

Abstract
Objective
Bilateral synchronous cortical activity occurs during sleep, attention, and seizures. Canonical models place the thalamus at the center of bilateral cortical synchronization because it generates bilateral sleep spindle oscillations and primarily generalized absence seizures. However, classical studies suggest that the corpus callosum mediates bilateral cortical synchronization.
Methods
We mapped the spread of right frontal lobe‐onset, focal to bilateral seizures in mice and modified it using chemo and optogenetic suppression of motor thalamic nucleus and corpus callosotomy.
Results
Seizures from the right cortex spread faster to the left cortex than to the left thalamus. The 2 thalami have minimal monosynaptic commissural connections compared to the massive commissure corpus callosum. Chemogenetic and closed‐loop optogenetic inhibition of the right ventrolateral thalamic nucleus did not alter inter‐hemispheric seizure spread. However, anterior callosotomy delayed bilateral seizure oscillations.
Interpretation
Thalamocortical oscillations amplify focal onset motor seizures, and corpus callosum spreads them bilaterally. ANN NEUROL 2022;91:682–696
Bilateral synchronous neuronal oscillations are essential for many states, such as sleep, wakefulness, bilateral motor coordination, attention, and interhemispheric spread of seizures. Spindle oscillations are one of the classic examples of bilateral synchronous neuronal oscillations that occur during sleep. 1 The last 4 decades of research showed that the thalamus plays a key role during these bilateral oscillations. 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 Neurons of the somatosensory cortex, GABAergic reticular thalamic nucleus (RTN), and excitatory thalamocortical neurons generate characteristic low‐frequency oscillations of 7 to 14 Hz that underlie sleep spindles. 1 , 10 , 11 , 12 A modification of this mechanism generates rhythmic spike‐and‐wave discharges that propagate and amplify primarily generalized absence seizures. 1 , 13
In 1952, Wilder Penfield proposed the centrencephalic model (Fig 1A). The model proposes that the bilateral spread of neuronal activity occurs through the diencephalon, which acts as a rhythm generator in the brain's center and controls both sides of the brain simultaneously. 5 Jasper and Kershman proposed the existence of a subcortical pacemaker. 6 Thalamocortical oscillations that underlie sleep spindles and primarily generalized absence seizures strongly supported the centrencephalic hypothesis. 1 , 6 Disconnection of thalamocortical neurons from the reticular thalamic inputs abolished bilateral spindles. 9 Pioneering studies that used optogenetic technique to inhibit the thalamic nuclei ipsilateral to the stroke‐induced seizure focus in a post‐stroke model of epilepsy interrupted focal‐onset, and bilateral tonic–clonic seizures, defining the thalamus as a choke point. 2 , 14 Stimulation of the intralaminar thalamus, such as the centromedian nucleus, also led to bilateral synchronization that remained even after a complete transection of the corpus callosum during primarily generalized absence seizures. 6
FIGURE 1.
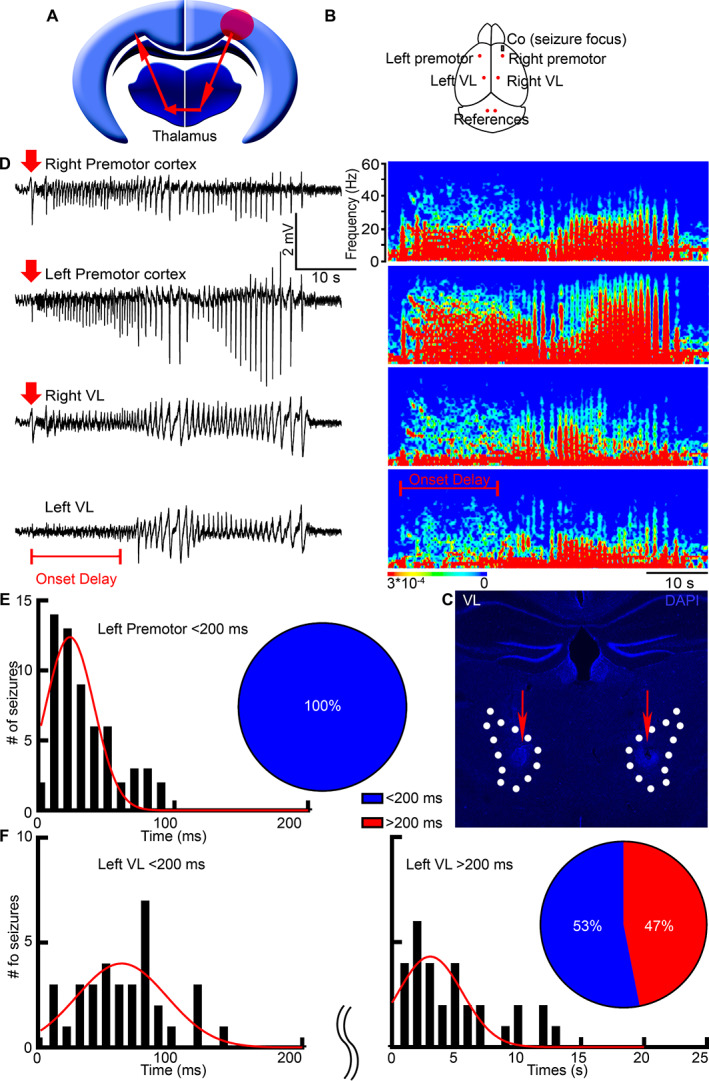
Seizures spread faster to the contralateral cortex than to the contralateral thalamus. (A) A schematic illustrates the classic centrencephalic model. Red arrows represent a bilateral spread of neuronal activity. The red circle is the seizure focus. (B) A schematic shows cobalt (grey rectangle) and LFP electrode placement (red dots). (C) We marked the electrode tips' location in the VL (dotted lines) by creating an electrical lesion at the end of the recordings (red arrows). (D) LFP microelectrodes record simultaneously from the bilateral premotor cortex and bilateral thalamic nucleus VL with the corresponding power spectrums. The seizure focus was in the right premotor cortex. Red arrows indicate seizure onset (amplitude twice the baseline); the red bar indicates seizure onset delay in the left VL. (E) Frequency distribution histogram shows seizure onset times in the left premotor cortex after seizure beginning in the right premotor cortex. Pie chart: 100% of seizures required < 200 ms (blue) to arrive at the left premotor cortex. (F) Frequency distribution histogram shows seizure onset times in the left VL. Pie chart: 53% of seizures required < 200 ms (blue) to arrive at the left VL from the right premotor cortex, and 47% required > 200 ms (red). LFP = local field potential; VL = ventrolateral.
In contrast, Roger Sperry, who studied patients that underwent a complete transection of the corpus callosum (split‐brain) to control intractable tonic–clonic seizures, showed that the corpus callosum is essential for interhemispheric transfer. 15 , 16 Sperry and colleagues found that despite a lack of obvious neurological or psychological dysfunction, the interhemispheric transfer of learning, memory, sensory, and motor functions was impaired. 15 Similarly, a complete transection of the corpus callosum abolished bilateral synchrony in primarily generalized penicillin epilepsy. 17 The lack of techniques made it difficult to separate the relative roles of the thalamus and corpus callosum in bilateral neuronal activity, so the uncertainty whether cortical synchronization occurs through the subcortical relays or the corpus callosum still remains.
Focal to bilateral seizures provide an excellent opportunity to test the mechanisms of bilateral synchrony because seizures originate at a defined focus and spread to both hemispheres. Several newly developed techniques allowed us to map bilateral seizure spread on a cellular level, visualize activated cells, and precisely silence specific brain structures. We disentangle the roles that the thalamus and corpus callosum play, elucidating the properties of these structures in bilateral spread of neuronal activity.
Methods
Animals
University of Virginia Animal Care and Use Committee approved all protocols. Transient Recombination in Active Populations second‐generation (TRAP2) mice were generated by crossing Fos2A−ICreERT2 (#030323) to Ai9 mice (#007909; Jackson Laboratories). 18 , 19 TRAP2 and C57Bl/6 (Charles River) mice (7–12 weeks) of both sexes were maintained on a 12 hour light/12 hour dark cycle and had ad libitum food and water. For genotyping, KAPA Biosystems kit was used.
