

Abstract
Sirtuin 5 (SIRT5) is a protein lysine deacylase enzyme that regulates diverse biology by hydrolyzing ϵ‐N‐carboxyacyllysine posttranslational modifications in the cell. Inhibition of SIRT5 has been linked to potential treatment of several cancers but potent compounds with activity in cells have been lacking. Here we developed mechanism‐based inhibitors that incorporate isosteres of a carboxylic acid residue that is important for high‐affinity binding to the enzyme active site. By masking of the tetrazole moiety of the most potent candidate from our initial SAR study, we achieved potent and cytoselective growth inhibition for the treatment of SIRT5‐dependent leukemic cancer cell lines in culture. Thus, we provide an efficient, cellularly active small molecule that targets SIRT5, which can help elucidate its function and potential as a future drug target. This work shows that masked isosteres of carboxylic acids are viable chemical motifs for the development of inhibitors that target mitochondrial enzymes, which may have applications beyond the sirtuin field.
Keywords: Bioisosteres, Enzyme Inhibitors, Prodrugs, SIRT5, Sirtuins
A SAR study of mechanism‐based inhibitors of the sirtuin 5 (SIRT5) hydrolase, containing isosteres of an essential carboxylic acid moiety, furnished heterocycles with excellent potency and slow, tight‐binding kinetics against the enzyme. Evaluation of selected compounds in cells revealed that transient masking of the heterocycle provided improved inhibitors with a high level of target engagement in cultured cells.

Introduction
The sirtuins are NAD+‐dependent lysine deacylases, which are conserved across all domains of life. Humans express seven isoforms (SIRT1–7), which are involved in the regulation of important biological pathways such as gene expression and metabolism.[ 1 , 2 ] All sirtuins share a fundamental NAD+‐dependent hydrolytic mechanism but the seven isoforms differ in substrate specificity, which is particularly manifested with respect to their recognition of various ϵ‐N‐acyllysine functionalities. These differences have been elucidated through substrate screening efforts and numerous X‐ray co‐crystal crystal structures.[ 3 , 4 , 5 , 6 ] Sirtuins 1–3 and 6 have exhibited preference for aliphatic acyl chains of different length ranging from ϵ‐N‐acetyllysine (Kac) to ϵ‐N‐myristoyllysine (Kmyr),[ 7 , 8 , 9 ] which is also reflected by hydrophobic binding pockets.[ 10 , 11 , 12 , 13 ] Moreover, SIRT1–3 has recently been reported to cleave both the d and l stereoisomers of ϵ‐N‐lactyllysine (Klac);[ 14 , 15 ] albeit, with lower efficiency than the zinc‐dependent histone deacetylases (HDACs) 1–3. [15] Sirtuin 4 has been reported to cleave negatively charged modifications, such as ϵ‐N‐methylglutaconyllysine (Kmgc) and ϵ‐N‐(3‐hydroxy‐3‐methylglutaryl)lysine (Khmg),[ 16 , 17 ] as well as ϵ‐N‐lipoyllysine (Klip) and ϵ‐N‐biotinyllysine (Kbio). [18] Sirtuin 7 has been reported to cleave long chain acyl groups[ 19 , 20 ] and ϵ‐N‐glutaryllysine (Kglut), [21] and has been reported to be activated by oligonucleotides.[ 19 , 22 ] The sirtuin with the most distinct structural features, governing substrate recognition in its binding pocket is SIRT5, which contains a Tyr102–Arg105 motif that interact with carboxylates in the substrate. As such, SIRT5 has a preference for acidic posttranslational modifications (PTMs) such as ϵ‐N‐malonyllysine (Kmal), ϵ‐N‐succinyllysine (Ksuc), and Kglut.[ 23 , 24 , 25 ]
Sirtuin 5 is one of three sirtuins (SIRT3–5) that primarily localize to the mitochondria and several mitochondrial proteins are subjected to SIRT5‐mediated deacylation.[ 26 , 27 , 28 ] For example, SIRT5 rescues the activity of superoxide dismutase 1 (SOD1) by desuccinylation of modified lysine residues to (1) eliminate reactive oxygen species (ROS) generated during oxidative phosphorylation, [29] (2) protect mitochondrial autophagy under starvation, [30] and (3) reduce macrophage‐induced inflammation in vivo. [31] Furthermore, SIRT5 has been identified in both the cytosol and nucleus, where it is proposed to desuccinylate modified proteins as well. [26] With an increasing number of reports implicating SIRT5 in pathogenesis, including as a liability in various cancers,[ 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 ] there is a growing need for effective inhibitors that can be applied in living cells.
