

Abstract
Fragment‐based drug discovery (FBDD) has a growing need for unique screening libraries. The cyclobutane moiety was identified as an underrepresented yet attractive three‐dimensional (3D) scaffold. Synthetic strategies were developed via a key 3‐azido‐cyclobutanone intermediate, giving potential access to a range of functional groups with accessible growth vectors. A focused set of 33 novel 3D cyclobutane fragments was synthesised, comprising three functionalities: secondary amines, amides, and sulfonamides. This library was designed using Principal Component Analysis (PCA) and an expanded version of the rule of three (RO3), followed by Principal Moment of Inertia (PMI) analysis to achieve both chemical diversity and high 3D character. Cis and trans ring isomers of library members were generated to maximise the shape diversity obtained, while limiting molecular complexity through avoiding enantiomers. Property analyses of the cyclobutane library indicated that it fares favourably against existing synthetic 3D fragment libraries in terms of shape and physicochemical properties.
Keywords: Fragment-based drug discovery (FBDD), Drug design, Library design, Cyclobutane, Three-dimensional (3D)
A 33‐member library with three‐dimensional cyclobutane fragments is described. Principle component analysis, the fragment rule‐of‐three, and principal moment of inertia analysis guided design to produce diastereomerically‐pure cis/trans isomers with three functional groups: amines, amides and sulfonamides. Calculated physicochemical and 3D properties of the library are deemed attractive for fragment screening.
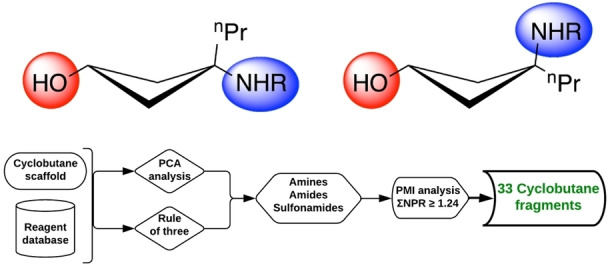
Introduction
Fragment‐based drug discovery (FBDD) has matured into a powerful approach to drug research. As of January 2022, six FDA‐approved drugs (Figure 1) and numerous clinical candidates have emerged from FBDD efforts.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 ] FBDD relies on small molecules of mass up to approximately 300 g mol−1, termed fragments. One key strength of FBDD is the superior sampling of chemical space that is achieved through the use of smaller molecules, particularly when compared to the larger compounds of around 500 g mol−1 from high‐throughput screening (HTS). This allows FBDD to deploy smaller library sizes of around 1000 members – a more accessible quantity for academic groups and smaller biotech companies.[ 11 , 12 ] Another key strength of FBDD is that it is agnostic of protein target, meaning any protein class can in principle be targeted by fragments. It has been shown that the low‐complexity fragment hits can be efficiently optimised into larger and more potent leads, highlighting the utility of fragments as valuable starting points in medicinal chemistry.[ 13 , 14 ]
Figure 1.

FDA‐approved drugs developed by FBDD (as of January 2022). Remnants of the original fragment hit(s) are highlighted in red (fragment growing approaches) and blue (fragment linking/merging approaches).[ 2 , 3 , 4 , 6 , 7 , 9 , 10 ]
The identification of appropriate fragment screening libraries is key to the success of FBDD. Historically, fragment libraries have been overwhelmingly populated by aromatic rings, as shown by a recent analysis of fragment‐protein crystal structures, [15] giving rise to mostly two‐dimensional (2D) fragments of low molecular complexity. [16] Lovering et al. demonstrated that the proportion of sp3‐hybridised carbon atoms increases as compounds progress through the drug discovery pipeline,[ 17 , 18 ] thereby suggesting that increased 3D character may offer a reduced rate of candidate attrition. It has been shown that 3D features of drug molecules can offer a wealth of potential benefits compared to their 2D counterparts, such as improved solubility, [19] superior metabolic stability, [20] as well as in achieving selectivity amongst closely‐related targets. [21] Within FBDD, in recent years there has been substantial interest in the design and synthesis of 3D fragments.[ 22 , 23 ] While the higher complexity of 3D fragments may reduce the chances of binding to a protein target, [16] the current scarcity of 3D fragments in screening collections might be a missed opportunity for FBDD. Often a limiting factor in FBDD, one crucial feature of any fragment hit is the ability to develop the molecule into a more potent lead molecule through the use of accessible growth vectors and associated chemistry. [24] This concept has been coined fragment ‘sociability’. [25] We believe that this can be achieved through the tailored synthesis of 3D fragments possessing accessible growth vectors – specifically, with the systematic control of regio‐ and stereo‐chemical outcomes. [26] Indeed, in recent years various synthetic strategies have been deployed for the generation of 3D fragments, [26] producing appreciable diversity in both shape and physicochemical properties. [23]
In 2014, Taylor et al. [27] showed that of the ring systems present in FDA‐approved drugs, the phenyl ring dominates the field, being almost ten times more prevalent than the second most utilised unit (pyridine). Fragments that contain these scaffolds have frequently been identified as hits and exploited in a significant number of successful fragment‐to‐lead campaigns. [28] Importantly, these two rings are planar 2D structures which only offer growth within the plane of the ring. Amongst saturated cycloalkanes, the cyclohexyl ring is the most widespread, with cyclopentyl and cyclopropyl units also showing popularity.[ 27 , 29 ] With far fewer entries, the cyclobutane ring proves to be a heavily underrepresented unit in marketed drugs and medicinal chemistry publications as a whole.[ 27 , 29 ] Indeed, the cyclobutane ring only appears in ten FDA‐approved drugs according to a DrugBank search in January 2022. [30] This scarcity may be attributed to 1) a lack of accessible synthetic methods (the most common method of cyclobutane generation is [2+2] photocycloadditions between olefins) [31] and 2) the perceived instability of cyclobutanes (ring strain of 26 kcal mol−1) compared to their homologues cyclopentane (6 kcal mol−1) and cyclohexane (no ring strain). [32] Interestingly, upon inspection of our own in‐house library of approximately 1,500 fragments and 15,000 drug‐like compounds, we found no examples in which a cyclobutane ring is the central scaffold. Similarly, we have shown that an isolated cyclobutane ring in synthetic 3D fragment libraries, if present, is most commonly used as a pendant aliphatic group or isostere of a geminal dimethyl unit and virtually never as a central scaffold. [23] In general, the strategy for cyclobutane inclusion in medicinal chemistry and FBDD appears to be that chemists tend to put ‘a ring on a molecule’ (as an aliphatic group), rather than ‘a ring in a molecule’ (as part of the framework).