Seizure Induction, Electroencephalogram, and Local Field Potential Electrophysiology
To induce frontal lobe focal to bilateral seizures, we implanted 1.7 mg of cobalt (Co) wire in the right premotor cortex (anterior posterior [AP] +2.60 mm, medial lateral [ML] −1.80 mm) in either TRAP2 or C57Bl/6 mice with 4 bilateral electroencephalography (EEG) electrodes and a reference under isoflurane anesthesia. 20 Mice were monitored continuously via video/EEG, and all of them developed seizures within 13 to 20 hours after cobalt insertion. Convulsions occurred at the start of the EEG onset because seizure focus was in the right premotor cortex. The 4‐hydroxytamoxifen (4‐OHT, 50 mg/kg s.c.) was injected within 90 min of a focal motor to bilateral tonic–clonic seizure, and mice were perfused 5 days after to allow tdTomato expression.
We recorded local field potentials (LFPs) with a custom‐made array of microelectrodes and analyzed LabChart 8.0 LFP data with Fast Fourier Transform, as previously described. 21 The 2 cm electrodes maintained 60 to 70 kΩ resistance and sampled at 10 kHz. The signal was amplified and filtered using a bandpass filter (1 Hz low pass and 5 kHz high pass) and a notch filter. We placed microelectrodes in the bilateral premotor cortex (AP −2.20 mm, ML ±1.80 mm) and bilateral ventrolateral cortex (VL; AP −1.30 mm, ML ±1.00 mm, dosal ventral [DV] −3.75 mm) with cobalt and 2 cerebellar references to record seizures (see Fig 1B). Seizure start was defined as a deflection of voltage trace at least twice the baseline. After recordings, the electrode position was marked by a single 40 μA, 0.75 ms pulse at 50 Hz for 30 seconds. The brains were sectioned at 40 μm, stained with DAPI, and imaged on Nikon Eclipse Ti‐S, 2x/0.45 NA. We discarded the data if the electrode was misplaced.
Immunohistochemistry, Microscopy, and Viral Injections
Mice were perfused with 4% PFA, sliced on a cryostat at 40 μm, and immunolabeled with rabbit anti‐NeuN (1:500, #24307; Cell Signaling), rabbit anti‐PV (1:500, #ab11427; Abcam), rat anti‐CTIP2 (1:500, #ab18465; Abcam), mouse anti‐MBP (1:500, #ab62631; Abcam), and secondary antibodies were Invitrogen, 1:500, as previously described. 21 Confocal images were taken on Nikon Eclipse Ti‐U at 10x/0.45 NA or 20x/0.95 NA, 1024 × 1024 or 512 × 512 frame size, 5 μm z‐interval. Myelinated axons were imaged on Leica SP8 STED at 93x/1.3 NA, glycerol. GFP FLEX callosum fibers were imaged on CSU‐W1 SoRa Yokogawa Spinning Disk Confocal, Nikon at 100x/1.35 NA, silicon, or 60x/1.49 NA, oil, 2304 × 2304, 0.5 μm step size. Excitation lasers were 405, 488, and 561. Imaris version 9.8 (Bitplane) was used for visualization and Adobe Photoshop CC for figure display.
We injected AAV9 CamKII0.4.eGFP.WPRE.rBG (#105541‐AAV9; Addgene), Cre‐driven AAV9 pCAG‐FLEX‐EGFP‐WPRE (#51502‐AAV9; Addgene), or green retrograde Lumafluor RetroBeads IX in the right premotor cortex of TRAP2 or C57Bl/6 mice (AP +2.6 mm, ML −1.8 mm, DV −0.5 mm; 100 nl) or in the right VL (anterior VL: AP −1.05 mm, posterior VL: AP: −1.40 mm, ML −1.00 mm, DV −3.70 mm) with Hamilton syringe (#7634‐01) and allowed 2 weeks for the viral expression.
Whole‐Cell Electrophysiology and Chemogenetics
We mixed AAV8 hSyn‐dF‐HA‐KORD‐IRES‐mCitrine (#65417‐AAV8; Addgene, 60 nl) and AAV9 CamKII.HI.GFP‐Cre.WPRE.SV40 (#105551‐AAV9; Addgene 60 nl) in 1:1 ratio and injected into the right VL (AP −1.30 mm, ML −1.00 mm, DV −3.70 mm, 120 nl) of C57Bl/6 mice and allowed 2 weeks for expression. We placed LFP microelectrodes in the bilateral premotor cortex and bilateral VL along with cobalt and recorded for 48 hours. Mice were injected either with salvinorin B (SALB; 10 mg/kg in DMSO) or saline s.c. at 20 hours after Co implantation at the peak of seizures. A blinded investigator counted the number of seizures and duration.
We confirmed the ability of KORD to generate SALB‐induced hyperpolarization using whole‐cell electrophysiology. The brains of C57Bl/6 KORD injected mice were immersed in ice‐cold oxygenated dissection buffer (4°C, 95% O2, and 5% CO2) containing (in mM) 65.5 NaCl, 2 KCl, 5 MgSO4, 1.1 KH2PO4, 1 CaCl2, 10 dextrose, and 113 sucrose (300 mOsm), and coronal thalamic slices (300 μm) were prepared with a vibratome (VT1200s; Leica). The slices were then placed in an interface chamber containing oxygenated artificial cerebrospinal fluid (aCSF) at 25°C and allowed to equilibrate for 30 minutes. The aCSF contained (in mM) 124 NaCl, 4 KCl, 1 MgCl2, 25.7 NaHCO3, 1.1 KH2PO4, 10 dextrose, and 2.5 CaCl2 (300 mOsm). The patch electrode was filled with an internal solution containing (in mM) 115 cesium methanesulfonate, 20 CsCl, 10 KCl, 10 HEPES, 0.5 sodium‐EGTA, 2.5 MgCl2, 5 Mg‐ATP, and 5 lidocaine, pH 7.3, 285mOsm. The mCitrine fluorescence positive neurons were current‐clamped. The 100 nM SALB in DMSO was bath applied for 10 minutes, and we tested the resting membrane potential to determine hyperpolarization.
Closed‐Loop Optogenetics
We injected C57Bl/6 mice with AAV9 CamKII‐ArchT‐GFP (#99039; Addgene, 120 nl) in the right VL and allowed 2 weeks for expression. Mice were then implanted with LFP microelectrodes in the bilateral premotor cortex and bilateral VL, cobalt, and an optic fiber in the right VL (AP −1.30 mm, ML −2.20 mm, DV −2.80 mm, at 21 degrees such that the probe did not intersect the right lateral ventricle). Our optogenetics setup consisted of fiber‐coupled LED light source (540 nm, 4.9 mW, 5 pulses in 500 ms [each pulse is 50 ms ON/50 ms OFF]) and polymer optical fiber 1,000 μm core, which was coupled to the polymer optical fiber 500 μm core via rotary joint (Prizmatix). The LED was externally triggered by a USB‐TTL interface based on Arduino UNO Rev3 board. The timing of pulses was controlled from MATLAB (2020a). The LFP signals were recorded using LabChart software (ADInstruments). We accessed LabChart functions from MATLAB and fetched LFP recording data in online mode. We developed a deep convolutional neural network‐based automated seizure detection model in MATLAB, which accepted single channel LFP measurements from the left motor cortex because right cortical LFP showed suppression of activity in a number of cases that could have affected the neural net output. The neural net continuously accessed data for 100 ms (1,000 samples at 10 kHz sampling frequency) in addition to 1.6384 s (16,384 samples) of the most recent data. The 100 ms data was then padded to the end of the 1.6384 s data. Later, the padded data was cropped to 16,384 samples that included the newly accessed 100 ms data. This formed a running window. Before feeding the data to the net, it was preprocessed in multiple stages to discard high‐frequency noise. The net output a scalar number corresponding to 16,384 samples, which was used to assess whether the 16,384 samples were a part of an ongoing seizure or not by using a manually set threshold −0.5. Once a seizure was detected, a signal from the USB‐TTL interface was promptly dispatched to activate transitor‐transitor logic (TTL) pulses.
Callosotomy
We used a custom‐made knife to cut the corpus callosum of C57Bl/6 mice. A small craniotomy was made at the posterior left cortex (AP −3.50 mm, ML ±0.50 mm), avoiding damaging the superior sagittal sinus to minimize bleeding. The knife was slowly advanced at an entrance angle of 52 degrees for 5 mm. The withdrawal angle was 54 degrees for 2.5 mm and changed back to 52 degrees for another 2.5 mm. Mice were also implanted with Co, 4 bilateral EEG electrodes, a reference, and were monitored for seizures via continuous video/EEG.
