

SUMMARY
Occupation of living space is one of the main driving forces of adaptive evolution, especially for aquatic plants whose leaves float on the water surface and thus have limited living space. Euryale ferox, from the angiosperm basal family Nymphaeaceae, develops large, rapidly expanding leaves to compete for space on the water surface. Microscopic observation found that the cell proliferation of leaves is almost completed underwater, while the cell expansion occurs rapidly after they grow above water. To explore the mechanism underlying the specific development of leaves, we performed sequences assembly and analyzed the genome and transcriptome dynamics of E. ferox. Through reconstruction of the three sub‐genomes generated from the paleo‐hexaploidization event in E. ferox, we revealed that one sub‐genome was phylogenetically closer to Victoria cruziana, which also exhibits gigantic floating leaves. Further analysis revealed that while all three sub‐genomes promoted the evolution of the specific leaf development in E. ferox, the genes from the sub‐genome closer to V. cruziana contributed more to this adaptive evolution. Moreover, we found that genes involved in cell proliferation and expansion, photosynthesis, and energy transportation were over‐retained and showed strong expression association with the leaf development stages, such as the expression divergence of SWEET orthologs as energy uploaders and unloaders in the sink and source leaf organs of E. ferox. These findings provide novel insights into the genome evolution through polyploidization, as well as the adaptive evolution regarding the leaf development accomplished through biased gene retention and expression sub‐functionalization of multi‐copy genes in E. ferox.
Keywords: adaptive evolution, paleo‐hexaploidization, aquatic plant, sub‐genome, Euryale ferox
Significance Statement
Through the separation, reconstruction and evolutionary dissection of the three sub‐genomes of Euryale ferox, we reveal that the three sub‐genomes have different origins, with one sub‐genome showing a closer phylogenetic relationship to Victoria cruziana, which shows a similar adult leaf development process as that in E. ferox. Further analysis revealed that while all three sub‐genomes promoted the evolution of the specific leaf development process in E. ferox, the genes from the sub‐genome closer to V. cruziana contributed more to this adaptive evolution.
Moreover, we found that genes involved in cell proliferation and expansion, photosynthesis, and energy transportation were over‐retained in multi‐copies and showed strong expression association with the different leaf development stages in E. ferox.
INTRODUCTION
Evolution is one of the most important topics in biological research, and the chief unifying theory linking research on the molecular, cellular and whole organism levels. Evolution is the major factor that determines the phenotype of organisms, and these phenotypes cannot be fully understood without the context of the ecological environment in which the organisms live, especially for plant species (Bradshaw, 1965). Most plants grow in certain habitats and are unable to move during their lifespan (Blanquart et al., 2013). Therefore, adaption to their habitats is vital for the survival and reproduction of plants. Adaptive evolution is established mainly through the force of natural selection applied by environmental conditions, resulting in flora with unique morphology and physiology within different ecosystems, which then leads to biodiversity on Earth (Cox et al., 2010). Therefore, plants provide a unique opportunity to study the mechanistic basis of adaptive evolution to diverse environmental conditions (Anderson et al., 2011).
Leaves show rich shape diversity in the plant kingdom. In addition to multiple roles, including roles as a major component of plant architecture and an interface for light capture, gas exchange and thermoregulation, the leaf is the major organ of adaptive evolution that is subject to natural selection (Chitwood and Sinha, 2016). Different ecological environments gave rise to different morphologies or developmental strategies in leaves. Aquatic plants live in water environments and face rather more complicated physicochemical conditions than terrestrial plants, including low carbon dioxide and oxygen, rapid light attenuation, and wave exposure (Titus and Urban, 2009). Through long‐term adaptation, aquatic plants evolved specific leaf shapes and thereby can be grouped into four types: floating‐leaved; submerged; free‐floating; and emergent plants (Chambers et al., 2008). These aquatic plants show a striking variability of leaf morphology, such as the leaf shape and size. The adaptive plasticity of leaves enables aquatic plants to effectively regulate water availability, lighting area, gas exchange and nutrient absorption, and ensure the smooth progress of growth and development (Nakayama et al., 2013; Kim et al., 2018). The specific morphology of leaves in aquatic plants provides a resource for the study of the adaptive evolution of plants to specific environments.
Euryale ferox Salisb. (2n = 58), also known as gorgon fruit or prickly waterlily, is an annual floating‐leaved macrophyte aquatic herb that belongs to the genus Euryale from the angiosperm basal plant family Nymphaeaceae (Bradshaw, 1965; Chen, 2007). Euryale ferox develops gigantic prickly floating leaves that can grow up to 2 m in diameter within a short time, and thus can quickly occupy a large area of water surface. The adult leaf of E. ferox has a specific underwater development stage. During this stage, the leaf wraps up like a fist, and grows larger and tighter over time. Then, as the leaf raises out of the water, it rapidly expands up to about 2 m in diameter. The wrapped leaf underwater cannot photosynthesize, and the energy required for its growth should be transported from the leaves floating on the surface of water. In this way, E. ferox adult leaves can grow efficiently underwater and quickly occupy living space for photosynthesis after reaching the surface of the water. However, the mechanism of adaptive evolution underlying the rapid growth of leaves underwater and their fast expansion when raising out of the water in E. ferox is unclear.
All plant species have evolved from ancient polyploidization (paleo‐polyploidization) events (Salse, 2012; Soltis et al., 2014; Murat et al., 2017). Polyploidization, also referred to as whole‐genome duplication, generates multiple sets of sub‐genomes in a new genome. These sub‐genomes are subject to sequence fractionation (gene loss), and return to diploid status (re‐diploidization) through genomic reshuffling following paleo‐polyploidization. The repeat cycles of polyploidization, fractionation and re‐diploidization facilitate the extensive evolution of genetic material at the whole‐genome level, and are involved in the adaptive evolution of plants to diverse environments (Ohno, 1970; Otto and Whitton, 2000; Cui et al., 2006; Soltis et al., 2009; Wood et al., 2009; Jiao et al., 2011; Cheng et al., 2018; Parks et al., 2018), the functional innovation of plants (Hancock, 2005; Van de Peer et al., 2009; Schranz and Mohammadin, 2012; Vekemans et al., 2012; Hughes et al., 2014; Renny‐Byfield and Wendel, 2014; Salman‐Minkov et al., 2016), and the flourishing of the bulk of species (Schranz and Mohammadin, 2012). Although sub‐genomes are fractionated and re‐arranged with each other in the re‐diploidized genome, fragments from different sub‐genomes are usually distinguishable. Based on the genomic block (GB) system (Schranz et al., 2006), diploid ancestors of paleo‐polyploids and the evolution following polyploidizations have been revealed in many plants including Brassica (Cheng et al., 2013) and Poaceae (Murat et al., 2010), which has contributed to gene function and trait dissection studies in corresponding species. As reported previously, E. ferox experienced a paleo‐hexaploidization event (Yang et al., 2020). Further studies on the evolution of the paleo‐hexaploidization event and the sub‐genomes in E. ferox will contribute to our understanding of polyploidization in the basal Nymphaeaceae plant family.
In this study, we inferred the ancestral diploid genome before the paleo‐hexaploidization of E. ferox, and investigated its evolution on the sub‐genome level during and after the ancient hexaploidization event, as well as the featured gene function innovations that were associated with its specific leaf development, through genome assembly and transcriptome profiling in E. ferox. The findings provided new insights into the adaptive evolution of plants through the merging of ancestral genomes, the subsequent biased gene retention, the expression divergence and neo‐functionalization of multi‐copy genes.
RESULTS
Euryale ferox experienced a paleo‐hexaploidization event with no sub‐genome dominance
In order to investigate the genomic basis underlying the adaptive evolution of E. ferox to the aquatic environment, we assembled its genome using the combined methods of Nanopore and Hi‐C sequencing (Figure S1; Table S1; see Experimental Procedures). The 29 pairs of chromosomes were identified by in situ hybridization in the E. ferox (Figure S2). K‐mer counting estimated that the genome size of E. ferox was ~716 Mb (Figure S3), and the final assembled genome for E. ferox was 748.26 Mb in size, with a contig N50 of 2.47 Mb, containing 37 870 genes (Figure S4; Tables 1, S2–S4). The genomic synteny analysis between the current assembled genome of the domesticated E. ferox and the previously reported one of the wild prickly E. ferox (Yang et al., 2020) showed overall good consistence, with 16 intra‐chromosome structural variations that were supported by the Hi‐C map in our genome (Figures S5–S7).
Table 1.
The assembly statistics of the Euryale ferox genome
| Type | Contig | Scaffold | ||
|---|---|---|---|---|
| Size (Mb) | Number | Size (Mb) | Number | |
| Maximum | 13.54 | 1 | 35.44 | 1 |
| N50 | 2.47 | 83 | 24.26 | 15 |
| N90 | 0.16 | 537 | 0.18 | 165 |
| Total length | 837.60 | 1462 | 837.64 | 1098 |
| Chromosomes | / | 713.55 (87.15%) | 29 | |
| Genes | / | 48 719 | ||
| TEs | 315.18 (38.49%) | / | ||
TE, transposable element.
It was found that E. ferox had experienced a whole‐genome triplication (WGT) event compared with Nymphaea colorata (Figure S8), which was consistent with the findings of a previous study (Yang et al., 2020). Using the SynOrths tool (Cheng et al., 2012), we identified 29 163 syntenic gene pairs corresponding to 29 163 and 13 519 non‐redundant genes in the genomes of E. ferox and N. colorata, respectively. These 29 163 genes were composed of 13 414 pairs of paralogous genes in E. ferox, indicating an extra polyploidization event in E. ferox. K s values between these paralogs suggested that the polyploidization event occurred ~17 million years ago (K s = 0.17) in E. ferox (Yang et al., 2020). For the 13 519 genes in N. colorata, 3866 genes retained one copy, 5181 retained two copies and 4117 retained all three copies in E. ferox. The retention rate was 2.02 copies per gene on average, which was much higher than that of the other species (Brassica rapa = 1.59, Lupinus albus = 1.45) that experienced WGT events in a comparable historical period (K s = 0.12 in L. albus, K s = 0.29 in B. rapa; Wang et al., 2011; Xu et al., 2020), suggesting that there was much less gene fractionation following WGT in E. ferox than in other species. Furthermore, based on these syntenic gene pairs, 151 genomic fragments in E. ferox were determined, which showed synteny to the genome of N. colorata (see Experimental Procedures). For each of the genomic fragments in N. colorata, there were three copies of syntenic fragments in the genome of E. ferox, indicating a WGT event in E. ferox after its divergence from N. colorata.
The ancestral diploid genome of E. ferox before WGT was inferred to have 12 chromosomes. Using the genome of N. colorata as a reference, we sorted the three copies of syntenic fragments in the genome of E. ferox. Along each of the 14 chromosomes of N. colorata, the number of break points in the syntenic fragments of E. ferox were counted. If a break point was present in more than one syntenic fragment, then the break point was most likely inherited from the ancestral genome as multi‐copies in the E. ferox genome through the WGT event. We found that there were 22 such break points, which divided the 14 chromosomes of N. colorata into 36 GBs (Figures 1a and S9a; Tables S5 and S6). Furthermore, we counted the number of GB associations in the syntenic fragments of E. ferox. Similarly, if an association of two GBs existed in more than one copy in these syntenic fragments, then we considered it as inherited from the ancestral genome. A total of 24 such GB associations were identified (Figure 1a; Table S7). These 24 GB associations fused the 36 GBs into 12 groups. With this information, we finally inferred that the diploid ancestor of E. ferox before its hexaploidization had 12 chromosomes (AKE, ancestral karyotype of E. ferox; Figures 1b and S9b).
Figure 1.
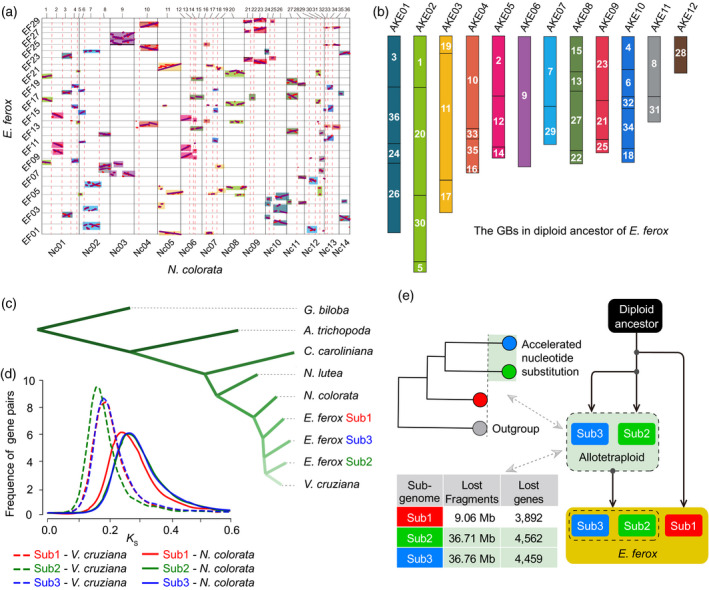
The dissection of the ancestral genome evolution of Euryale ferox.
(a) Dot‐plotting of syntenic genes between the genomes of E. ferox and Nymphaea colorata. The dotted red lines indicate the break points in syntenic fragments of E. ferox genome compared with that of N. colorata; the rectangles of the same color indicate that these fragments are associated with each other in the diploid ancestral genome of E. ferox.
(b) The karyotype of the diploid ancestral genome of E. ferox. The numbers on the bars are the IDs of corresponding genomic blocks (GBs).
(c) Phylogenetic tree of four Nymphaeaceae species and the three E. ferox sub‐genomes, as well as two species, Ginkgo biloba and Amborella trichopoda, that were used as outgroups.
(d) Frequency distribution of K s values between orthologous genes of pairwise genomes/sub‐genomes.
