

Abstract
Primary ciliary dyskinesia (PCD) can be defined as a multiorgan ciliopathy with a dominant element of chronic airway disease affecting the nose, sinuses, middle ear, and in particular, the lower airways. Although most patients with PCD are diagnosed during preschool years, it is obvious that the chronic lung disease starts its course already from birth. The many faces of the clinical picture change, as does lung function, structural lung damage, the burden of infection, and of treatment throughout life. A markedly severe neutrophil inflammation in the respiratory tract seems pervasive and is only to a minimal extent ameliorated by a treatment strategy, which is predominantly aimed at bacterial infections. An ever‐increasing understanding of the different aspects, their interrelationships, and possible different age courses conditioned by the underlying genotype is the focus of much attention. The future is likely to offer personalized medicine in the form of mRNA therapy, but to that end, it is of utmost importance that all patients with PCD be carefully characterized and given a genetic diagnosis. In this narrative review, we have concentrated on lower airways and summarized the current understanding of the chronic airway disease in this motile ciliopathy. In addition, we highlight the challenges, gaps, and opportunities in PCD lung disease research.
Keywords: airway disease, chronic, ciliary, dyskinesia, Primary
1. PRIMARY CILIARY DYSKINESIA—A CHRONIC RESPIRATORY MOTILE CILIOPATHY
Primary ciliary dyskinesia (PCD) can be defined as a multiorgan ciliopathy with a rather dominant element of chronic respiratory or airway disease affecting the nose, sinuses, middle ear, and pulmonary airways (Wallmeier et al., 2020). Other organs typically affected by dysfunctional cilia are the general random lateralization of the thoracic and abdominal organs leading to situs inversus or ambiguous position of the heart and possible congenital cardiac defects (Shapiro et al., 2014), the reproductive functions, and in rarer cases, the brain in the form of hydrocephalus (Wallmeier et al., 2020).
Furthermore, PCD is both genetically and clinically complex but also phenotypically very heterogeneous when it comes to severity despite everyone sharing the same pathophysiology of abnormal motile ciliary structure and function causing impaired mucociliary clearance, which, in the respiratory system, results in a vicious cycle of recurrent but probably rather chronic inflammation and suppurative sinopulmonary infections and bronchiectasis (Figure 1) (Leigh et al., 2019; Wallmeier et al., 2020).
FIGURE 1.
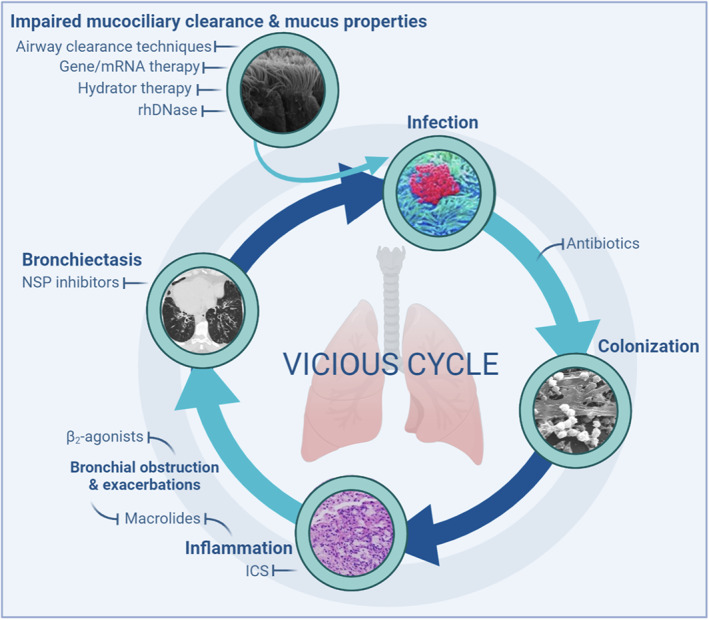
The vicious cycle of primary ciliary dyskinesia lung disease including current treatments with and without evidence and future possibilities for treatments. Abnormal ciliary function impairs mucociliary clearance and constitute along with abnormal mucus properties the initial link in the cycle predisposing individuals to microbial infections. Airway clearance techniques, hydrator therapies, and rhDNase may potentially increase the clearance, while gene/mRNA therapy could potentially restore ciliary function. Antibiotics are the primary treatment to prevent persistent colonization, while macrolides and inhaled corticosteroids (ICS) may reduce inflammation and and exacerbations, and β2‐agonists ameliorate bronchial obstruction. Neutrophil serine protease (NSP) inhibitors are potential treatments for reduction of inflammation and exacerbations in patients with bronchiectasis
PCD probably occurs in all ethnic groups around the globe but is best described in the western parts of the world, mainly due to the required significantly resource‐intensive diagnostics (Lucas et al., 2017; Shapiro et al., 2016). Traditionally, global prevalence is stated to be probably at least 1:10,000 (Wallmeier et al., 2020), but a new publication describes a general incidence of 1 out of 7.7554 people with surprisingly frequent incidence in people of African descent (Hannah et al., 2022). Such prevalence would mean that there are approximately 45,000 people with PCD among EU citizens alone.
The primary goal of this narrative review is to highlight significant features of chronic lower respiratory disease in patients with PCD. This will include clinical, pathophysiological, and histopathological consequences of dysfunctional cilia in addition to airway microbiology, evolution of pulmonary function, PCD lung structural damage, and treatment aspects. Further, aspects of health care provision, research opportunities and future perspectives, and way forward will be covered, while aspects such as diagnostics, upper airway disease, and disease in other organs are not further covered.
2. CLINICAL ASPECTS OF CHRONIC AIRWAY DISEASE IN PRIMARY CILIARY DYSKINESIA
Symptom pattern recognition is key when faced with a term infant with respiratory distress, a child with chronic wet cough or an adult with bronchiectasis. The characteristic clinical features of PCD may vary depending on the patient's age (Table 1), but common to all are a chronic persistent wet cough that began in infancy or early childhood, repeated episodes of bronchitis or pneumonia, a chronic persistently blocked or runny nose from infancy, or early childhood and chronic sinusitis(Leigh et al., 2016). The eventual development of bronchiectasis is almost universal and may be severe enough to cause chronic respiratory failure with the need for lung transplantation (Hayes, Reynolds, & Tumin, 2016). Severity of lung disease is primarily related to genotype (Davis et al., 2019); other factors associated with poorer lung function include low BMI in childhood (Goutaki et al., 2017), mannose binding lectin deficiency (Videbaek et al., 2019), chronic infection with Pseudomonas aeruginosa (Shah et al., 2016), being an adult female (Frija‐Masson et al., 2017), and late diagnosis (Shah et al., 2016), a feature of PCD and in part a consequence of low health care expenditure on diagnostic facilities (Kuehni et al., 2010).
TABLE 1.
Features of airway disease in different age groups of patients with primary cilia dyskinesia (PCD)
| Neonate | Preschool child | Older child/adolescent | Adult | |
|---|---|---|---|---|
| Respiratory distress | + | |||
| Atelectasis | + | ++ | ++ | ++ |
| Daily wet cough | +/− | +++ | +++ | +++ |
| Blocked, runny nose | +/− | +++ | +++ | +++ |
| Nasal polyps | − | − | + | ++ |
| Sinusitis | − | +/− | ++ | +++ |
| Chronic otitis media with effusion | +/− | +++ | ++ | + |
| Pneumonia | + | ++ | + | + |
| Bronchiectasis | − | + | ++ | +++ |
| Empyema | − | − | + | + |
| Hemoptysis | − | − | + | + |
| Lung abscess | − | − | − | + |
2.1. Clinical features in the term neonate
Respiratory distress occurs in most term neonates with PCD (Goutaki et al., 2020). The onset is typically around 12 hr of age, later than the most common cause of respiratory distress in term neonates, “wet lung” or transient tachypnoea of the newborn (TTN). The neonate with PCD, compared with term infants with respiratory distress due to other causes, is more likely to need oxygen supplementation exceeding 48 hr, noninvasive respiratory support in the form of CPAP, to be treated for pneumonia and develop lobar atelectasis visible on chest X‐ray that can be slow to resolve (Mullowney et al., 2014). The presence of situs inversus is a further clue to the diagnosis, but this may not be sufficient to alert the clinician with little experience or knowledge of PCD. Anecdotally, TTN is the most usual diagnosis given to the neonate with PCD, who is discharged often without follow‐up. Once home, parents may voice their concerns about persistent rapid breathing, a continuously rattly chest, and a blocked or runny nose they have to drip with saline, to the community health services and be falsely reassured that their child is healthy. A daily wet cough is not always a prominent feature during early life.
2.2. Clinical features in the infant and young child
Respiratory infections are common in normal healthy children and children with PCD, but it is the persistence of a daily wet cough and nasal congestion throughout the year that typifies the child with PCD (Goutaki et al., 2016). The cough can be spontaneous, provoked by physical activity, laughter or crying but unlike asthma, and tends not to occur at night unless the child has an intercurrent infection. Audible wheeze may be heard and the administration of inhaled steroids for suspected asthma that has no impact on symptoms is common, while antibiotic treatment may improve but not abolish the wet cough. Sometimes, particularly if the child is the parents' first, they may never notice or attach any significance to the cough because it has always been there. Persistent nasal stuffiness and discharge, which can be anything from watery and clear to thick and yellow, is almost universal. Hearing loss due to chronic middle ear effusion is common, occurring in up to 80% (Goutaki et al., 2016) and may prompt referral to an ear nose and throat specialist for grommet insertion before a diagnosis of PCD has been considered, thus risking persistent aural discharge and permanent perforation. Recurrent ear infections do occur but are a less sensitive and specific feature of PCD (Goutaki et al., 2016). Physical examination including auscultation of the chest may be unremarkable in a baseline state of health or rhonci, wheeze, and crackles may be heard. It is useful to ask the child to cough or to perform a forced expiratory manoeuver or “huff” while palpating the chest with the palm of the hand to detect the presence of secretions. Lung Clearance Index (LCI) derived from Multiple Breath Washout and spirometry, if the child is able to perform the latter, may be normal or show an obstructive pattern (Kinghorn et al., 2020). Atelectasis may be visible on chest X‐ray and bronchiectasis with a predilection for the lower lobes may be visible on CT scan in children as young as 3 years (Brown, Pittman, Leigh, Fordham, & Davis, 2008), although CT scanning at this age is not routine. Haemophilus influenzae, Moraxella catarrhalis, Staphylococcus aureus, and Streptococcus pneumoniae are the most common airway bacteria isolated but infection with P. aeruginosa (PA) does occur in this age group and should always be cultured for. Laryngeal aspirates for culture are preferable to cough swabs.
2.3. Clinical features in the older child and adolescent
The daily wet cough has by now usually become productive, enabling color and quantity of sputum to be described. Nasal stuffiness and discharge persist, while hearing loss tends to improve. Respiratory symptoms begin to have a significant impact on quality of life, particularly in relation to limitation of vigorous physical activity and being unable to keep up with peers. The need to expectorate sputum can become both a nuisance by interrupting physical activity and an embarrassment, leading some teenagers to suppress cough and under‐report their symptoms (Behan, Rubbo, Lucas, & Dunn Galvin, 2017). Acute respiratory infections remain prevalent. On average, a child already diagnosed with PCD will be treated with 2.6 courses of antibiotics per year (Rubbo et al., 2020). Loss of lung function, already moderately severe by age 6 years (Marthin, Petersen, Skovgaard, & Nielsen, 2010), continues at least until the age of 15 years (Rubbo et al., 2020). A quarter of children admitted to hospital for treatment of an infective exacerbation fail to regain their baseline lung function 3 months later (Sunther, Bush, Hogg, McCann, & Carr, 2016). The likelihood of airway infection with PA increases with age, both in the sinuses and lower airways, particularly in the adolescent (Alanin, Johansen, et al., 2015), but H. influenzae remains the dominant pathogen. Bronchiectasis will be present on CT scan in the majority of patients as they approach adulthood (Magnin et al., 2012).