We believe that the cyclobutane motif offers several distinct advantages as a scaffold for medicinal chemistry. A detailed comparison between the cyclobutane ring and various cycloaliphatic rings as well as 4‐membered heterocycles revealed that the cyclobutane ring compares favourably in terms of physicochemical properties and metabolic stability. [33] Cycloaliphatic rings can also serve as modest bioisosteric replacements of the phenyl ring, with the cyclobutane ring boasting improved physicochemical advantages over the cyclopentane and cyclohexane rings, respectively. [34] Another attractive application that the cyclobutane motif boasts is in the rigidification of acyclic propyl chains, such as in the case of the histamine H3 receptor antagonists/inverse agonists [35] and retinoic acid‐ related orphan receptor γt (RORγt) inverse agonists [36] – both cases in which an improved affinity was achieved. Additionally, the available carbocentric (carbon atom‐based) growth vectors from the cyclobutane ring, which provide access to different molecular geometries, brand the motif particularly ‘sociable.’[ 25 , 37 ] Therefore, we postulated that it may be beneficial to begin with cyclobutane fragments at the screening level. Indeed, Osberger et al. recently disclosed their approach encompassing the generation of all‐syn cyclobutane‐based heterobicyclic fragments (Figure 2A). [38] In our efforts, we set out to explore complementary chemistries to unlock the 3D cyclobutane fragment scaffold and yield both spatially‐ and chemically‐diverse fragments to target unique chemical space for use in FBDD. Herein, we describe the shape‐guided computational design and synthesis of an achiral yet diastereomerically‐pure focused 33‐member cyclobutane fragment library.
Figure 2.

A) General structure and features of cyclobutane fragments in the recent literature [38] (left panel) compared to the approach in this work (right panel). B) The general design strategy of the cyclobutane‐based fragment library, showing the accessible growth vectors.
Results and Discussion
Design of synthesis approach
We envisioned a synthetic strategy that allows for the diversification of a common cyclobutane‐based intermediate with access to multiple stereoisomers, either by a level of diastereoselectivity and/or the possibility to separate stereoisomers. This diversification could be achieved through the generation of bifunctional intermediates, such as those offered by enone derivatives. [39] Enones are biselectrophiles, providing the opportunity to incorporate vectors by addition of different nucleophiles upon its alkene and ketone functionalities, with a concomitant switch from sp2 to sp3 hybridisation. These substitutions generate quaternary centres which may offer benefits in medicinal chemistry and aid in accessing three‐dimensional chemical space.[ 17 , 40 , 41 ] In the specific case of cyclobutenones, such a strategy allows for symmetrisation of the cyclobutane ring and the opportunity to access diastereoisomers without generating enantiomers (Figure 2B), a common complication associated with 3D fragment synthesis. That is, it is commonplace for fragments to be produced as racemates, albeit often as pure diastereomers. Molecular complexity is lowered by circumventing enantiomers (in contrast to the case of desymmetrisation of cyclobutanes), [42] a significant feature relating back to the effect of higher complexity potentially resulting in lower hit rates of fragment screening. [16] The nucleophiles to be added can be diverse, yielding a strategy for the synthesis of achiral yet diastereomeric fragments with a cyclobutane core. These additions, together with the implicit possibility to vary the 3‐alkyl chain (R), equip us with methodology with which to derivatise at two carbon atoms, and thus grow along three carbocentric vectors. This was shown by Chessari et al. [37] to be an important feature in developing a fragment so as to not lose, during growth, the strong hydrogen bond interactions that drive binding events. Similarly, it is important to keep fragments small, so as to facilitate additional molecular mass during the fragment‐to‐lead process.
Proof‐of‐concept validation of synthesis route
As proof‐of‐concept of the general route design (Figure 2B), we selected Nu1 to be an ammonia equivalent, and Nu2 as a hydride anion equivalent. In order to avoid the instability of 3‐amino‐cyclobutanones, [43] which are subject to ring‐opening, the azide group was used as surrogate to install the masked amine functionality. Our route did lead to substantial safety considerations: 1) small organic azides that violate the rule of six [44] bear an explosion risk; 2) small electrophiles such as cyclobutenones can be toxic, especially when of considerable volatility. To address these risks, we selected the n‐propyl chain as R.
The route (Scheme 1) began with a reported thermal [2+2] cycloaddition between 1‐pentyne 1 and a ketene (made in situ from Cl3CCOCl using a Zn−Cu couple) to yield dichlorocyclobutenone 2. [45] This was followed by reductive dehalogenation upon treatment with Zn in THF/satd. aq. NH4Cl [46] to give cyclobutenone 3. Biselectrophile 3 could undergo subsequent nucleophilic additions on two positions. In the exemplified route, we focused on Michael addition of the N3 − anion using NaN3 with aq. HCl in DCM to afford highly versatile azido‐ketone intermediate 4. Though examples of Michael additions to 3‐unsubstituted cyclobutenones and their activated iminium analogues are known, [47] to the best of our knowledge, we report here the first example of azide addition to a cyclobutenone. The ketone group of 4 was reduced using NaBH4 in EtOH to yield the cyclobutanol 5, giving rise to a mixture of cis/trans ring isomers in a ratio of ca. 7 : 3 respectively, as observed by 1H NMR analysis of the crude material. A preliminary screening of additional reducing agents and conditions was conducted (with the n‐butyl analogue of 4) in order to obtain a greater diastereoselectivity (Table S1, Supporting Information), however NaBH4 was deemed optimal in balancing yield and d. r. This observation of significant cis‐diastereoselectivity is in alignment with literature precedent on the reduction of 3‐substituted cyclobutanones, [48] postulated to proceed via the lower energy TS cis as a result of the cyclobutanone ring puckering to decrease torsional strain arising from eclipsed interactions, favouring an antifacial attack upon the carbonyl group. The mixture of diastereomers 5 was then subjected to PhCOCl to produce the benzoate esters of each ring isomer (6 and 7), which contain a UV‐chromophore, boast increased lipophilicity to ease purification and are separable by flash chromatography. The azide moiety in individual diastereomers 6 and 7 was then reduced using Pd/C/H2, providing both the cis and trans isomers of aminobenzoates 8 and 9 which serve as common intermediates for library enumeration.
Scheme 1.