(e) Pattern diagram of the two‐step process for the hexaploidization of E. ferox. [Colour figure can be viewed at wileyonlinelibrary.com]
There is no sub‐genome dominance among the three sub‐genomes in E. ferox. Using the ancestral genome as the reference, we sorted the 151 genomic fragments of E. ferox. For each of the 12 AKECs (AKE chromosome), we reconstructed three copies using these genomic fragments of E. ferox, corresponding to the three sub‐genomes that merged in the WGT event. We then found that there was no significant bias of the number of genes retained among the three copies of each of the reconstructed AKECs (Figure S10; Table S8), and there was also no significant difference among the three copies in unit of the 36 GBs (Table S9). Instead, the variations of gene density in local genomic regions were caused by the loss of large genomic fragments (Figure S11; Table S10). Meanwhile, there was no significant bias in gene expression dominance toward one copy of the reconstructed AKECs (Figure S12; Table S11). These data together indicated no genome‐wide sub‐genome dominance in the paleo‐hexaploidized genome of E. ferox.
One sub‐genome in Euryale ferox is phylogenetically closer to Victoria cruziana
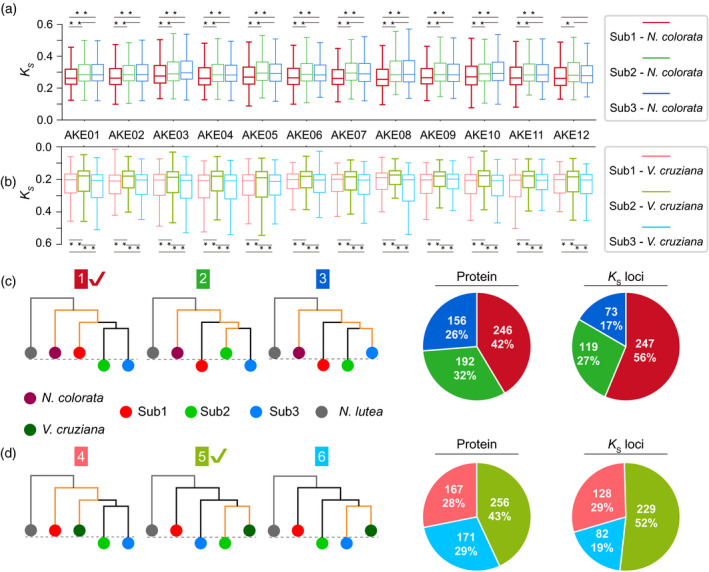
Euryale ferox experienced a specific hexaploidization process in the Nymphaeaceae. The leaves of Nymphaeaceae species are varied in size, with E. ferox and V. cruziana evolving larger leaves, and N. colorata and others developing small leaves. A previous study showed that V. cruziana and N. colorata had relatively close relationships to E. ferox compared with the other two Nymphaeaceae species (Cabomba caroliniana and Nymphaea lutea; Zhang et al., 2020). To further investigate the phylogenetic relationships among V. cruziana, N. colorata and the three sub‐genomes in E. ferox, we determined syntenic orthologous genes between the E. ferox sub‐genomes and each of V. cruziana and N. colorata. For each of the 12 AKECs of E. ferox, we found that there was always a copy of the reconstructed AKEC that showed the closest relationship to N. colorata (significantly smaller K s values) compared with the other two copies of the AKEC (Figure 2a; Table S12). This pattern was also true when the K s values were compared for each of the GBs (Table S13). Furthermore, there was also a copy of each AKEC that showed the closest relationship to V. cruziana compared with that of the other two copies of the AKEC (Figure 2b; Table S14), and the same was true at the GB level (Table S15). These data together suggested that one sub‐genome was evolutionarily closer to N. colorata, while another sub‐genome was closer to V. cruziana. Based on these results, we grouped the one copy of 12 AKECs that showed a closer relationship to N. colorata into sub‐genome 1 (Sub1, 9280 genes), the other copy of 12 AKECs that showed a closer relationship to V. cruziana into sub‐genome 2 (Sub2, 8601 genes), and the last copy of 12 AKECs into sub‐genome 3 (Sub3, 8717 genes; Table S17).
Figure 2.
The separation of the three sub‐genomes in Euryale ferox.
Boxplots of K s values on syntenic genes between Nymphaea colorata and E. ferox (a), as well as between N. colorata and E. ferox (b) for each ancestral karyotype of E. ferox (AKE) chromosome (AKEC). Variance tests were performed with * denoting P < 0.05 and ** denoting P < 0.01. The six candidate topologies of the phylogenetic tree, with the three topologies in (c) showing the phylogenetic tree on syntenic genes between the three sub‐genomes of E. ferox and the genome of N. colorata, as well as the pie diagrams for the corresponding tree (the same color) on the left. The three trees in (d) show the phylogenetic tree on orthologous genes between the three sub‐genomes of E. ferox and the genome of Victoria cruziana, as well as the pie diagrams for the corresponding tree. [Colour figure can be viewed at wileyonlinelibrary.com]
We further looked into the phylogenetic relationships among the three sub‐genomes of E. ferox, N. colorata and V. cruziana to investigate why Sub1 and Sub2 are closer to N. colorata and V. cruziana, respectively. First, 12 phylogenetic trees were built based on the orthologous gene groups of 12 individual AKECs, with N. lutea as outgroup (Figure S13). Intriguingly, all of the 12 trees supported that N. colorata clearly diverged from the diploid ancestor shared by V. cruziana and sub‐genomes of E. ferox. Meanwhile, 10 trees indicated the divergence of V. cruziana and Sub2 occurred later than Sub2 and Sub3. Furthermore, the same result was obtained with the phylogenetic tree built by 222 single‐copy gene families among the three sub‐genomes of E. ferox and the four Nymphaeaceae species (N. colorata, V. cruziana, C. caroliniana and N. lutea), as well as two outgroup species Ginkgo biloba and Amborella trichopoda (Figure 1c).
Aside from the phylogenetic relationships observed in these trees, we noticed that N. colorata was always closer with the Sub1 than with the Sub2 and Sub3 through evaluating the distances of phylogenetic branches. To further validate the differences in phylogenetic distances, we utilized an individual‐gene‐based tree method, in which the phylogenetic trees were constructed for each syntenic orthologous group using the protein sequence. Considering the potential interference of other homologous genes (showing sequence similarity with the syntenic orthologous/paralogous group) on the phylogenetic relationships of polyploid species observed in a previous study (Meng et al., 2020), we filtered out those genes having considerable homologs in these analyzed genomes, and obtained 439 syntenic orthologous gene groups for further analysis (see Experimental Procedures). As shown in Figure 2(c,d), there were six candidates for the phylogenetic trees. The proportions of individual trees supporting each candidate were counted and shown in Figure 2(c,d). Among them, trees 1 and 5 had the largest proportions (42% and 56%), which strongly suggest that Sub2 and Sub3 experienced an acceleration of nucleotide substitution compared with Sub1, while V. cruziana is closer to Sub2 than the other two sub‐genomes. We further repeated this analysis using the K S loci of gene sequences rather than the protein sequences, and consistent results were obtained (Table S16). These results together suggested that the hexaploidization occurred through a two‐step process (Figure 1e). In the first step, after the divergence of Sub2 and V. cruziana, Sub2 and Sub3 merged and formed an intermediate allotetraploid. The tetraploid then experienced sequence fractionation and relaxed selection at an accelerated rate of nucleotide substitution as reported in a previous study (Zhang et al., 2021). In the second step, the third sub‐genome Sub1 merged with the intermediate tetraploid of Sub2 and Sub3. The three sub‐genomes then experienced a further round of sequence fractionation and an accelerated rate of nucleotide substitution, and re‐diploidized into the genome of E. ferox. The two‐step polyploidization of the E. ferox genome was further supported by the size of the lost fragments and genes in the three sub‐genomes, with Sub2 and Sub3 losing significantly more genomic fragments (36.71 and 36.76 Mb sequences, respectively) and genes (4562 and 4459 genes, respectively) than Sub1 (9.06 Mb sequences and 3892 genes; Figure S11; Tables S10 and S17).
Differences in cell proliferation between early adult and adult leaves of Euryale ferox
Three types of leaf shapes (small peltate, medium round and large round) develop in three continuous growing phases (juvenile, early adult and adult, respectively) in E. ferox (Figure 3a). The growing period of small peltate leaves is ~7 days to a maximum size of ~6 cm; medium round leaves grow for ~18 days and grow to a maximum size of ~50 cm; and large round leaves develop to a maximum size of ~200 cm within ~20 days (Figure S14). The early adult and adult leaves are the main leaf form of E. ferox, sharing the same round shape and comparable growing time, while showing significant differences in the size of their finalized leaves. More interestingly, compared with the successive growth of early adult leaves from a small leaf bud to a round leaf on the surface of the water (Figure S14a,b), the growth period of adult leaves is composed of two stages: the underwater and above‐water development stages (Figure S14c). For the underwater stage, the leaf develops from a small ball‐like organ, then grows larger into a tightly wrapped‐up leaf ball, which continuously grows to ~25 cm in size. In the next above‐water stage, the leaf ball raises from the water, rapidly unfolds into a flattened leaf and expands to ~100 cm in size within 1 day, and finally develops to a maximum size of ~200 cm within 10 days (Figures 3b and 4d). It is clear that the development of adult leaves in E. ferox adopts a distinctive strategy compared with that of the early adult leaves.
Figure 3.
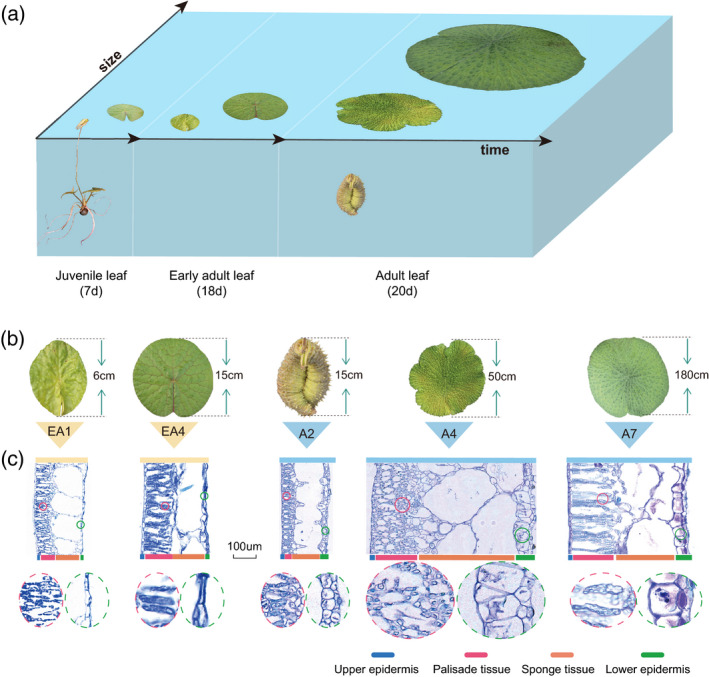
The main development process and cytological observations of Euryale ferox leaves.
(a) Different development stages of E. ferox leaves.
(b) The E. ferox early adult leaves (EA1 and EA4) and adult leaves (A2, A4, and A7).
(c) The paraffin sections (PS) of E. ferox early adult leaves (EA1 and EA4) and adult leaves (A2, A4 and A7). Differently colored bars denote the different parts of the leaf; photos inside the ellipse are the enlarged threefold views of the palisade and lower epidermis. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 4.
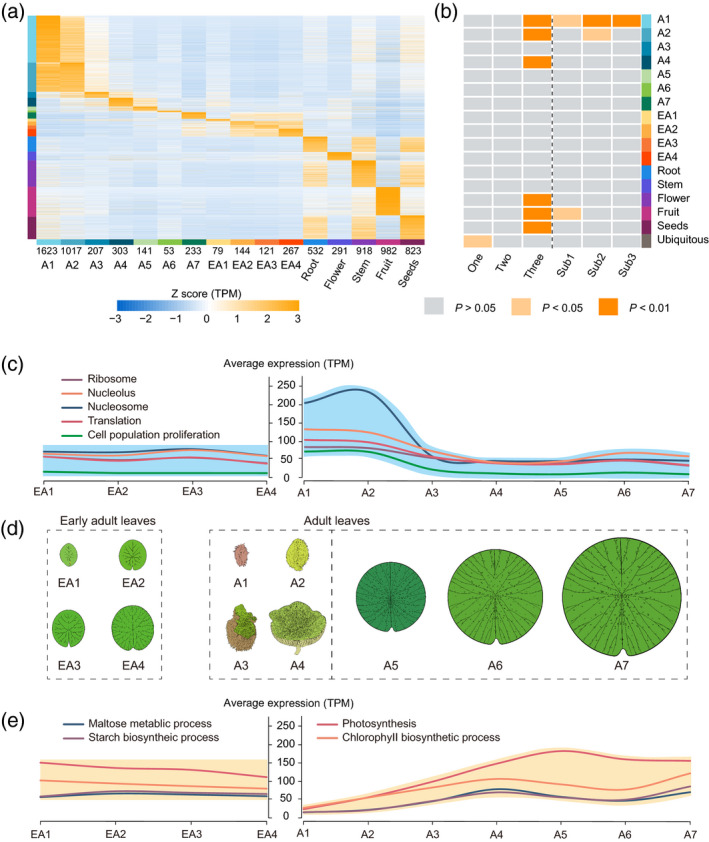
Expression atlases of Euryale ferox leaves in different developmental stages and other organs.
(a) Expression profiles of sample‐specific genes in E. ferox. Genes are listed along the y‐axis; samples are listed along the x‐axis. The numbers at the bottom of the x‐axis indicate the number of sample‐specific genes in each sample. Bars on the left of the heatmap show the sample‐specific genes, which correspond to the samples in the same colors on the bottom of the x‐axis.
(b) The heatmap shows the enrichment levels of sample‐specific genes and ubiquitous genes (y‐axis) in different sets of genes as listed in the x‐axis; one, two and three represent the copy number of polyploidized genes; Sub1, Sub2 and Sub3 represent the sub‐genomes to which the genes belong.