Statistics
We analyzed data using Prism 9 and presented them as mean ± SEM, where n is the number of animals or the number of seizures followed by the number of animals. Results were considered statistically significant for p < 0.05, where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. All data were tested for normality. Statistical analysis comparing 2 groups was performed by paired 2‐tailed t test or Kolmogorov–Smirnov test. Statistical analysis comparing multiple groups was performed by one‐way analysis of variance (ANOVA) with multiple comparisons and Bonferroni's correction, one‐way ANOVA with Tuckey test, Kruskal‐Wallis test with Dunn's multiple comparison test, Brown Forsythe ANOVA test with Welch's correction for variance, and Dunnett's multiple comparison test, as appropriate.
Results
Seizures Arrive faster to the Contralateral Cortex than to the Contralateral Thalamus
In this study, we compared the interhemispheric spread of seizures between the corpus callosum and the thalamus in focal onset seizures. To initiate frontal lobe onset, focal to bilateral tonic–clonic seizures, we placed 1.7 mg of cobalt in the right (ipsilateral) premotor cortex of C57Bl/6 mice as previously described. 20 , 21 To determine if the thalamus allows bilateral spread of neuronal activity, we mapped the temporal sequence of activation of the left cortex and left thalamus during bilateral seizure spread from the right cortex. To measure seizure onset latencies, we recorded LFPs bilaterally from the premotor cortex and VL motor thalamic nucleus simultaneously (see Fig 1B). We selected the motor thalamic nucleus VL because of its extensive reciprocal projections to the motor cortex 22 and ability to block after‐discharges in the motor cortex. 23 Custom‐made array of microelectrodes recorded with 65 kΩ resistance at a 10 kHz sampling rate, providing millisecond precision. A current was applied at the electrode tips at the end of the recordings to mark their location (see Fig 1C). We discarded the recording data if any electrode was placed incorrectly.
Surprisingly, right motor cortex seizures spread faster to the contralateral cortex than to the contralateral thalamus (see Fig 1D). This suggests that the bilateral seizure spread depends on extra‐thalamic mechanisms contrary to what the centrencephalic theory would predict (see Fig 1A). We recorded 64 seizures from 16 mice and found that all seizures arrived from the right cortex to the contralateral left cortex within 25 ms delay (25.49 ± 3.00 ms; see Fig 1E), which corresponds to the expected callosal transmission time. On the other hand, 47% of all seizures arrived at the left thalamus with a 4‐second delay (4.47 ± 0.65 s, n = 30 seizures from 11 mice; see Fig 1F), and the rest (53%) took approximately 60 ms (59.36 ± 5.61ms, n = 34 seizures from 13 mice; see Fig 1F).
Gamma oscillations, frequencies between 30 and 90 Hz, were still present in the left VL before seizure onset there (see Fig 1D, left VL power spectrum, gamma frequencies are in light blue). Gamma oscillations are thought to play a role in attention selection, memory, and cognition, and its presence before seizure onset in the left VL suggests that the cortex is generating these gamma frequencies. This is consistent with the previous literature that states it is unlikely for thalamocortical neurons to generate theta (4–8 Hz, red) and gamma rhythms concurrently, whereas the layered structure of the cortex generates delta (0–4 Hz, red) and theta rhythms from the deep layer 5 and gamma oscillations from the superficial layer 2/3 simultaneously. 24
The Ipsilateral Thalamus Expresses More c‐Fos Positive Neurons than the Contralateral Thalamus, with Only Minimal Monosynaptic Connections between the Two
We next determined whether the thalamus has bilateral neuronal activation during focal to bilateral seizures. We used second‐generation activity reporter TRAP2 mice that allowed us to visualize activated neurons. These transgenic mice express Cre under the immediate early gene promoter c‐Fos, and injection of 4‐OHT within 90 minutes of a focal to bilateral tonic–clonic seizure translocates Cre into the nucleus, allowing expression of tdTomato. Non‐active cells were not labeled with tdTomato because they did not express Cre in the 90 minutes prior to 4‐OHT injection. We serially sectioned the brain at 40 μm to analyze the pattern of tdTomato expression.
We found more robust tdTomato expression in the right thalamus ipsilateral to the seizure focus but not contralateral (Fig 2A). The right motor ventrolateral (VL) and ventromedial (VM), as well as the centromedian (CM) and mediodorsal (MD) nuclei were tdTomato positive, cellular activation of which we quantified previously. 21 The right RTN also expressed more tdTomato than the left (see Fig 2B). The seizure onset delay in the left thalamus made the overall seizure duration in the left thalamus shorter (see Fig 1D) and not sufficient for c‐Fos stimulation, explaining minimal tdTomato expression there (see Fig 2A). To verify that the observed thalamic activation was due to seizures and not due to motor activity or cortical injury, we implanted steel wire instead of cobalt and injected 4‐OHT 18 hours after surgery. These mice did not develop seizures and did not show thalamic activation (see Fig 2C).
FIGURE 2.
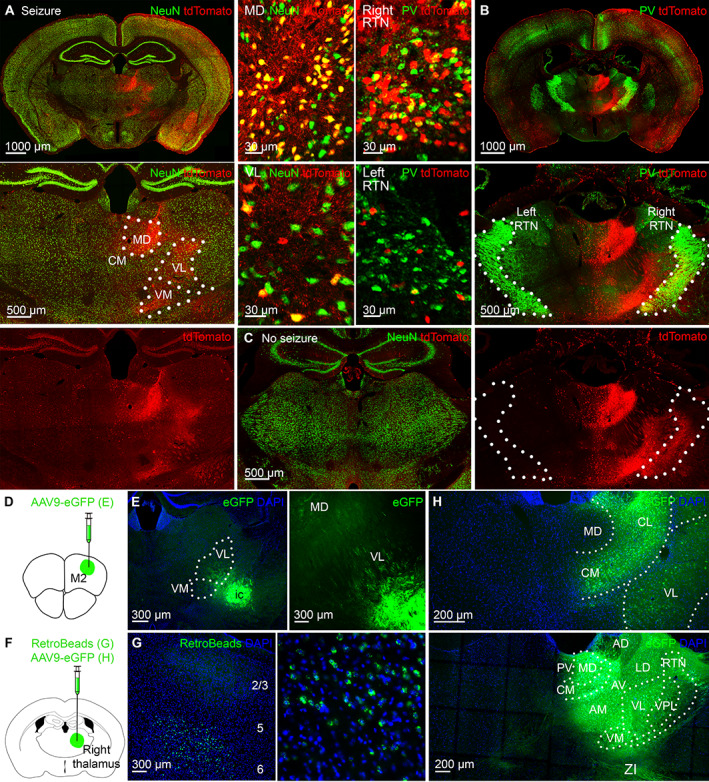
The activated ipsilateral thalamus sends minimal projections to the contralateral thalamus. (A) Only the right thalamus strongly expressed tdTomato. Dotted lines indicate nuclear boundaries. On the right are enlarged images of cellular tdTomato expression in the MD and VL immunolabeled for NeuN (green). (B) The right reticular thalamic nucleus (RTN) expressed more tdTomato than the left. RTN is immunolabeled for parvalbumin (PV, green). On the left are enlarged images of cellular tdTomato expression in the right and left RTN. (C) Minimal tdTomato expression is in the thalamus of steel wire implanted mice without seizures. (D) AAV9‐eGFP injection is in the right premotor cortex for E. (E) The right motor cortex projects to the right motor thalamic nuclei VM and VL. (F) AAV9 eGFP or RetroBeads injection is in the right VL for G and H. (G) The right motor cortex sends projections to the right thalamus from deep cortical layers 5 and 6 after retrograde RetroBeads injection in the right VL as indicated in F. (H) The right thalamic nuclei have minimal monosynaptic projections to the left thalamic nuclei. CM = centromedian; MD = mediodorsal; VL = ventrolateral; VM = ventromedial.
To determine if seizures utilize naturally existing circuit during the pathological state, we injected the AAV9 eGFP virus (AAV9‐CamKII0.4.eGFP.WPRE.rBG) in the right premotor cortex to label its direct anatomical projections (see Fig 2D). The right VM, VL, CM, and MD expressed GFP (see Fig 2E), the thalamic nuclei that expressed the most tdTomato during seizures (see Fig 2A). We then injected RetroBeads (a retrograde tracer) in the right VL nucleus (see Fig 2F); these were transported to deep layer 6 of the right premotor cortex (see Fig 2G), confirming the previous report that it is the deep layers of the motor cortex that project to the motor thalamus. 25 Direct anatomic connections between the seizure focus and the right thalamus explain the immediate seizure onset in the right VL (see Fig 1D). Seizures utilize direct anatomic connections of the seizure focus to propagate the pathological state.