Inspired by the substrate specificity of SIRT5,[ 24 , 42 , 43 ] we and others have reported on mechanism‐based inhibitors of SIRT5 that contained carboxylate moieties to mimic the preferred substrate of the enzyme.[ 44 , 45 , 46 , 47 , 48 ] Recently, one of our lead compounds (1) [44] was developed into an ethyl ester prodrug (1‐Et), which was shown to exhibit efficacy against cultured cancer cells (see Figure 1 for structures). [41] These investigations indicated that the carboxylate moiety of the parent compounds was a major liability with respect to cellular entry. Whereas the simple ester prodrugs are likely hydrolyzed by various esterases once inside the cell. In medicinal chemistry, an alternative strategy to prodrug formation is the use of bioisosteres, which may mimic the interactions of a certain functional group (here a carboxylate), while imposing improved properties with respect to for example degradation, clearance, and cellular entry. Here, we report a substantial structure–activity relationship (SAR) study of a series of inhibitors, containing isosteres of the carboxylic acid moiety that is important for ligand affinity to the SIRT5 active site, which produced inhibitors that were equipotent or slightly more potent than the parent compound. Cell penetration was then assessed by applying the chloroalkane penetration assay (CAPA)[ 49 , 50 , 51 , 52 ] and target engagement was evaluated by cellular thermal shift assays, revealing similar behavior of the carboxylic acid‐containing parent compound and its most potent new analogs. Thus, we show that masking of the tetrazole moiety of the most potent compound can provide improved target engagement in cells to furnish a cellularly active tool compound. Finally, we demonstrate that cultured SIRT5‐dependent cancer cells (e.g., SKM‐1) are susceptible to the masked tetrazole compound at concentrations where limited effect is observed against HEK293T cells.
Figure 1.

Structure–activity relationship of SIRT5 inhibitors containing carboxylic acid isosteres. A) Potencies for inhibition of the deglutarylase activity of recombinant SIRT5 (150 nM) against Ac‐LGKglut‐AMC substrate (50 μM) are given as mean IC50 values±standard deviation (SD) or %‐inhibition at denoted concentrations. B) Docking poses of compounds 1 and 24, respectively (SIRT5, pdb: 6EQS; see also Supporting Figure S2). C) Selectivity of compounds 1, 16, and 24 across SIRTs 1–3, 5, and 6 measured at an inhibitor concentration of 1 μM. The ϵ‐N‐acyllysine residue in each substrate is indicated for the individual sirtuin. Data are based on at least two individual assays performed in duplicate. See the Supporting Information Figure S1 for dose‐response curves, Table S1 for inhibitory potencies, and Table S2 for full selectivity profiling. NI = no inhibition.