(c) The average expression level of differentially expressed genes (DEGs) related to photosynthesis between early adult and adult leaves.(d) The morphology of adult and early adult leaves of E. ferox. EA1–EA4 are early adult leaves; A1–A3 are adult leaves underwater; A4 is the adult leaf just rising from the water; and A5–A7 are leaves above‐water.
(e) The average expression level of DEGs related to cell proliferation between early adult and adult leaves. [Colour figure can be viewed at wileyonlinelibrary.com]
In order to explore the differences during leaf development between early adult and adult leaves of E. ferox at the cellular level, we observed their leaf tissues using paraffin sectioning (PS). The early adult leaf was sampled at four time‐series points (EA1–EA4), while the adult leaf was sampled at seven time‐series points, with three points (A1–A3) for the underwater stage, one point for when the leaf had just risen from the water (A4), and three points (A5–A7) for the above‐water stage (Figure 4d). For the early adult leaf, it was found that the cell proliferation rate decreased along with leaf development, while the cell expansion rate and airspace were increased. The activity of cell proliferation and expansion in the early adult leaf was maintained throughout the whole development process (Figures 3c and S15). In comparison, the cell proliferation activity was extremely high during the underwater stage of the adult leaf. As shown in Figures 3(c) and S15, stacking and squeezing cells occurred in palisade and spongy tissues at stage A2, and reached the peak when the leaf rose from the water. At A4–A7, along with the unfolding of the leaf, the cells of palisade and spongy tissues were rapidly expanded. Generally, cell proliferation was completed at the underwater stage and cell expansion occurred at the above‐water stage. In addition, the airspace of the aerenchyma grew larger along with the development of the leaf.
The internal structure of leaf cells was further investigated using transmission electron microscopy (TEM; Figure S16). For the early adult leaf, the palisade tissue cells in EA1 were arranged tightly and neatly. After the leaf was mature at EA4, the cells became larger and loosely arranged. The cytoplasm contained chloroplasts, mitochondria, starch granules and other organelles from EA1 to EA4. For the adult leaf, the palisade tissue cells in A2 were also arranged tightly and neatly, but the cytoplasm did not contain chloroplasts and starch granules. At A3, as the cells proliferated, the palisade tissue cells became smaller and more numerous, and the nucleus was larger and located at the center of the cell. The cytoplasm contained many chloroplasts, mitochondria and other organelles. After the leaf reached maturity (A6 and A7), the cell volume became larger, the nucleus was smaller, and the number of various organelles was reduced; only chloroplasts were present and starch grains were squeezed to the edge of the cell. There were 5–10 chloroplasts in each palisade tissue cell, which were distributed around the cell with an elliptical shape. These results observed by PS and TEM were consistent at different developmental stages of the corresponding leaf in E. ferox.
Unbalanced contribution of the three sub‐genomes to the evolution of leaf development
It was observed that the leaf development process in E. ferox was similar to that of V. cruziana. Both species develop large leaves. It would be interesting to investigate whether there are differences in the contribution of sub‐genomes to the evolution of leaf morphogenesis, as E. ferox Sub1 is phylogenetically closer to N. colorata and Sub2 is closer to V. cruziana. We performed mRNA‐seq on all seven time‐points (A1–A7) covering the whole development process of the adult leaf, and the four time‐points (EA1–EA4) for the early adult leaf, as well as on the root, stem, flower, fruit and seed organs. To better annotate the gene functions, we identified the specifically expressed genes in each of these developmental time‐points (A1–A7, EA1–EA4) or in other organs of E. ferox, and marked them with the function of corresponding sample identity using a method reported previously (Julca et al., 2021). The specificity measure (SPM) was calculated to identify sample‐specific or ubiquitously expressed genes (see Experimental Procedures). The results showed that A1 and A2 had more specifically expressed genes, followed by the fruit (Figure 4a). We then used the sample identity to annotate each of these specifically expressed genes; for example, if a gene was specifically expressed in flowers, we annotated it as a flower‐specific gene.
Using the gene annotation results, we found that A1, A2, A4, as well as the flowers, fruit and seeds were significantly enriched in genes that retained all three copies from the WGT event (Figure 4b; Table S19). Conversely, the ubiquitous genes were mainly enriched in genes that only retained single copies. The results indicated that the multi‐copy genes retained from the WGT event were important to the evolution of these organs (the adult leaf, flower, fruit and seed) in E. ferox. Moreover, we found that sub‐genomes Sub2 and Sub3 had more genes that were enriched in A1‐specific genes, and sub‐genome Sub2 had more genes that were enriched in A2‐specific genes (Figure 4b). In detail, we identified 298 A1‐specific genes that were located in Sub2 or Sub3 rather than in Sub1. These 298 genes were enriched in cytokinesis‐related functions, such as cytokinesis, microtubule binding and the regulation of meristem growth (Table S20), as well as in the function of protein serine/threonine kinase (12 PID genes). These results together indicated that both the WGT event and the sub‐genome that were closer to V. cruziana were important to the evolution of leaf development in E. ferox, further suggesting that the specific process of leaf development may have initially formed before the divergence of Sub2/3 and V. cruziana, and then contributed to the evolution of large leaf in both E. ferox and V. cruziana.
Adaptive pathways underlie the specific evolution of Euryale ferox leaf development
We further explored the gene pathways involved in the adult leaf development of E. ferox by comparing the mRNA‐seq data of A1–A7 with those of EA1–EA4. A total of 27 069 differentially expressed genes (DEGs) were identified across A1–A7. The tool Mfuzz (Futschik and Carlisle, 2005) was then used to cluster these DEGs based on their expression patterns. The results showed that these genes were separated into nine clusters (Figure S17a). Among them, cluster 9 showed a pattern of higher gene expression in the underwater stage (A1–A3) than in the above‐water stage (A5–A7), and these genes were enriched in functions including those related to phytohormones, cell population proliferation, nucleolus, nucleosome and ribosome (Figures 4c and S17b). Meanwhile, cluster 9 retained more multi‐copy genes—the average number of gene copies was 2.3, and genes in cluster 9 were under stronger selection pressure (cluster 9: K a/K s ≈ 0.20; other clusters: K a /K s ≈ 0.22–0.26; Table S21). In contrast to cluster 9, cluster 8 showed a pattern of higher gene expression in the above‐water stage than in the underwater stage, and the genes of cluster 8 were enriched in the functions of photosynthesis, substance and energy metabolism, and light harvesting (Figures 4e and S17b).
We further investigated the differences in gene expression between the adult and early adult leaves. Genes with Gene Ontology (GO) functions that were enriched in the DEGs of adult leaves were considered. For the 1567 DEGs that were involved in cell proliferation and were strongly upregulated in the underwater development stage of adult leaves, 1079 of them showed lower expression levels in the early adult leaf. Furthermore, most of these DEGs showed stable expression (74.54% showed no significant expression changes across EA1–EA4) in the early adult leaf (Figure 4c; Table S22). The gene expression pattern was consistent with the rapid cell division observed by PS and TEM during the underwater development of the adult leaf. Meanwhile, for the material‐ and energy metabolism‐related genes that were strongly upregulated in the above‐water stage of adult leaf, most of them also showed stable expression (89.70% showed no significant expression changes across EA1–EA4) in the early adult leaf (Figure 4e; Table S22). These results together indicated that the development model of adult leaf underwent specific adaptive evolution was different from that of the development of the early adult leaf in E. ferox.
Phytohormone gene pathways
Phytohormones are important signal substances that regulate the development, physiological, growth and reproduction of plants (Santner and Estelle, 2009; Durbak et al., 2012). In E. ferox, it was found that genes involved in leaf development showed significant enrichment in phytohormone signaling pathways.
For the auxin pathway, we found that the genes encoding the auxin response factors (ARFs) were highly expressed in the underwater stage of E. ferox. ARFs are key transcription factors that regulate the expression of auxin response genes (Tiwari et al., 2003; Chandler, 2016). In previous studies, 23 ARFs were identified in Arabidopsis (Guilfoyle et al., 1998). Through the HMM profile and BLASTP searching (see Experimental Procedures), we found a total of 13 and 52 ARF genes in N. colorata and E. ferox, respectively (Table S23). The ARF gene family was significantly expanded (P = 2.87 × 10−27) in E. ferox compared with N. colorata. More importantly, the expression levels of 34 out of the 52 ARF genes were found to be upregulated in the underwater development stage of adult leaves (Table S24). In addition, other phytohormone‐related genes including the gibberellin insensitive dwarf (GID) in the gibberellin pathway, histidine kinase (HK) in the cytokinin pathway, and TCH4 in the brassinosteroid pathway were also found expanded and highly expressed in the underwater stage of E. ferox (Figures S18–S20; Tables S25 and S26). The expression pattern of these expanded genes was consistent with the rapid cell proliferation and expansion that occurred during the underwater development of adult leaves.
Cell division‐ and expansion‐related genes
Twenty gene families were reported in Arabidopsis that were involved in leaf expansion through the regulation of cell expansion or cell division, including growth regulation factor (GRF) and cyclin D3 (CYCD3) genes (Gonzalez et al., 2012; Figure 5a). These 20 gene families have 83 and 232 orthologous genes in N. colorata and E. ferox, respectively (Figure S21; Tables S23 and S24), showing significant expansion in E. ferox (P = 4.02 × 10−60). These genes also showed higher retention ratios (2.52 copies per gene on average) compared with the genome‐wide background (2.02 copies).
Figure 5.
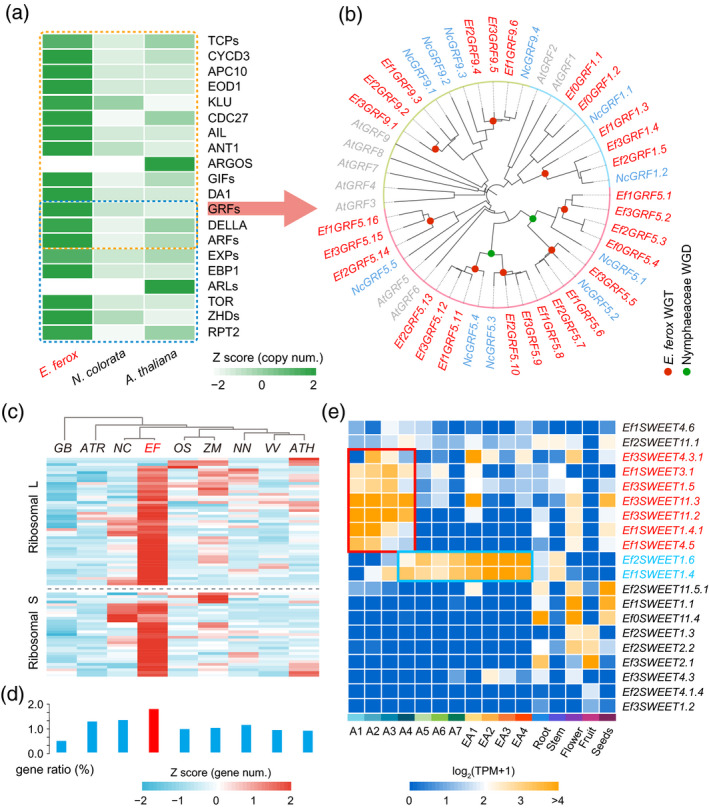
Gene families associated with the specific development of leaves in Euryale ferox.
(a) The expansion of gene families related to cell expansion and division in E. ferox, compared with that in Nymphaea colorata and Arabidopsis.
(b) Phylogenetic tree of GRF genes in E. ferox, N. colorata and Arabidopsis. Red denotes E. ferox genes, green denotes N. colorata genes, and gray denotes Arabidopsis genes.
(c) Comparisons of the number of ribosomal protein (RP) gene families in Ginkgo biloba (Gb), Amborella trichopoda (Atr), N. colorata (Nc), E. ferox (Ef), Oryza sativa (Os), Zea mays (Zm), Nelumbo nucifera (Nn), Vitis vinifera (Vv) and Arabidopsis (Ath).
(d) Ratio of the number of RP genes to the number of total genes in the corresponding genome.
(e) Expression atlases of SWEET genes in the leaves of different developmental stages and other organs of E. ferox. [Colour figure can be viewed at wileyonlinelibrary.com]
The GRF gene family plays an important regulatory role in the growth and development of leaves, and they regulate leaf size through the increase or decrease of cell numbers and size (Kim et al., 2003; Horiguchi et al., 2005). The nine Arabidopsis GRFs have 11 and 27 orthologous genes in the genomes of N. colorata and E. ferox, respectively. Among the 27 GRFs in E. ferox, 24 were triplicated (through WGT) from eight of the 11 N. colorata GRFs. As shown in Figure 5(b), the phylogenetic tree indicated that there were 15 orthologous genes of AtGRF5 in E. ferox that were expanded by WGT. Twenty‐three out of the 27 GRFs were found to be highly expressed in the underwater stage of adult leaves in E. ferox, while only three (Ef2GRF5.14, Ef3GRF5.15 and Ef1GRF5.16) of these 23 genes were continuously and stably expressed in the early adult leaf. These results together suggest that GRF genes play important roles in the rapid growth of E. ferox adult leaves during the underwater stage.
The CYCD3 genes are prime candidates for integrating cell division in leaves and lateral organ development (Dewitte et al., 2007). There are three members of CYCD3 (CYCD3;1, CYCD3;2 and CYCD3;3) in Arabidopsis. The AtCYCD3 gene had three and six orthologs in N. colorata and E. ferox, respectively (Figure S22). The six EfCYCD3s consisted of three copies of two (NC9G0092580 and NC3G0223880) out of the three NCCYCD3 genes. All six EfCYCD3s were found to be specifically expressed to higher levels in the underwater development stage of the adult leaf, which was similar to that of most GRFs, indicating the important function of EfCYCD3 in the rapid cell division during the early development of E. ferox adult leaves.