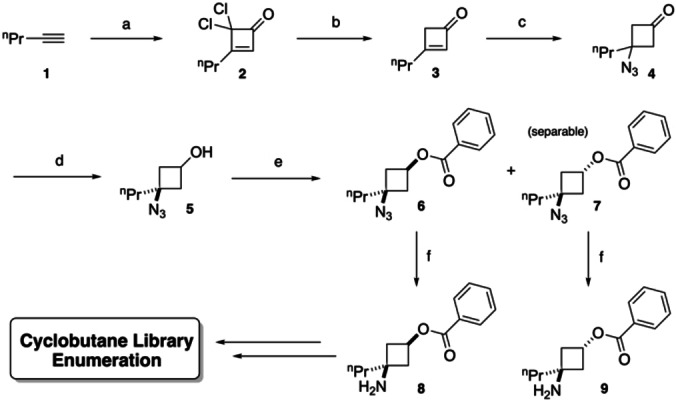
Synthetic route of common intermediates for library enumeration. Conditions: a) Cl3CCOCl (2.0 eq), Zn−Cu (3.0 eq), Et2O, 10–15 °C, 3 h, ∼41 %. b) Zn (4.0 eq), THF:satd. aq. NH4Cl 7 : 3, rt, 1 h, ∼86 %. c) NaN3 (3.0 eq), 37 % aq. HCl (2.0 eq), Et3N (1.0 eq), DCM, rt, 18 h, ∼61 %. d) NaBH4 (3.0 eq), EtOH, 0 °C to rt, 2 h, 82 % (d. r. 2 : 1 cis:trans from NMR analysis of crude product). e) BzCl (1.2 eq), Et3N (1.3 eq), DMAP (1.0 eq), DCM, rt, 18 h: 63 % (cis, 6)+18 % (trans, 7). f) H2, 5 % wt Pd/C, MeOH, rt, 18 h, cis (8): 94 %, trans (9): 91 %. Due to volatility and/or safety concerns, for steps a, b and c the products were not isolated in neat form and yields are extrapolated from molar ratios by NMR analysis.
Design, synthesis and properties of the library
With aminobenzoates 8 and 9 at hand for diversification, the stage was set for computer‐aided design of a concise fragment library. In an analysis by Giordanetto et al. [15] which compared the features of fragment hits within the protein data bank (PDB) to larger ligands such as those arising from HTS or natural product/biomimetics, it was noted that alcohol and amide functionalities are the most attractive in terms of their capability of acting as both a hydrogen bond donor (HBD) and hydrogen bond acceptor (HBA). This dual role makes these groups particularly valuable for interaction sampling during the screening process, as hydrogen bonds are often one of the most robust binding anchors between protein residues and a ligand. [15] Consequently, we chose to keep the secondary alcohol intact for all of our fragments, and prioritised amide fragments for enumeration. In order to increase the chemical diversity of our library, we also included amines and sulfonamides in our design – both groups which can form hydrogen bonds, as well as potential electrostatic charges upon (de)protonation within an appropriately acidic or basic protein environment, respectively, thus offering potential new interactions.
The selection of compounds to be synthesised within the three compound classes – amines, amides, and sulfonamides – was made using the combinatorial library platform of Molecular Operating Platform (MOE, version 2018, by Chemical Computing Group), using the amine intermediate (8 or 9) as one defined reagent and performing in‐silico coupling reactions. This was carried out for both cis and trans isomers, in order to maximise spatial diversity. The database used was the default provided by Chemical Computing Group, merged with our in‐house database of reagents. For each of the six cyclobutanol cores, we enumerated 50 potential fragments and selected compounds to synthesise based on 1) fragment properties; 2) 3D shape as assessed by Principal Moment of Inertia (PMI) analysis, the use of which [49] in selecting fragments for synthesis was first proposed by O'Brien and co‐workers. [50] The analysis concerning fragment properties was conducted using the software's default Principal Component Analysis (PCA) to maximise diversity within the following parameters: number of H‐bond acceptors (a_acc); number of aromatic atoms (a_aro); number of H‐bond donor atoms (a_don); number of heavy atoms (a_heavy); number of rotatable bonds (b_rotN); Log Octanol/Water partition coefficient (SlogP); topological polar surface area (TPSA) (Å2); and molecular weight (Weight). The following descriptor filters were applied as guided by the ‘Rule of three (RO3) [51] ’: a_acc≤3, a_don≤3, a_heavy≤19, b_rotN≤5, SlogP≤3,TPSA≤70 Å2, Weight<300 g mol−1, (number of chiral centres) chiral=0. Concerning 3D shape analysis by PMI analysis, it was proposed by Firth et al. [52] that a ΣNPR≥1.07 is deemed sufficiently off the rod‐disk axis to be classified as 3D, where ΣNPR is the sum value of the normalised PMI ratios (x‐ and y‐axis) of a given point. As such, we prioritised the virtual structures with the greatest deviation from the rod‐disk axis in our selection. For each class, compounds were ranked using ΣNPR values as a measure of three‐dimensionality. A ΣNPR value of ≥1.24 (well above the commonly used value of 1.07)[ 23 , 52 ] was chosen as a cut‐off to ensure that only those fragments with the highest three‐dimensionality were selected for synthesis while maintaining a focused library of modest size. The workflow did not suggest any trans‐sulfonamides that satisfied the ΣNPR≥1.24 cut‐off. For the current study, we have chosen to use an expanded version of the RO3, and allow both an increased number of rotatable bonds (from three to five), and an increased TPSA limit (from 60 to 70 Å2). The former is due to the inherent number of rotatable bonds (3) present within the common core of the fragment set as a result of the selection of n‐propyl as R for safety reasons (vide supra) and it was therefore necessary to increase the limit to five to facilitate diversification at the amine handle. The adjustment of TPSA allows for increased chemical diversity in the case of the more polar sulfonamides. Both of these parameters are in fact known for being the more flexible parameters of those defined in the preceding section.[ 23 , 53 , 54 ]
Scheme 2 shows the synthetic approaches toward the three classes. Amine fragments 10–11 were synthesised via indirect reductive amination [55] – first pre‐forming the imine in dry MeOH, followed by reduction with NaBH4. This step was done in the absence of AcOH catalyst, as we found that its addition led to substantial amounts of dialkylated product, likely due to the highly nucleophilic nature of the cyclobutylamine moiety. Amine synthesis was completed by ester hydrolysis. For 10 a and 11 c, additional deprotection of the N‐Boc group with TFA was necessary. Amide fragments 12–13 were generated from the corresponding acid chloride, prepared from the parent carboxylic acid upon treatment with (COCl)2 or SOCl2, and subsequent ester hydrolysis. Sulfonamide fragments 14 were made using the corresponding sulfonyl chloride. Interestingly, in the case of the sulfonamides, it was noted that subsequent hydrolysis of the benzoate ester did not proceed with LiOH but required NaOH.
Scheme 2.