Next, we wanted to determine if the right and left thalami are connected to each other via extensive intrathalamic commissural connections. To trace the anatomic projections from the right to left thalamus, we injected the AAV9 eGFP virus in the right thalamus (see Fig 2F). Surprisingly, we found minimal viral expression in the left thalamus (see Fig 2H), indicating that the right thalamus is very weakly connected to the left via direct monosynaptic connections and further explaining seizure onset delay in the left thalamus (see Fig 1D).
Seizures Still Spread Bilaterally after Chemogenetic Inhibition of the Ipsilateral Motor Thalamus
We next inhibited the right (ipsilateral) motor thalamus to determine if seizures would still spread bilaterally, using KORD/SALB chemogenetics, ĸ‐opioid receptor DREADD. We injected AAV8 hSyn‐dF‐HA‐KORD‐IRES‐mCitrine and AAV9 CamKII.HI.GFP‐Cre.WPRE.SV40 in 1:1 ratio in the right VL of C57Bl/6 mice (Fig 3A) and allowed 2 weeks for expression before cobalt and LFP microelectrode placement. We selected the right motor thalamic nucleus VL because this nucleus was strongly activated during frontal lobe seizures (see Fig 2A). Its principal thalamic neurons give rise to thalamocortical oscillations, and VL blockade by cooling abolished seizures originating in the premotor cortex. 23
FIGURE 3.
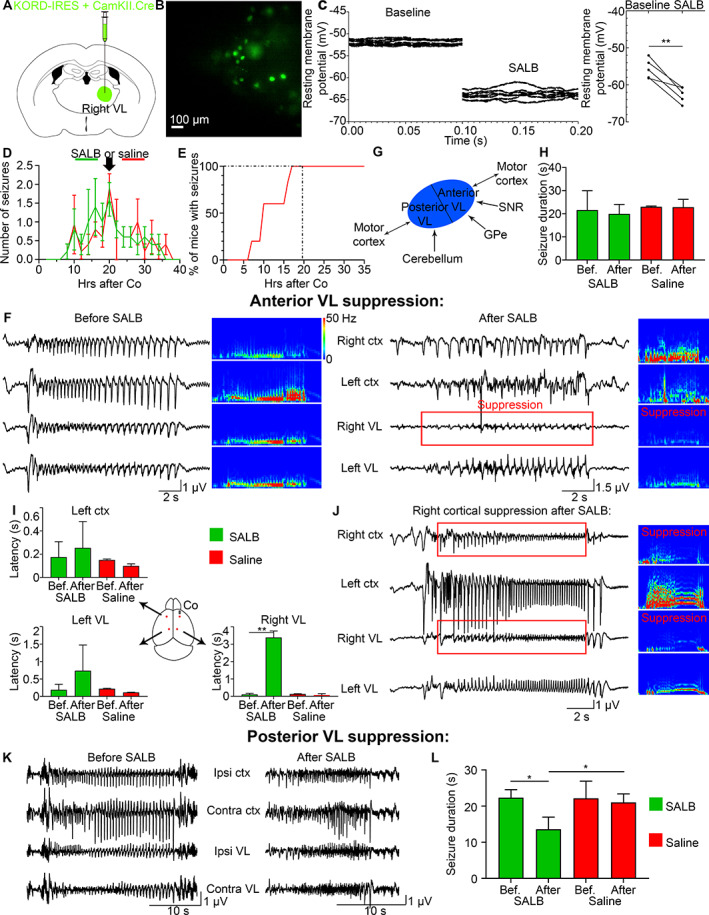
Chemogenetic suppression of the ipsilateral VL does not change the onset latency in the contralateral VL or contralateral cortex. (A) KORD/CamKII.Cre was injected in the right VL. (B) Patch‐clamp recordings were done on KORD‐expressing neurons in the right VL. (C) Resting membrane potential (mV) of transduced cells before and after SALB application, where the left graph is repeated recordings from a single cell, and the right graph is the average for 5 mice. (D) Mean number of seizures in C57Bl/6 mice after cobalt insertion in C57Bl/6 mice that were injected with SALB (green) or saline (red) at 20 hours after Co (black arrow) at the peak of seizures. (E) One hundred percent of all mice developed seizures by 20 hours after Co. (F) LFPs of a seizure recorded before and after SALB injection. Power was suppressed in the right VL (red box). (G) A schematic illustrates anatomical projections to the anterior and posterior VL. (H) Mean seizure duration (seconds) remained the same before (16–20 hours after Co) and after (20–24 hours after Co) SALB or saline injection. (I) Seizure onset was delayed only in the right VL after SALB injection but not in the left cortex or left VL. (J) Right cortex was suppressed after right VL suppression (in 6 out of 11 mice). (K, L) Seizure duration decreased after posterior VL suppression. Co = cobalt; VL = ventrolateral; SALB = salvinorin B.
We first confirmed KORD's ability to generate SALB‐induced hyperpolarization using whole‐cell patch‐clamp recordings in thalamic slices. Bath application of 100 nM SALB in DMSO led to hyperpolarization of transduced VL neurons (see Fig 3B,C, from −55.65 ± 1.176 mV to −62.60 ± 0.9522 mV, p = 0.0050, paired t test, n = 5 cells from 5 mice). We next tested it in vivo. We administered SALB (10 mg/kg) or saline systemically in KORD injected C57Bl/6 mice at 20 hours after cobalt placement at the peak of seizures (see Fig 3D), the time point when 100% of mice developed seizures (see Fig 3E). We found power suppression in the right VL in SALB but not in the saline injected mice (see Fig 3F).
We investigated the effects of anterior and posterior VL suppression on bilateral seizure spread. Both anterior and posterior VL receive projections from the motor cortex. Anterior VL also receives projections from the globus pallidus internus and substantia nigra reticulata. In contrast, the posterior VL receives projections from 4 deep cerebellar nuclei (the dentate, interposed anterior/posterior, and fastigial nuclei; see Fig 3G).
We first evaluated the effects of suppressing anterior VL activity on seizure duration, frequency, and bilateral spread. Anterior VL suppression did not shorten seizures (see Fig 3H, one‐way ANOVA with multiple comparisons and Bonferroni's correction for repeated data measurements, p = 0.91 (4 groups; 6 pairs); before SALB: 21.61 ± 3.75 s, after SALB: 19.95 ± 2.04 s; before saline: 23.03 ± 0.23 s, and after saline: 22.83 ± 1.97 s). Anterior VL suppression did not reduce the number of seizures (one‐way ANOVA with Tukey test, p = 0.69; before SALB: 13 seizures, after SALB: 11; before saline: 9, after saline: 10; n = 5 mice). Surprisingly, seizures still spread to the left cortex after chemogenetic suppression of the right thalamus (see Fig 3F). Despite right VL power suppression during the seizure, the onset latency from the right cortex to the left cortex and to the left VL did not change, whereas seizure onset latency to the right VL increased (see Fig 3I, Left ctx: Kruskal‐Wallis test with Dunn's multiple comparison test, p = 0.29; before SALB: 175.0 ± 59.29 ms, after SALB: 254.8 ± 113.0 ms; before saline: 150.6 ± 5.63 ms, after saline: 99.75 ± 10.31 ms; left VL: Kruskal‐Wallis test with Dunn's multiple comparison test, p = 0.26; before SALB: 192.3 ± 70.35 ms, after SALB: 739.1 ± 368.2 ms; before saline: 222.8 ± 10.25 ms, after saline: 118.2 ± 3.17 ms; right VL: Brown Forsythe ANOVA test with Welch's correction for variance and Dunnett's multiple comparison test, p = 0.0050; before SALB: 113.8 ± 21.56 ms, after SALB: 3084.0 ± 437.1 ms, p = 0.0020; before saline: 135.6 ± 20.63 ms, after saline: 102.1 ± 27.08 ms; after SALB, after saline, p = 0.024; n = 5 mice). We saw no change in seizure behavior after the right VL suppression. SALB did not produce an anticonvulsant effect. In some seizures (55%), seizure amplitude was suppressed in the right motor cortex simultaneously with the right VL suppression (see Fig 3J), which is consistent with the ipsilateral thalamocortical oscillations. Decrease in seizure amplitude during the VL suppression (and the corresponding decrease in lower frequencies associated with thalamocortical oscillations) indicates that the thalamus acts as an amplifier.