2.4. Clinical features in the adult
Patients presenting for the first time as adults under suspicion of having PCD may have no knowledge of their neonatal history but are likely to report frequent attacks of bronchitis and sinusitis that began in childhood and admission to hospital for recurrent pneumonia. They will complain of chronic, daily productive cough; many will experience exercise limitation, and some will be too unwell to work full time (Lucas et al., 2015). Empyema complicating pneumonia, lung abscess, and hemoptysis that may be life threatening all occur with increasing prevalence with increasing age, but their description is limited to a handful of case reports. The true prevalence is unknown. Persistent rhinosinusitis, often associated with a poor sense of smell, can be particularly troublesome, heavily impacting their quality of life. About 75% of men will be infertile, depending on which gene is mutated; the majority of women can conceive naturally (Vanaken et al., 2017). Lung function is usually obstructive and may be severely reduced (Shah et al., 2016) especially in women and independently of their PA status (Frija‐Masson et al., 2017). Lung function may improve or stabilize in patients diagnosed as adults or continue to deteriorate (Marthin et al., 2010). Respiratory failure with the need for supplemental oxygen or even lung transplantation may ensue. The most commonly isolated organism is PA, chronic infection occurring in nearly 50%. CT evidence of bronchiectasis will be found in 95% (Figure 2) (Shah et al., 2016).
FIGURE 2.
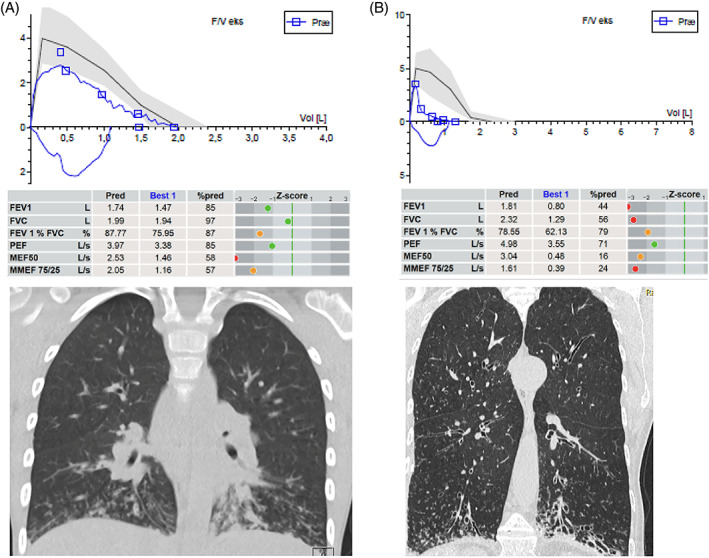
Lung function and structural damage in two patients with primary cilia dyskinesia (PCD) at opposite ends of the age spectrum. Upper parts show flow‐volume curves from forced expiratory maneuvers with airway flow on the Y‐axis in liters per seconds (L/s) and volume on x‐axis in liters (L). Blue loops are the patients' actual performances, while the black line is the predicted/expected ideal flow‐volume loop (gray zone the reference interval). Tables in the middle displays predicted and best performance values of forced expired volume in first second (FEV1), forced expired vital capacity (FVC), FEV1/FVC%, peak expired flow (PEF), maximal (mid‐)expiratory flow at 50% of expired FVC, and maximal mid‐expiratory flow between 25 and 75% of expired FVC (MMEF 75/25). Z‐scores are displayed as green dots when values are within normal range etc. HRCT scans in the bottom show mucus plugging and “tree in bud” appearance to the left, while the right image demonstrates bilateral severe and multiple bronchiectasis at multiple locations but as typically seen in PCD preferentially at the bottom of the lungs. Notice also emphysematic appearance. A is a 9‐year‐old boy recently diagnosed demonstrating He demonstrates extreme difficulty in performing a smooth flow‐volume curve—note the jagged appearance. This is due to his mucus‐filled airways appearing on HRCT as white lines with budding—tree in bud. Later on, his lung function and HRCT improved significantly by treatment. B panel is a 72‐year‐old female with PCD just keeping an FEV1 above 40% of predicted
3. PATHOPHYSIOLOGICAL, STRUCTURAL, AND HISTOPATHOLOGICAL CONSEQUENCES OF PRIMARY CILIARY DYSKINESIA IN THE LOWER AIRWAYS
In disease conditions, histological changes in the four layers—mucosa, submucosa, musculo‐cartilaginous layer, and adventitia—of the airway wall vary according to the underlying disease, and modifications in all layers act concurrently to accentuate the impact of the airway caliber on lung function (Fayon & Beaufils, 2021).
In PCD, in contrast to CF and asthma, the pathophysiological and histological changes and presence in different compartments of chronic airway inflammation have been far less investigated, restricted to relatively small numbers of children (Bush et al., 2006; Ratjen et al., 2016; Zihlif, Paraskakis, Lex, Van de Pohl, & Bush, 2005). Thus, our knowledge concerning inflammatory processes in the mucus, as well as the extent of these in the airway wall including remodeling, throughout the respiratory system is extremely limited in PCD. However a few studies are found: Children with chronic inflammatory lung disease already have increased smooth muscle mass present in the airway wall as demonstrated in an endobronchial biopsy study accomplished in children with asthma, CF, and non‐CF bronchiectasis of whom a significant number had PCD (Regamey et al., 2008).
Additionally, reticular basement membrane (RBM) thickening is a feature of PCD and was significantly related to numbers of neutrophils in lavage fluid in a comparative study of endobronchial biopsy and BAL in 24 children with CF, 11 with PCD, 15 with asthma, and in 19 control subjects (Koucký et al., 2020). In fact, in PCD and CF, RBM thickness was similar to that seen in asthma.
Assessment of induced sputum in a small cohort of 20 stable children with PCD revealed a significantly higher sputum neutrophil count, (median 70.3%; IQR, 55.3–78%), compared to healthy controls (median, 27%; IQR, 24.5–33%; p < .004) but no correlation with lung function (FEV1 percent). Due to sputum processing methodology, cytokine measures such as interleukin‐8 and other proinflammatory cytokines of interest were not performed, and contemporaneous sputum cultures were all negative (Zihlif et al., 2005).
A comprehensive comparison of biophysical properties of sputum in age, and pulmonary function–matched PCD and CF children showed no difference between the two diseases although some evidence of more airway inflammation in PCD as manifested by IL‐8 levels (Bush et al., 2006).
Airway remodeling using both broncho alveolar lavage fluid and endobronchial biopsies was investigated in a large group of children with CF, disease controls including a small number of children with PCD and children with chronic respiratory symptoms (CRSs) investigated for recurrent infection and/or cough, and control children with no lower airway symptoms. Matrix constituents, glycosaminoglycan, elastin, and collagen concentrations were similar in PCD and CF and in both significantly higher than in the CRS and control groups. RBM thickness did not differ between PCD (4.6 mm), CF (5.9 mm), and CRS (4.6 mm) and was only significantly thicker in CF patients compared to controls (4.0 mm, p < .01) (Hilliard et al., 2007).
In a study investigating whether patients with PCD and CF differed in inflammatory response in the airways during pulmonary exacerbation, as assessed in sputum samples, a higher neutrophil burden was found in PCD compared with CF patients although bacterial load was higher in patients with CF. Neutrophil elastase (NE) activity was similar in PCD and CF at the time of exacerbation while a reduction in NE activity was achieved by antibiotic therapy in PCD but not in CF patients (Ratjen et al., 2016).
4. AIRWAY MICROBIOLOGY OF PATIENTS WITH PRIMARY CILIARY DYSKINESIA
Haemophilus influenzae is the most frequently isolated pathogen in children with PCD where other common isolated pathogens also include, Streptococcus pneumoniae, Moraxella catarrhalis, and Staphylococcus aureus (Wijers, Chmiel, & Gaston, 2017). When reaching adulthood, these pathogens tend to decrease in prevalence and P. aeruginosa takes over and becomes the dominant airway pathogen in PCD (Shah et al., 2016; Wijers et al., 2017).
Airway PA infection is common in people with PCD, and reported prevalences range from 27% to 44% (Alanin, Johansen, et al., 2015; Cohen‐Cymberknoh et al., 2017; Frija‐Masson et al., 2017; Maglione et al., 2014; Noone et al., 2004; Piatti et al., 2020; Pifferi et al., 2012; Shah et al., 2016).
Lung PA infection in PCD has been associated with worse CT scan scores (Cohen‐Cymberknoh et al., 2017; Piatti et al., 2020). PA impact on lung function has been reported with mixed findings since association between PA infection and poorer lung function has been reported in some studies (Boon et al., 2014; Cohen‐Cymberknoh et al., 2017; Frija‐Masson et al., 2017), whereas other authors report no correlation between PA infection and FEV1 (Maglione et al., 2014; Pifferi et al., 2012) as well as no significant longitudinal lung function decline in adult PA‐infected patients compared to noninfected (Shah et al., 2016).
In studies of PA in the paranasal sinuses, the same PA clones have been found to exist in the sinuses and the lower airways (Arndal et al., 2020) suggesting that the sinuses can act as a bacterial reservoir for PA lung infection (Alanin, Johansen, et al., 2015) as previously shown for CF (Aanæs, 2013; Hansen et al., 2012). Hence, eradication strategy for chronic lung infection in PCD should also include assessment of the paranasal sinuses with a view to eradicate bacterial sinus infection.
The concern for PA in PCD health care is reflected by the PA eradication strategy across European PCD centers, where the vast majority of pediatric centers choose to treat PA in children irrespective of lung function (Crowley, Holgersen, & Nielsen, 2019) and is also reflected in the recently published international consensus report within BEAT‐PCD where 20 statements were produced with the goal being to increase attention and improve infection prevention and control in people with PCD (Marthin et al., 2021). In this document, statements directly addressed PA infection in terms of early identification of isolates and definition of chronicity, early treatment, as well as segregational aspects in hospital settings to avoid patient‐to‐patient transmission (Marthin et al., 2021). The adoption of a modified version (Alanin, Nielsen, et al., 2015) of the original Leeds criteria established for CF (Lee, Brownlee, Conway, Denton, & Littlewood, 2003) was proposed to define chronicity of PA in PCD and the culture airway secretion samples from patients at least 4 times annually (Marthin et al., 2021) was recommended in attempt to better uncover and prevent chronicity of PA in PCD.
4.1. Nontuberculous mycobacteria
NTM in PCD is rarely reported and prevalence in one study reported as low as 3.3% (Shah et al., 2016). For the treatment of NTM in PCD, the BEAT‐PCD IP&C Consensus statement has suggested to adopt the criteria from the ATS/ERS/ESCMID/IDSA guideline (Daley et al., 2020; Marthin et al., 2021).
4.2. Achromobacter species
Knowledge of prevalence and impact on Achromobacter species (AS) infection is sparse (Marthin et al., 2021). However, AS infection may be underdiagnosed in PCD and may need increased awareness in PCD health care providers. In a recent single‐center retrospective study including 136 patients (up to 18 years of observation) with PCD, persistent Achromobacter infections with a median length of 6.6 years were found in 38% of patients and led to a significant number of antibiotic treatments (Holgersen, Marthin, Johansen, & Nielsen, 2021).
4.3. Fungal infections and allergic bronchopulmonary aspergillosis
Overall, fungal infections in PCD are not well‐described. Likewise, ABPA in PCD is limited to a few case reports (Sehgal, Dhooria, Bal, & Agarwal, 2015; Shah et al., 2016; Sharma, Sharma, Bondi, & Sharma, 2005) but is described with severe clinical and radiographic manifestations and need for long‐term treatment, thus underlining the need for increased awareness of ABPA in PCD.