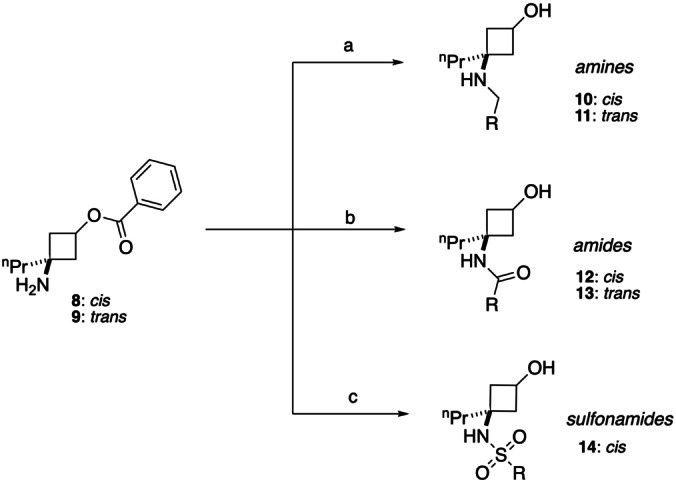
Synthetic route for generation of designed cyclobutane fragments. General conditions: a) i) RCHO (0.97 eq), MeOH, rt, 18 h. ii) NaBH4 (3.0 eq), 0 °C to rt, 1 h. iii) LiOH.H2O (4.0 eq), MeOH:THF 7 : 3, rt, 1–2 h, 21–63 %. b) i) RCOCl (1.2 eq, from RCOOH and (COCl)2 or SOCl2), Et3N (1.2 eq), DCM, 0 °C to rt, 18–44 h. ii) LiOH.H2O (4.0 eq), MeOH:THF 7 : 3, rt, 1–2 h, 19–65 %. c) i) RSO2Cl (1.2 eq), Et3N (1.2 eq), DCM, 0 °C to rt. ii) NaOH (4.0 eq), MeOH:THF 7 : 3, rt, 1–2 h, 13–37 %. For compounds 10 a and 11 c, an additional step with TFA was performed for cleavage of the N‐Boc group.
In total, the cyclobutane library comprised 12 amines (10 a–g and 11 a–e), 18 amides (12 a–h and 13 a–j), and three sulfonamide fragments (14 a–c), all novel and with defined stereochemistry (Table 1). Our workflow produced one fragment (12 d, as an E/Z‐mixture) containing an α,β‐unsaturated ketone unit rendering it an electrophilic fragment with potential to act as a Michael acceptor. This could serve as a covalent fragment, alongside other well‐known acrylamide warheads[ 56 , 57 ] and our previously published cyclobutenaminone fragments, which have been shown to exhibit covalent inhibition of bacterial enzymes. [58] A PMI plot was generated for the synthesised library set (Figure 3), with points labelled according to their substituent geometry (cis=triangles; trans=squares) and functional group (amine=blue; amide=green; sulfonamide=red). Interestingly, of the seven pairs of cis/trans analogues amongst the collection (10 a/11 c, 10 e/11 d, 12 a/13 a, 12 b/13 b, 12 e/13 d, 12 f/13 h, 12 h/13 g), there appears to be a marginally higher ΣNPR value for the trans isomer, with differences ranging from Δ ΣNPR=0.00–0.05 (mean average 0.02). This may suggest that for an isolated cyclobutane ring, 1,3‐trans‐substitutions may be more likely to access more spherical molecules compared to their 1,3‐cis‐counterparts, albeit that the Δ ΣNPR differences are minor. This observation differs from that of Osberger et al, [38] in which the bicyclic 1,2,3‐trisubstituted all‐syn cyclobutanes possess lower ΣNPR scores compared to their corresponding 3‐anti‐epimers in PMI analysis. Also in slight contrast, our workflow did not produce any trans‐sulfonamides. It is, however, conformation that defines molecular shape, and these observations together highlight how different approaches to the design of 3D fragments can often be complementary. To understand how the cyclobutane library compares to other synthetic 3D fragment collections in a general sense, we overlaid 897 fragments (grey spots) extracted from 25 recent publications that describe novel 3D fragment libraries recently reviewed and analysed by us. [23] The cyclobutane library boasts a higher average ΣNPR value of 1.29, compared to 1.20 for the literature fragments, although notably this is by design in that only the highest ΣNPR‐scoring compounds were synthesised in our current study, resulting in a narrower distribution of molecular shapes. Although fraction sp3 (Fsp3), as defined by Yan and Gasteiger, [59] is not necessarily a good measure of 3D shape,[ 50 , 52 ] this simple metric remains widely used amongst the FBDD community and it is notable that our cyclobutane library possesses an average Fsp3 value of 0.65 – substantially higher than the suggested cut‐off of Fsp3≥0.45 for three‐dimensionality. [60]
Table 1.
Synthesised cis/trans‐cyclobutanol fragments, sorted according to functional group. (amines=blue; amides=green; sulfonamides=red).
|
Cis scaffold |
Trans scaffold |
|---|---|
|
| |
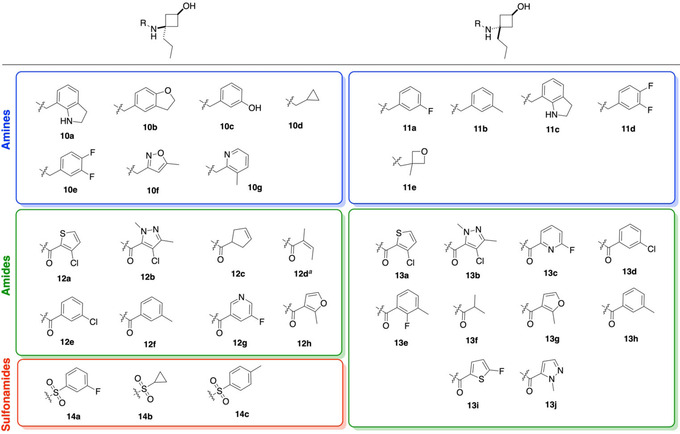
Compound 12 d was isolated as a mixture of E/Z‐isomers in a ratio of 3 : 7.
Figure 3.
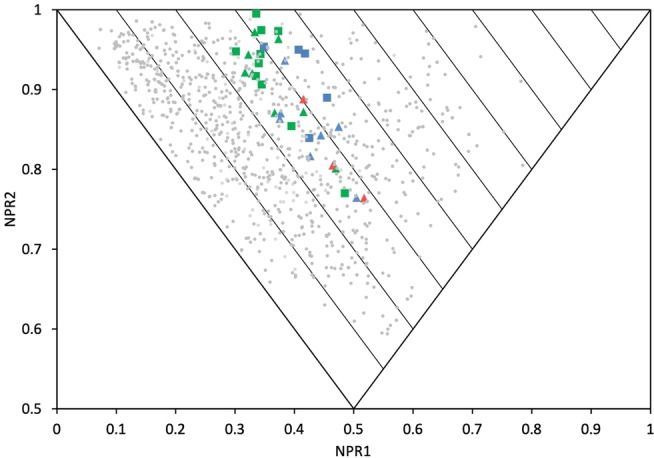
PMI plot of the cyclobutane fragment library. Cis‐isomers are depicted as triangles and trans‐isomers as squares. Points are coloured according to functional group: amines (blue), amides (green) and sulfonamides (red). Grey points correspond to 897 fragments extracted from 25 recent publications according to the workflow described in a recent 3D fragment analysis. [23]
We calculated common physicochemical properties of our library (Table S2 – Supporting Information). We expressed the values as mean averages (Figure 4A) and generated a radar plot to visualise their distribution – comparing this to the RO3 (Figure 4B, D). It can be seen that in terms of physicochemical properties, the cyclobutane library largely mimics the distribution of properties described in the RO3, with the ranges (dark grey) of most parameters not differing substantially from the average value (blue line). There is substantial room for fragment growth following a potential screening hit, with only 17.3 heavy atoms on average per molecule.