When we inhibited the posterior VL, the activity amplitude in the right VL did not change (see Fig 3K). However, although the overall seizure duration decreased by almost half, the total number of seizures doubled (see Fig 3L, Seizure duration: Brown Forsythe ANOVA test with Dunnett's multiple comparison test, p = 0.035; before SALB: 22.35 ± 1.28 s, after SALB: 13.60 ± 1.68 s, p = 0.024; before saline: 22.17 ± 2.74 s, after saline: 21.03 ± 1.35 s; after SALB, after saline, p = 0.048; Seizure number: one‐way ANOVA with Tukey test, p = 0.0084, before SALB: 11 seizures, after SALB: 17; before saline: 8, after saline: 8; p = 0.013; n = 5 mice). Shortening of seizures in the absence of motor thalamic activity and increase in seizure duration in the presence of VL further indicates that the thalamus exacerbates seizures, amplifying their duration.
Seizures Spread Bilaterally after Closed‐Loop Optogenetic Inhibition of the Ipsilateral Motor Thalamus
Because chemogenetics does not allow precise temporal control over the VL inhibition and requires administration of SALB systemically, we suppressed VL activity using closed‐loop optogenetics. We injected AAV9 CamKII‐ArchT‐GFP in the right anterior VL (Fig 4A) that produced hyperpolarization in transduced neurons when illuminated with 540 nm green light. Two weeks after viral injection, we placed cobalt, 4 LFP microelectrodes in the bilateral premotor cortex and VL, as well as an optic fiber in the right VL (see Fig 4B). We developed a deep convolutional neural net model in MATLAB for online seizure detection in a fully automated way. We designed a regression‐based learning algorithm and trained it using a training dataset that included seizures against a baseline, ictal and pre‐ictal spikes, noise, and artifacts. The neural net continuously fetched a window of the LFP data without interrupting the LFP recording, transferred the data to the neural net, generated a score for a running window of data, and compared it to a threshold (set at −0.5) to activate a train of light pulses, whenever a seizure was detected. In 5 mice, we interrupted 27 seizures (the light started to trigger after 20 hours from Co insertion) and left 18 seizures uninterrupted (before 20 hours after Co) to obtain the same mouse internal control.
FIGURE 4.
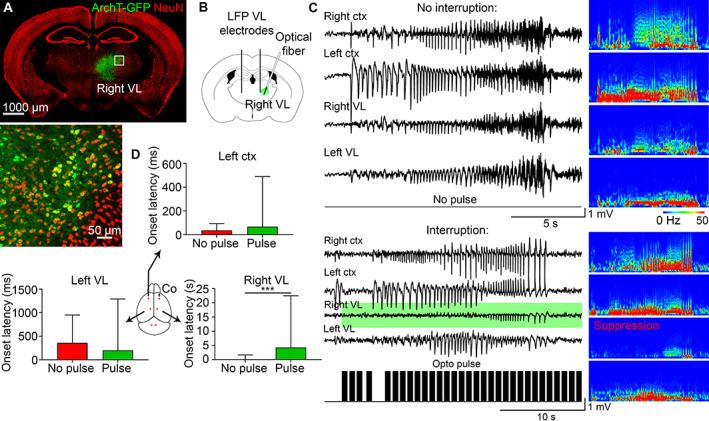
Closed‐loop optogenetic inhibition of the ipsilateral VL does not change the onset latency in the contralateral VL or contralateral cortex. (A) ArchT‐GFP expressed in the right VL. (B) AAV9 CamKII‐ArchT‐GFP was injected in the right VL. (C) Seizures that were uninterrupted (no pulse) and interrupted (with opto pulse [black bars], green background in the right VL) in the same mouse with the corresponding power spectrums. (D) Seizure onset was delayed only in the right VL after pulse activation, whereas onset latency in the left cortex or left VL did not change. LFP = local field potential; VL = ventrolateral.
Seizures still spread to the contralateral cortex and contralateral VL after closed‐loop optogenetic inhibition of the ipsilateral VL activity (see Fig 4C), supporting the chemogenetics data. The power spectrum validated VL suppression compared to uninterrupted control seizures (see Fig 4C). Seizure onset latency from the right cortex to the right VL increased (see Fig 4D, control seizure onset latency (no pulse): median 50 ms, 90th percentile 4.7 s; interrupted seizure (pulse): median 1.4 s, 90th percentile 18.8 s; p = 0.0003, Kolmogorov‐Smirnov test). In contrast, seizure onset latency in the left cortex remained the same (see Fig 4D; no pulse: median 50 ms, 90th percentile 100 ms; pulse: median 50 ms, 90th percentile 155 ms; p = 0.129, Kolmogorov‐Smirnov test) with a slightly longer latency in the left VL (no pulse: median 200 ms, 90th percentile 1.25 s; pulse: median 100 ms, 90th percentile 2.6 s; p = 0.56, Kolmogorov‐Smirnov test).
Focal to Bilateral Seizures Activate the Corpus Callosum
We propose that the corpus callosum allows the bilateral spread of the focal to bilateral tonic–clonic seizures, whereas the thalamus amplifies them (see Fig 5A). The LFP recordings confirmed that 100% of all seizures required approximately 25 ms to arrive at the left cortex from the right cortex, whereas half of all seizures either needed 4 seconds or 60 ms to arrive at the left VL (see Fig 1E,F).
FIGURE 5.
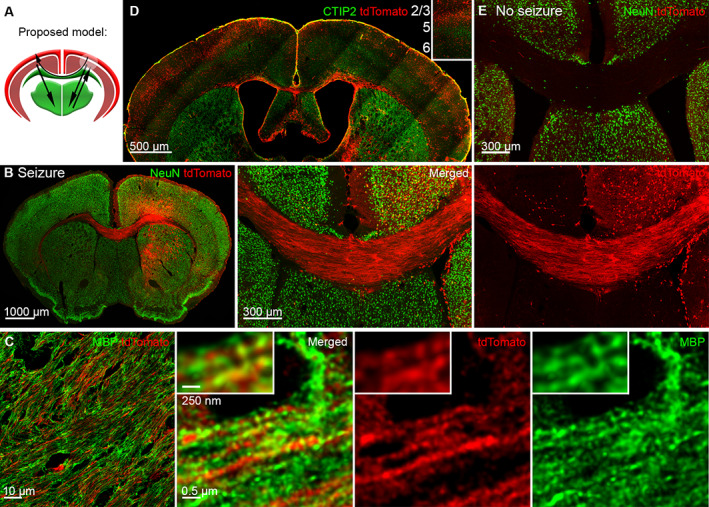
Focal to bilateral seizures activate the corpus callosum. (A) The proposed model of focal to bilateral seizures, where black arrows indicate seizure spread across the corpus callosum and to the thalamus, while a white circle in the right cortex indicates the seizure focus. (B) The corpus callosum strongly expressed tdTomato after focal to bilateral tonic‐clonic seizures. (C) SP8 super‐resolution Lightning microscopy images tdTomato positive axons expressing myelin basic protein (MBP; green) in the corpus callosum. (D) Strong activation of layers 2 and 3 in the right and left cortex, immunolabeled for CTIP2 (green) that marks layers 5 and 6, separating superficial from deep cortical layers. (E) Steel wire implanted control mice without seizures did not express tdTomato in the corpus callosum.
In activity reporter TRAP2 mice, we found that the whole corpus callosum extensively expressed tdTomato from anterior to posterior sections (see Fig 5B), indicating its activation during focal to bilateral seizures. To verify that these were axons, we used an antibody against myelin basic protein (MBP). SP8 super‐resolution lightning microscopy revealed myelin spiraling around tdTomato positive corpus callosum axons (see Fig 5C). Eighty percent of the callosal axons originate from superficial layers 2 and 3, 26 and we found strong activation of the superficial cortical layers 2 and 3 in both right and left cortex (see Fig 5D). Control steel wire implanted mice without seizures had no callosal activation (see Fig 5E).
Seizure Focus Sends Extensive Projections Contralaterally across the Corpus Callosum
To trace efferent projections of the seizure focus, we injected AAV9 GFP virus in TRAP2 mice at the same coordinates in the right premotor cortex 2 weeks before cobalt placement and injected 4‐OHT within 90 minutes after focal to bilateral tonic–clonic seizure to label anatomic connections and seizure activated cells at the same time. The corpus callosum strongly expressed AAV9 GFP and tdTomato (Fig 6A), confirming that seizures follow direct anatomic projections of the seizure focus across the corpus callosum to spread contralaterally.
FIGURE 6.