4.4. Burkholderia cepacia
Concern for Burkholderia cepacia (BC) infection in PCD is primarily driven by a concern derived from CF experience, as BC has only been rarely described in PCD, and no studies of PCD and B. cepacia infection were identified in our literature search. In the author's own experience, one case of BC infection in an adolescent patient with PCD led to a temporary but severe decline in lung function, returning however to base line after long‐term antibiotic treatment (personal communication, case not published).
5. RADIOLOGICAL AND STRUCTURAL DAMAGE THROUGH ALL AGE GROUPS
Most of the information about PCD structural lung damage is derived from smaller predominantly retrospective cross‐sectional studies using findings from high‐resolution chest computed tomography (HRCT). The characteristic HRCT changes typically encompass atelectasis, bronchiectasis, bronchial wall thickening, and mucous plugging (Tadd et al., 2019). Most scans are obtained for clinical indications with standardization of neither imaging technique nor PCD‐specific scoring systems, and consequently, due to the lack of the latter, modified versions of different CF related scoring scales are employed (Gahleitner et al., 2021). However, it has been clearly shown that assessment of PCD structural changes should not rely on CF‐derived scoring systems since structural changes identified on CT scans in PCD are not identical to those previously described in patients with CF (Tadd et al., 2019). As an example, a higher frequency of extensive tree‐in‐bud pattern of mucus plugging, bronchocoeles or nodules, thickening of interlobar and intralobular septa, and atelectasis or collapse of the whole lobes were commonly seen in PCD but not in patients with CF.
Taking these relative weaknesses and premises into account, for patients with PCD, a correlation has been found between age and severity of the total score thus demonstrating clear progression of PCD structural lung changes over time (Boon et al., 2015; Magnin et al., 2012; Shah et al., 2016). In fact, in a small case series, it has been shown that children with PCD as young as 33 months of age have demonstrable structural damage in the form of bronchiectasis (Brown et al., 2008).
Several groups have assessed the association of PCD structural lung disease on HRCT with lung function parameters (spirometry, LCI) in children with PCD (Davis et al., 2015; Santamaria et al., 2008) (Boon et al., 2015; Maglione, Montella, & Santamaria, 2012; Magnin et al., 2012; Shah et al., 2016). In a longitudinal prospective study conducted by Magnin et al. (2012) in which 20 children with a definitive diagnosis of PCD, aged less than 15 years at entry, and with at least 8 years of follow‐up had at least two pairs of concurrent CT and lung function tests available. CT scores increased with age and were negatively correlated to PaO2, FVC, FEV1, and FEF25–75% longitudinal changes. Bronchiectasis was present in 70% of the first CTs but appeared over time in all patients. Other studies have found similar associations and importantly demonstrating that bronchiectasis is already seen in 33% of preschool children, 56–71% of older children, and 95%) of adults (Kennedy et al., 2007; Maglione et al., 2012; Noone et al., 2004; Santamaria et al., 2008).
Despite the aforementioned lack of validation of scoring systems for PCD, a few studies have compared CT scores in patients with PCD to patients with CF and showed no differences in the global Brody score (Gahleitner et al., 2021). One study even found that bronchial wall thickening, bronchiectasis, mucus plugging, atelectasis, and air trapping were even more common in patients with PCD. A significantly higher subscore for severity of collapse or consolidation in PCD compared to CF was reported by Maglione, Bush, et al. (2012). Further, lower and middle lobes were more frequently affected in PCD compared to the upper lobes as typically seen in CF (Kennedy et al., 2007; Shah et al., 2016).
Very interestingly, an observational study examined the association between PCD genotype (including hallmark Transmission Electron Microscopy ‐ TEM) and PCD structural lung disease on HRCT in children 5–11 years of age (Davis et al., 2015) and found that those with mutations in CCDC39 and CCDC40 (including all IDA/MTD defects) had more lobes with bronchiectasis and alveolar consolidation than those in the ODA and ODA/IDA defect groups.
Future studies of PCD structural lung disease should, as far as possible, ensure a well‐validated scoring system that reflects the domains of structural damage that predominate in PCD (Tadd et al., 2019), and which has similarly been developed for CF (Rosenow et al., 2015). In addition to this, given the rarity of PCD and the often quite extensive structural changes, it is important to ensure an impartial and blinded centralized service for the often resource‐intensive analyses spent on scoring scans from multicenter studies and where also the interobserver consistency is guaranteed. At the same time, consideration should be given to introducing a standard operating procedure for the actual acquisition of the scans during both inspiration and expiration, so‐called volume or spirometry‐controlled CT (Kongstad et al., 2013) to achieve the best possible chance of assessing interindividual and intraindividual longitudinal differences.
6. PULMONARY FUNCTION—EVOLUTION FROM INFANCY TO OLDER AGE GROUPS
One of the hallmarks of PCD is affected lung function, which in general, is found to be reduced when compared to age‐related healthy peers (Halbeisen et al., 2018). In most patients, signs of reduced lung function are present during childhood, and hence the progression of lung function is closely monitored by structural and functional assessments. Due to the advancements in diagnostic methods, primarily by genetic analysis, patients are now diagnosed younger than before, and different methods are used to monitor the course of lung function according to age.
6.1. Methods for assessment of lung function
Spirometry is the gold‐standard for the assessment of lung function and widely used, while Multiple Breath Washout (MBW) is considered a more sensitive method with minimal cooperation requirements and hence is feasible to perform in all age groups, including infants.
6.1.1. Multiple breath washout
Multiple Breath Washout is a gas‐washout technique describing gas‐clearance curves and ventilation distribution during tidal breathing (Robinson, Goldman, & Gustafsson, 2009). The parameter most often reported is the Lung Clearance Index (LCI), typically reported as LCI2.5 describing the ventilation distribution at 2.5% (1/40) of the initial concentration of the inert tracer gas being used. The method has long been known, but the application of it has only recently gained ground and most studies including patients with PCD are recent. The evidence for its clinical utility remains preliminary, with the technique mostly being performed in tertiary centers.
6.1.2. Spirometry
Spirometry is the main method used to describe lung function in both clinical and research settings. In general, the method is used from school‐age and upwards. The main parameters reported are the forced expiratory volume during the first second (FEV1) and the functional vital capacity (FVC), both reported either as percent predicted or as z‐scores (Halbeisen et al., 2019).
6.2. Lung function assessed by Multiple Breath Washout
LCI has been found to be correlated with FEV1 in patients with PCD (Boon et al., 2015; Nyilas et al., 2015), although contrary results exist (Green et al., 2012; Irving et al., 2013). However, several studies have found MBW to be a more sensitive method than spirometry in detecting early impairments of lung function. To our knowledge, all studies report higher proportions of patients with abnormal LCI compared to FEV1, with reports ranging from abnormal LCI in 59–85% of patients as opposed to abnormal FEV1 in 24–52% of patients in the same cohort (Boon et al., 2015; Green et al., 2012; Kinghorn et al., 2020; Nyilas et al., 2018; Singer et al., 2021). A probable explanation for some of the difference in reported proportions may be due to different age limits of the cohorts studied. It is worth noting that most of the studies were performed in children and young adults and none included patients >41 years of age.
Only a few studies have examined the evolution of LCI over time. Recently, Singer et al found LCI and FEV1 to worsen during pulmonary exacerbations and predicted an 87% increased risk of exacerbation when LCI changed from normal to abnormal (Singer et al., 2021). Further, they found differences in recovery, where improvements were seen in FEV1 4 months later, while this was not the case for LCI. They found no trend in LCI or FEV1 over an average of 1 year's observation. Similar results have been reported in a small study (n = 12) by Irving et al who compared baseline LCI values to values obtained at a five‐year follow‐up and found an overall nonsignificant change in LCI (Irving et al., 2017). To the contrary, Kobbernagel et al found a minimal, but significant worsening of LCI during a one‐year observation period, while FEV1 remained unchanged (Kobbernagel et al., 2019). With only a modest number of studies assessing the course of LCI over time, LCI trends in PCD are largely unknown, and more research is warranted to assess the natural evolution.
6.3. Lung function assessed by spirometry
In a meta‐analysis by Halbeisen et al, reported FEV1 varied among included studies, possibly in part explained by differences in genetics, inclusion criteria and performance and evaluation of the lung functions measurements (Halbeisen et al., 2019). Overall, the mean FEV1 percent predicted ranged from 51–96% with a weighted mean of 75% (95% CI 69–80%). In a subgroup analysis stratifying for age, they found mean FEV1 percent predicted ranging from 44–79% (weighted mean 63%, 95% CI 57–69%) in subjects ≥18 years of age and 73–96% (weighted mean 81%, 95% CI 78–83%) in subjects <18 years of age. They were not able to perform a meta‐analysis on longitudinal trends due to differences in designs and follow‐up periods. Likewise, a large study on the international PCD (iPCD) cohort, including 991 patients of all age‐groups including 21 centers, found FEV1 to be reduced in all age groups, best in children 6–9 years of age, and with an overall FEV1 lower than the reference across all countries (Halbeisen et al., 2018).
Newer studies assessing the course of FEV1 over time have found an annual decline in FEV1 percent predicted ranging from −0.89 to −0.49 percentage points per year (Davis et al., 2018; Halbeisen et al., 2018; Magnin et al., 2012; Marthin et al., 2010; Shah et al., 2016). However, in a long‐term observational study by Marthin et al, large variations in trajectories are reported. They found that most patients (57%) remained stable while 10% improved more than 10 percentage points and 34% lost more than 10 percentage points during the median 9.5 years of observation (Marthin et al., 2010). More recently, numerous studies have attempted to associate different rates of lung function decline to the ultrastructural composition of cilia determined by the genotype present, and new genotype–phenotype relationships are starting to emerge. Examples of lung function tests in patients at opposite ends of the age spectrum can be seen in Figure 2.
6.4. Lung function in association to genotypes and ciliary ultrastructure
To our knowledge, only one study performed by Irving et al has examined MBW indices in relation to ciliary ultrastructure in PCD (Irving et al., 2018). They found LCI to be significantly worse in patients with microtubular defects (MTDs), when compared to subjects with normal ultrastructure (when assessed by transmission electron microscopy), subjects with outer dynein arm defects (ODA), and subjects with combined inner and outer dynein arm defects (ODA/IDA). These results are consistent with other studies examining lung function by spirometry in relation to ultrastructure. Results from the iPCD cohort, with data from multiple countries, found FEV1 to differ among groups with different ciliary ultrastructure, among whom subjects with MTD defects had a worse lung function (Halbeisen et al., 2018). Other approaches were included by Shoemark et al who used a topological data analysis approach clustering on FEV1 to assess lung function associations with genotype (Shoemark et al., 2021). The cluster with the most reduced FEV1 consisted primarily of subjects with mutations in N‐DRC or axonemal ruler genes (CCDC39, CCDC40, CCDC65, DRC1) while the cluster with the best FEV1 mainly consisted of subjects with DNAH11 mutations (normal ultrastructure on electron microscopy). Subjects with DNAH5 mutations, corresponding to ODA defects, were more diverse and scattered among clusters without any characteristic centering, that is, presented with highly variable FEV1.
Similar results in longitudinal trends have been found by several studies (Davis et al., 2019; Pifferi et al., 2020; Shah et al., 2016). Among these, Davis et al found a worse decline in FEV1 percent predicted among children and adolescents with MTD/IDA defects when compared to subjects with isolated ODA defects (−1.11 opposed to −0.73 percentage points per year) (Davis et al., 2019). Shah et al report similar findings in adults, with an annual decline in FEV1 percent predicted of −0.75 percentage points in subjects with MTD defects compared to −0.51 in a combined group of subjects with ODA and ODA/IDA defects and −0.13 in subjects with normal ultrastructure (as assessed by electron microscopy) (Shah et al., 2016).