Figure 4.
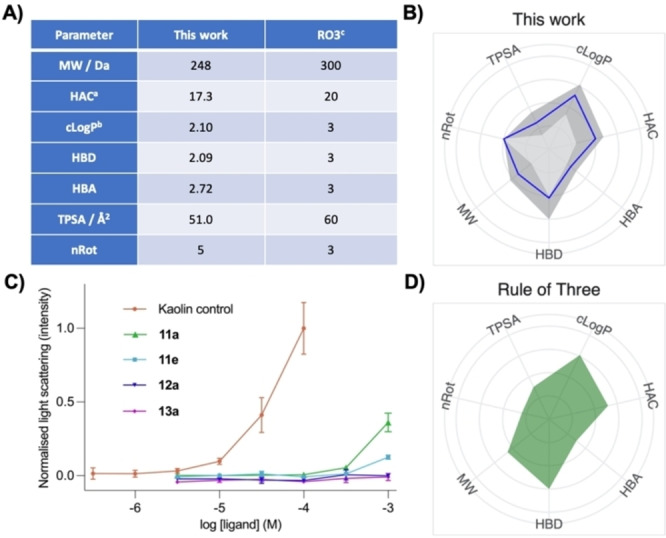
Calculated and experimental properties of the cyclobutane fragment library. (A) Calculated physicochemical properties, expressed as mean averages, and the rule of three. (B and D) Radar plots showing the distribution of properties of the cyclobutane library and of the rule of three. The data were generated using MOE or PipeLine Pilot with the neutral chemical species and values are given in Table S2 (Supporting Information). Axes were scaled as follows: [23] cLogP: [−1.9;4.5], HAC: [7;27], HBA: [0;8], HBD: [0;4], MW: [95;455 Da], nRot: [0;10], TPSA:[10;140 Å2]. Mean average is depicted by the blue line. Ranges, defined by the minimum and maximum values, are defined by the grey areas. The rule of three (accompanied by HAC=20) is exemplified as the first entry to provide context for scale interpretation. a Heavy atom count (HAC) has been included as an additional useful metric. b cLogP was calculated as SLogP. c An adjusted nRot count was used in our work – see main text. (C) Nephelometry analysis of selected fragments at increasing concentrations and of kaolin (control suspension) in HBSS buffer containing 1 % DMSO. Fragments tested comprise the ones with the lowest cLogD (0.28, 11 e), the median cLogD (1.85, 11 a), and the highest cLogD (2.82, diastereomers 12 a and 13 a). Data points represent the mean±SD of values measured in triplo normalised to kaolin control. Inflection points are qualitatively deemed to be a sign of aggregation – this is quantified using 3 times the standard deviation of the average blank measurements (see Figure S1, Supporting Information) and represents a cut‐off above which aggregation begins to occur.
Finally, we used nephelometry to determine kinetic solubility in buffer.[ 61 , 62 ] We subjected the compounds with the lowest (11 e), median (11 a) and highest (diastereomers 12 a and 13 a) cLogD values to this technique (Figure 4C). cLogD values were used over cLogP values to account for ionisable groups in aqueous buffer at pH 7.4. The results showed that the two compounds with the highest cLogD value (2.82), 12 a and 13 a (cis and trans diastereomers, respectively), produced no sign of aggregation up to a concentration of at least 1.0 mM. This suggests that these amides are highly soluble in aqueous buffer, and do not suffer from precipitation issues at high concentrations which are often required in FBDD screening methods. Interestingly, the compounds with lower cLogD values showed aggregation at still high yet slightly reduced concentrations. That is, the median cLogD (1.85) compound 11 a showed no aggregation up to 100 μM, and the lowest cLogD (0.28) compound, 11 e, performed well until 300 μM. This observation may seem counterintuitive given their lower cLogD values, but both being protonated amines (as opposed to the neutral 12 a and 13 a) it is postulated that this effect could result from their amphiphilic properties.
In all, different approaches to fragment design offer distinct features, often complementing one another. Whilst fragment libraries may differ in physicochemical properties, it is also important to consider the shape diversity (vide supra), as well as ensure that fragments can be developed should they arise as a hit from a screening cascade – relating back to the concept of fragment ‘sociability.’ [25] It is our belief that the cyclobutane library performs well in terms of physicochemical and 3D properties, as well as possessing multiple accessible growth vectors, thus producing attractive 3D fragments for FBDD screening campaigns.
Conclusion
The cyclobutane moiety is underrepresented in FBDD, yet may offer distinct advantages in terms of physicochemical properties, stereochemistry and growth vectors. We have developed a proof‐of‐concept synthesis strategy for the generation of diverse cyclobutane‐based 3D fragments. The described chemistry gives access to single diastereomers of cis and trans 1,3‐substituted key intermediates, whilst avoiding chirality. We have exemplified this approach through the construction of a library of 33 novel fragments, based around a common 3‐propyl‐3‐azido‐cyclobutanone intermediate, and achieved chemical diversity through synthesis of three classes – amines, amides and sulfonamides. Using PCA and PMI analyses to steer our library design, both chemical as well as shape diversity was achieved, with control over stereochemistry and two synthetic handles accessible for further elaboration. The shape and physicochemical properties are overall well‐behaved. Trans‐cyclobutane fragments were found to have marginally higher ΣNPR scores compared to their cis‐counterparts, albeit not profound. Nephelometry measurements on selected members suggest solubility up to concentrations of at least 100 μM, with amides showing no signs of aggregation at 1.0 mM. To gauge their utility in FBDD screening campaigns, we aim to test the described cyclobutane fragments against a variety of biological targets in due course.
Experimental Section
Computational Protocol
The selection of compounds to be synthesised was made using the combinatorial library platform of Molecular Operating Platform (MOE, version 2018, by Chemical Computing Group), using the amine intermediate (8 or 9) as one defined reagent and coupling with appropriate reagents. The database used was the default provided by Chemical Computing Group, merged with our in‐house database of reagents. The analysis was conducted using the software's default Principal Component Analysis (PCA) to maximise diversity within the following criteria: number of H‐bond acceptors (a_acc); number of aromatic atoms (a_aro); number of H‐bond donor atoms (a_don); number of heavy atoms (a_heavy); number of rotatable bonds (b_rotN); Log Octanol/Water partition coefficient (SlogP); topological polar surface area (TPSA) (Å2); and molecular weight (Weight). The following descriptor filters were applied as guided by the ‘Rule of three (RO3) [51] ’: a_acc≤3, a_don≤3, a_heavy≤19, b_rotN≤5, SlogP≤3,TPSA≤70 Å2, Weight<300 g mol−1, chiral=0. Duplicates were then removed using a unique entry filter, and a visual inspection was carried out upon the remaining molecules in terms of what was deemed synthetically feasible using the described route. For each functional group interconversion, a Principal Moment of Inertia (PMI) analysis was conducted using PipeLine Pilot 8.5.0.200 (2011, Accelrys Software Inc.) for conformational analysis using the BEST method in Catalyst using the Rel option (maximum 255 conformers per compound), with a preceding wash step at pH=7.4.