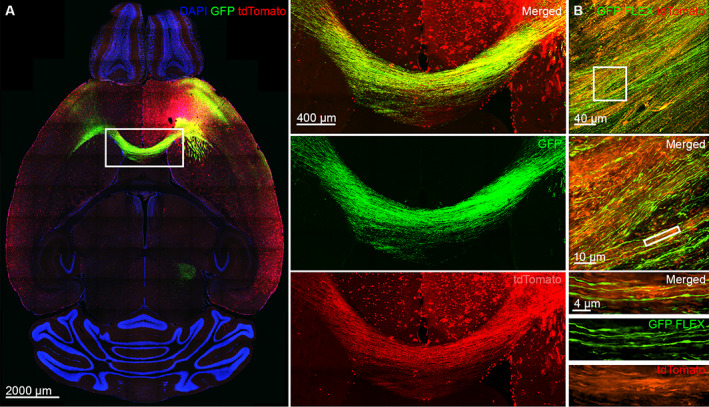
The seizure focus sends extensive direct projections across the corpus callosum contralaterally. (A) The tdTomato and AAV9 GFP colocalized in the axons of the corpus callosum that connects the seizure focus with the contralateral premotor cortex. The white rectangle area is magnified on the right. (B) Top: A confocal image of Cre‐driven AAV9 GFP expression in the corpus callosum of TRAP2 mice. Middle: Super‐resolution SoRa image of the callosal axons expressing Cre‐driven AAV9 GFP with a magnified view.
To further confirm that seizures propagate through the corpus callosum, we injected Cre‐dependent AAV9 virus (AAV9 pCAG‐FLEX‐EGFP‐WPRE) in TRAP2 mice (which express Cre under c‐Fos regulation), so that only seizure‐activated neurons and axons would express eGFP. The corpus callosum strongly expressed Cre‐dependent GFP from the anterior to posterior sections (see Fig 6B). Super‐resolution microscopy confirmed that tdTomato positive axons of the corpus callosum colocalized with GFP (see Fig 6B).
Anterior Callosotomy Prevents Bilateral Seizure Spread
We next transected the corpus callosum to determine if it would stop bilateral seizure spread. A thin knife was inserted in the left posterior cortex and advanced anteriorly to avoid damaging the superior sagittal sinus (Fig 7A,B). The genu and the body of the corpus callosum were cut on the left side as the knife was removed, the cobalt was inserted, the EEG electrodes were placed in the bilateral anterior and posterior cortices (see Fig 7C), and the mice were continuously monitored for seizures via video/EEG.
FIGURE 7.
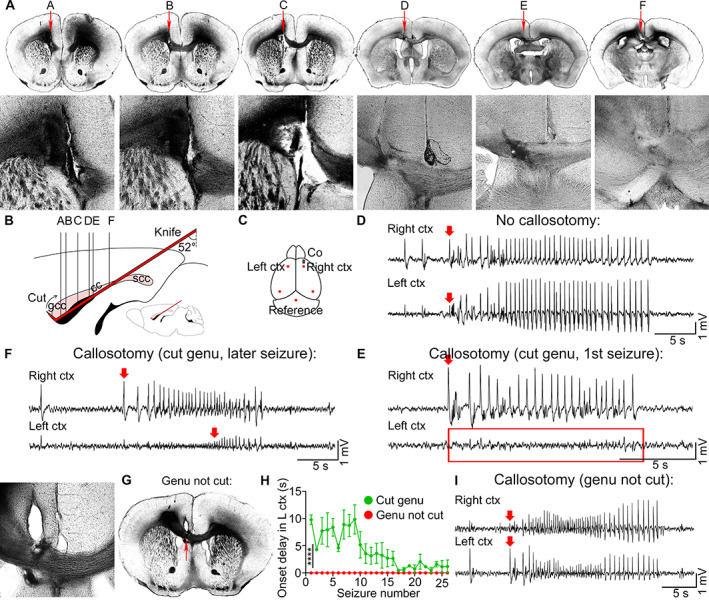
Anterior callosotomy prevents bilateral seizure spread. (A) Sequential light images of the coronal sections show callosotomy (red arrows, magnified views) from anterior to posterior cortex (A–F, relative brain locations in B). (B) A schematic of the callosotomy, sagittal view, illustrates knife placement (red), cut genu of the corpus callosum (gcc), and uncut splenium (scc). (C) Cobalt and EEG electrodes (red dots) were positioned in the right and left anterior and posterior cortices. (D) EEG of a seizure that was recorded in mice without callosotomy. Red arrows indicate seizure onset. (E) Representative EEG of a first seizure that was recorded after callosotomy in mice with cut genu with no seizure recorded in the left premotor cortex (red rectangle). (F) Onset oflater seizures was delayed in mice with cut genu. (G) Light image of the callosotomy where only the body of the corpus callosum was cut and the genu remained intact (red arrow). (H) Seizure onset delay (seconds) in the left premotor cortex (L ctx) after callosotomy in mice with cut genu (green) and in mice with body callosotomy and intact genu (red). (I) Mice with body callosotomy and intact genu had immediate seizure onset in the left premotor cortex just like the mice without callosotomy. EEG = electroencephalography.
Corpus callosotomy abolished bilateral seizure spread compared to mice without callosotomy (see Fig 7D,E), indicating that the corpus callosum is sufficient for bilateral spread of neuronal activity. Seizures from the ipsilateral cortex arrived later to the contralateral cortex, as indicated by a seizure onset delay on EEG (see Fig 7E), corresponding to the callosal transmission time after the genu of the corpus callosum was cut. However, as the number of seizures that the lesioned mice experienced increased, seizure onset delay in the left hemisphere became shorter, but seizures still arrived contralaterally later than in mice without callosotomy (see Fig 7F,D; cut genu, mean seizure onset delay in the left cortex: 3.8 ± 0.56 s, n = 5 mice).
In the instances when the genu was not severed but the body of the corpus callosum was (see Fig 7G), the mice experienced immediate contralateral seizure onset just like the mice without callosotomy (see Fig 7I,H), suggesting that it is the genu of the corpus callosum that is essential for focal to bilateral seizure spread (genu not cut, mean seizure onset delay in the left cortex: 0.0 ± 0.0 s, Kolmogorov‐Smirnov test, n = 3 mice).
Discussion
Our studies re‐examined the role of the “centrencephalon” and corpus callosum in the bilateral spread of cortical activity, a debate going back more than 5 decades. We find for the first time that thalamocortical oscillations and seizure spread via the corpus callosum occur in parallel and propose that the corpus callosum allows bilateral seizure spread, whereas the thalamus amplifies seizures. Our results reflect the parallel nature of cortical and thalamic circuits that process lateralized sensory and motor information as well as the interlobar integration of this sensory and motor information in the cortex. Seizures hijack normal circuits, providing the pathway for the spread of pathological activity. Understanding the relative contributions of the thalamus and corpus callosum provides insights into exacerbation of focal to bilateral seizures as well as the integrative function of the cortex of bilateral neuronal activity.
Local field potential recordings showed that focal to bilateral seizures arrived at the contralateral cortex faster than at the contralateral thalamus. The 2 thalami have minimal direct monosynaptic commissural connections, whereas the corpus callosum massively projects contralaterally. We propose that seizures spread between the 2 thalami by engaging the cortico‐cortical commissure, the corpus callosum. Previous studies optogenetically inhibited the right ventral posterolateral nucleus/ventral posteromedial nucleus (VPM/VPL) thalamic nuclei during stroke‐induced seizures in the right somatosensory cortex, which sends extensive anatomic projections to the right VPM/VPL, and stopped bilateral seizure spread. 2 We inhibited the right motor nucleus of the thalamus because it receives strong projections from the right motor cortex, 22 the seizure focus in our case. Surprisingly, seizures still spread contralaterally. Our study did not reproduce the results of Paz et al 2 study due to differences in seizure focus location. They located the seizure focus in the somatosensory cortex, which sends strong reciprocal connections with the sensory thalamus. In 2 examples presented, the suppression of thalamic activity suppressed the seizure in the ipsilateral cortex. In contrast, suppression of motor thalamic nuclei did not wholly suppress seizures in the motor cortex. Perhaps the motor cortex has a different relationship with the motor thalamus than the somatosensory cortex and thalamus. We next developed a fully automated deep convolutional neural network‐based seizure detection system in MATLAB for closed‐loop optogenetic inhibition of the right motor thalamus. Seizure spread to the left cortex was not altered, confirming our chemogenetic findings. Anterior callosotomy, on the other hand, abolished contralateral spread, indicating that the corpus callosum is sufficient for interhemispheric cortical synchronization.
Classically, primarily generalized seizures engage the networks of both sides of the brain at the onset. In contrast, secondarily generalized seizures originate at a focus and are thought to access the circuit for generalized seizures, 27 which causes bilateral spread. However, the term secondary generalized is confusing because it conflates the ideas of interlobar and interhemispheric spread. Although the name of “secondary generalized” seizures has recently been changed to “focal to bilateral,” 28 the idea that secondarily and primarily generalized seizure access to the thalamocortical oscillatory circuit is required for bilateral spread still remains. 2 , 8 , 9 , 27 , 29 We could not confirm the existence of a single seizure choke point and demonstrate that neuronal circuits generating seizures with focal cortical onset with bilateral spread are more complex than previously proposed.