Overall, these results point in a direction of a relationship between MTD, especially caused by CCDC39 and CCDC40 mutations, and both an earlier and faster decline in lung function as well as a relationship among patients with a normal (non‐diagnostic) ultrastructure, for example, caused by DNAH11 mutations, and a less affected lung function.
7. TREATMENT WITH AND WITHOUT EVIDENCE—CHALLENGES AND FUTURE POSSIBILITIES
The key to a direct specific treatment of patients with PCD lies in being able to restore ciliary activity in the airways to a level that can ensure normal or near‐normal mucociliary clearance. Until then, all types of treatments that directly or indirectly otherwise improve mucociliary clearance and reduce the secondary inflammatory destructive processes caused by the primary basic defect and secondarily of various infections will be paramount.
Due to a lack of evidence base, pharmacotherapeutic treatment varies widely between sites and countries in Europe (Strippoli et al., 2012) and is currently predominantly used for averting and managing disease complications. Guidelines are mainly based on expert opinion and extrapolation from non‐PCD bronchiectasis guidelines (Barbato et al., 2009; Shapiro et al., 2016).
In practice, this means that many infants, schoolchildren, and adults, depending on the experience of the PCD expert, are treated empirically with hydrator or airway clearance therapy in the form of inhaled hypertonic saline (HS) with some albeit weak evidence (Paff et al., 2017), inhaled recombinant human Dornase Alfa (rhDNase), varying composition of medication with documented effect registered for use in asthma or COPD in terms of bronchodilators, inhaled corticosteroids and others, and not least antibiotics for respiratory infections, which are indisputably indicated and effective but often based on different principles and standards as well as different perceptions of which bacteria are important as mentioned previously (Barbato et al., 2009; Crowley et al., 2019; Marthin et al., 2021; Shapiro et al., 2016; Strippoli et al., 2012).
Various consensus guidelines and reviews have already addressed recommendations for current treatment standards (Barbato et al., 2009; Lucas et al., 2017; Shapiro et al., 2016; Strippoli et al., 2012; Wallmeier et al., 2020). It is the authors' clear belief that all empirically applied treatment for PCD should be subjected to testing in randomized controlled trials which of course should also apply to future pharmacotherapeutic products. At the same time, we have no doubt that future treatments should, as far as possible, address both the upper and lower airways in accordance with the united airway nature of the disease.
7.1. Current treatments—Looking for higher evidence
7.2. Lower airway clearance therapy
Chest physiotherapy applying airway clearance techniques is an indispensable, intuitively understandable, and very principled part of the treatment of PCD and used in all centers (Lucas, Alanin, et al., 2017; Strippoli et al., 2012), and this despite the fact that the evidence is not impressive. Mucus retention promotes bacterial infections and inflammation, which in turn can lead to pulmonary exacerbations and bronchiectasis as highlighted earlier. Treatment options that are used to improve mucociliary clearance in PCD include the use of hydrator therapy and mucolytics.
7.3. Hypertonic saline and ENaC inhibitor
Inhaled HS improves airway clearance by hydrating the viscous airway secretions and by stimulating cough (Donaldson et al., 2006; King, Dasgupta, Tomkiewicz, & Brown, 1997; Tarran et al., 2001). A Cochrane Review concluded that frequency of pulmonary exacerbations in adolescents and adults and quality of life of adult patients with CF are improved by inhaled HS (Wark & McDonald, 2003). The evidence is quite inconsistent concerning non‐CF bronchiectasis (Anuradha, Gunathilaka, & Wickramasinghe, 2021; Donaldson et al., 2006; Kellett, Redfern, & Niven, 2005; Nicolson et al., 2012), but a well‐designed 52‐week trial on the effect of HS on exacerbation frequency in adults is underway (Bradley et al., 2019). Concerning PCD, the effect of daily inhaled HS compared to isotonic saline for 12 weeks was evaluated in a randomized double‐blind, placebo‐controlled cross‐over study in 20 adults (Paff et al., 2017). The study showed a minor increase in the primary outcome measure quality of life total score using St Georges Respiratory Questionnaire SGRQ but without clinically important difference or statistical significance. HS accomplished a clinically and statistically significant but small improvement on the QOL‐Bronchiectasis Health Perception scale. HS was safe and well tolerated (Paff et al., 2017). Of note, QOL PCD did not exist at the time of this study. Overall, the results of the study must be taken with caution. Indeed, of the 20 randomized patients only 16 were included in the per protocol study and as such an underpowered study. Thus, it seems likely that patients with PCD can benefit from HS inhalations, but larger sufficiently powered studies are urgently needed.
A quite interesting and somewhat different method of increasing mucus hydration was studied in the CLEAN‐PCD (Clearing Lungs with ENaC Inhibition in Primary Ciliary Dyskinesia) study (https://clinicaltrials.gov/ct2/show/NCT02871778, accessed on 8 Feb 2022). This was an international phase 2 cross‐over RCT investigating safety and effect on the quality of life, and on Percent Predicted Forced Expiratory Volume in 1 Second (ppFEV1) of an epithelial sodium channel (ENaC) inhibitor (VX‐371) with and without Ivacaftor, a CFTR potentiator, or HS. ENaC inhibitors are thought to improve airway hydration and mucociliary clearance by blocking the reabsorption of sodium in the airway surface liquid. The results of this study are presently being analyzed.
7.4. Recombinant human Dornase Alfa
rhDNase is an enzyme which, by nebulization, is known to reduce the abnormal viscoelastic mucus in patients with CF by breaking the long extracellular DNA molecules into minor fragments, thus improving clearance of sputum from the airways. Inhaled rhDNase is extremely effective in both improving lung function and reducing pulmonary exacerbations in CF in trials lasting from 1 month to 2 years (Yang & Montgomery, 2021), whereas, and not entirely explicably, inhalations were not effective in patients with non‐CF bronchiectasis, or even potentially harmful by increasing exacerbations and worsening lung function (O'Donnell, Barker, Ilowite, & Fick, 1998; Wills et al., 1996). A finding that in theory could be explained by DNase causing a reduction in the number of neutrophil extracellular traps beneficial in general as part of the innate immune system as previously indicated (Wallmeier et al., 2020). Although rhDNase is used to a minimal extent in centers that treat patients with PCD (Strippoli et al., 2012), this is currently not recommended in expert guidelines because of the lack of RCTs in patients with PCD. Well powered trials studying the effectiveness of rhDNase specifically in patients with PCD are required despite these discouraging results and theories on the specific mechanisms by which DNase might be detrimental.
7.5. Antibiotic treatment—In general
A large part of PCD management deals with microbial surveillance based on culture results and type of bacterial pathogen and requires regular airways secretion sampling using various methods and antibiotic treatment with oral, inhaled, and intravenous antibiotics, depending on absence or presence of clinical symptoms and their severity (Barbato et al., 2009; Lucas, Alanin, et al., 2017; Shapiro et al., 2016). Most children without chronic and simple occasional infections are treated with, for example, broad‐spectrum oral antibiotics such as amoxicillin with clavulanic acid or equivalent for 14 days to target the common respiratory pathogens (Shapiro et al., 2016).
The IP&C (Marthin et al., 2021) consensus statement advises to treat regardless of symptoms in case positive culture for PA, methicillin‐resistant S. aureus (MRSA) or BC complex appears in cultures.
Infection with PA should give rise to special caution since it may become chronic, thus risking a decline in lung function and worsening of PCD structural lung changes (Cohen‐Cymberknoh et al., 2017; Piatti et al., 2020). Unfortunately, evidence for the management of PA infection in PCD is also worryingly deficient, and consequently, treatment shows considerable variation across European centers and therefore calls for controlled clinical trials to clarify best strategy (Crowley et al., 2019). Regardless of this variability, it is important to recognize that PA simultaneously infects both upper and lower airways in patients with PCD, while treatment often needs to be targeting both sinuses and lungs (Arndal et al., 2020).
7.6. Antibiotic treatment—Maintenance therapy—Evidence based in PCD
Prophylaxis with oral antibiotics is debatable, but they are used in some centers. Prolonged macrolide use has been studied in many other chronic respiratory diseases characterized by neutrophilic inflammation. Both pulmonary exacerbations and FEV1 improved consistently in CF over 6 months treatment (Mayer‐Hamblett et al., 2018; Southern, Barker, Solis‐Moya, & Patel, 2012), and further prolonged treatment from 6 to 12 months showed similar results in patients with non‐CF bronchiectasis (Altenburg et al., 2013; Wong et al., 2012).
Macrolides, and specifically azithromycin, have in addition to their antibacterial properties, also anti‐inflammation properties and efficacy with respect to quorum sensing inhibition (Southern et al., 2012; Wilms, Touw, Heijerman, & van der Ent, 2012; Wong et al., 2012), whereby they may reduce biofilm growth, an important and problematic feature of PA infection (Kanoh & Rubin, 2010).
The BESTCILIA consortium successfully conducted a randomized, multicenter, double‐blind, placebo‐controlled phase III trial on the efficacy and safety of azithromycin maintenance therapy over 6 months in 90 patients with PCD. Patients used azithromycin 3 times a week or a placebo. Azithromycin was well tolerated and halved both the rate of exacerbations and detected pathogenic bacterial species and showed good tolerability (Kobbernagel et al., 2016; Kobbernagel et al., 2020). Thus, this is the first randomized controlled trial on pharmacotherapy to provide evidence‐based treatment for patients with PCD.
7.7. Bronchodilators (B2‐agonists) and inhaled corticosteroids (ICS)
B2‐agonists are often prescribed to patients with PCD who present with a recurrent wheeze (Dehlink et al., 2018; Strippoli et al., 2012), but they may theoretically also improve mucus clearance in patients with PCD and provide at least transient improvement in lung function (Hellinckx, Demedts, & De Boeck, 1998) when needed and in fact this is useful not only in patients with severely reduced lung function, for example, before exercise, during exacerbations, or during treatment with inhaled antibiotics that may cause chemical bronchitis (personal observation). ICS are not recommended for PCD unless concurrent asthma is diagnosed (Barbato et al., 2009). A cross‐sectional single‐center study to determine whether ICS prescriptions were targeted appropriately reported that 35% of patients (n = 99) with PCD were prescribed high‐dose (median daily dose = 500 microgram beclomethasone) ICS of whom two‐thirds received fixed‐dose combination with long‐acting beta agonists (LABA) (Dehlink et al., 2018). About one‐third of the patients had >12% reversibility of airway obstruction had FeNO typically within the normal range, while LCI was often abnormal in those on ICS. The authors concluded that ICS were not being used rationally and rightfully emphasized that ICS prescription is inappropriate in PCD unless type 2 airway inflammation is present. However, it is not unlikely that the use of ICS in patients with PCD among different centers in Europe remains at the same level as reported 10 years ago (Strippoli et al., 2012). RCTs are needed to determine whether ICS might be indicated without convincing demonstration of eosinophilic inflammation, specifically in patients with PCD.
7.8. Treatment specifically directed against neutrophilic inflammation
Patients with PCD, as previously specified, like other in patients with non‐CF bronchiectasis, are typically characterized by pulmonary neutrophilic inflammation. A treatment that directly aims at reducing this type of inflammation might also benefit patients with PCD. A study to assess the efficacy, safety, and tolerability of Brensocatib in patients with non‐CF Bronchiectasis (ASPEN, ClinicalTrials.gov Identifier: NCT04594369) is currently recruiting. Brensocatib (INS1007) is an oral reversible inhibitor of dipeptidyl peptidase 1 (DPP‐1), an enzyme responsible for the activation of neutrophil serine proteases.
7.9. Treatments in their infancy or in pipeline
Classical gene therapy, mRNA‐based therapy, and read‐through therapy.