Nephelometry Protocol
In transparent flat‐bottom 96‐well plates, compounds were placed at different concentrations in triplo (10−3 M, 10−3.5 M, 10−4 M, 10−4.5 M, 10−5 M, 10−5.5 M and two blanks) in HBBS buffer with 1 % DMSO for 6 h before the measurement. A Kaolin dispersion was used as a positive control [63] in each plate at different concentrations (10−4 M, 10−4.5 M, 10−5 M, 10−5.5 M, 10−6 M and 10−6.5 M) under the same conditions as test compounds. Nephelometry measurements were performed with a NEPHELO star Plus (BMG Labtech, Germany) with the following settings: measurement interval time 1 s, laser intensity 80 %, beam focus 2.5 mm, Orbital shaking mode at 200 rpm for 10 s before measuring. Results were analysed using GraphPad Prism 9 software, plotting all available data points and plotting mean and standard deviation values in a line chart compared to Kaolin control.
General Procedure A – Reductive Amination
To a solution of cis‐ or trans‐amine 8 or 9 (1.0 eq) in dry MeOH (0.30 M) containing a spatula of 3 Å molecular sieves at rt was added the corresponding aldehyde (0.97 eq). The reaction mixture was stirred for 18 h. The mixture was cooled to 0 °C before portion wise addition of NaBH4 (3.0 eq). The mixture was stirred for 2 min. The ice bath was removed and the reaction mixture was stirred for a further 1 h. The volatiles were removed in vacuo and the residue was partitioned between EtOAc and satd. aq. NaHCO3. The aqueous phase was extracted with DCM containing 5 % CF3CH2OH. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was used in the next step without additional purification. To a solution of crude benzoate ester in a 7 : 3 vol mixture of THF : MeOH (0.050 M) was added a suspension of LiOH.H2O (4.0 eq) in H2O (3.0 mL). The mixture was stirred for 2 h at rt. The volatiles were removed in vacuo and the residue was partitioned between EtOAc and satd. aq. Na2CO3. The aqueous phase was extracted with DCM containing 5 % CF3CH2OH. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification was conducted as specified to afford the cyclobutanol product.
General Procedure B – Amide Formation
To a solution of the corresponding carboxylic acid (1.70 mmol) in DCM (0.30 M) containing a spatula of 3 Å molecular sieves was added (COCl)2 (2.04 mmol) and one drop of DMF. The reaction mixture was stirred at rt until the carboxylic acid was no longer observed (reaction progress was monitored by quenching a sample of reaction mixture with MeOH and conducting TLC analysis). The mixture was filtered and the filtrate concentrated in vacuo to afford the corresponding acid chloride, part of which was used directly in the amide‐forming reaction without further analysis. The acid chloride (1.2 eq) and Et3N (1.2 eq) were added to a solution of cis‐ or trans‐amine 8 or 9 (1.0 eq) in DCM (0.30 M) containing a spatula of 3 Å molecular sieves at 0 °C. The mixture was allowed to warm to rt and stirred for the specified time. The mixture was filtered and the filtrate was partitioned between EtOAc and satd. aq. Na2CO3. The aqueous phase was extracted with DCM containing 5 % CF3CH2OH. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was used in the next step without additional purification. To a solution of crude benzoate ester in a 7 : 3 vol mixture of THF : MeOH (0.050 M) was added a suspension of LiOH.H2O (4.0 eq) in H2O (3.0 mL). The mixture was stirred for 2 h at rt. The volatiles were removed in vacuo and the residue was partitioned between EtOAc and satd. aq. Na2CO3. The aqueous phase was extracted with DCM containing 5 % CF3CH2OH. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification was conducted as specified to afford the cyclobutanol product.
General Procedure C – Sulfonamide Formation
To a solution of cis‐ or trans‐amine 8 or 9 (1.0 eq) in DCM (0.30 M) containing a spatula of 3 Å molecular sieves at 0 °C was added the corresponding sulfonyl chloride (1.2 eq) and Et3N (1.2 eq). The reaction mixture was allowed to warm to rt and stirred for the specified time. The mixture was filtered and the filtrate was partitioned between EtOAc and satd. aq. Na2CO3 solution. The aqueous phase was extracted with DCM containing 5 % CF3CH2OH. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was used in the next step without additional purification. To a solution of crude benzoate ester in a 7 : 3 vol mixture of THF : MeOH (0.050 M) was added a suspension of LiOH.H2O (4.0 eq) in H2O (3.0 mL). The mixture was stirred for 2 h at rt. The volatiles were removed in vacuo and the residue was partitioned between EtOAc and satd. aq. Na2CO3. The aqueous phase was extracted with DCM containing 5 % CF3CH2OH. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification was conducted as specified to afford the cyclobutanol product.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was funded by the European Union's Framework Programme for Research and Innovation Horizon 2020 (2014–2020) under the Marie‐Skłodowska‐Curie grant agreement number 675899 (“Fragment based drug discovery Network, FRAGNET”) and under the grant agreement number 777828 (“The best online drug discovery platform. Building the Ultimate chemical database for drug discovery”). We thank Robert Kiss and Attila Wootsch for helpful discussions and support. We would like to thank Bas de Boer, Florian van Vugt, Emylie Nguyen and Fariëlle Boldewijn for their involvement in synthesis. We are grateful to Hans Custers for HRMS and nephelometry measurements, Tom Dekker for assistance with radar plots and nephelometry, and Elwin Janssen for assistance with NMR analysis.