The remaining families of 20 gene families showed a similar pattern of retaining more gene copies following fractionation after the WGT event, which supported the fact that WGT played an important role in the evolution of rapid cell proliferation and thus the formation of large leaves in E. ferox. However, there were two exceptions in these gene families. One is ARGOS, which functions in cell proliferation, and the other is ARGOS‐LIKE (ARL), which is involved in the growth of large cells (Hu et al., 2003, 2006). ARGOS and ARL had no orthologous genes in either N. colorata or E. ferox, which suggested that the two gene families may not exist or were lost in the angiosperm base family Nymphaeaceae.
Ribosome protein genes
The genes for most ribosomal proteins (RPs) appear to be evolutionarily conserved among species (Zheng et al., 2016), and RPs that play important roles in ribosome biogenesis and translation have also been found to be involved in leaf development (Van Lijsebettens et al., 1994; Ramakrishnan and White, 1998; Naora and Naora, 1999; Ito et al., 2000; Maguire and Zimmermann, 2001; Yao et al., 2008; Horiguchi et al., 2011; Wang et al., 2013). A total of 665 RP genes from 81 gene families were identified in the genome of E. ferox by the PFAM database (El‐Gebali et al., 2019). Among them, 32 of 81 RP gene families were small subunit proteins, while the other 49 were large subunit proteins (Figure S23; Table S25). In comparison to the 665 RPs in E. ferox, there were fewer RPs in Arabidopsis (275), N. colorata (412), G. biloba (193), Oryza sativa (270), Zea mays (329), Nelumbo nucifera (267), Vitis vinifera (233) and A. trichopoda (213), indicating that RPs were largely expanded in E. ferox (Figure 5c,d). Moreover, the calculated value of K a/K s (0.12) was significantly lower than that of the genome‐wide level (0.25), indicating that RPs were under purifying selection in E. ferox. Furthermore, transcriptome analysis showed that ~70% of RPs (466 out of 665) were highly expressed in the underwater development of adult leaves, implying that transcription was strongly activated in this stage (Figure S23). However, in early adult leaves, most of the RP genes (94%) were expressed at lower level, which further indicated the important function of RPs in the rapid growth of adult leaves during underwater development (Table S25).
Energy transportation genes
Considering that the ball‐like wrapped leaf in the underwater stage cannot produce energy through photosynthesis itself, it was hypothesized that the above‐water adult leaf and the wrapped underwater leaf might serve as the source and sink tissues, respectively, and the transportation of carbon assimilates from the photosynthesizing leaf above‐water is vital to the rapid growth of the underwater leaf. We observed that photosynthesis‐related genes including light‐harvesting chlorophyll a/b‐binding (LHC), phosphoribulokinase (PRK), glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and phosphoglycerate kinase (PGK; Allen et al., 1981; Hariharan et al., 1998; Pfannschmidt, 2003; Klimmek et al., 2006; Gurrieri et al., 2021) were expanded in the genome of E. ferox and were highly expressed in above‐water rather than the underwater stage, suggesting the importance of these genes in the enhanced photosynthesis and energy supply in E. ferox (Figures S24 and S25; Table S26).
More importantly, it was found that the energy transportation might be facilitated by the neo‐functionalization of the multi‐copy SWEET genes. The SWEET proteins play important roles in the transportation of sucrose, glucose and fructose in plants (Baker et al., 2012; Chen et al., 2012). The SWEETs had 17 members in Arabidopsis. There were 12 and 31 members of SWEET in N. colorata and E. ferox, respectively. Interestingly, among the seven EfSWEET1s in E. ferox, two (Ef2SWEET1.6 and Ef1SWEET1.4) were highly expressed in the above‐water stage of the adult leaf, which may function in the phloem loading of energy substances, while seven genes (Ef3SWEET4.3.1, Ef1SWEET3.1, Ef3SWEET1.5, Ef3SWEET11.3, Ef3SWEET11.2, Ef1SWEET1.4.1 and Ef1SWEET4.5) were highly expressed in the underwater stage of the adult leaf (Figures 5e and S26), which may function in the unloading of bulk energy substances into the sink organ underwater after their transportation from the source organ above‐water.
DISCUSSION
The basal Nymphaeaceae family is phylogenetically and evolutionarily important for angiosperm species. There are many aquatic plants in Nymphaeaceae. Aquatic plants have evolved a variety of survival modes and diversified leaf shapes to adapt to the aquatic environment. The Nymphaeaceae species E. ferox has evolved large, rapidly growing and expanding leaves to occupy the water surface as light‐harvesting space, making E. ferox a representative species for studying the adaptive evolution of Nymphaeaceae aquatic plants to the aquatic environment. In order to investigate the mechanism underlying its specific leaf development, we surveyed the whole development process of E. ferox leaves, then also dissected the E. ferox WGT event and the evolution of multi‐copy genes after its divergence from N. colorata through genome assembly and transcriptome profiling. We revealed the evolutionary origins of the three sub‐genomes from the WGT event, as well as the biased retention of genes after WGT and the adaptive evolution of important gene pathways that were associated with the evolution of the specific leaf development strategy of E. ferox.
The systematic study of features of the paleo‐hexaploidization event in an aquatic plant genome from the basal Nymphaeaceae family adds greatly to our understanding of the effects of paleo‐polyploidization on the genome evolution and function innovation of plant species. Through the genome assembly and the comparative genomic analysis of E. ferox, the WGT event of E. ferox was extensively dissected. Deducing the diploid ancestor species has always been a challenge in polyploid study. The GB system is a good framework to perform comparative genomic analysis, which has been found to be helpful in the determination of diploid ancestors for Brassicaceae and other plant systems (Wang et al., 2011; Xu et al., 2020). In the current study, through the genomic synteny analysis between N. colorata and E. ferox, we identified the syntenic fragments between the two genomes. Following this step, the genomic fragments were used as the basic units in the analysis of the genome rearrangement of E. ferox after its WGT event. These basic units were then determined as GBs for the comparative genomic analysis in Nymphaeaceae. With the construction of the Nymphaeaceae GB system, the diploid ancestral karyotype before the WGT event of E. ferox was inferred, and it had 12 chromosomes (Table 2). The GB system of Nymphaeaceae and the ancestral karyotype of E. ferox are valuable resources for comparative genomic research and evolutionary studies in the plant family Nymphaeaceae.
Table 2.
The Euryale ferox AKECs and the information for 36 GBs using the Nymphaea colorata genome as the reference
| AKEC | GB | Nymphaea colorata reference | ||
|---|---|---|---|---|
| Chromosome | Start gene | End gene | ||
| AKE01 | 3 | NC01 | NC1G0135590 | NC1G0065420 |
| 36 | NC14 | NC14G0010460 | NC14G0175320 | |
| 24 | NC10 | NC10G0038650 | NC10G0233820 | |
| 26 | NC10 | NC10G0266740 | NC10G0042960 | |
| AKE02 | 1 | NC01 | NC1G0091250 | NC1G0101960 |
| 20 | NC08 | NC8G0271420 | NC8G0299260 | |
| 30 | NC12 | NC12G0062040 | NC12G0188260 | |
| 5 | NC01 | NC1G0178350 | NC1G0177200 | |
| AKE03 | 19 | NC07 | NC7G0031640 | NC7G0308530 |
| 11 | NC05 | NC5G0051240 | NC5G0158220 | |
| 17 | NC07 | NC7G0277000 | NC7G0279380 | |
| AKE04 | 10 | NC04 | NC4G0199180 | NC4G0236840 |
| 33 | NC13 | NC13G0195510 | NC13G0293560 | |
| 35 | NC13 | NC13G0307330 | NC13G0242890 | |
| 16 | NC07 | NC7G0293160 | NC7G0291830 | |
| AKE05 | 2 | NC01 | NC1G0101990 | NC1G0103200 |
| 12 | NC06 | NC6G0257870 | NC6G0156440 | |
| 14 | NC06 | NC6G0155490 | NC6G0257980 | |
| AKE06 | 9 | NC03 | NC3G0204700 | NC3G0229120 |
| AKE07 | 7 | NC02 | NC2G0006350 | NC2G0126710 |
| 29 | NC12 | NC12G0093270 | NC12G0094900 | |
| AKE08 | 15 | NC06 | NC6G0154420 | NC6G0265400 |
| 13 | NC06 | NC6G0156920 | NC6G0154510 | |
| 27 | NC11 | NC11G0117150 | NC11G0017360 | |
| 22 | NC09 | NC9G0234650 | NC12G0095810 | |
| AKE09 | 23 | NC09 | NC9G0276790 | NC9G0174200 |
| 21 | NC09 | NC9G0111540 | NC10G0248070 | |
| 25 | NC10 | NC10G0232680 | NC10G0248740 | |
| AKE10 | 4 | NC01 | NC1G0065430 | NC1G0178020 |
| 6 | NC02 | NC2G0041910 | NC2G0005680 | |
| 32 | NC13 | NC13G0196600 | NC13G0195510 | |
| 34 | NC13 | NC13G0140990 | NC13G0029800 | |
| 18 | NC07 | NC7G0062930 | NC7G0031650 | |
| AKE11 | 8 | NC02 | NC2G0126810 | NC2G0057050 |
| 31 | NC12 | NC12G0188810 | NC12G0183750 | |
| AKE12 | 28 | NC11 | NC11G0058340 | NC11G0138570 |
AKEC, ancestral karyotype of Euryale ferox (AKE) chromosome; GB, genomic block.
The three E. ferox sub‐genomes generated through the WGT event show different histories of divergence. Based on the diploid ancestor, we reconstructed three copies of chromosomes for each of the 12 AKECs. However, it is difficult to group the three copies of each of the 12 AKECs into three sets of sub‐genomes. Previously, the differences in gene density among the different copies of AKECs were used to separate multi‐copy AKECs into sub‐genomes (Wang et al., 2011; Xu et al., 2020). However, gene density showed no significant difference for the three copies of AKECs in E. ferox. Therefore, we used an alternative method based on the evolutionary distance between sub‐genomes and the closely‐related species of E. ferox. Fortunately, we found that N. colorata and V. cruziana, which were closely related to E. ferox, had relatively closer relationships to sub‐genomes Sub1 and Sub2, respectively. Based on this finding, we successfully separated the three copies of AKECs and grouped them into three sub‐genomes. The reconstruction of the three sub‐genomes in E. ferox then facilitated further sub‐genome‐related evolutionary and comparative analyses.
Sub‐genome dominance refers to the unequal evolution of sub‐genomes generated by allo‐polyploidization (Ohno, 1970; Otto and Whitton, 2000). This phenomenon has been found in many paleo‐allopolyploid genomes (Wang et al., 2011; Sankoff and Zheng, 2012; Xu et al., 2020), but not in pumpkin, pear or Camelina sativa (Douglas et al., 2015; Sun et al., 2017; Li et al., 2019). Based on the three sub‐genomes reconstructed, we were able to perform sub‐genome comparison in E. ferox and found that there was no genome‐wide dominance phenomenon. No significant differences were observed in gene fractionation, gene expression or transposable element (TE) distribution among the three sub‐genomes (Figures S11 and S27). Furthermore, the three sub‐genomes retained more copies of duplicated genes compared with that of the other ancient polyploidized plants whose WGT events occurred in a comparable time period (Wang et al., 2011; Xu et al., 2020). The much lower gene fractionation may be due to the lack of sub‐genome dominance. The E. ferox genome that showed no sub‐genome dominance provided an important control for the study of sub‐genome dominance.
With the findings of this study, we hypothesized that the allo‐hexaploidization of E. ferox occurred through a two‐step process (Figure 2e). The two‐step polyploidization was supported by the fact that Sub2 and Sub3 lost more genomic fragments (in two steps) than Sub1 (only one step). Considering that we have not observed the overall sub‐genome dominance among the three sub‐genomes, the two‐step hexaploidization may not be necessary for the formation of sub‐genome dominance (Cheng et al., 2014). Furthermore, as the V. cruziana shows similar leaf development to that of E. ferox, while it is different in N. colorata, it seems that the specific leaf development might have initially formed after the divergence of N. colorata and the ancestor of V. cruziana, but before the divergence of sub‐genomes Sub2 and Sub3. Sub2 and Sub3, which are closer to N. cruziana than Sub1, may have developed a similar leaf development strategy to that of N. cruziana. This possibility is supported by the findings that Sub2 and Sub3 have contributed more to genes involved in the development of the adult leaf during its underwater stage.
Euryale ferox has evolved a specific development strategy for leaves to rapidly occupy the water surface. In order to reveal the mechanism underlying the rapid growth and expansion of E. ferox leaves, we surveyed the whole development cycle of the leaf through cytological observation. The whole development cycle of the early adult leaf of E. ferox is relatively moderate, and the leaf grows up to ~50 cm in diameter in about 18 days. However, the adult leaf expands rapidly after it raises from the water and reaches a full size of up to ~200 cm in diameter within 10 days. The rapid expansion of the leaf on the surface of water resembles a sheet of shriveled paper being smoothed out, suggesting that the proliferation of leaf cells has been completed before the expansion of the leaf when the leaf raises out of the water. This is supported by the observation of the number of leaf cells at the different development stages of E. ferox leaves.