7.10. Classical gene therapy
With an increasingly successful uncovering of genotypes causing PCD, it is not unlikely that personalized medicine in the form of targeted gene therapy has come a big step closer. So far, three studies (ex vivo human or animal cell cultures) have been published describing the partial restoration of ciliary function in ciliopathies using classical gene therapy (Chhin et al., 2009; Ostrowski et al., 2014) and one study using gene editing (Lai et al., 2016). Further review of this interesting research area is outside the scope of this publication but can be sought in a recent review (Paff, Omran, Nielsen, & Haarman, 2021).
7.11. mRNA‐based therapy
mRNA‐based therapies are exciting alternatives to gene therapies using a further downstream approach with the potential to treat a wide collection of diseases. This approach does not cause any alterations in genomic DNA and effects are reversible. mRNA therapy encoding proteins in PCD are currently being explored by ETHRIS (Planegg, Germany). mRNA therapy has hitherto not been feasible as a therapeutic agent because it activates the immune system (the mechanism of action utilized in the COVID vaccine) and in addition is highly unstable. To be functional, mRNA must enter the target cells of interest by crossing the cell membrane, which requires a unique vector for safe transportation into the cells. ETHRIS has developed two technology platforms to enable the use of mRNAs as a therapeutic agent: The Stabilized Non‐Immunogenic mRNA (SNIM® RNA) platform as well as the complementary proprietary technologies for SNIM® RNA delivery. The ETH42 program aims to provide full‐length mRNA to restore CCDC40 protein (https://ethris.com/technology/ accessed on 8 Feb 2022) in patients with PCD. Ten percent of patients with PCD have biallelic disease causing variants in CCDC40 a subgroup of patients, as previously mentioned, shown to be at risk of a more severe disease course in more domains than other groups. The first results appear promising while the program is still in a preclinical stage.
ReCode Therapeutics similarly claims to have created a platform of nonviral lipid nanoparticles (LNPs), enabling delivery of medicines of different types (mRNA, tRNA, CRISPR) safely and effectively to treat different respiratory diseases. A program toward rescuing ciliary defect in patients with PCD with biallelic variants in DNAI1—representing approximately 8–10% of patients—is in preclinical phase. However, in a recent presentation (https://recodetx.com/wp-content/uploads/PCDposter_210721-v4-mh.pdf accessed 8 Feb 2022), newly translated DNAI1 protein was shown to be incorporated throughout the ciliary axoneme of ex vivo cultures of primary human respiratory epithelial cells and could be detected for 2 weeks after treatment. LNP‐formulated DNAI1 mRNA delivered as an aerosol rescued ciliary function in cell‐based PCD models despite the presence of mucus.
7.12. Read‐through therapy
Nonsense mutations resulting in a premature termination codon (PTC) and thus translation into nonfunctional protein have been identified as disease causing in approximately one third of patients with PCD (Paff et al., 2018). Currently, in vitro studies on PTC therapeutics for PCD are being performed by Eloxx Pharmaceuticals (Waltham, United States of America (https://www.eloxxpharma.com/ accessed 8 Feb 2022), in collaboration with Amsterdam University Medical Center (The Netherlands) and University Medical Center Utrecht (The Netherlands) (Paff et al., 2021).
8. FUTURE PERSPECTIVES—THE WAY FORWARD
New drugs with a direct effect on the underlying primary ciliary defect, for example by restoring ciliary function, are obviously a very high priority, but drugs with a positive indirect effect on mucociliary clearance are also of great importance, while the development of the evidence base for drugs already widely used in patients with PCD also deserves attention (Figure 1).
With only a single real evidence‐based pharmaceutical treatment offering for patients with PCD (Kobbernagel et al., 2020), there is a huge need to fill this gap. Fortunately, there is currently a great deal of interest from various pharmaceutical companies to develop new drugs and perform RCTs focusing on PCD chronic airway disease. mRNA therapy, as discussed above, is currently in particular focus and may ultimately lead to personalized medicine.
The rapid development in genetics in general and PCD in particular has led to a large proportion of patients with PCD in principle in the future being able to be diagnosed via genetic testing, although this cannot stand alone so far.
More intense focus on adult PCD is warranted since there is a diagnostic gap for PCD in adulthood (Ardura‐Garcia et al., 2020), most likely because these adult patients are followed mistakenly for other diagnoses such as asthma, COPD, or non‐CF bronchiectasis. Once a patient is mistakenly diagnosed this may obstruct further recognition of clinical PCD symptoms and work up for PCD. Correct PCD diagnosis is hence not only an issue in infants and young children but remains a challenge across all age groups. In adult patients with non‐CF bronchiectasis PCD should always be considered as a possible underlying explanation.
Not only could the huge development in genetic diagnosis lead to further early, perhaps neonatal, diagnosis but also ensure a rapid response with personal medication, for example, mRNA therapy, thereby completely avoiding the development of chronic destructive PCD airway disease.
International research collaboration in PCD is instrumental for success and gained momentum in 2007, when several meetings of international PCD experts were organized on the basis of an ERS PCD Task Force, resulting in a highly cited guideline (Barbato et al., 2009). Like other rare diseases, it is impossible to make much progress without international collaboration between dedicated researchers, as exemplified by the explosion in research activities and publications that has since resulted from BESTCILIA and BEAT PCD funded by the European Union. These activities have also fostered the iPCD cohort (Goutaki et al., 2017) and the international PCD registry (Werner et al., 2016), which is growing both in terms of the number of patients enrolled and associated key data, and increasingly represents a goldmine for researchers in the field.
With the establishment of ERN‐LUNG (https://ern-lung.eu), PCD has the chance for further momentum in health care and QOL improvement and research. ERN‐LUNG is a network of European healthcare providers that aims to ensure and promote excellence in care and research for the benefit of patients with rare lung diseases. The vision is to be a European center of knowledge for rare lung diseases with the aim of reducing morbidity and mortality from such diseases in people of all ages. ERN‐LUNG consists of 9 core networks representing the diversity of diseases and conditions affecting the lungs (https://ern-lung.eu/governance). The current structure of the network has evolved since its creation in 2017 and will continue to do so with better and more inclusive geographical coverage. The ERN‐LUNG PCD Core network currently consists of 26 centers in 18 countries (https://ern-lung.eu/reference-centers-2/), and 9 new centers from 6 new countries are expected to soon contribute to further expansion of the ERN‐LUNG PCD Core network. The ERN‐LUNG PCD CORE network aims to optimize current efforts to improve PCD patient care. ERN‐LUNG has been evaluated by the European Commission and in December 2016, the European Reference Network (ERN) Board of Member States approved ERN‐LUNG as one of 24 ERNs for rare diseases. ERNs are virtual cross‐border healthcare networks that provide rare disease clinicians with a centralized platform across borders to share knowledge and experiences with the objectives of improving patients' access to diagnosis, care, and treatment, establishing common rare disease databases, developing clinical guidelines, thereby facilitating the mobility of expertise rather than patients themselves.
Within the framework of the ERN‐LUNG PCD Core network, it was decided a few years ago to establish a PCD CTN strongly inspired by the successful corresponding CTN for CF. The disease‐specific CTN for PCD was established in 2020 and consists of 22 clinical trial sites in 12 countries in Europe, which together are HCPs for >3,000 people with PCD >1,600 adults and > 1,400 children. However, the CTN will continue to grow in numbers, both in number of sites and number of patients with PCD potentially available for participation in trials.
The overall objective of the PCD‐CTN is to intensify clinical research by primarily encouraging and contributing to the initiation of RCTs in PCD, thereby bringing new medicines to patients as soon as possible. Thus, the PCD CTN aims to increase clinical and translational research and promote the development of evidence‐based treatments, including new therapies for PCD. To achieve these goals, better access for pharmaceutical companies and investigator‐initiated research projects to genetically well‐characterized patient populations is ensured through a network of clinical trial sites, all of which also refer to the international PCD registry (Werner et al., 2016). Through the structures of the established PCD‐CTN, clinical trials can be successfully planned and implemented, while achieving sufficient numbers of patients and thus the necessary strength of the research projects. To achieve these goals, the network also promotes strong collaboration with patient organizations and pharmaceutical companies.
Geno–phenotype associations are just like natural history studies essential for the understanding of disease mechanisms and degrees of severity and progression—made possible by the international PCD registry (Werner et al., 2016)—but require capital and resources to be able to maintain the value in the registers. Both patients, health care providers, pharmaceutical companies, and PCD researchers have a common interest in such registers functioning optimally, which is why a joint effort for the basic funding must be ensured in a collaboration, regardless of whether the benefits for the individual stakeholders are different.