D. J. Hamilton, M. Beemsterboer, C. M. Carter, J. Elsayed, R. E. M. Huiberts, H. F. Klein, P. O'Brien, I. J. P. de Esch, M. Wijtmans, ChemMedChem 2022, 17, e202200113.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Bollag G., Tsai J., Zhang J., Zhang C., Ibrahim P., Nolop K., Hirth P., Nat. Rev. Drug Discovery 2012, 11, 873–886. [DOI] [PubMed] [Google Scholar]
- 2. Kim A., Cohen M. S., Expert Opin. Drug Discovery 2016, 11, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Souers A. J., Leverson J. D., Boghaert E. R., Ackler S. L., Catron N. D., Chen J., Dayton B. D., Ding H., Enschede S. H., Fairbrother W. J., Huang D. C. S., Hymowitz S. G., Jin S., Khaw S. L., Kovar P. J., Lam L. T., Lee J., Maecker H. L., Marsh K. C., Mason K. D., Mitten M. J., Nimmer P. M., Oleksijew A., Park C. H., Park C. M., Phillips D. C., Roberts A. W., Sampath D., Seymour J. F., Smith M. L., Sullivan G. M., Tahir S. K., Tse C., Wendt M. D., Xiao Y., Xue J. C., Zhang H., Humerickhouse R. A., Rosenberg S. H., Elmore S. W., Nat. Med. 2013, 19, 202–208. [DOI] [PubMed] [Google Scholar]
- 4. Murray C. W., Newell D. R., Angibaud P., MedChemComm 2019, 10, 1509–1511. [Google Scholar]
- 5. Zhang C., Ibrahim P. N., Zhang J., Burton E. A., Habets G., Zhang Y., Powell B., West B. L., Matusow B., Tsang G., Shellooe R., Carias H., Nguyen H., Marimuthu A., Zhang K. Y. J., Oh A., Bremer R., Hurt C. R., Artis D. R., Wu G., Nespi M., Spevak W., Lin P., Nolop K., Hirth P., Tesch G. H., Bollag G., Proc. Natl. Acad. Sci. USA 2013, 110, 5689–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tap W. D., Wainberg Z. A., Anthony S. P., Ibrahim P. N., Zhang C., Healey J. H., Chmielowski B., Staddon A. P., Cohn A. L., Shapiro G. I., Keedy V. L., Singh A. S., Puzanov I., Kwak E. L., Wagner A. J., Von Hoff D. D., Weiss G. J., Ramanathan R. K., Zhang J., Habets G., Zhang Y., Burton E. A., Visor G., Sanftner L., Severson P., Nguyen H., Kim M. J., Marimuthu A., Tsang G., Shellooe R., Gee C., West B. L., Hirth P., Nolop K., van de Rijn M., Hsu H. H., Peterfy C., Lin P. S., Tong-Starksen S., Bollag G., N. Engl. J. Med. 2015, 373, 428–437. [DOI] [PubMed] [Google Scholar]
- 7.D. A. Erlanson, “Fragments in the clinic: 2021 edition,” can be found under http://practicalfragments.blogspot.com/2021/11/fragments-in-clinic-2021-edition.html, 2021. (accessed 31st January 2022).
- 8. Ostrem J. M., Peters U., Sos M. L., Wells J. A., Shokat K. M., Nature 2013, 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lanman B. A., Allen J. R., Allen J. G., Amegadzie A. K., Ashton K. S., Booker S. K., Chen J. J., Chen N., Frohn M. J., Goodman G., Kopecky D. J., Liu L., Lopez P., Low J. D., Ma V., Minatti A. E., Nguyen T. T., Nishimura N., Pickrell A. J., Reed A. B., Shin Y., Siegmund A. C., Tamayo N. A., Tegley C. M., Walton M. C., Wang H.-L., Wurz R. P., Xue M., Yang K. C., Achanta P., Bartberger M. D., Canon J., Hollis L. S., McCarter J. D., Mohr C., Rex K., Saiki A. Y., San Miguel T., Volak L. P., Wang K. H., Whittington D. A., Zech S. G., Lipford J. R., Cee V. J., J. Med. Chem. 2020, 63, 52–65. [DOI] [PubMed] [Google Scholar]
- 10. Schoepfer J., Jahnke W., Berellini G., Buonamici S., Cotesta S., Cowan-Jacob S. W., Dodd S., Drueckes P., Fabbro D., Gabriel T., Groell J.-M., Grotzfeld R. M., Hassan A. Q., Henry C., Iyer V., Jones D., Lombardo F., Loo A., Manley P. W., Pellé X., Rummel G., Salem B., Warmuth M., Wylie A. A., Zoller T., Marzinzik A. L., Furet P., J. Med. Chem. 2018, 61, 8120–8135. [DOI] [PubMed] [Google Scholar]
- 11. Erlanson D. A., Fesik S. W., Hubbard R. E., Jahnke W., Jhoti H., Nat. Rev. Drug Discovery 2016, 15, 605–619. [DOI] [PubMed] [Google Scholar]
- 12. Keserű G. M., Erlanson D. A., Ferenczy G. G., Hann M. M., Murray C. W., Pickett S. D., J. Med. Chem. 2016, 59, 8189–8206. [DOI] [PubMed] [Google Scholar]
- 13. Erlanson D. A., de Esch I. J. P., Jahnke W., Johnson C. N., Mortenson P. N., J. Med. Chem. 2020, 63, 4430–4444. [DOI] [PubMed] [Google Scholar]
- 14. Jahnke W., Erlanson D. A., de Esch I. J. P., Johnson C. N., Mortenson P. N., Ochi Y., Urushima T., J. Med. Chem. 2020, 63, 15494–15507. [DOI] [PubMed] [Google Scholar]
- 15. Giordanetto F., Jin C., Willmore L., Feher M., Shaw D. E., J. Med. Chem. 2019, 62, 3381–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hann M. M., Leach A. R., Harper G., J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [DOI] [PubMed] [Google Scholar]
- 17. Lovering F., Bikker J., Humblet C., J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 18. Lovering F., MedChemComm 2013, 4, 515–519. [Google Scholar]
- 19. Liu K. G., Kim J.-I., Olszewski K., Barsotti A. M., Morris K., Lamarque C., Yu X., Gaffney J., Feng X.-J., Patel J. P., Poyurovsky M. V., J. Med. Chem. 2020, 63, 5201–5211. [DOI] [PubMed] [Google Scholar]
- 20. Zhao Y., Yu S., Sun W., Liu L., Lu J., McEachern D., Shargary S., Bernard D., Li X., Zhao T., Zou P., Sun D., Wang S., J. Med. Chem. 2013, 56, 5553–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson J. A., Nicolaou C. A., Kirberger S. E., Pandey A. K., Hu H., Pomerantz W. C. K., ACS Med. Chem. Lett. 2019, 10, 1648–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morley A. D., Pugliese A., Birchall K., Bower J., Brennan P., Brown N., Chapman T., Drysdale M., Gilbert I. H., Hoelder S., Jordan A., Ley S. V., Merritt A., Miller D., Swarbrick M. E., Wyatt P. G., Drug Discovery Today 2013, 18, 1221–1227. [DOI] [PubMed] [Google Scholar]
- 23. Hamilton D. J., Dekker T., Klein H. F., Janssen G. V., Wijtmans M., O'Brien P., de Esch I. J. P., Drug Discovery Today Technol. 2020, 38, 77–90. [DOI] [PubMed] [Google Scholar]
- 24. Murray C. W., Rees D. C., Angew. Chem. Int. Ed. 2016, 55, 488–492; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 498–503. [Google Scholar]
- 25. St Denis J. D., Hall R. J., Murray C. W., Heightman T. D., Rees D. C., RSC Med. Chem. 2021, 12, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caplin M. J., Foley D. J., Chem. Sci. 2021, 12, 4646–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taylor R. D., MacCoss M., Lawson A. D. G., J. Med. Chem. 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]
- 28. De Esch I. J. P., Erlanson D. A., Jahnke W., Christopher N., Walsh L., J. Med. Chem. 2021, 65, 1, 84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Serafini M., Cargnin S., Massarotti A., Tron G. C., Pirali T., Genazzani A. A., J. Med. Chem. 2021, 64, 4410–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.“Drug bank online” can be found under https://go.drugbank.com/structures/search/small_molecule_drugs/structure#results, 2022. (accessed 31st January 2022).