The complexity may come from the layered organization of the motor cortex that allows independent, parallel transfer of neuronal activity to the contralateral cortex and ipsilateral subcortical structures. Eighty percent of all callosal axons originate in layers 2 and 3 principal pyramidal neurons, 26 whereas layer 5 pyramidal tract neurons (PT‐type) and layer 6 corticothalamic neurons (CT‐type) project to the subcortical structures ipsilaterally. 25 , 30 Therefore, inhibiting the right motor thalamus affects right thalamocortical projections to layers 5 and 6 of the motor cortex, whereas seizure still spreads bilaterally via layers 2 and 3. We reveal that right and left cortico‐thalamic circuits operate independently of each other (in parallel) and are actually synchronized by cortico‐cortical connections arising from layers 2 and 3 through the corpus callosum. Studies in infant sleep EEG further suggest a role for corpus callosum in synchronizing thalamocortical sleep spindles. The posterior part of the corpus callosum, the splenium, is myelinated by the sixth month of age, whereas the genu of the corpus callosum does not myelinate until the eighth month. 31 Coincidentally, mature spindle activity reaches the peak between 3 and 9 months of age. 32
We selected kappa‐opioid receptor‐based DREADD over clozapine N‐oxide‐based chemogenetics because CNO is reverse metabolized to clozapine, 33 a dopamine 2 receptor antagonist that increases susceptibility to seizures, 34 , 35 compared to inert SALB that opens GIRK channels to induce hyperpolarization. 36 , 37 However, chemogenetics still has temporal limitations; systemic injection of SALB inhibits a structure long before seizure onset, which potentially could have additional biological effects. In addition, activity was not sufficiently inhibited by chemogenetics because gamma frequencies still remained after the right VL suppression (see Fig 3F). To more precisely control the activity, we used optogenetics. Instead of devising a customized rule‐based seizure detection or a hardware‐based implementation, 2 , 38 we for the first time integrated a deep convolutional neural regressor network to detect seizures in the closed‐loop optogenetic system. Except for the optogenetic toolbox and Arduino TTL pulse generator, our system does not require additional hardware, such as a digital signal processor chip. The network is rigorously trained on seizure LFP against that of a baseline, high‐frequency bursts, noise due to mouse movements, acquisition noise, and single spikes. The system advantages are configuring desired probing (ON and OFF) pattern, stoppage/delayed start of pulses, integration of multichannel features, and low‐latency adaptive triggering unlike any hardware‐based implementation. When using different EEG/LFP acquisition systems, transfer learning on our network can easily be performed as well.
We found that the amplitude and duration of a seizure are amplified by the VL thalamus. Seizure amplitude was suppressed at the motor cortex without any change in seizure duration during anterior VL inhibition. The seizure duration at the motor cortex was reduced upon silencing the posterior motor thalamus with a rise in the frequency of seizures. These findings demonstrate that the VL thalamus amplifies seizures. Anatomic tracing suggests that layers 5 and 6 of the motor cortex innervates the VL neurons, whereas VL sends glutamatergic projections to layer 1 and layers 2 and 3 of the motor cortex. 22 , 39 , 40 Inhibiting VL activity disabled this path, thereby ceasing the amplification of a seizure.
Studies of primarily generalized absence seizures showed that stimulation of the intralaminar thalamus was sufficient for bilateral synchronization even after a complete section of the corpus callosum. 6 We did not find evidence for that. Although we could not exclude the possibility that the brainstem might be responsible for contralateral seizure spread, there are limited anatomic and functional evidence for bilateral seizure spread via the brainstem. For example, the 2 pyramids containing the cortex's motor fibers decussate in the medulla oblongata and descend via the spinal tract. In addition, extrapyramidal tracts in the brainstem carry motor fibers to target lower motor neurons in the spinal cord like the rubrospinal tract that originates in the red nucleus. Finally, polysynaptic connections via reticular formation could connect back to the contralateral cortex. This polysynaptic circuit complex going through the reticular formation would cause a long delay in seizure spread to the contralateral cortex. However, we observed only a 25 ms delay (see Fig 1E). When we cut the genu of the corpus callosum, there was no ictal activity in the contralateral (left) cortex. If the ipsilateral (right) cortical discharge spread via the brainstem to the contralateral cortex, then the effect of callosotomy would not be observed, as shown in Figure 7E. Other studies of the intralaminar thalamus showed desynchronized activation of the right and left centromedian nucleus during nonconvulsive and tonic–clonic generalized seizures, 41 supporting the idea of independent operation of the thalamocortical circuits in each hemisphere and their cortical integration through the anterior corpus callosum. In later years, Penfield considerably modified the centrencephalic hypothesis to account for the work of Roger Sperry on patients with split brain. 42
Hughlings Jackson first pointed the role of the cerebral cortex in generating convulsions (Jacksonian March). 43 Wilder Penfield and Herbert Jasper made extensive recordings from the motor cortex confirming it the site of origin of bilateral convulsions. 5 , 44 Extensive literature and clinical practice show that corpus callosotomy effectively treat medically refractory generalized epilepsy, drop attacks, Lennox–Gastaut syndrome, infantile spasms, and other generalized seizures. 45 , 46
Future therapies could focus on targeting the neocortical neurons and circuits. Currently, anterior thalamic deep brain stimulation is performed on patients with intractable epilepsy, but it is not always effective. In fact, only 16% of patients report seizure‐free interval of at least 6 months, 35.5% develop device‐related serious adverse side effects, 37.3% report depression, and 27.3% memory impairment. 47 , 48 Callosotomies were previously used to treat refractory generalized epilepsy but are less common in current clinical practice. Selective lesions of the callosal fibers that connect the seizure focus for example through the development of noninvasive focused ultrasound and deep brain stimulation to modulate cortico‐cortical projections could reduce generalized tonic–clonic seizures and sudden unexpected death in epilepsy (SUDEP) and serve as alternatives to thalamic stimulation.
In summary, our findings emphasize that the cortex transmits seizures to the contralateral side via the corpus callosum, even when thalamic input is suppressed, emphasizing that the cortical integration is independent of thalamic input.
Author Contributions
A.B., T.B., S.S., and J.K. contributed to conception and design of the study. A.B., T.B., S.S., and H.S. contributed to acquisition and analysis of data. A.B., T.B., S.S., and J.K. contributed to drafting the text or preparing the figures.
Potential Conflicts of Interest
The authors report no conflicts of interests.
Acknowledgments
Super‐resolution SP8 STED, Leica, and CSU‐W1 SoRa Yokogawa Spinning Disk Confocal, Nikon imaging was performed at the University of Virginia W.M. Keck Center for Cellular Imaging, which is supported by NIH‐OD025156. The authors also thank John Williamson, Suchitra Joshi, and the rest of the Kapur laboratory for valuable comments on this study. This work was supported by the National Institute of Health (R37 NS119012, R01NS120945, and RO1 NS040337 to J.K.) and the UVA Brain Institute.
Contributor Information
Anastasia Brodovskaya, Email: ab9mc@virginia.edu.
Tamal Batabyal, Email: tb2ea@virginia.edu.
Shinnosuke Shiono, Email: s.shiono1604@gmail.com.
Huayu Sun, Email: hs4kn@virginia.edu.
Jaideep Kapur, Email: jk8t@virginia.edu.
Data Availability Statement
All datasets and codes are available upon request from the Lead Contact.