Results and Discussion
With compound 1 as the starting point, we first directed our attention to investigating whether bioisosteres of its carboxylic acid moiety could be developed that would retain potency against SIRT5. Inspired by previously reported isosteres, we synthesized the series of compounds 2–27, containing a variety of different functional groups with varying ability to form hydrogen bonds and engage in electrostatic interactions with the target. The inhibitory potencies of all compounds against the lysine deglutarylase activity of recombinant SIRT5 were then determined (Figure 1A; for syntheses see Supporting Schemes S2–S13). The aliphatic analog 2 and alcohol‐containing analog 3 did not show substantial inhibition of SIRT5 even at 100 μM. The amide analog 4, on the other hand, exhibited some potency, but with an IC50 value two orders of magnitude higher than for compound 1. The sulfonates (5, 6), sulfonamide (7), phosphonate (8), boronates (9–11), and fluorophenols (12,13) all exhibited poor inhibition of SIRT5. Among the series of heterocycles in our array of compounds (14–26), on the other hand, the 1,2,4‐oxadiazol‐5(4H)‐one (16) and the tetrazole (24) exhibited similar IC50 values to the parent compound (1), while the 2‐hydroxy‐isoxazole (22) also had a sub‐micromolar IC50 value (Figure 1). Finally, substituting the carboxylic acid for a hydroxamic acid (27) resulted in substantial loss of potency. Interestingly, the pK a values of 16 and 24 were estimated to be in the same range as 1 (Supporting Table S1), indicating that an electrostatic interaction may be required for potent inhibition of SIRT5 by mechanism‐based inhibitors of this type. In addition, the substantial difference in potency between 23 and 24 indicates that both length and flexibility within the lysine side chain modification is important for high‐affinity binding of the isosteres (Figure 1) as also previously shown for carboxylates.[ 43 , 44 ]
During the course of our investigations, the laboratory of Meier independently investigated the substitution of the carboxylic acid moiety in Kmal for a tetrazole moiety, to provide an isosteric element that does not undergo decarboxylation. [53] In that work, the modification was connected to lysine through a native amide bond to provide a substrate mimic that was recognized by both anti‐Kmal antibody and SIRT5 enzyme. Because the native amide bond gives rise to a more flexible structure than the thioureas in our study and because the malonyl analog is shorter than both the modifications in 23 and 24, we think that these observations provide even further support that both distance and flexibility of the PTM are important for binding to SIRT5. Furthermore, computational docking of compound 24 to SIRT5 provided an excellent overlay with the carboxylate‐containing inhibitor bound in the X‐ray co‐crystal structure of SIRT5, indicating interaction of the tetrazole moiety with the Tyr102–Arg105 motif in the active site (Figure 1B and Supporting Figure S2).
Finally, we validated the ability of the most potent inhibitors (1, 16, and 24) to inhibit recombinant SIRT5 in a different assay format, relying on longer peptide substrates without fluorescent labels (Supporting Figure S3), and tested their selectivity across other members of the SIRT family, using recombinant SIRT1–3 and SIRT6 (Figure 1C; see Supporting Table S2 for additional examples). Compounds 1, 16, and 24 all exhibited high selectivity for SIRT5 at 1 μM concentration; however, at higher concentration, compound 16 showed some inhibition of SIRT1 (Supporting Table S2).
We have previously demonstrated that the use of IC50 values as the sole determination of potency can be misleading for mechanism‐based sirtuin inhibitors, because these types of inhibitors may display time‐dependent inhibition profiles.[ 44 , 54 ] Generally, SIRT inhibitors relying on a thiocarbonyl moiety are believed to gain their potency through formation of stalled intermediates with ADP‐ribose from the NAD+ co‐substrate in the active sites of the enzymes. This mechanism, in turn, has been shown to result in slow, tight‐binding inhibition kinetics rather than fast‐on‐fast‐off kinetics, which is a prerequisite for estimating K i values by the Cheng–Prusoff equation. Furthermore, despite a relatively low K M value for the applied substrate in our initial screen (K M=20±1.9 μM, Supporting Figure S4 and Table S3), we noted that the most potent compounds had reached stoichiometric inhibition (i.e., IC50 values in the same range as the enzyme concentration). Therefore, we turned to a continuous SIRT5 inhibition assay format that was previously used to determine the slow, tight‐binding kinetics of compound 1, [44] enabling more meaningful estimation of the potencies of compounds 16, 22, and 24.