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
Nielsen, K. G. , Holgersen, M. G. , Crowley, S. , & Marthin, J. K. (2022). Chronic airway disease in primary ciliary dyskinesia—spiced with geno–phenotype associations. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190C:20–35. 10.1002/ajmg.c.31967
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Aanæs, K. (2013). Bacterial sinusitis can be a focus for initial lung colonisation and chronic lung infection in patients with cystic fibrosis. Journal of Cystic Fibrosis, 12(Suppl 2), S1–S20. 10.1016/S1569-1993(13)00150-1 [DOI] [PubMed] [Google Scholar]
- Alanin, M. C. , Johansen, H. K. , Aanaes, K. , Høiby, N. , Pressler, T. , Skov, M. , … von Buchwald, C. (2015). Simultaneous sinus and lung infections in patients with primary ciliary dyskinesia. Acta Oto‐Laryngologica, 135(1), 58–63. 10.3109/00016489.2014.962185 [DOI] [PubMed] [Google Scholar]
- Alanin, M. C. , Nielsen, K. G. , von Buchwald, C. , Skov, M. , Aanaes, K. , Høiby, N. , & Johansen, H. K. (2015). A longitudinal study of lung bacterial pathogens in patients with primary ciliary dyskinesia. Clinical Microbiology and Infection, 21(12), 1093.e1–7. 10.1016/j.cmi.2015.08.020 [DOI] [PubMed] [Google Scholar]
- Altenburg, J. , de Graaff, C. S. , Stienstra, Y. , Sloos, J. H. , van Haren, E. H. , Koppers, R. J. , … Boersma, W. G. (2013). Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non‐cystic fibrosis bronchiectasis: The BAT randomized controlled trial. JAMA, 309(12), 1251–1259. 10.1001/jama.2013.1937 [DOI] [PubMed] [Google Scholar]
- Anuradha, K. W. D. A. , Gunathilaka, P. K. G. , & Wickramasinghe, V. P. (2021). Effectiveness of hypertonic saline nebulization in airway clearance in children with non‐cystic fibrosis bronchiectasis: A randomized control trial. Pediatric Pulmonology, 56(2), 509–515. 10.1002/ppul.25206 [DOI] [PubMed] [Google Scholar]
- Ardura‐Garcia, C. , Goutaki, M. , Carr, S. B. , Crowley, S. , Halbeisen, F. S. , Nielsen, K. G. , … Kuehni, C. E. (2020). Registries and collaborative studies for primary ciliary dyskinesia in Europe. ERJ Open Research, 6(2), 5–20. 10.1183/23120541.00005-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndal, E. , Johansen, H. K. , Haagensen, J. A. J. , Bartell, J. A. , Marvig, R. L. , Alanin, M. , … von Buchwald, C. (2020). Primary ciliary dyskinesia patients have the same P. aeruginosa clone in sinuses and lungs. The European Respiratory Journal, 55(1), 1901472. 10.1183/13993003.01472-2019 [DOI] [PubMed] [Google Scholar]
- Barbato, A. , Frischer, T. , Kuehni, C. E. , Snijders, D. , Azevedo, I. , Baktai, G. , … Bush, A. (2009). Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. The European Respiratory Journal, 34(6), 1264–1276. 10.1183/09031936.00176608 [DOI] [PubMed] [Google Scholar]
- Behan, L. , Rubbo, B. , Lucas, J. S. , & Dunn Galvin, A. (2017). The patient's experience of primary ciliary dyskinesia: A systematic review. Quality of Life Research, 26(9), 2265–2285. 10.1007/s11136-017-1564-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon, M. , Smits, A. , Cuppens, H. , Jaspers, M. , Proesmans, M. , Dupont, L. J. , … De Boeck, K. (2014). Primary ciliary dyskinesia: Critical evaluation of clinical symptoms and diagnosis in patients with normal and abnormal ultrastructure. Orphanet Journal of Rare Diseases, 9, 11. 10.1186/1750-1172-9-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon, M. , Vermeulen, F. L. , Gysemans, W. , Proesmans, M. , Jorissen, M. , & De Boeck, K. (2015). Lung structure‐function correlation in patients with primary ciliary dyskinesia. Thorax, 70(4), 339–345. 10.1136/thoraxjnl-2014-206578 [DOI] [PubMed] [Google Scholar]
- Bradley, J. M. , Anand, R. , O'Neill, B. , Ferguson, K. , Clarke, M. , Carroll, M. , … CLEAR, S. G. (2019). A 2 × 2 factorial, randomised, open‐label trial to determine the clinical and cost‐effectiveness of hypertonic saline (HTS 6%) and carbocisteine for airway clearance versus usual care over 52 weeks in adults with bronchiectasis: A protocol for the CLEAR clinical trial. Trials, 20(1), 747. 10.1186/s13063-019-3766-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, D. E. , Pittman, J. E. , Leigh, M. W. , Fordham, L. , & Davis, S. D. (2008). Early lung disease in young children with primary ciliary dyskinesia. Pediatric Pulmonology, 43(5), 514–516. 10.1002/ppul.20792 [DOI] [PubMed] [Google Scholar]
- Bush, A. , Payne, D. , Pike, S. , Jenkins, G. , Henke, M. O. , & Rubin, B. K. (2006). Mucus properties in children with primary ciliary dyskinesia: Comparison with cystic fibrosis. Chest, 129(1), 118–123. 10.1378/chest.129.1.118 [DOI] [PubMed] [Google Scholar]
- Chhin, B. , Negre, D. , Merrot, O. , Pham, J. , Tourneur, Y. , Ressnikoff, D. , … Bouvagnet, P. (2009). Ciliary beating recovery in deficient human airway epithelial cells after lentivirus ex vivo gene therapy. PLoS Genetics, 5(3), e1000422. 10.1371/journal.pgen.1000422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen‐Cymberknoh, M. , Weigert, N. , Gileles‐Hillel, A. , Breuer, O. , Simanovsky, N. , Boon, M. , … Kerem, E. (2017). Clinical impact of Pseudomonas aeruginosa colonization in patients with primary ciliary dyskinesia. Respiratory Medicine, 131, 241–246. 10.1016/j.rmed.2017.08.028 [DOI] [PubMed] [Google Scholar]
- Crowley, S. , Holgersen, M. G. , & Nielsen, K. G. (2019). Variation in treatment strategies for the eradication of Pseudomonas aeruginosa in primary ciliary dyskinesia across European centers. Chronic Respiratory Disease, 16, 1479972318787919. 10.1177/1479972318787919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley, C. L. , Iaccarino, J. M. , Lange, C. , Cambau, E. , Wallace, R. J. , Andrejak, C. , … Winthrop, K. L. (2020). Treatment of nontuberculous mycobacterial pulmonary disease: An official ATS/ERS/ESCMID/IDSA clinical practice guideline. Clinical Infectious Diseases, 71(4), 905–913. 10.1093/cid/ciaa1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, S. D. , Ferkol, T. W. , Rosenfeld, M. , Lee, H. S. , Dell, S. D. , Sagel, S. D. , … Leigh, M. W. (2015). Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. American Journal of Respiratory and Critical Care Medicine, 191(3), 316–324. 10.1164/rccm.201409-1672OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, S. D. , Rosenfeld, M. , Lee, H. S. , Ferkol, T. W. , Sagel, S. D. , Dell, S. D. , … Genetic Disorders of Mucociliary Clearance Consortium . (2018). Primary ciliary dyskinesia: Longitudinal study of lung disease by ultrastructure defect and genotype. American Journal of Respiratory and Critical Care Medicine, 199, 190–198. 10.1164/rccm.201803-0548OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, S. D. , Rosenfeld, M. , Lee, H. S. , Ferkol, T. W. , Sagel, S. D. , Dell, S. D. , … Leigh, M. W. (2019). Primary ciliary dyskinesia: Longitudinal study of lung disease by ultrastructure defect and genotype. American Journal of Respiratory and Critical Care Medicine, 199(2), 190–198. 10.1164/rccm.201803-0548OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehlink, E. , Richardson, C. , Marsh, G. , Lee, K. , Jamalzadeh, A. , Bush, A. , … Carr, S. B. (2018). Are inhaled corticosteroids prescribed rationally in primary ciliary dyskinesia. The European Respiratory Journal, 51(3), 1702221. 10.1183/13993003.02221-2017 [DOI] [PubMed] [Google Scholar]
- Donaldson, S. H. , Bennett, W. D. , Zeman, K. L. , Knowles, M. R. , Tarran, R. , & Boucher, R. C. (2006). Mucus clearance and lung function in cystic fibrosis with hypertonic saline. The New England Journal of Medicine, 354(3), 241–250. 10.1056/NEJMoa043891 [DOI] [PubMed] [Google Scholar]
- Fayon, M. , & Beaufils, F. (2021). The lower respiratory airway wall in children in health and disease. ERJ Open Res, 7(3), 874–2020. 10.1183/23120541.00874-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frija‐Masson, J. , Bassinet, L. , Honoré, I. , Dufeu, N. , Housset, B. , Coste, A. , … Maître, B. (2017). Clinical characteristics, functional respiratory decline and follow‐up in adult patients with primary ciliary dyskinesia. Thorax, 72(2), 154–160. 10.1136/thoraxjnl-2015-207891 [DOI] [PubMed] [Google Scholar]
- Gahleitner, F. , Thompson, J. , Jackson, C. L. , Hueppe, J. F. , Behan, L. , Dehlink, E. , … Rubbo, B. (2021). Lower airway clinical outcome measures for use in primary ciliary dyskinesia research: A scoping review. ERJ Open Research, 7(4), 320–2021. 10.1183/23120541.00320-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutaki, M. , Halbeisen, F. S. , Barbato, A. , Crowley, S. , Harris, A. , Hirst, R. A. , … Kuehni, C. E. (2020). Late diagnosis of infants with PCD and neonatal respiratory distress. Journal of Clinical Medicine, 9. 10.3390/jcm9092871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutaki, M. , Halbeisen, F. S. , Spycher, B. D. , Maurer, E. , Belle, F. , Amirav, I. , … French, R. C. F. R. L. D. (2017). Growth and nutritional status, and their association with lung function: A study from the international primary ciliary dyskinesia cohort. The European Respiratory Journal, 50(6), 1701659. 10.1183/13993003.01659-2017 [DOI] [PubMed] [Google Scholar]
- Goutaki, M. , Maurer, E. , Halbeisen, F. S. , Amirav, I. , Barbato, A. , Behan, L. , … Genetic, D. O. M. C. C. (2017). The international primary ciliary dyskinesia cohort (iPCD cohort): Methods and first results. The European Respiratory Journal, 49(1), 1601181. 10.1183/13993003.01181-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutaki, M. , Meier, A. B. , Halbeisen, F. S. , Lucas, J. S. , Dell, S. D. , Maurer, E. , … Kuehni, C. E. (2016). Clinical manifestations in primary ciliary dyskinesia: Systematic review and meta‐analysis. The European Respiratory Journal, 48(4), 1081–1095. 10.1183/13993003.00736-2016 [DOI] [PubMed] [Google Scholar]
- Green, K. , Buchvald, F. F. , Marthin, J. K. , Hanel, B. , Gustafsson, P. M. , & Nielsen, K. G. (2012). Ventilation inhomogeneity in children with primary ciliary dyskinesia. Thorax, 67(1), 49–53. 10.1136/thoraxjnl-2011-200726 [DOI] [PubMed] [Google Scholar]
- Halbeisen, F. S. , Goutaki, M. , Spycher, B. D. , Amirav, I. , Behan, L. , Boon, M. , … Kuehni, C. E. (2018). Lung function in patients with primary ciliary dyskinesia: An iPCD cohort study. The European Respiratory Journal, 52(2), 1801040. 10.1183/13993003.01040-2018 [DOI] [PubMed] [Google Scholar]
- Halbeisen, F. S. , Jose, A. , de Jong, C. , Nyilas, S. , Latzin, P. , Kuehni, C. E. , & Goutaki, M. (2019). Spirometric indices in primary ciliary dyskinesia: Systematic review and meta‐analysis. ERJ Open Res, 5(2), 231–2018. 10.1183/23120541.00231-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah, W. B. , Seifert, B. A. , Truty, R. , Zariwala, M. A. , Ameel, K. , Zhao, Y. , … Gaston, B. (2022). The global prevalence and ethnic heterogeneity of primary ciliary dyskinesia gene variants: A genetic database analysis. The Lancet Respiratory Medicine. 10.1016/S2213-2600(21)00453-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, S. K. , Rau, M. H. , Johansen, H. K. , Ciofu, O. , Jelsbak, L. , Yang, L. , … Molin, S. (2012). Evolution and diversification of Pseudomonas aeruginosa in the paranasal sinuses of cystic fibrosis children have implications for chronic lung infection. The ISME Journal, 6(1), 31–45. 10.1038/ismej.2011.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, D. , Reynolds, S. D. , & Tumin, D. (2016). Outcomes of lung transplantation for primary ciliary dyskinesia and Kartagener syndrome. The Journal of Heart and Lung Transplantation, 35(11), 1377–1378. 10.1016/j.healun.2016.08.025 [DOI] [PubMed] [Google Scholar]