References
- 1. Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science 1993;262:679–685. [DOI] [PubMed] [Google Scholar]
- 2. Paz JT, Davidson TJ, Frechette ES, et al. Closed‐loop optogenetic control of thalamus as a new tool to interrupt seizures after cortical injury. Nat Neurosci 2013;16:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blumenfeld H. The thalamus and seizures. Arch Neurol 2002;59:135–137. [DOI] [PubMed] [Google Scholar]
- 4. Huguenard JR, Prince DA. Basic mechanisms of epileptic discharges in the thalamus. In: Steriade M, Jones EG, McCormick DA, eds. Thalamus. Amsterdam, Netherlands: Elsevier, 1997:295‐330. [Google Scholar]
- 5. Penfield W, Jasper HH. Epilepsy and the Functional Anatomy of the Human Brain. Boston: Little, Brown, 1954. [Google Scholar]
- 6. Hunter J, Jasper HH. Effects of thalamic stimulation in unanaesthetised animals. Electroencephalogr Clin Neurophysiol 1949;1:305–324. [DOI] [PubMed] [Google Scholar]
- 7. Steriade M, Deschenes M. The thalamus as a neuronal oscillator. Brain Res Rev 1984;8:1–63. [DOI] [PubMed] [Google Scholar]
- 8. Gloor P, Avoli M, Kostopoulos G. Thalamocortical relationships in generalized epilepsy with bilaterally synchronous spike‐and‐wave discharge. In: Avoli M, Gloor P, Kostopoulos G, Naquet R, eds. Generalized Epilepsy. Boston: Birkhäuser, 1990:190‐212. [Google Scholar]
- 9. Steriade M, Deschenes M, Domich L, Mulle C. Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J Neurophysiol 1985;54:1473–1497. [DOI] [PubMed] [Google Scholar]
- 10. Cain SM, Snutch TP. Voltage‐gated calcium channels in epilepsy. In: Noebels JL, Avoli M, Rogawski MA, eds. Jasper's Basic Mechanisms of the Epilepsies. 4th ed. New York: Oxford University Press, Inc., 2012. [Google Scholar]
- 11. Steriade M, Domich L, Oakson G. Reticularis thalami neurons revisited: activity changes during shifts in states of vigilance. J Neurosci 1966;6:68–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Steriade M, Domich L, Oakson G, Deschfines M. The deafferented reticular thalamic nucleus generates spindle rhythmicity. J Neurophysiol 1987;57:260–273. [DOI] [PubMed] [Google Scholar]
- 13. Huguenard JR, Coulter DA, Prince DA. Physiology of thalamic relay neurons: properties of calcium currents involved in burst‐firing. In: Avoli M, Gloor P, Kostopoulos G, Naquet R, eds. Generalized Epilepsy. Boston: Birkhäuser, 1990:181‐189. [Google Scholar]
- 14. Paz JT, Huguenard JR. Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat Neurosci 2015;18:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sperry RW, Gazzaniga MS, Bogen J. Interhemispheric relationships: the neocortical commissures; syndromes of hemisphere disconnection. In: Vinken PJ, Bruyn GW, eds. Disorders of Speech, Perception and Symbolic Behavior. Handbook of Clinical Neurology. Vol 4. Amsterdam, Netherlands:North‐Holland Publishing Co., 1969:273‐290. [Google Scholar]
- 16. Sperry RW. Hemisphere deconnection and unity in conscious awareness. Am Psychol 1968;23:723–733. [DOI] [PubMed] [Google Scholar]
- 17. Press R, York N, Musgrave J, Gloor P. The role of the corpus callosum in bilateral interhemispheric synchrony of spike and wave discharge in feline generalized penicillin epilepsy. Epilepsia 1980;21:369–378. [DOI] [PubMed] [Google Scholar]
- 18. Guenthner CJ, Miyamichi K, Yang HH, et al. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 2013;78:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Allen WE, DeNardo LA, Chen MZ, et al. Thirst‐associated preoptic neurons encode an aversive motivational drive. Science 2017;357:1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh T, Joshi S, Williamson JM, Kapur J. Neocortical injury‐induced status epilepticus. Epilepsia 2020;61:2811–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brodovskaya A, Shiono S, Kapur J. Activation of the basal ganglia and indirect pathway neurons during frontal lobe seizures. Brain 2021;144:2074–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hooks BM, Mao T, Gutnisky DA, et al. Organization of cortical and thalamic input to pyramidal neurons in mouse motor cortex. J Neurosci 2013;33:748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gasteiger EL, Albowitz B, Barken FM. Interictal afterdischarge in focal penicillin epilepsy: block by thalamic cooling. Exp Neurol 1985;88:349–359. [DOI] [PubMed] [Google Scholar]
- 24. Hunt MJ, Kopell NJ, Traub RD, Whittington MA. Aberrant network activity in schizophrenia. Trends Neurosci 2017;40:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shepherd GM, Rowe TB. Neocortical lamination: insights from neuron types and evolutionary precursors. Front Neuroanat 2017;11:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fame RM, Macdonald JL, Macklis JD. Development, specification, and diversity of callosal projection neurons. Trends Genet 2011;34:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Westbrook GL. Seizures and epilepsy. In: Kandel ER, Schwartz JH, Jessell TM, et al., eds. Principles of Neural Science. New York: McGraw‐Hill, 2013:1116‐1139. [Google Scholar]
- 28. Falco‐Walter JJ, Scheffer IE, Fisher RS. The new definition and classification of seizures and epilepsy. Epilepsy Res 2018;139:73–79. [DOI] [PubMed] [Google Scholar]
- 29. Englot DJ, Morgan VL, Chang C. Impaired vigilance networks in temporal lobe epilepsy: mechanisms and clinical implications. Epilepsia 2020;61:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gerfen CR, Michael E, Jayaram C. Long distance projections of cortical pyramidal neurons. J Neurosci Res 2018;96:1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krupa K, Bekiesinska‐Figatowska M. Congenital and acquired abnormalities of the corpus callosum: a pictorial essay. Biomed Res Int 2013;2013:265619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tanguay P, Ornitz E, Kaplan A, Bozzo E. Evolution of sleep spindles in childhood. Electroencephalogr Clin Neurophysiol 1975;38:175–181. [DOI] [PubMed] [Google Scholar]
- 33. Manvich DF, Webster KA, Foster SL, et al. The DREADD agonist clozapine N‐oxide (CNO) is reverse‐metabolized to clozapine and produces clozapine‐like interoceptive stimulus effects in rats and mice. Sci Rep 2018;8:3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koch‐Stoecker S. Antipsychotic drugs and epilepsy: indications and treatment guidelines. Epilepsia 2002;43:19–24. [DOI] [PubMed] [Google Scholar]
- 35. Alper K, Schwartz KA, Kolts RL, Khan A. Seizure incidence in psychopharmacological clinical trials: an analysis of Food and Drug Administration (FDA) summary basis of approval reports. Biol Psychiatry 2007;62:345–354. [DOI] [PubMed] [Google Scholar]
- 36. Whissell PD, Tohyama S, Martin LJ. The use of DREADDs to deconstruct behavior. Front Genet 2016;7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roth BL. DREADDs for neuroscientists. Neuron 2016;89:683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mickle AD, Won SM, Noh KN, et al. A wireless closed‐loop system for optogenetic peripheral neuromodulation. Nature 2019;565:361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bosch‐Bouju C, Hyland BI, Parr‐Brownlie LC. Motor thalamus integration of cortical, cerebellar and basal ganglia information: implications for normal and parkinsonian conditions. Front Comput Neurosci 2013;7:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McFarland NR, Haber SN. Thalamic relay nuclei of the basal ganglia form both reciprocal and nonreciprocal cortical connections, linking multiple frontal cortical areas. J Neurosci 2002;22:8117–8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Velasco M, Velasco F, Velasco AL, et al. Epileptiform EEG activities of the centromedian thalamic nuclei in patients with intractable partial motor, complex partial, and generalized seizures. Epilepsia 1989;30:295–306. [DOI] [PubMed] [Google Scholar]
- 42. Jasper HH. Historical introduction. In: Avoli M, Gloor P, Kostopoulos G, Naquet R, eds. Generalized Epilepsy: Neurobiological Approaches. Boston: Birkhäuser, 1990:1‐15. [Google Scholar]
- 43. Hughlings JJ. The Lumleian lectures on convulsive seizures. Lancet 1890;135:785–788. [Google Scholar]
- 44. Penfield W, Rasmussen T. The Cerebral Cortex of Man: A Clinical Study of Localization of Function. New York: Macmillan, 1950. [Google Scholar]
- 45. Graham D, Tisdall MM, Gill D. Corpus callosotomy outcomes in pediatric patients: a systematic review. Epilepsia 2016;57:1053–1068. [DOI] [PubMed] [Google Scholar]
- 46. Badger CA, Lopez AJ, Heuer G, Kennedy BC. Systematic review of corpus callosotomy utilizing MRI guided laser interstitial thermal therapy. J Clin Neurosci 2020;76:67–73. [DOI] [PubMed] [Google Scholar]
- 47. Doležalová I, Kunst J, Kojan M, et al. Anterior thalamic deep brain stimulation in epilepsy and persistent psychiatric side effects following discontinuation. Epilepsy Behav Rep 2019;12:100344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salanova V, Witt T, Worth R, et al. Long‐term efficacy and safety of thalamic stimulation for drug‐resistant partial epilepsy. Neurology 2015;84:1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets and codes are available upon request from the Lead Contact.