Transcription profiling analysis on different leaf development stages and the comparison between early adult and adult leaves in E. ferox, as well as the gene copy number analysis, found that genes related to pathways in phytohormones, cell division and expansion, ribosome proteins, and photosynthesis and energy transportation were likely coordinated to contribute to the adaptive evolution of E. ferox leaves to the aquatic environment. For example, we found that the gene families related to cell division and cell expansion, such as EXP, GRF, CYCD3 and TCP, were largely expanded in E. ferox and were significantly upregulated in the underwater development of the adult leaf (Table S23), suggesting their important roles in this process. Moreover, in the vegetative development stage of plants, young leaves and roots are major sink organs (Wardlaw, 1990). During the underwater development of the E. ferox adult leaf, it wraps up like a ball and does not carry out photosynthesis, which makes it a typical sink organ. In comparison, the whole growth process of the early adult leaf is above the water, and it plays a role as a source organ to provide energy for the development of the adult leaf underwater. Previous studies found that the plant SWEET gene family was mainly involved in the sugar transport between organelles, cells and organs, especially in the sugar transport from source to sink (Chen et al., 2012). SWEET transporters not only mediate sugar loading, but also are major sugar unloaders, importing energy substances from source tissues to sinks (Chen, 2014; Lin et al., 2014; Yang et al., 2018; Ho et al., 2019). SWEET genes are largely expanded in E. ferox (Figure S26). Interestingly, two out of seven copies of the gene EfSWEET1 and five other SWEET orthologs were significantly upregulated in the underwater sink organ—the ball‐like adult leaf (Figure 5e)—while the other two copies of EfSWEET1 were highly expressed in the source organs—the above‐water adult leaf and the early adult leaf. These findings suggest that the multi‐copies of EfSWEET1 and the other SWEET genes have contributed to the formation of the specific development strategy of E. ferox leaves through expression and function divergence. The example of EfSWEET1 genes supports that the hexaploidization of E. ferox provided the multi‐copy gene materials for the function innovation and adaptive evolution of the specific leaf development that allowed E. ferox to be competitive in the water environment.
The dissection of the ancestral genome evolution and the transcription features of the specific leaf development in E. ferox revealed the genomic basis and the coordination of multiple gene pathways (Figure 6) underlying the rapid growth and expansion of the large adult leaves of E. ferox. These findings are valuable to the study of adaptive evolution for not only aquatic plants, but also for other organisms living in specific environments.
Figure 6.
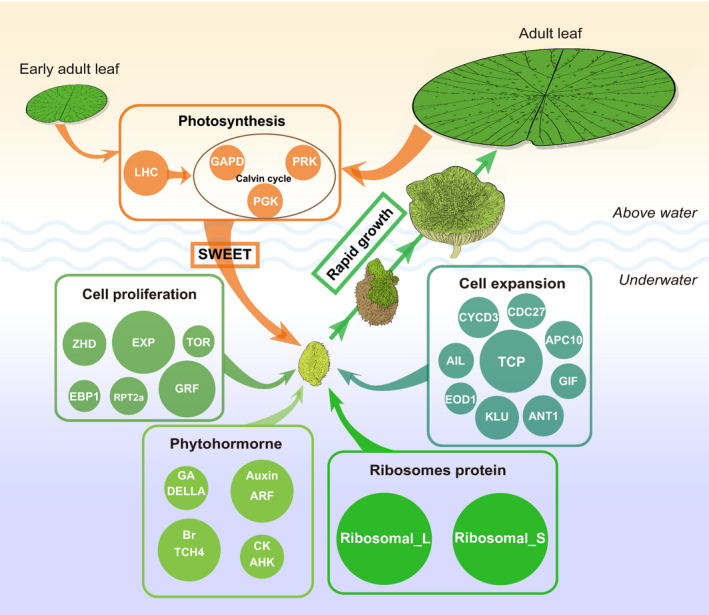
The model of coordinated gene pathways underlying the adaptive evolution of specific strategy in Euryale ferox leaf development.There are five main gene pathways involved in the leaf development of E. ferox. (i) Photosynthesis. Early adult leaves and adult leaves above water provide the energy for the development of adult leaves underwater through SWEET genes. (ii) Cell proliferation. This process occurs and is completed at the underwater development stage of adult leaves, which lays the foundation for rapid expansion of leaves after they rise above the water. (iii) Cell expansion. The high expression of cell expansion‐related genes occurs at the underwater stage, and is completed at the above‐water stage. (iv) Phytohormones and (v) ribosome proteins (RPs). These genes are related to different phytohormones (Auxin, GA, Br and CK) and RPs that are involved in leaf development and occur at the underwater stage of adult leaves. These genes were over‐retained through paleo‐hexaploidization and became involved in the regulation of E. ferox leaf development. Different sizes of circles represent the copy number of genes. LHC, light‐harvesting chlorophyll a/b‐binding; PRK, phosphoribulokinase; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; PGK, phosphoglycerate kinase; APC10, anaphase promoting complex10; GIF, grf‐interacting factor; CDC27, cell division cycle protein 27; TCP, teosinte branched1/cycloidea/pcf; GRF, growth‐regulating factor; ANT, aintegumenta; AIL, aintegumenta‐like1; CYCD3, cyclin d3; EOD1, enhancer of da1‐1; ARF, auxin response factor; KLU, kluh; EXP, expansin; EBP1, eRBb‐3 epidermal growth factor receptor binding protein; RPT2a, regulatory particle aaa‐atpase 2a; TOR, target of rapamycin; ZHD, zinc finger homeodomain; BR, brassinosteroids; GA, gibberellins; HK, histidine kinase; CK, cytokinin. [Colour figure can be viewed at wileyonlinelibrary.com]
EXPERIMENTAL PROCEDURES
Anatomical structure analysis
Tissue samples from leaves of different developmental stages were cut into small pieces (2–3 mm). A phosphate buffer solution (pH 7.2) containing glutaraldehyde (2.5%) was used to immobilize the tissue structures. The slices were initially deparaffinized in xylene and then dehydrated through an ethanol series. Then, the slices were stained with 0.1% (w/v) toluidine blue O (TBO). The section observations and image acquisition were conducted via light microscope, and the number of cells was measured by Image J (Schindelin et al., 2015).
TEM experiment
Leaf samples were immobilized in 0.1 m phosphate buffer (pH 7.0) containing 2% glutaraldehyde at 4°C for 4 h. After 30 min rinses in 0.1 m phosphate buffer (pH 7.0), samples were fixed overnight at 4°C in 1% osmium tetroxide, washed three times in 0.1 m phosphate buffer (pH 7.0), and rinsed three times with ddH2O water. Samples were dehydrated in a gradient ethanol series (50%, 70% and 90% once each, then three times in 100%) at 4°C, followed by a three times rinse in 100% 1,2‐epoxypropane. Sections 0.07 m thick were obtained using a Leica EM UC6 ultramicrotome (Vienna, Austria). Sections were observed with a HITACHI 7800 TEM (Hitachi, Japan).
Fluorescence in situ hybridization
The fluorescence in situ hybridization method for the mitotic metaphase of E. ferox was conducted as described by a previous study (Tang et al., 2014). The telomere‐specific probe was (TTTAGGGTTTAGGGTTTAGGG).
Genome sequencing and assembly
We produced 139.02 Gb (~160 ×) of Nanopore reads data (Table S1), used Canu v1.8 (Koren et al., 2017) to correct the clean data, and then used SMARTdenovo to assemble the reads based on the corrected data. Nanopore reads were used to perform three rounds of genome polishing with Racon (Li and Durbin, 2009), and Illumina data were used to performed three rounds of polishing with Pilon v1.22 (Walker et al., 2014). The clean Hi‐C reads, accounting for 80 × coverage of the E. ferox genome, were truncated at the putative Hi‐C junctions, and the resulting trimmed reads were aligned to the assembly results with BWA ‐v 0.7.17‐r1188 (Li and Durbin, 2009). Only uniquely aligned paired‐end reads with mapping quality > 20 were retained for further analysis. Invalid read pairs were filtered by HiC‐Pro v2.8.1 (Servant et al., 2015). The 58.99% of unique mapped read pairs were valid interaction pairs, and were used to correct, cluster, order and orientate the scaffolds onto chromosomes with LACHESIS (parameters: CLUSTER_MIN_RE_SITES 101, CLUSTER_MAX_LINK_DENSITY 2, ORDER_MIN_N_RES_IN_TRUN 60, ORDER_MIN_N_RES_IN_SHREDS 58; Burton et al., 2013). Placement and orientation errors were manually adjusted using Juicebox (Robinson et al., 2018). A total of 858.55 Mb of sequences was mounted on 29 chromosomes, accounting for 97.75% of the genome assembly (Figure S9). Among the localized chromosomes, the total sequence length capable of determining the sequence direction was 748.21 Mb, accounting for 87.15% of the total length of the anchored chromosome sequences (Table S2).
Evaluation of the chromosome structures
To examine the quality of the assembled E. ferox chromosome karyotypes, genomic synteny analysis was performed between the E. ferox genome obtained here and the previously published E. ferox (prickly waterlily) genome (Figure S5; Yang et al., 2020). To validate the potential structural variation, our E. ferox genome was adjusted according to the karyotype of the previous E. ferox genome and then checked whether the Hi‐C heat map was reasonable.
Repetitive element prediction
Two softwares, LTR_FINDER v1.07 (Xu and Wang, 2007) and RepeatScout v1.0.5 (Price et al., 2005), based on the principles of structure prediction and de novo prediction, respectively, were used to build a library of repeated sequences for the E. ferox genome. The library was classified by PASTEClassifier (Hoede et al., 2014) and was further merged with the Repbase (Jurka et al., 2005) database to obtain the final library. The RepeatMasker v 4.0.6 (Tarailo‐Graovac and Chen, 2009; parameter: ‐nolow ‐no_is ‐norna ‐engine wublast) was used to mask the repeated sequences of the genome based on the library.
Gene prediction and annotation
Three different strategies including ab initio prediction, homologous prediction and transcriptome prediction were used for the gene prediction. Specifically, we used Genscan (Burge and Karlin, 1997), Augustus v2.4 (Stanke and Waack, 2003), GlimmerHMM v3.0.4 (Majoros et al., 2004), GeneID v1.4 (Cochrane et al., 2016) and SNAP (Korf, 2004) to perform ab initio prediction. The protein sequences of five species including V. vinifera (Jaillon et al., 2007), Z. mays (Schnable et al., 2009), Arabidopsis (Kaul et al., 2000), N. nucifera (Gui et al., 2018) and O. sativa (Ouyang et al., 2007) were used for homology‐based prediction through GeMoMa v1.3.1 (Keilwagen et al., 2016, 2018). HISAT2 v2.1.0 (Kim et al., 2015) and Stringtie v1.2.3 (Pertea et al., 2015) were used to assemble reference‐based transcripts, and the protein coding regions were predicted by TransDecoder v2.0 and GeneMarkS‐T v5.1 (Tang et al., 2015). PASA v2.0.2 (Campbell et al., 2006) was used to predict unigene sequences from the transcripts assembled without reference based on transcriptome data using the Trinity v 2.9.0 (Haas et al., 2013). Finally, EVM v1.1.1 (Haas et al., 2008) was used to integrate the results from the above three methods, and PASA was used to modify the prediction results (Figure S10; Table S3).
Determination of syntenic gene pairs and fragments
The tool SynOrths (Cheng et al., 2012) with default parameters was used to perform syntenic gene identification. Syntenic gene pairs distributed continuously along chromosomes in the two genomes were considered as ancestral fragments inherited by the two species. Based on this principle, we identified large‐scale genomic fragments under synteny between the N. colorata and E. ferox by linking adjacent syntenic gene pairs. Due to the potential local structural variation and the errors of genome assembly in either or both genomes, local syntenic gene pairs may not be distributed immediately adjacent to other syntenic genes in one or both genomes. Therefore, if two pairs of syntenic genes were interrupted by less than 50 genes or had a distance less than 300 kb, they were grouped into one pair of syntenic fragments. Based on syntenic gene pairs between E. ferox and N. colorata, 168 syntenic fragments were identified in E. ferox. These syntenic fragments corresponded to 91.83% and 79.66% of E. ferox and N. colorata genomes, respectively.
Definition of GBs in the Nymphaeaceae
Two types of genomic breakages separated the E. ferox genome into 168 fragments. The first type was caused by the different genomic order between the N. colorata and the E. ferox ancestral diploid genome before the WGT, i.e. the loss of synteny between these two species, which was caused by the genomic reshuffling, occurred pre the WGT event of E. ferox. Therefore, this type of breakage should exist as two or three copies that were triplicated and inherited through the WGT into the three sub‐genomes and interrupted by randomly‐ and independently‐occurring fusion events in no more than one sub‐genome. The second type of breakage occurred in the E. ferox genome after the WGT; because such breakages occurred randomly and independently in the three sub‐genomes, they should exist as one copy in one of the sub‐genomes. A total of 22 break points were identified as the first type of breakage (Table S6). These 22 break points and 13 separations of 11 chromosomes divided the N. colorata genome into 36 GBs (Figures 2a and S13a).
Deciphering the ancestral diploid genome of Euryale ferox
The syntenic fragments between the N. colorata genome and E. ferox genome were compared, and genomic fusions that existed in E. ferox but not in the N. colorata genome were screened. Similar to the genomic breakages, if two or three sub‐genomes had the same association of GBs, we considered that the GB association had existed in the ancestral diploid genome of E. ferox. In other words, the associated GBs exist on the same chromosome of the ancestral diploid genome (AKEC). The fusion of GBs occurred randomly and independently in the three sub‐genomes after the WGT, so the probability was low for a novel fusion of GB association to have occurred in more than one sub‐genome. A total of 24 such GB associations were found (Table S7), which assigned the 36 GBs in the E. ferox genome into 12 groups corresponding to the 12 AKECs of the E. ferox ancestral diploid genome (Figure 1b).
Reconstruction of three sub‐genomes chromosomes in Euryale ferox
Each sub‐genome of E. ferox contained 36 GBs, which were able to be further assembled into 12 AKECs. Therefore, according to the deduced 12 AKECs of the diploid ancestor of E. ferox, the three sets of AKECs for three sub‐genomes were reconstructed based on two main principles: (i) the block inside a chromosome should not have overlapping and redundant fragments; (ii) each block should be rearranged as little as possible according to the position of the chromosome.
Reconstruction of three sub‐genomes in Euryale ferox
The K s value between orthologous gene pairs of E. ferox, N. colorata and V. cruziana were used to represent the divergence time. The K s values were grouped separately for each set of E. ferox AKECs, and the F‐test was then performed with the K s values of the three copies of each AKEC. For each copy of the 12 AKECs, there was always one copy of the AKECs that had a significantly close relationship with N. colorata, and one copy that had a significantly close relationship with V. cruziana (Figure 3a,b). Then, the copy of AKECs that were close to N. colorata were grouped as sub‐genome Sub1, the copy of AKECs that were close to V. cruziana were grouped as sub‐genome Sub2, and the remaining chromosomes were grouped as sub‐genome Sub3.