- Hellinckx, J. , Demedts, M. , & De Boeck, K. (1998). Primary ciliary dyskinesia: Evolution of pulmonary function. European Journal of Pediatrics, 157(5), 422–426. 10.1007/s004310050843 [DOI] [PubMed] [Google Scholar]
- Hilliard, T. N. , Regamey, N. , Shute, J. K. , Nicholson, A. G. , Alton, E. W. , Bush, A. , & Davies, J. C. (2007). Airway remodelling in children with cystic fibrosis. Thorax, 62(12), 1074–1080. 10.1136/thx.2006.074641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgersen, M. G. , Marthin, J. K. , Johansen, H. K. , & Nielsen, K. G. (2021). A retrospective review of Achromobacter species and antibiotic treatments in patients with primary ciliary dyskinesia. Chronic Respiratory Disease, 18, 14799731211061600. 10.1177/14799731211061600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving, S. , Carr, S. , Hogg, C. , Loebinger, M. , Shoemark, A. , & Bush, A. (2017). Lung clearance index (LCI) is stable in Most primary ciliary dyskinesia (PCD) patients managed in a specialist Centre: A pilot study. Lung, 195(4), 441–443. 10.1007/s00408-017-0022-5 [DOI] [PubMed] [Google Scholar]
- Irving, S. , Dixon, M. , Fassad, M. R. , Frost, E. , Hayward, J. , Kilpin, K. , … Bush, A. (2018). Primary ciliary dyskinesia due to microtubular defects is associated with worse lung clearance index. Lung, 196(2), 231–238. 10.1007/s00408-018-0086-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving, S. J. , Ives, A. , Davies, G. , Donovan, J. , Edey, A. J. , Gill, S. S. , … Bush, A. (2013). Lung clearance index and high‐resolution computed tomography scores in primary ciliary dyskinesia. American Journal of Respiratory and Critical Care Medicine, 188(5), 545–549. 10.1164/rccm.201304-0800OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoh, S. , & Rubin, B. K. (2010). Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clinical Microbiology Reviews, 23(3), 590–615. 10.1128/CMR.00078-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellett, F. , Redfern, J. , & Niven, R. M. (2005). Evaluation of nebulised hypertonic saline (7%) as an adjunct to physiotherapy in patients with stable bronchiectasis. Respiratory Medicine, 99(1), 27–31. 10.1016/j.rmed.2004.05.006 [DOI] [PubMed] [Google Scholar]
- Kennedy, M. P. , Noone, P. G. , Leigh, M. W. , Zariwala, M. A. , Minnix, S. L. , Knowles, M. R. , & Molina, P. L. (2007). High‐resolution CT of patients with primary ciliary dyskinesia. AJR. American Journal of Roentgenology, 188(5), 1232–1238. 10.2214/AJR.06.0965 [DOI] [PubMed] [Google Scholar]
- King, M. , Dasgupta, B. , Tomkiewicz, R. P. , & Brown, N. E. (1997). Rheology of cystic fibrosis sputum after in vitro treatment with hypertonic saline alone and in combination with recombinant human deoxyribonuclease I. American Journal of Respiratory and Critical Care Medicine, 156(1), 173–177. 10.1164/ajrccm.156.1.9512074 [DOI] [PubMed] [Google Scholar]
- Kinghorn, B. , McNamara, S. , Genatossio, A. , Sullivan, E. , Siegel, M. , Bauer, I. , … Pittman, J. (2020). Comparison of Multiple Breath Washout and spirometry in children with primary ciliary dyskinesia and cystic fibrosis and healthy controls. Annals of the American Thoracic Society, 17(9), 1085–1093. 10.1513/AnnalsATS.201905-375OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobbernagel, H. E. , Buchvald, F. F. , Haarman, E. G. , Casaulta, C. , Collins, S. A. , Hogg, C. , … Nielsen, K. G. (2016). Study protocol, rationale and recruitment in a European multi‐Centre randomized controlled trial to determine the efficacy and safety of azithromycin maintenance therapy for 6 months in primary ciliary dyskinesia. BMC Pulmonary Medicine, 16(1), 104. 10.1186/s12890-016-0261-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobbernagel, H. E. , Buchvald, F. F. , Haarman, E. G. , Casaulta, C. , Collins, S. A. , Hogg, C. , … Nielsen, K. G. (2020). Efficacy and safety of azithromycin maintenance therapy in primary ciliary dyskinesia (BESTCILIA): A multicentre, double‐blind, randomised, placebo‐controlled phase 3 trial. The Lancet Respiratory Medicine, 8(5), 493–505. 10.1016/S2213-2600(20)30058-8 [DOI] [PubMed] [Google Scholar]
- Kobbernagel, H. E. , Green, K. , Ring, A. M. , Buchvald, F. F. , Rosthøj, S. , Gustafsson, P. M. , & Nielsen, K. G. (2019). One‐year evolution and variability in multiple‐breath washout indices in children and young adults with primary ciliary dyskinesia. European Clinical Respiratory Journal, 6(1), 1591841. 10.1080/20018525.2019.1591841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongstad, T. , Buchvald, F. F. , Green, K. , Lindblad, A. , Robinson, T. E. , & Nielsen, K. G. (2013). Improved air trapping evaluation in chest computed tomography in children with cystic fibrosis using real‐time spirometric monitoring and biofeedback. Journal of Cystic Fibrosis, 12(6), 559–566. 10.1016/j.jcf.2013.05.012 [DOI] [PubMed] [Google Scholar]
- Koucký, V. , Uhlík, J. , Hoňková, L. , Koucký, M. , Doušová, T. , & Pohunek, P. (2020). Ventilation inhomogeneity and bronchial basement membrane changes in chronic neutrophilic airway inflammation. Chest, 157(4), 779–789. 10.1016/j.chest.2019.10.023 [DOI] [PubMed] [Google Scholar]
- Kuehni, C. E. , Frischer, T. , Strippoli, M. P. , Maurer, E. , Bush, A. , Nielsen, K. G. , … A. Barbato for the ERS Task Force on Primary Ciliary Dyskinesia in Children . (2010). Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. The European Respiratory Journal, 36(6), 1248–1258. 10.1183/09031936.00001010 [DOI] [PubMed] [Google Scholar]
- Lai, M. , Pifferi, M. , Bush, A. , Piras, M. , Michelucci, A. , di Cicco, M. , … Pistello, M. (2016). Gene editing of DNAH11 restores normal cilia motility in primary ciliary dyskinesia. Journal of Medical Genetics, 53(4), 242–249. 10.1136/jmedgenet-2015-103539 [DOI] [PubMed] [Google Scholar]
- Lee, T. W. , Brownlee, K. G. , Conway, S. P. , Denton, M. , & Littlewood, J. M. (2003). Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. Journal of Cystic Fibrosis, 2(1), 29–34. 10.1016/S1569-1993(02)00141-8 [DOI] [PubMed] [Google Scholar]
- Leigh, M. W. , Ferkol, T. W. , Davis, S. D. , Lee, H. S. , Rosenfeld, M. , Dell, S. D. , … Knowles, M. R. (2016). Clinical features and associated likelihood of primary ciliary dyskinesia in children and adolescents. Annals of the American Thoracic Society, 13(8), 1305–1313. 10.1513/AnnalsATS.201511-748OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh, M. W. , Horani, A. , Kinghorn, B. , O'Connor, M. G. , Zariwala, M. A. , & Knowles, M. R. (2019). Primary ciliary dyskinesia (PCD): A genetic disorder of motile cilia. Translational Science of Rare Diseases, 4(1–2), 51–75. 10.3233/TRD-190036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, J. S. , Alanin, M. C. , Collins, S. , Harris, A. , Johansen, H. K. , Nielsen, K. G. , … Walker, W. T. (2017). Clinical care of children with primary ciliary dyskinesia. Expert Review of Respiratory Medicine, 11(10), 779–790. 10.1080/17476348.2017.1360770 [DOI] [PubMed] [Google Scholar]
- Lucas, J. S. , Barbato, A. , Collins, S. A. , Goutaki, M. , Behan, L. , Caudri, D. , … Kuehni, C. E. (2017). European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. The European Respiratory Journal, 49(1), 1601090. 10.1183/13993003.01090-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, J. S. , Behan, L. , Dunn Galvin, A. , Alpern, A. , Morris, A. M. , Carroll, M. P. , … Quittner, A. L. (2015). A quality‐of‐life measure for adults with primary ciliary dyskinesia: QOL‐PCD. The European Respiratory Journal, 46(2), 375–383. 10.1183/09031936.00216214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglione, M. , Bush, A. , Montella, S. , Mollica, C. , Manna, A. , Esposito, A. , & Santamaria, F. (2012). Progression of lung disease in primary ciliary dyskinesia: Is spirometry less accurate than CT. Pediatric Pulmonology, 47(5), 498–504. 10.1002/ppul.21569 [DOI] [PubMed] [Google Scholar]
- Maglione, M. , Bush, A. , Nielsen, K. G. , Hogg, C. , Montella, S. , Marthin, J. K. , … Santamaria, F. (2014). Multicenter analysis of body mass index, lung function, and sputum microbiology in primary ciliary dyskinesia. Pediatric Pulmonology, 49(12), 1243–1250. 10.1002/ppul.22984 [DOI] [PubMed] [Google Scholar]
- Maglione, M. , Montella, S. , & Santamaria, F. (2012). Chest CTs in primary ciliary dyskinesia: Not too few, but not too many. Pediatric Pulmonology, 47(8), 733–735. 10.1002/ppul.22589 [DOI] [PubMed] [Google Scholar]
- Magnin, M. L. , Cros, P. , Beydon, N. , Mahloul, M. , Tamalet, A. , Escudier, E. , … Blanchon, S. (2012). Longitudinal lung function and structural changes in children with primary ciliary dyskinesia. Pediatric Pulmonology, 47(8), 816–825. 10.1002/ppul.22577 [DOI] [PubMed] [Google Scholar]
- Marthin, J. K. , Lucas, J. S. , Boon, M. , Casaulta, C. , Crowley, S. , Destouches, D. M. S. , … Nielsen, K. G. (2021). International BEAT‐PCD consensus statement for infection prevention and control for primary ciliary dyskinesia in collaboration with ERN‐LUNG PCD Core network and patient representatives. ERJ Open Research, 7(3), 301–2021. 10.1183/23120541.00301-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthin, J. K. , Petersen, N. , Skovgaard, L. T. , & Nielsen, K. G. (2010). Lung function in patients with primary ciliary dyskinesia: A cross‐sectional and 3‐decade longitudinal study. American Journal of Respiratory and Critical Care Medicine, 181(11), 1262–1268. 10.1164/rccm.200811-1731OC [DOI] [PubMed] [Google Scholar]
- Mayer‐Hamblett, N. , Retsch‐Bogart, G. , Kloster, M. , Accurso, F. , Rosenfeld, M. , Albers, G. , … OPTIMIZE, S. G. (2018). Azithromycin for early pseudomonas infection in cystic fibrosis. The OPTIMIZE randomized trial. American Journal of Respiratory and Critical Care Medicine, 198(9), 1177–1187. 10.1164/rccm.201802-0215OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullowney, T. , Manson, D. , Kim, R. , Stephens, D. , Shah, V. , & Dell, S. (2014). Primary ciliary dyskinesia and neonatal respiratory distress. Pediatrics, 134(6), 1160–1166. 10.1542/peds.2014-0808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolson, C. H. , Stirling, R. G. , Borg, B. M. , Button, B. M. , Wilson, J. W. , & Holland, A. E. (2012). The long term effect of inhaled hypertonic saline 6% in non‐cystic fibrosis bronchiectasis. Respiratory Medicine, 106(5), 661–667. 10.1016/j.rmed.2011.12.021 [DOI] [PubMed] [Google Scholar]
- Noone, P. G. , Leigh, M. W. , Sannuti, A. , Minnix, S. L. , Carson, J. L. , Hazucha, M. , … Knowles, M. R. (2004). Primary ciliary dyskinesia: Diagnostic and phenotypic features. American Journal of Respiratory and Critical Care Medicine, 169(4), 459–467. 10.1164/rccm.200303-365OC [DOI] [PubMed] [Google Scholar]
- Nyilas, S. , Bauman, G. , Pusterla, O. , Sommer, G. , Singer, F. , Stranzinger, E. , … Latzin, P. (2018). Structural and functional lung impairment in primary ciliary dyskinesia. Assessment with magnetic resonance imaging and Multiple Breath Washout in comparison to spirometry. Annals of the American Thoracic Society, 15(12), 1434–1442. 10.1513/AnnalsATS.201712-967OC [DOI] [PubMed] [Google Scholar]
