for structural variant calling in case 19.
Figure S2. Complex translocation involving chromosome 11, 12 and 21 resolved by OGM (case 3).
Figure S3. Clone size of low level chromosomal abnormalities.
Table S1. Standard testing pathway in B‐ and T‐ALL at diagnosis.
Table S2. Regions with known submicroscopic copy number changes in ALL.
Table S3. Number of structural variants retained with each filter step for 41 ALL cases.
Table S4. Optical genome mapping quality parameters of 41 ALL cases.
Table S5. The full results for all techniques including the standard genetic testing pathway and the optical genome mapping.
Table S6. Comparison of OGM and MLPA.
Data Availability Statement
Original data and protocols are available after specific request.