Compounds 22 and 24 behaved as slow, tight‐binding inhibitors like parent carboxylic acid‐containing compound 1, with K i values in the low nanomolar range for both 1 and 24 and somewhat higher at around 100 nM for compound 22 (Figure 2 and Supporting Figure S3). The oxadiazolone‐containing analog 16 exhibited slow‐binding kinetics according to mechanism A, also with a K i value in the low nanomolar range (Figure 2). The less potent analogs 4 and 27 were also evaluated in the continuous assay and displayed slow‐binding kinetics according to mechanism A with K i values in the micromolar range. Their potencies, on the other hand, were in good agreement with the IC50 values determined by end‐point assays, as would be expected (Supporting Figure S5). Taken together, these assays show that the simple IC50 determinations from end‐point assays were useful as a screening platform but did not provide accurate affinities for the more potent compounds that exhibit longer residence times in the enzyme active site.
Figure 2.
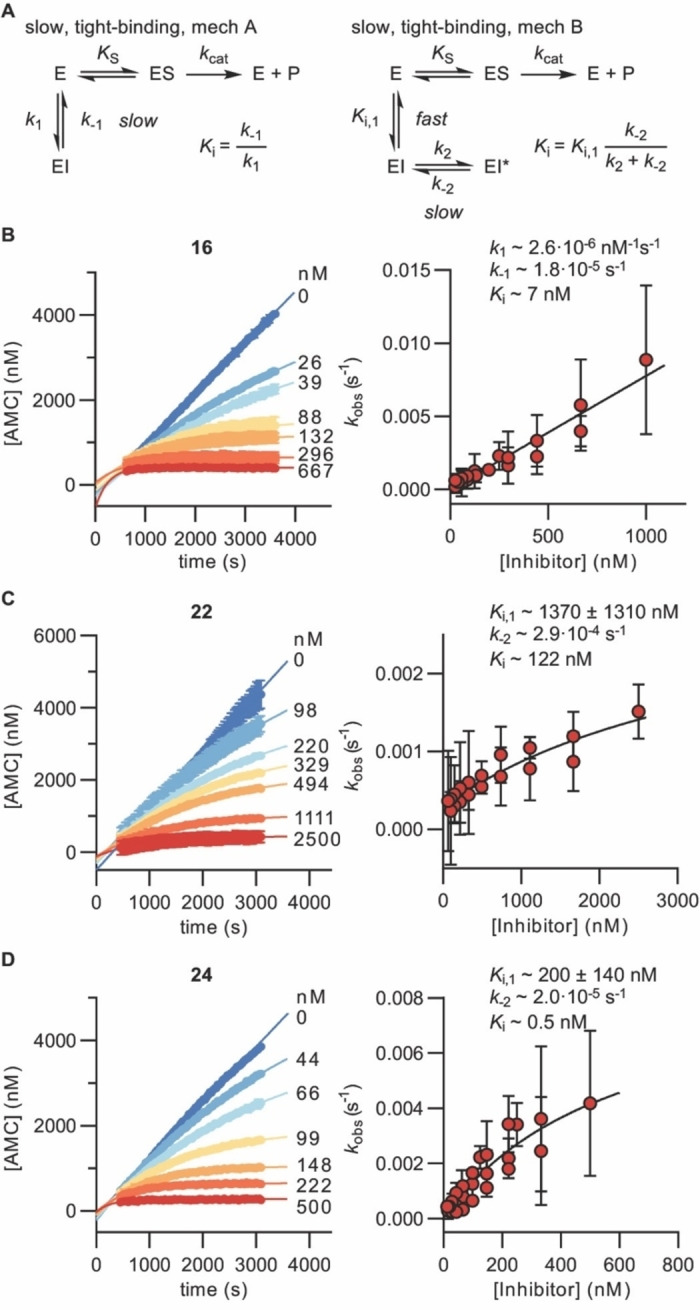
Kinetics of SIRT5 inhibition by compounds 16, 22, and 24. A) Common mechanisms of slow‐binding inhibitor kinetics with associated equilibrium and rate constants. B)–D) Sample rate experiment curves and plots showing the dependence of k obs on inhibitor concentration for compound 16 (mechanism A) as well as compounds 22 and 24 (mechanism B). Concentrations of inhibitor for each experiment indicated on the right side of the curve. Continuous assays were performed with SIRT5 (80 nM), NAD+ (500 μM), Ac‐LGKglu‐AMC (40 μM) as substrate, trypsin (1.70 ng×μL−1). See Supporting Figure S3 and Table S4 for additional details and complete data fitting.