- 31. Poplata S., Tröster A., Zou Y.-Q., Bach T., Chem. Rev. 2016, 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wiberg K. B., Rappoport Z., Liebman J. F., in The Chemistry of Cyclobutanes, John Wiley and Sons Inc, 2005. [Google Scholar]
- 33. Bauer M. R., Di Fruscia P., Lucas S. C. C., Michaelides I. N., Nelson J. E., Storer R. I., Whitehurst B. C., RSC Med. Chem. 2021, 12, 448–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Subbaiah M. A. M., Meanwell N. A., J. Med. Chem. 2021, 64, 14046–14128. [DOI] [PubMed] [Google Scholar]
- 35. Wijtmans M., Denonne F., Célanire S., Gillard M., Hulscher S., Delaunoy C., Van houtvin N., Bakker R. A., Defays S., Gérard J., Grooters L., Hubert D., Timmerman H., Leurs R., Talaga P., de Esch I. J. P., Provins L., MedChemComm 2010, 1, 39. [Google Scholar]
- 36. Kono M., Ochida A., Oda T., Imada T., Banno Y., Taya N., Masada S., Kawamoto T., Yonemori K., Nara Y., Fukase Y., Yukawa T., Tokuhara H., Skene R., Sang B.-C., Hoffman I. D., Snell G. P., Uga K., Shibata A., Igaki K., Nakamura Y., Nakagawa H., Tsuchimori N., Yamasaki M., Shirai J., Yamamoto S., J. Med. Chem. 2018, 61, 2973–2988. [DOI] [PubMed] [Google Scholar]
- 37. Chessari G., Grainger R., Holvey R. S., Ludlow R. F., Mortenson P. N., Rees D. C., Chem. Sci. 2021, 12, 11976–11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Osberger T. J., Kidd S. L., King T. A., Spring D. R., Chem. Commun. 2020, 56, 7423–7426. [DOI] [PubMed] [Google Scholar]
- 39. Chen P. H., Dong G., Chem. A Eur. J. 2016, 22, 18290–18315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clemons P. A., Wilson J. A., Dančík V., Muller S., Carrinski H. A., Wagner B. K., Koehler A. N., Schreiber S. L., Proc. Natl. Acad. Sci. USA 2011, 108, 6817–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Talele T. T., J. Med. Chem. 2020, 63, 13291–13315. [DOI] [PubMed] [Google Scholar]
- 42. Sietmann J., Wiest J. M., Angew. Chem. Int. Ed. 2020, 59, 6964–6974; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7028–7038. [Google Scholar]
- 43. Huisgen R., Mayr H., J. Chem. Soc. Chem. Commun. 1976, 55–56. [Google Scholar]
- 44.“Information on Azide Compounds – Stanford Environmental Health & Safety,” can be found under https://ehs.stanford.edu/reference/information-azide-compounds, (accessed 3rd November 2021).
- 45. Ammann A. A., Rey M., Dreiding A. S., Helv. Chim. Acta 1987, 70, 321–328. [Google Scholar]
- 46. Hekmatshoar R., Sajadi S., Heravi M. M., J. Chin. Chem. Soc. 2008, 55, 616–618. [Google Scholar]
- 47. Lumbroso A., Catak S., Sulzer-Mossé S., De Mesmaeker A., Tetrahedron Lett. 2015, 56, 2397–2401. [Google Scholar]
- 48. Deraet X., Voets L., Van Lommel R., Verniest G., De Proft F., De Borggraeve W., Alonso M., J. Org. Chem. 2020, 85, 7803–7816. [DOI] [PubMed] [Google Scholar]
- 49. Sauer W. H. B., Schwarz M. K., J. Chem. Inf. Comput. Sci. 2003, 43, 987–1003. [DOI] [PubMed] [Google Scholar]
- 50. Downes T. D., Jones S. P., Klein H. F., Wheldon M. C., Atobe M., Bond P. S., Firth J. D., Chan N. S., Waddelove L., Hubbard R. E., Blakemore D. C., De Fusco C., Roughley S. D., Vidler L. R., Whatton M. A., Woolford A. J.-A., Wrigley G. L., O'Brien P., Chem. A Eur. J. 2020, 26, 8969–8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Congreve M., Carr R., Murray C., Jhoti H., Drug Discovery Today 2003, 8, 876–877. [DOI] [PubMed] [Google Scholar]
- 52. Firth N. C., Brown N., Blagg J., J. Chem. Inf. Model. 2012, 52, 2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Köster H., Craan T., Brass S., Herhaus C., Zentgraf M., Neumann L., Heine A., Klebe G., J. Med. Chem. 2011, 54, 7784–7796. [DOI] [PubMed] [Google Scholar]
- 54. Jhoti H., Williams G., Rees D. C., Murray C. W., Nat. Rev. Drug Discovery 2013, 12, 644–644. [DOI] [PubMed] [Google Scholar]
- 55. Abdel-Magid A. F., Carson K. G., Harris B. D., Maryanoff C. A., Shah R. D., J. Org. Chem. 1996, 61, 3849–3862. [DOI] [PubMed] [Google Scholar]
- 56. Gehringer M., Laufer S. A., J. Med. Chem. 2019, 62, 5673–5724. [DOI] [PubMed] [Google Scholar]
- 57. Ábrányi-Balogh P., Petri L., Imre T., Szijj P., Scarpino A., Hrast M., Mitrović A., Fonovič U. P., Németh K., Barreteau H., Roper D. I., Horváti K., Ferenczy G. G., Kos J., Ilaš J., Gobec S., Keserű G. M., Eur. J. Med. Chem. 2018, 160, 94–107. [DOI] [PubMed] [Google Scholar]
- 58. Hamilton D. J., Ábrányi-Balogh P., Keeley A., Petri L., Hrast M., Imre T., Wijtmans M., Gobec S., De Esch I. J. P., Keserű G. M., Pharmaceuticals 2020, 13, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yan A., Gasteiger J., J. Chem. Inf. Comput. Sci. 2003, 43, 429–434. [DOI] [PubMed] [Google Scholar]
- 60. Tajabadi F. M., Campitelli M. R., Quinn R. J., Springer Sci. Rev. 2013, 1, 141–151. [Google Scholar]
- 61. Bevan C. D., Lloyd R. S., Anal. Chem. 2000, 72, 1781–1787. [DOI] [PubMed] [Google Scholar]
- 62. Hoelke B., Gieringer S., Arlt M., Saal C., Anal. Chem. 2009, 81, 3165–3172. [DOI] [PubMed] [Google Scholar]
- 63. Roessler W. G., Brewer C. R., Appl. Microbiol. 1967, 15, 1114–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.