- Nyilas, S. , Schlegtendal, A. , Yammine, S. , Casaulta, C. , Latzin, P. , & Koerner‐Rettberg, C. (2015). Further evidence for an association between LCI and FEV1 in patients with PCD. Thorax, 70(9), 896. 10.1136/thoraxjnl-2015-207206 [DOI] [PubMed] [Google Scholar]
- O'Donnell, A. E. , Barker, A. F. , Ilowite, J. S. , & Fick, R. B. (1998). Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase study group. Chest, 113(5), 1329–1334. 10.1378/chest.113.5.1329 [DOI] [PubMed] [Google Scholar]
- Ostrowski, L. E. , Yin, W. , Patel, M. , Sechelski, J. , Rogers, T. , Burns, K. , … Olsen, J. C. (2014). Restoring ciliary function to differentiated primary ciliary dyskinesia cells with a lentiviral vector. Gene Therapy, 21(3), 253–261. 10.1038/gt.2013.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paff, T. , Daniels, J. M. , Weersink, E. J. , Lutter, R. , Vonk Noordegraaf, A. , & Haarman, E. G. (2017). A randomised controlled trial on the effect of inhaled hypertonic saline on quality of life in primary ciliary dyskinesia. The European Respiratory Journal, 49(2), 1601770. 10.1183/13993003.01770-2016 [DOI] [PubMed] [Google Scholar]
- Paff, T. , Kooi, I. E. , Moutaouakil, Y. , Riesebos, E. , Sistermans, E. A. , Daniels, H. J. M. A. , … Micha, D. (2018). Diagnostic yield of a targeted gene panel in primary ciliary dyskinesia patients. Human Mutation, 39(5), 653–665. 10.1002/humu.23403 [DOI] [PubMed] [Google Scholar]
- Paff, T. , Omran, H. , Nielsen, K. G. , & Haarman, E. G. (2021). Current and future treatments in primary ciliary dyskinesia. International Journal of Molecular Sciences, 22(18). 10.3390/ijms22189834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti, G. , De Santi, M. M. , Farolfi, A. , Zuccotti, G. V. , D'Auria, E. , Patria, M. F. , … Ambrosetti, U. (2020). Exacerbations and Pseudomonas aeruginosa colonization are associated with altered lung structure and function in primary ciliary dyskinesia. BMC Pediatrics, 20(1), 158. 10.1186/s12887-020-02062-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifferi, M. , Bush, A. , Mariani, F. , Piras, M. , Michelucci, A. , Cangiotti, A. , … Peroni, D. (2020). Lung function longitudinal study by phenotype and genotype in primary ciliary dyskinesia. Chest, 158(1), 117–120. 10.1016/j.chest.2020.02.001 [DOI] [PubMed] [Google Scholar]
- Pifferi, M. , Bush, A. , Pioggia, G. , Caramella, D. , Tartarisco, G. , Di Cicco, M. , … Boner, A. (2012). Evaluation of pulmonary disease using static lung volumes in primary ciliary dyskinesia. Thorax, 67(11), 993–999. 10.1136/thoraxjnl-2011-200137 [DOI] [PubMed] [Google Scholar]
- Ratjen, F. , Waters, V. , Klingel, M. , McDonald, N. , Dell, S. , Leahy, T. R. , … Grasemann, H. (2016). Changes in airway inflammation during pulmonary exacerbations in patients with cystic fibrosis and primary ciliary dyskinesia. The European Respiratory Journal, 47(3), 829–836. 10.1183/13993003.01390-2015 [DOI] [PubMed] [Google Scholar]
- Regamey, N. , Ochs, M. , Hilliard, T. N. , Mühlfeld, C. , Cornish, N. , Fleming, L. , … Davies, J. C. (2008). Increased airway smooth muscle mass in children with asthma, cystic fibrosis, and non‐cystic fibrosis bronchiectasis. American Journal of Respiratory and Critical Care Medicine, 177(8), 837–843. 10.1164/rccm.200707-977OC [DOI] [PubMed] [Google Scholar]
- Robinson, P. D. , Goldman, M. D. , & Gustafsson, P. M. (2009). Inert gas washout: Theoretical background and clinical utility in respiratory disease. Respiration, 78(3), 339–355. 10.1159/000225373 [DOI] [PubMed] [Google Scholar]
- Rosenow, T. , Oudraad, M. C. , Murray, C. P. , Turkovic, L. , Kuo, W. , de Bruijne, M. , … Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST, C. F.) . (2015). PRAGMA‐CF. a quantitative structural lung disease computed tomography outcome in young children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine, 191(10), 1158–1165. 10.1164/rccm.201501-0061OC [DOI] [PubMed] [Google Scholar]
- Rubbo, B. , Best, S. , Hirst, R. A. , Shoemark, A. , Goggin, P. , Carr, S. B. , … English, N. C. P. C. D. M. S. (2020). Clinical features and management of children with primary ciliary dyskinesia in England. Archives of Disease in Childhood, 105(8), 724–729. 10.1136/archdischild-2019-317687 [DOI] [PubMed] [Google Scholar]
- Santamaria, F. , Montella, S. , Tiddens, H. A. W. M. , Guidi, G. , Casotti, V. , Maglione, M. , & de Jong, P. A. (2008). Structural and functional lung disease in primary ciliary dyskinesia. Chest, 134(2), 351–357. 10.1378/chest.07-2812 [DOI] [PubMed] [Google Scholar]
- Sehgal, I. S. , Dhooria, S. , Bal, A. , & Agarwal, R. (2015). Allergic bronchopulmonary aspergillosis in an adult with Kartagener syndrome. BML Case Reports, 2015, bcr2015211493. 10.1136/bcr-2015-211493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, A. , Shoemark, A. , MacNeill, S. J. , Bhaludin, B. , Rogers, A. , Bilton, D. , … Loebinger, M. R. (2016). A longitudinal study characterising a large adult primary ciliary dyskinesia population. The European Respiratory Journal, 48(2), 441–450. 10.1183/13993003.00209-2016 [DOI] [PubMed] [Google Scholar]
- Shapiro, A. J. , Davis, S. D. , Ferkol, T. , Dell, S. D. , Rosenfeld, M. , Olivier, K. N. , … Genetic, D. O. M. C. C. (2014). Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: Insights into situs ambiguus and heterotaxy. Chest, 146(5), 1176–1186. 10.1378/chest.13-1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, A. J. , Zariwala, M. A. , Ferkol, T. , Davis, S. D. , Sagel, S. D. , Dell, S. D. , … Genetic, D. O. M. C. C. (2016). Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatric Pulmonology, 51(2), 115–132. 10.1002/ppul.23304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, B. , Sharma, M. , Bondi, E. , & Sharma, M. (2005). Kartagener's syndrome associated with allergic bronchopulmonary aspergillosis. MedGenMed, 7(2), 25 Retrieved from https://pubmed.ncbi.nlm.nih.gov/16369404 [PMC free article] [PubMed] [Google Scholar]
- Shoemark, A. , Rubbo, B. , Legendre, M. , Fassad, M. R. , Haarman, E. G. , Best, S. , … Lucas, J. S. (2021). Topological data analysis reveals genotype‐phenotype relationships in primary ciliary dyskinesia. The European Respiratory Journal, 58(2), 2002359. 10.1183/13993003.02359-2020 [DOI] [PubMed] [Google Scholar]
- Singer, F. , Schlegtendal, A. , Nyilas, S. , Vermeulen, F. , Boon, M. , & Koerner‐Rettberg, C. (2021). Lung clearance index predicts pulmonary exacerbations in individuals with primary ciliary dyskinesia: A multicentre cohort study. Thorax, 76(7), 681–688. 10.1136/thoraxjnl-2020-215504 [DOI] [PubMed] [Google Scholar]
- Southern, K. W. , Barker, P. M. , Solis‐Moya, A. , & Patel, L. (2012). Macrolide antibiotics for cystic fibrosis. Cochrane Database of Systematic Reviews, 11, CD002203. 10.1002/14651858.CD002203.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strippoli, M. P. , Frischer, T. , Barbato, A. , Snijders, D. , Maurer, E. , Lucas, J. S. , … ERS Task Force on primary ciliary dyskinesia in children . (2012). Management of primary ciliary dyskinesia in European children: Recommendations and clinical practice. The European Respiratory Journal, 39(6), 1482–1491. 10.1183/09031936.00073911 [DOI] [PubMed] [Google Scholar]
- Sunther, M. , Bush, A. , Hogg, C. , McCann, L. , & Carr, S. B. (2016). Recovery of baseline lung function after pulmonary exacerbation in children with primary ciliary dyskinesia. Pediatric Pulmonology, 51(12), 1362–1366. 10.1002/ppul.23479 [DOI] [PubMed] [Google Scholar]
- Tadd, K. , Morgan, L. , Rosenow, T. , Schultz, A. , Susanto, C. , Murray, C. , & Robinson, P. (2019). CF derived scoring systems do not fully describe the range of structural changes seen on CT scans in PCD. Pediatric Pulmonology, 54(4), 471–477. 10.1002/ppul.24249 [DOI] [PubMed] [Google Scholar]
- Tarran, R. , Grubb, B. R. , Parsons, D. , Picher, M. , Hirsh, A. J. , Davis, C. W. , & Boucher, R. C. (2001). The CF salt controversy: In vivo observations and therapeutic approaches. Molecular Cell, 8(1), 149–158. 10.1016/s1097-2765(01)00286-6 [DOI] [PubMed] [Google Scholar]
- Vanaken, G. J. , Bassinet, L. , Boon, M. , Mani, R. , Honoré, I. , Papon, J. F. , … Christin‐Maitre, S. (2017). Infertility in an adult cohort with primary ciliary dyskinesia: Phenotype‐gene association. The European Respiratory Journal, 50(5), 1700314. 10.1183/13993003.00314-2017 [DOI] [PubMed] [Google Scholar]
- Videbaek, K. , Buchvald, F. , Holgersen, M. G. , Henriksen, A. , Eriksson, F. , Garred, P. , & Nielsen, K. G. (2019). The impact of mannose‐binding lectin polymorphisms on lung function in primary ciliary dyskinesia. Pediatric Pulmonology, 54(8), 1182–1189. 10.1002/ppul.24346 [DOI] [PubMed] [Google Scholar]
- Wallmeier, J. , Nielsen, K. G. , Kuehni, C. E. , Lucas, J. S. , Leigh, M. W. , Zariwala, M. A. , & Omran, H. (2020). Motile ciliopathies. Nature Reviews Disease Primers, 6(1), 77. 10.1038/s41572-020-0209-6 [DOI] [PubMed] [Google Scholar]
- Wark, P. A. , & McDonald, V. (2003). Nebulised hypertonic saline for cystic fibrosis. Cochrane Database System Review, 1, CD001506. 10.1002/14651858.CD001506 [DOI] [PubMed] [Google Scholar]
- Werner, C. , Lablans, M. , Ataian, M. , Raidt, J. , Wallmeier, J. , Große‐Onnebrink, J. , … Omran, H. (2016). An international registry for primary ciliary dyskinesia. The European Respiratory Journal, 47(3), 849–859. 10.1183/13993003.00776-2015 [DOI] [PubMed] [Google Scholar]
- Wijers, C. D. , Chmiel, J. F. , & Gaston, B. M. (2017). Bacterial infections in patients with primary ciliary dyskinesia: Comparison with cystic fibrosis. Chronic Respiratory Disease, 14(4), 392–406. 10.1177/1479972317694621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills, P. J. , Wodehouse, T. , Corkery, K. , Mallon, K. , Wilson, R. , & Cole, P. J. (1996). Short‐term recombinant human DNase in bronchiectasis. Effect on clinical state and in vitro sputum transportability. American Journal of Respiratory and Critical Care Medicine, 154(2 Pt 1), 413–417. 10.1164/ajrccm.154.2.8756815 [DOI] [PubMed] [Google Scholar]
- Wilms, E. B. , Touw, D. J. , Heijerman, H. G. , & van der Ent, C. K. (2012). Azithromycin maintenance therapy in patients with cystic fibrosis: A dose advice based on a review of pharmacokinetics, efficacy, and side effects. Pediatric Pulmonology, 47(7), 658–665. 10.1002/ppul.21620 [DOI] [PubMed] [Google Scholar]
- Wong, C. , Jayaram, L. , Karalus, N. , Eaton, T. , Tong, C. , Hockey, H. , … Ashton, T. (2012). Azithromycin for prevention of exacerbations in non‐cystic fibrosis bronchiectasis (EMBRACE): A randomised, double‐blind, placebo‐controlled trial. Lancet, 380(9842), 660–667. 10.1016/S0140-6736(12)60953-2 [DOI] [PubMed] [Google Scholar]
- Yang, C. , & Montgomery, M. (2021). Dornase Alfa for cystic fibrosis. Cochrane Database of Systematic Reviews, 3, CD001127. 10.1002/14651858.CD001127.pub5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zihlif, N. , Paraskakis, E. , Lex, C. , Van de Pohl, L. A. , & Bush, A. (2005). Correlation between cough frequency and airway inflammation in children with primary ciliary dyskinesia. Pediatric Pulmonology, 39(6), 551–557. 10.1002/ppul.20202 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.