Determination of single‐copy gene families
To avoid false‐positive results from alternative splicing, transcript‐based proteins need to be de‐redundant. We used cd‐hit v4.6 (−c 0.9 ‐aS 0.9 ‐d 0; Fu et al., 2012) to remove the potential redundant transcript‐based proteins of V. cruziana, C. caroliniana, Nymphaea advena, N. lutea and Nymphaea thermarum (data from previous study; Zhang et al., 2020). OrthoMCL (Li et al., 2003; https://github.com/apetkau/orthomcl‐pipeline) was used to cluster the orthologous groups of the E. ferox sub‐genomes, N. colorata genome, G. biloba genome, A. trichopoda genome, V. cruziana transcriptome, C. caroliniana transcriptome, N. advena transcriptome, N. lutea transcriptome and N. thermarum transcriptome. We then selected orthologous groups with one gene per species/sub‐genome as single‐copy gene families. Finally, a total of 222 single‐copy gene families were identified and then used to construct a phylogenetic tree.
K a, K s, K a/K s analysis and phylogenetic trees construction
The K a, K s and K a/K s values of gene pairs were calculated by paraAt v2.0 (Zhang et al., 2012; parameters ‘‐f axt ‐m mafft’) and KaKs_Calculator v2.0 (Wang et al., 2010; parameters ‘‐m NG’). For the phylogenetic tree construction of Nymphaeaceae species, we aligned the 222 single‐copy gene family sequences with mafft v7.458 (Katoh et al., 2005; default parameters), trimmed them with trimAl v1.4.rev15 (Capella‐Gutierrez et al., 2009; parameters ‘‐automated1’), connected the trimmed sequences of each species, and then used RAxML v8.2.12 (Stamatakis, 2014; parameters ‘‐f a ‐N 100 ‐m PROTGAMMAJTT’) to construct the phylogenetic tree. Furthermore, the phylogenetic tree of each AKEC was built by orthologous gene groups among sub‐genomes of E. ferox, N. lutea, N. colorata and V. cruziana, using the same methods as for the tree of Nymphaeaceae species.
Moreover, paralogous genes among three sub‐genomes of E. ferox and their corresponding syntenic orthologous genes in N. lutea, N. colorata and V. cruziana were combined to form the orthologous groups. Orthologous groups were filtered by the number of their homologous (non‐synteny) genes (Blastp: E‐value > 1 × 10−20) in the E. ferox genome. Only orthologous groups that have no more than three homologs were kept for further analysis. After the strict filtering, 439 orthologous groups were obtained. For each orthologous group, the protein and K S loci of gene sequence were used to build a phylogenetic tree following the aforementioned method. In total, 439 individual phylogenetic trees were built. For each tree, we determined the sub‐genome, the paralogous gene from which was closest to N. colorata or V. cruziana, according to the branch length of the tree. Finally, the number of each trees were counted and summarized.
Comparison of dominant expression between paralogous gene pairs
The transcriptome data of six tissues (root, stem, leaf, flower, fruit and seed) were used to identify the dominantly expressed paralogous genes in E. ferox. The DEGs among the three sub‐genomes were calculated by DESeq2 v1.26.0 (Anders and Huber, 2010; ¦log2FoldChange¦ > 1, Padj < 0.05). The dominantly expressed genes were counted based on the number in the three copies of each AKEC. Whether the number of dominantly expressed gene differed in three copies of AKECs were identified by χ2 test (P‐value < 0.05).
TE distribution in neighboring regions of Euryale ferox genes
We used a 100‐bp sliding window with a 10‐bp step moving across the 5′ and 3′ flanking regions of genes to calculate the TE density. In each 100‐bp window, we calculated the density of the TE sequence and then averaged the density across each sub‐genome gene of the E. ferox genome. The averaged values were plotted as the TE density in the flanking region of these sub‐genomes of E. ferox.
Identification of gene families
The protein sequences of gene families in Arabidopsis were compared with E. ferox genes and N. colorata genes by Blastp with E‐value > 1 × 10−5. Then, the gene family candidate protein sequences were used to build the phylogenetic tree with Phylogeny of MEGAX (parameters ‘Method: Neighbor, Test: Bootstrap, No. of bootstrap Replications: 100, Model: Poisson model, Rates among Sites: Uniform Rates, Gaps Data Treatment: Pairwise deletion.’; Kumar et al., 2018). The homologous genes with Arabidopsis genes that were on the same branch were retained. The RP gene family were identified by the hmmscan in HMMER v3.2.1 (Potter et al., 2018) with PFAM database (El‐Gebali et al., 2019). The phylogenetic trees were visualized by figtree (https://github.com/rambaut/figtree).
RNA‐seq data analysis
The transcriptome data were mapped to the E. ferox genome using HISAT2 v2.1.0 (Kim et al., 2015; default parameters). The number of reads on each gene were counted by featureCounts v1.6.0 (Liao et al., 2014; parameters ‘‐p ‐F GTF’). The gene expression levels in all the samples were calculated using transcripts per kilobase of exon model per million mapped reads (TPM).
Analyses of time‐series gene expression profiles
Only genes with the sum of TPM values ≥ 1 across the time‐series data were retained for the following analyses. DEGs identified between at least two periods were considered as genes with fluctuating expression. These genes were clustered according to their time‐series gene expression patterns using the R package Mfuzz v2.46.0 (parameter ‘min.mem = 0.4’; Futschik and Carlisle, 2005).
Identifying tissue‐specific genes
Tissue‐specific genes based on expression data were detected by calculating the SPM, using a similar method described in a previous study (Julca et al., 2021). For each gene, we calculated the average TPM value in each sample. Then, the SPM value of a gene in a sample was computed by dividing the average TPM in the sample by the sum of the average TPM values of all samples. The SPM value ranged from 0 (a gene was not expressed in a tissue) to 1 (a gene was fully tissue‐specific). We sorted all SPM values from largest to smallest, and selected the top 5% as tissue‐specific genes.
Enrichment analysis of gene sets
The KEGG and GO annotations of E. ferox were identified by the KEGG database (Kanehisa and Goto, 2000) and GO database (Dimmer et al., 2012), respectively. Function enrichment analysis for target genes was performed by the R package clusterProfiler (Yu et al., 2012). For the tissue‐specific gene enrichment, E. ferox genes were divided into three gene sets according to two classification methods: the first was based on which sub‐genome the genes located (Sub1, Sub2, Sub3); the second was based on the copy number of genes (one, two, three) in three sub‐genomes. The tissue‐specific gene enrichment analysis was conducted using a right‐sided hypergeometric test followed by the Benjamini–Hochberg (BH) correction. The enriched clusters with a P‐value < 0.05 were considered statistically significant.
Analyses of expression profiles of cell proliferation and photosynthesis‐related genes
The genes related to cell proliferation and photosynthesis were identified by GO annotation. The genes annotated as ‘GO:0006412: translation’, ‘GO:0005840: ribosome’, ‘GO:0005730: nucleolus’, ‘GO:0000786: nucleosome’ and ‘GO:0008283: cell proliferation’ were selected as proliferation‐related genes. The genes annotated as ‘GO:0000023: maltose metabolic process’, ‘GO:0019252: starch biosynthetic process’, ‘GO:0015979: photosynthesis’ and ‘GO:0015995: chlorophyll biosynthetic process’ were selected as photosynthesis‐related genes. The average expression of all genes in each GO term was then calculated for different periods.
CONFLICT OF INTERESTS
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
LL, XC and FC conceived the project; LZ, KZ, PW, FC and YF performed the data analysis; YY, FS, AL and YZ prepared plant materials; PW, XX, SZ and KF conceived and designed the experimental design; PW, LZ and FC wrote the article.
Supporting information
Figure S1. Hi‐C‐based clustering of the E. ferox chromosomes.
Figure S2. Visualization of metaphase chromosomes of E. ferox after fluorescence in situ hybridization.
Figure S3. E. ferox genome size and heterozygosity estimation results.
Figure S4. Characterization of the E. ferox genome with circos plotting.
Figure S5. Chromosomal synteny comparison between two E. ferox genomes, the green circles mark the break points of chromosome structural variants.
Figure S6. Hi‐C‐heatmap of our E. ferox genome before and after adjustment according to the prick waterlily genome.
Figure S7. Hi‐C‐heatmap of our E. ferox genome before and after adjustment according to the prick waterlily genome.
Figure S8. Frequency distribution of K s values between homologs genes.
Figure S9. Genome evolution trajectory of E. ferox.
Figure S10. Sub‐genome features observed in the paleo‐allopolyploidy genome of E. ferox.
Figure S11. E. ferox diploid ancestor karyotype and three sub‐genomes.
Figure S12. Differential expression of paralogous gene pairs.
Figure S13. Phylogenetic trees of 12 AKECs.
Figure S14. E. ferox early adult leaves of different stage.
Figure S15. (a,b) The paraffin section of E. ferox early adult leaves (EA1–EA4) and adult leaves cross‐section (A2–A7). (c,d) The cell number of E. ferox different leaf tissues at different development stage.
Figure S16. The ultrastructure of E. ferox palisade tissue cell early adult leaves (EA1–EA4) and adult leaves (A2–A7).
Figure S17. Clustering and GO enrichment analyses of differentially expressed genes at adult leaves.
Figure S18. Expression of HK gene family of E. ferox.
Figure S19. Expression of GID, DELLA and PIF3 gene families of E. ferox.
Figure S20. Expression of TCH4 gene family of E. ferox.
Figure S21. Expression of cell expansion and cell proliferation relation of gene family of E. ferox.
Figure S22. The CYCD3 gene family.
Figure S23. Gene families for ribosomal protein.
Figure S24. Expression of LHC gene family of E. ferox.
Figure S25. Expression of PRK, PGK and GAPDH gene families of E. ferox.
Figure S26. Phylogenetic tree of SWEET gene homologous in E. ferox, N. colorata and A. thaliana.
Figure S27. The difference in average TE density in the vicinity regions of paralogous genes from the three sub‐genomes of E. ferox.
Table S1.The statistics of Nanopore reads
Table S2. The length and the number of contigs in each chromosome of E. ferox
Table S3. The statistics of genes in each prediction process
Table S4. The statistics of repeat sequence
Table S5. The 36 genomic blocks (GB) and their intervals in genome of N. colorata
Table S6. GB break points in E. ferox genome
Table S7. The GB associations in the genome of E. ferox
Table S8. The P‐values of χ2 test on the number of genes retained between sub‐genomes in each ancestral chromosome
Table S9. The P‐values of χ2 test on the number of genes retained between pairs of sub‐genomes in each genomic block
Table S10. The large fragments missing in sub‐genomes of E. ferox
Table S11. Number of paralog gene pairs differentially expressed in six different tissues
Table S12. Testing for significant differences in KS values between N. colorata and E. ferox sub‐genomes
Table S13. Testing for significant differences in KS values between N. colorata and E. ferox sub‐genomes in each genomic block
Table S14. Testing for significant differences in KS values between V. cruziana and E. ferox sub‐genomes
Table S15. Testing for significant differences in KS values between V. cruziana and E. ferox sub‐genomes in each genomic block
Table S16. The number distribution of the six topologies in the 12 ancestral chromosomes
Table S17. The three sub‐genomes identified in E. ferox genome
Table S18. Testing for significant differences in KS values between sub‐genomes of E. ferox
Table S19. Enrichment of tissue‐specific genes
Table S20. GO function enrichment of SSMG
Table S21. Polyploidy gene retention and selective pressure of cluster genes of E. ferox
Table S22. Expression of different functional genes in early adult and adult leaf
Table S23. The number and id of genes for cell division and expansion in A. thaliana, E. ferox and N. colorata
Table S24. Expression of gene families associated with cell division expansion in E. ferox
Table S25. Expression of gene families associated with phytohormone, ribosomal protein and photosynthesis in E. ferox
ACKNOWLEDGEMENTS
The authors gratefully thank Professor Xiaowu Wang (Chinese Academy of Agricultural Sciences, China) for the helpful advice and discussion of this manuscript. This work was supported by the National Natural Science Foundation of China (grant number: 31902002), the China Agriculture Research System (grant number: CARS‐24), China Postdoctoral Science Foundation (grant number: 2020 T130706), and the Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences, and the Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, P.R. China, Jiangsu seed industry revitalization ‘Jie Bang Gua Shuai’ project (grant number: JBGS [2021]017).
Linked article: This paper is the subject of a Research Highlight article. To view this Research Highlight article visit https://doi.org/10.1111/tpj.15777.
Contributor Information
Xuehao Chen, Email: xhchen@yzu.edu.cn.
Feng Cheng, Email: chengfeng@caas.cn.
Liangjun Li, Email: ljli@yzu.edu.cn.
DATA AVAILABILITY STATEMENT
Nanopore whole‐genome sequencing data, Illumina data, Hi‐C data and transcriptome data have been deposited to the NCBI Sequence Read Archive (SRA) as BioProject PRJNA769254 and PRJNA769560. The genome assembly sequences and gene annotations have been deposited in the Genome Warehouse in the BIG Data Center under accession number GWHBFHH00000000.
REFERENCES
- Allen, J.F. , Bennett, J. , Steinback, K.E. & Arntzen, C.J. (1981) Chloroplast protein phosphorylation couples plastoquinone redox state to distribution of excitation energy between photosystems. Nature, 291, 25–29. [Google Scholar]
- Anders, S. & Huber, W. (2010) Differential expression analysis for sequence count data. Genome Biology, 11, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, J.T. , Willis, J.H. & Mitchell‐Olds, T. (2011) Evolutionary genetics of plant adaptation. Trends in Genetics, 27, 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, R.F. , Leach, K.A. & Braun, D.M. (2012) SWEET as sugar: new sucrose effluxers in plants. Molecular Plant, 5, 766–768. [DOI] [PubMed] [Google Scholar]
- Blanquart, F. , Kaltz, O. , Nuismer, S.L. & Gandon, S. (2013) A practical guide to measuring local adaptation. Ecology Letters, 16, 1195–1205. [DOI] [PubMed] [Google Scholar]
- Bradshaw, A.D. (1965) Evolutionary significance of phenotypic plasticity in plants. Advances in Genetics, 13, 115–155. [Google Scholar]
- Burge, C. & Karlin, S. (1997) Prediction of complete gene structures in human genomic DNA. Journal of Molecular Biology, 268, 78–94. [DOI] [PubMed] [Google Scholar]
- Burton, J.N. , Adey, A. , Patwardhan, R.P. , Qiu, R.L. , Kitzman, J.O. & Shendure, J. (2013) Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology, 31, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, M.A. , Haas, B.J. , Hamilton, J.P. et al . (2006) Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics, 7, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutierrez, S. , Silla‐Martinez, J.M. & Gabaldon, T. (2009) trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics, 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers, P.A. , Lacoul, P. , Murphy, K.J. & Thomaz, S.M. (2008) Global diversity of aquatic macrophytes in freshwater. Hydrobiologia, 595, 9–26. [Google Scholar]
- Chandler, J.W. (2016) Auxin response factors. Plant, Cell & Environment, 39, 1014–1028. [DOI] [PubMed] [Google Scholar]
- Chen, L.Q. (2014) SWEET sugar transporters for phloem transport and pathogen nutrition. The New Phytologist, 201, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Chen, L.‐Q. , Qu, X.‐Q. , Hou, B.‐H. , Sosso, D. , Osorio, S. , Fernie, A.R. et al . (2012) Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science, 335, 207–211. [DOI] [PubMed] [Google Scholar]
- Chen, Z.J. (2007) Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annual Review of Plant Biology, 58, 377–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. , Mandakova, T. , Wu, J. , Xie, Q. , Lysak, M.A. & Wang, X. (2013) Deciphering the diploid ancestral genome of the Mesohexaploid Brassica rapa. Plant Cell, 25, 1541–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. , Wu, J. , Cai, X. , Liang, J. , Freeling, M. & Wang, X. (2018) Gene retention, fractionation and subgenome differences in polyploid plants. Nature plants, 4, 258–268. [DOI] [PubMed] [Google Scholar]
- Cheng, F. , Wu, J. , Fang, L. & Wang, X. (2012) Syntenic gene analysis between Brassica rapa and other Brassicaceae species. Frontiers in Plant Science, 3, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. , Wu, J. & Wang, X. (2014) Genome triplication drove the diversification of brassica plants. Horticulture Research, 1, 14024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood, D.H. & Sinha, N.R. (2016) Evolutionary and environmental forces sculpting leaf development. Current Biology, 26, R297–R306. [DOI] [PubMed] [Google Scholar]
- Cochrane, G. , Karsch‐Mizrachi, I. , Takagi, T. & International Nucleotide Sequence Database Collaboration . (2016) The international nucleotide Sequence database collaboration. Nucleic Acids Research, 44, D48–D50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, J. , Schubert, A.M. , Travisano, M. & Putonti, C. (2010) Adaptive evolution and inherent tolerance to extreme thermal environments. BMC Evolutionary Biology, 10, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, L. , Wall, P.K. , Leebens‐Mack, J.H. , Lindsay, B.G. , Soltis, D.E. , Doyle, J.J. et al . (2006) Widespread genome duplications throughout the history of flowering plants. Genome Research, 16, 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewitte, W. , Scofield, S. , Alcasabas, A.A. , Maughan, S.C. , Menges, M. , Braun, N. et al . (2007) Arabidopsis CYCD3 D‐type cyclins link cell proliferation and endocycles and are rate‐limiting for cytokinin responses. Proceedings of the National Academy of Sciences of the United States of America, 104, 14537–14542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer, E.C. , Huntley, R.P. , Alam‐Faruque, Y. , Sawford, T. , O'Donovan, C. , Martin, M.J. et al . (2012) The UniProt‐GO annotation database in 2011. Nucleic Acids Research, 40, D565–D570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas, G.M. , Gos, G. , Steige, K.A. , Salcedo, A. , Holm, K. , Josephs, E.B. et al . (2015) Hybrid origins and the earliest stages of diploidization in the highly successful recent polyploid Capsella bursa‐pastoris. Proceedings of the National Academy of Sciences of the United States of America, 112, 2806–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbak, A. , Yao, H. & McSteen, P. (2012) Hormone signaling in plant development. Current Opinion in Plant Biology, 15, 92–96. [DOI] [PubMed] [Google Scholar]
- El‐Gebali, S. , Mistry, J. , Bateman, A. , Eddy, S.R. , Luciani, A. , Potter, S.C. et al . (2019) The Pfam protein families database in 2019. Nucleic Acids Research, 47, D427–d432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L. , Niu, B. , Zhu, Z. , Wu, S. & Li, W. (2012) CD‐HIT: accelerated for clustering the next‐generation sequencing data. Bioinformatics, 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futschik, M.E. & Carlisle, B. (2005) Noise‐robust soft clustering of gene expression time‐course data. Journal of Bioinformatics and Computational Biology, 3, 965–988. [DOI] [PubMed] [Google Scholar]
- Gonzalez, N. , Vanhaeren, H. & Inze, D. (2012) Leaf size control: complex coordination of cell division and expansion. Trends in Plant Science, 17, 332–340. [DOI] [PubMed] [Google Scholar]
- Gui, S. , Peng, J. , Wang, X. , Wu, Z. , Cao, R. , Salse, J. et al . (2018) Improving Nelumbo nucifera genome assemblies using high‐resolution genetic maps and BioNano genome mapping reveals ancient chromosome rearrangements. The Plant Journal, 94, 721–734. [DOI] [PubMed] [Google Scholar]
- Guilfoyle, T.J. , Ulmasov, T. & Hagen, G. (1998) The ARF family of transcription factors and their role in plant hormone‐responsive transcription. Cellular and Molecular Life Sciences, 54, 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurrieri, L. , Fermani, S. , Zaffagnini, M. , Sparla, F. & Trost, P. (2021) Calvin‐Benson cycle regulation is getting complex. Trends in Plant Science, 26, 898–912. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Papanicolaou, A. , Yassour, M. , Grabherr, M. , Blood, P.D. , Bowden, J. et al . (2013) De novo transcript sequence reconstruction from RNA‐seq using the trinity platform for reference generation and analysis. Nature Protocols, 8, 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. et al . (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology, 9, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, J.F. (2005) Contributions of domesticated plant studies to our understanding of plant evolution. Annals of Botany, 96, 953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan, T. , Johnson, P.J. & Cattolico, R.A. (1998) Purification and characterization of phosphoribulokinase from the marine chromophytic alga Heterosigma carterae. Plant Physiology, 117, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, L.‐H. , Klemens, P.A.W. , Neuhaus, H.E. , Ko, H.‐Y. , Hsieh, S.‐Y. & Guo, W.‐J. (2019) SlSWEET1a is involved in glucose import to young leaves in tomato plants. Journal of Experimental Botany, 70, 3241–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoede, C. , Arnoux, S. , Moisset, M. , Chaumier, T. , Inizan, O. , Jamilloux, V. et al . (2014) PASTEC: an automatic transposable element classification tool. PLoS One, 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi, G. , Kim, G.T. & Tsukaya, H. (2005) The transcription factor AtGRF5 and the transcription coactivator AN3 regulate cell proliferation in leaf primordia of Arabidopsis thaliana. Plant Journal, 43, 68–78. [DOI] [PubMed] [Google Scholar]
- Horiguchi, G. , Mollá‐Morales, A. , Pérez‐Pérez, J.M. , Kojima, K. , Robles, P. , Ponce, M.R. et al . (2011) Differential contributions of ribosomal protein genes to Arabidopsis thaliana leaf development. The Plant Journal, 65, 724–736. [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Poh, H.M. & Chua, N.‐H. (2006) The Arabidopsis ARGOS‐LIKE gene regulates cell expansion during organ growth. Plant Journal, 47, 1–9. [DOI] [PubMed] [Google Scholar]
- Hu, Y.X. , Xie, O. & Chua, N.H. (2003) The Arabidopsis auxin‐inducible gene ARGOS controls lateral organ size. Plant Cell, 15, 1951–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, T.E. , Langdale, J.A. & Kelly, S. (2014) The impact of widespread regulatory neofunctionalization on homeolog gene evolution following whole‐genome duplication in maize. Genome Research, 24, 1348–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, T. , Kim, G.T. & Shinozaki, K. (2000) Disruption of an Arabidopsis cytoplasmic ribosomal protein S13‐homologous gene by transposon‐mediated mutagenesis causes aberrant growth and development. Plant Journal, 22, 257–264. [DOI] [PubMed] [Google Scholar]
- Jaillon, O. , Aury, J.M. , Noel, B. , Policriti, A. , Clepet, C. , Casagrande, A. et al . (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature, 449, 463–467. [DOI] [PubMed] [Google Scholar]
- Jiao, Y. , Wickett, N.J. , Ayyampalayam, S. , Chanderbali, A.S. , Landherr, L. , Ralph, P.E. et al . (2011) Ancestral polyploidy in seed plants and angiosperms. Nature, 473, 97–100. [DOI] [PubMed] [Google Scholar]
- Julca, I. , Ferrari, C. , Flores‐Tornero, M. , Proost, S. , Lindner, A.‐C. , Hackenberg, D. et al . (2021) Comparative transcriptomic analysis reveals conserved programmes underpinning organogenesis and reproduction in land plants. Nature Plants, 7, 1143–1159. [DOI] [PubMed] [Google Scholar]
- Jurka, J. , Kapitonov, V.V. , Pavlicek, A. , Klonowski, P. , Kohany, O. & Walichiewicz, J. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research, 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kanehisa, M. & Goto, S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Kuma, K. , Toh, H. & Miyata, T. (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Research, 33, 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul, S. , Koo, H.L. , Jenkins, J. , Rizzo, M. , Rooney, T. , Tallon, L.J. et al . (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408, 796–815. [DOI] [PubMed] [Google Scholar]
- Keilwagen, J. , Hartung, F. , Paulini, M. , Twardziok, S.O. & Grau, J. (2018) Combining RNA‐seq data and homology‐based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen, J. , Wenk, M. , Erickson, J.L. , Schattat, M.H. , Grau, J. & Hartung, F. (2016) Using intron position conservation for homology‐based gene prediction. Nucleic Acids Research, 44, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Landmead, B. & Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nature Methods, 12, 357–U121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Joo, Y. , Kyung, J. , Jeon, M. , Park, J.Y. , Lee, H.G. et al . (2018) A molecular basis behind heterophylly in an amphibious plant, Ranunculus trichophyllus. PLoS Genetics, 14, e1007208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.H. , Choi, D.S. & Kende, H. (2003) The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant Journal, 36, 94–104. [DOI] [PubMed] [Google Scholar]
- Klimmek, F. , Sjodin, A. , Noutsos, C. , Leister, D. & Jansson, S. (2006) Abundantly and rarely expressed Lhc protein genes exhibit distinct regulation patterns in plants. Plant Physiology, 140, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B.P. , Berlin, K. , Miller, J.R. , Bergman, N.H. & Phillippy, A.M. (2017) Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Research, 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. & Tamura, K. (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. & Durbin, R. (2009) Fast and accurate short read alignment with burrows‐wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J., Jr. & Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Research, 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Qiao, X. , Yin, H. , Zhou, Y. , Dong, H. , Qi, K. et al . (2019) Unbiased subgenome evolution following a recent whole‐genome duplication in pear (Pyrus bretschneideri Rehd.). Horticulture Research, 6, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Smyth, G.K. & Shi, W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin, I.W. , Sosso, D. , Chen, L.Q. , Gase, K. , Kim, S.G. , Kessler, D. et al . (2014) Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature, 508, 546–549. [DOI] [PubMed] [Google Scholar]
- Maguire, B.A. & Zimmermann, R.A. (2001) The ribosome in focus. Cell, 104, 813–816. [DOI] [PubMed] [Google Scholar]
- Majoros, W.H. , Pertea, M. & Salzberg, S.L. (2004) TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Meng, F. , Pan, Y. , Wang, J. , Yu, J. , Liu, C. , Zhang, Z. et al . (2020) Cotton duplicated genes produced by polyploidy show significantly elevated and unbalanced evolutionary rates, overwhelmingly perturbing gene tree topology. Frontiers in Genetics, 11, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat, F. , Armero, A. , Pont, C. , Klopp, C. & Salse, J. (2017) Reconstructing the genome of the most recent common ancestor of flowering plants. Nature Genetics, 49, 490–496. [DOI] [PubMed] [Google Scholar]
- Murat, F. , Xu, J.H. , Tannier, E. , Abrouk, M. , Guilhot, N. , Pont, C. et al . (2010) Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution. Genome Research, 20, 1545–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama, H. , Nakayama, N. , Nakamasu, A. , Sinha, N. & Kimura, S. (2013) Toward elucidating the mechanisms that regulate heterophylly. Plant Morphology, 24, 57–63. [Google Scholar]
- Naora, H. & Naora, H. (1999) Involvement of ribosomal proteins in regulating cell growth and apoptosis: translational modulation or recruitment for extraribosomal activity? Immunology and Cell Biology, 77, 197–205. [DOI] [PubMed] [Google Scholar]
- Ohno, S. (1970) Evolution by gene duplication. London: George Allen and Unwin. [Google Scholar]
- Otto, S.P. & Whitton, J. (2000) Polyploid incidence and evolution. Annual Review of Genetics, 34, 401–437. [DOI] [PubMed] [Google Scholar]
- Ouyang, S. , Zhu, W. , Hamilton, J. , Lin, H. , Campbell, M. , Childs, K. et al . (2007) The TIGR Rice genome annotation resource: improvements and new features. Nucleic Acids Research, 35, D883–D887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, M.B. , Nakov, T. , Ruck, E.C. , Wickett, N.J. & Alverson, A.J. (2018) Phylogenomics reveals an extensive history of genome duplication in diatoms (Bacillariophyta). American Journal of Botany, 105, 330–347. [DOI] [PubMed] [Google Scholar]
- Pertea, M. , Pertea, G.M. , Antonescu, C.M. , Chang, T.‐C. , Mendell, J.T. & Salzberg, S.L. (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology, 105, 330–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfannschmidt, T. (2003) Chloroplast redox signals: how photosynthesis controls its own genes. Trends in Plant Science, 8, 33–41. [DOI] [PubMed] [Google Scholar]
- Potter, S.C. , Luciani, A. , Eddy, S.R. , Park, Y. , Lopez, R. & Finn, R.D. (2018) HMMER web server: 2018 update. Nucleic Acids Research, 46, W200–w204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A.L. , Jones, N.C. & Pevzner, P.A. (2005) De novo identification of repeat families in large genomes. Bioinformatics, 21, I351–I358. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan, V. & White, S.W. (1998) Ribosomal protein structures: insights into the architecture, machinery and evolution of the ribosome. Trends in Biochemical Sciences, 23, 208–212. [DOI] [PubMed] [Google Scholar]
- Renny‐Byfield, S. & Wendel, J.F. (2014) Doubling down on genomes: polyploidy and crop plants. American Journal of Botany, 101, 1711–1725. [DOI] [PubMed] [Google Scholar]
- Robinson, J.T. , Turner, D. , Durand, N.C. , Thorvaldsdóttir, H. , Mesirov, J.P. & Aiden, E.L. (2018) Juicebox. Js provides a cloud‐based visualization system for hi‐C data. Cell Systems, 6, 256–258.e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman‐Minkov, A. , Sabath, N. & Mayrose, I. (2016) Whole‐genome duplication as a key factor in crop domestication. Nature plants, 2, 16115. [DOI] [PubMed] [Google Scholar]
- Salse, J. (2012) In silico archeogenomics unveils modern plant genome organisation, regulation and evolution. Current Opinion in Plant Biology, 15, 122–130. [DOI] [PubMed] [Google Scholar]
- Sankoff, D. & Zheng, C. (2012) Fractionation, rearrangement and subgenome dominance. Bioinformatics, 28, i402–i408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santner, A. & Estelle, M. (2009) Recent advances and emerging trends in plant hormone signalling. nature, 459, 1071–1078. [DOI] [PubMed] [Google Scholar]
- Schindelin, J. , Rueden, C.T. , Hiner, M.C. & Eliceiri, K.W. (2015) The ImageJ ecosystem: an open platform for biomedical image analysis. Molecular Reproduction and Development, 82, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable, P.S. , Ware, D. , Fulton, R.S. , Stein, J.C. , Wei, F. , Pasternak, S. et al . (2009) The B73 maize genome: complexity, diversity, and dynamics. Science, 326, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Schranz, M.E. , Lysak, M.A. & Mitchell‐Olds, T. (2006) The ABC's of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends in Plant Science, 11, 535–542. [DOI] [PubMed] [Google Scholar]
- Schranz, M.E. , Mohammadin, S. & Edger, P.P. (2012) Ancient whole genome duplications, novelty and diversification: the WGD radiation lag‐time model. Current Opinion in Plant Biology, 15, 147–153. [DOI] [PubMed] [Google Scholar]
- Servant, N. , Varoquaux, N. , Lajoie, B.R. , Viara, E. , Chen, C.J. , Vert, J.P. et al . (2015) HiC‐pro: an optimized and flexible pipeline for hi‐C data processing. Genome Biology, 16, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis, D.E. , Albert, V.A. , Leebens‐Mack, J. , Bell, C.D. , Paterson, A.H. , Zheng, C. et al . (2009) Polyploidy and angiosperm diversification. American Journal of Botany, 96, 336–348. [DOI] [PubMed] [Google Scholar]
- Soltis, D.E. , Visger, C.J. & Soltis, P.S. (2014) The polyploidy revolution then… And now: Stebbins revisited. American Journal of Botany, 101, 1057–1078. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. & Waack, S. (2003) Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics, 19, 215–225. [DOI] [PubMed] [Google Scholar]
- Sun, H. , Wu, S. , Zhang, G. , Jiao, C. , Guo, S. , Ren, Y. et al . (2017) Karyotype stability and unbiased fractionation in the paleo‐allotetraploid Cucurbita genomes. Molecular Plant, 10, 1293–1306. [DOI] [PubMed] [Google Scholar]
- Tang, S. , Lomsadze, A. & Borodovsky, M. (2015) Identification of protein coding regions in RNA transcripts. Nucleic Acids Research, 43, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z.X. , Li, M. , Chen, L. , Wang, Y.Y. , Ren, Z.L. & Fu, S.L. (2014) New types of wheat chromosomal structural variations NE in derivatives of wheat‐Rye hybrids. PLoS One, 9, e110282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo‐Graovac M, Chen N. 2009. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics, 25, 4.10.1–4.10.14. [DOI] [PubMed] [Google Scholar]
- Titus, J.E. & Urban, R.A. (2009) Aquatic plants: a general introduction. In: Likens, G.E. (Ed.) Encyclopedia of Inland Waters. Oxford: Academic Press, pp. 43–51. [Google Scholar]
- Tiwari, S.B. , Hagen, G. & Guilfoyle, T. (2003) The roles of auxin response factor domains in auxin‐responsive transcription. Plant Cell, 15, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer, Y. , Maere, S. & Meyer, A. (2009) The evolutionary significance of ancient genome duplications. Nature Reviews. Genetics, 10, 725–732. [DOI] [PubMed] [Google Scholar]
- Van Lijsebettens, M. , Vanderhaeghen, R. , De Block, M. , Bauw, G. , Villarroel, R. & Van Montagu, M. (1994) An S18 ribosomal protein gene copy at the Arabidopsis PFL locus affects plant development by its specific expression in meristems. The EMBO Journal, 13, 3378–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekemans, D. , Proost, S. , Vanneste, K. , Coenen, H. , Viaene, T. , Ruelens, P. et al . (2012) Gamma paleohexaploidy in the stem lineage of core eudicots: significance for MADS‐box gene and species diversification. Molecular Biology and Evolution, 29, 3793–3806. [DOI] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. et al . (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Zhang, Y. , Zhang, Z. , Zhu, J. & Yu, J. (2010) KaKs_Calculator 2.0: a toolkit incorporating gamma‐series methods and sliding window strategies. Genomics, Proteomics & Bioinformatics, 8, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Wang, H. , Wang, J. , Sun, R. , Wu, J. , Liu, S. et al . (2011) The genome of the mesopolyploid crop species Brassica rapa. Nature Genetics, 43, 1035–1039. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Hou, J. , Lu, L. , Qi, Z. , Sun, J. , Gao, W. et al . (2013) Small ribosomal protein subunit S7 suppresses ovarian tumorigenesis through regulation of the PI3K/AKT and MAPK pathways. PLoS One, 8, e79117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw, I.F. (1990) Tansley review no. 27 the control of carbon partitioning in plants. The New Phytologist, 116, 341–381. [DOI] [PubMed] [Google Scholar]
- Wood, T.E. , Takebayashi, N. , Barker, M.S. , Mayrose, I. , Greenspoon, P.B. & Rieseberg, L.H. (2009) The frequency of polyploid speciation in vascular plants. Proceedings of the National Academy of Sciences, 106, 13875–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , Zhang, Q. , Yuan, W. , Xu, F. , Muhammad Aslam, M. , Miao, R. et al . (2020) The genome evolution and low‐phosphorus adaptation in white lupin. Nature Communications, 11, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. & Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Research, 35, W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Luo, D. , Yang, B. , Frommer, W.B. & Eom, J.S. (2018) SWEET11 and 15 as key players in seed filling in rice. The New Phytologist, 218, 604–615. [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Sun, P. , Lv, L. , Wang, D. , Ru, D. , Li, Y. et al . (2020) Prickly waterlily and rigid hornwort genomes shed light on early angiosperm evolution. Nature Plants, 6, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, Y. , Ling, Q. , Wang, H. & Huang, H. (2008) Ribosomal proteins promote leaf adaxial identity. Development, 135, 1325–1334. [DOI] [PubMed] [Google Scholar]
- Yu, G. , Wang, L.‐G. , Han, Y. & He, Q.‐Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. Omics‐a Journal of Integrative Biology, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Chen, F. , Zhang, X. , Li, Z. , Zhao, Y. , Lohaus, R. et al . (2020) The water lily genome and the early evolution of flowering plants. Nature, 577, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Shen, Q. , Leng, L. , Zhang, D. , Chen, S. , Shi, Y. et al . (2021) Incipient diploidization of the medicinal plant perilla within 10,000 years. Nature Communications, 12, 5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Xiao, J.F. , Wu, J.Y. , Zhang, H.Y. , Liu, G.M. , Wang, X.M. et al . (2012) ParaAT: a parallel tool for constructing multiple protein‐coding DNA alignments. Biochemical and Biophysical Research Communications, 419, 779–781. [DOI] [PubMed] [Google Scholar]
- Zheng, M. , Wang, Y. , Liu, X. , Sun, J. , Wang, Y. , Xu, Y. et al . (2016) The RICE MINUTE‐LIKE1 (RML1) gene, encoding a ribosomal large subunit protein L3B, regulates leaf morphology and plant architecture in rice. Journal of Experimental Botany, 67, 3457–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Hi‐C‐based clustering of the E. ferox chromosomes.
Figure S2. Visualization of metaphase chromosomes of E. ferox after fluorescence in situ hybridization.
Figure S3. E. ferox genome size and heterozygosity estimation results.
Figure S4. Characterization of the E. ferox genome with circos plotting.
Figure S5. Chromosomal synteny comparison between two E. ferox genomes, the green circles mark the break points of chromosome structural variants.
Figure S6. Hi‐C‐heatmap of our E. ferox genome before and after adjustment according to the prick waterlily genome.
Figure S7. Hi‐C‐heatmap of our E. ferox genome before and after adjustment according to the prick waterlily genome.
Figure S8. Frequency distribution of K s values between homologs genes.
Figure S9. Genome evolution trajectory of E. ferox.
Figure S10. Sub‐genome features observed in the paleo‐allopolyploidy genome of E. ferox.
Figure S11. E. ferox diploid ancestor karyotype and three sub‐genomes.
Figure S12. Differential expression of paralogous gene pairs.
Figure S13. Phylogenetic trees of 12 AKECs.
Figure S14. E. ferox early adult leaves of different stage.
Figure S15. (a,b) The paraffin section of E. ferox early adult leaves (EA1–EA4) and adult leaves cross‐section (A2–A7). (c,d) The cell number of E. ferox different leaf tissues at different development stage.
Figure S16. The ultrastructure of E. ferox palisade tissue cell early adult leaves (EA1–EA4) and adult leaves (A2–A7).
Figure S17. Clustering and GO enrichment analyses of differentially expressed genes at adult leaves.
Figure S18. Expression of HK gene family of E. ferox.
Figure S19. Expression of GID, DELLA and PIF3 gene families of E. ferox.
Figure S20. Expression of TCH4 gene family of E. ferox.
Figure S21. Expression of cell expansion and cell proliferation relation of gene family of E. ferox.
Figure S22. The CYCD3 gene family.
Figure S23. Gene families for ribosomal protein.
Figure S24. Expression of LHC gene family of E. ferox.
Figure S25. Expression of PRK, PGK and GAPDH gene families of E. ferox.
Figure S26. Phylogenetic tree of SWEET gene homologous in E. ferox, N. colorata and A. thaliana.
Figure S27. The difference in average TE density in the vicinity regions of paralogous genes from the three sub‐genomes of E. ferox.
Table S1.The statistics of Nanopore reads
Table S2. The length and the number of contigs in each chromosome of E. ferox
Table S3. The statistics of genes in each prediction process
Table S4. The statistics of repeat sequence
Table S5. The 36 genomic blocks (GB) and their intervals in genome of N. colorata
Table S6. GB break points in E. ferox genome
Table S7. The GB associations in the genome of E. ferox
Table S8. The P‐values of χ2 test on the number of genes retained between sub‐genomes in each ancestral chromosome
Table S9. The P‐values of χ2 test on the number of genes retained between pairs of sub‐genomes in each genomic block
Table S10. The large fragments missing in sub‐genomes of E. ferox
Table S11. Number of paralog gene pairs differentially expressed in six different tissues
Table S12. Testing for significant differences in KS values between N. colorata and E. ferox sub‐genomes
Table S13. Testing for significant differences in KS values between N. colorata and E. ferox sub‐genomes in each genomic block
Table S14. Testing for significant differences in KS values between V. cruziana and E. ferox sub‐genomes
Table S15. Testing for significant differences in KS values between V. cruziana and E. ferox sub‐genomes in each genomic block
Table S16. The number distribution of the six topologies in the 12 ancestral chromosomes
Table S17. The three sub‐genomes identified in E. ferox genome
Table S18. Testing for significant differences in KS values between sub‐genomes of E. ferox
Table S19. Enrichment of tissue‐specific genes
Table S20. GO function enrichment of SSMG
Table S21. Polyploidy gene retention and selective pressure of cluster genes of E. ferox
Table S22. Expression of different functional genes in early adult and adult leaf
Table S23. The number and id of genes for cell division and expansion in A. thaliana, E. ferox and N. colorata
Table S24. Expression of gene families associated with cell division expansion in E. ferox
Table S25. Expression of gene families associated with phytohormone, ribosomal protein and photosynthesis in E. ferox
Data Availability Statement
Nanopore whole‐genome sequencing data, Illumina data, Hi‐C data and transcriptome data have been deposited to the NCBI Sequence Read Archive (SRA) as BioProject PRJNA769254 and PRJNA769560. The genome assembly sequences and gene annotations have been deposited in the Genome Warehouse in the BIG Data Center under accession number GWHBFHH00000000.