Next, we proceeded to evaluate the effect of the carboxylic acid isosteres on the overall ability of the compounds to penetrate the cellular plasma membrane. We decided to apply the recently described CAPA procedure. [50] Previous SAR studies have shown that the N‐terminal functionality of the inhibitor scaffold can be substituted relatively flexibly without significant loss of potency. [44] We therefore attached the chloroalkane (CA) tag at the N‐termini through an amide linkage. Compounds 28, 28‐Et, and 29–31 were synthesized (Supporting Schemes S14 and S15) and their ability to enter cells in culture was analyzed in a HaloTag‐expressing HeLa cell line (Figure 3). The cell penetration profiles of the CA‐tagged compounds were analyzed by fitting the dose‐response data to sigmoidal curves. The concentration at which 50 % cell penetration occurs is termed the CP50 value, which we used to compare the cell penetrating properties of the compounds. As positive control compound, we used tryptophan α‐N‐acylated with the same chloroalkane (CA‐Trp‐OH) to benchmark the degree of penetration of our compounds (Figure 3B and C).
Figure 3.
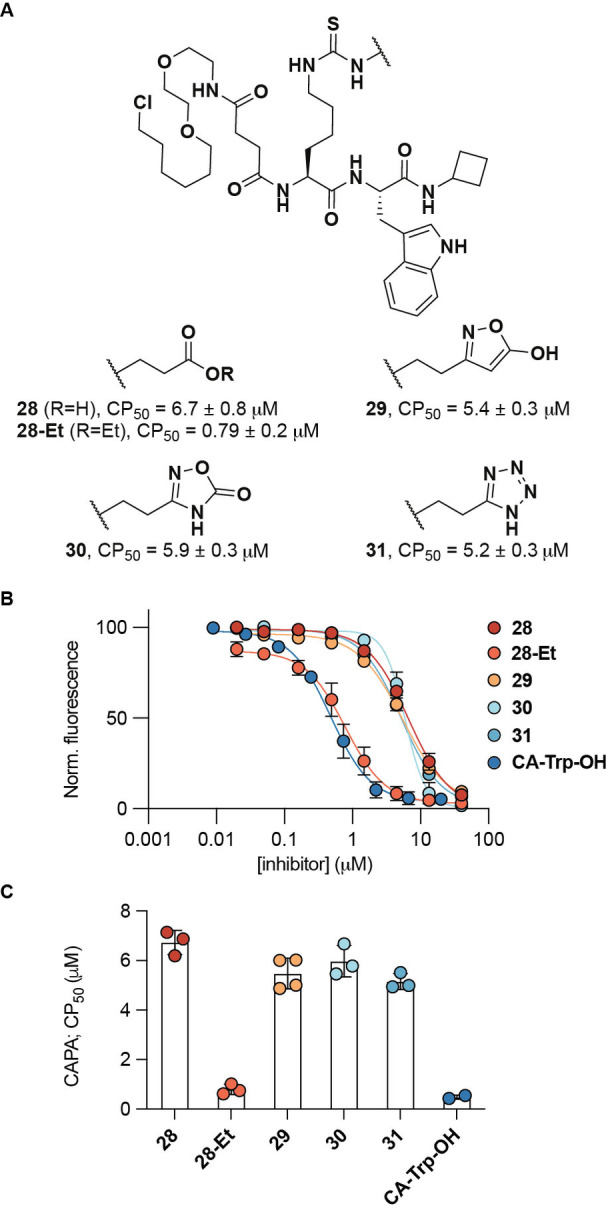
Cell penetration profiling using CAPA. A) Structure of synthesized chloroalkane probes and their mean CP50 values±SD (n=3). B) CAPA results for compounds 28–31 and α‐N‐“chloroalkane”tryptophan CA‐Trp‐OH as the positive control after 4 hours of treatment with inhibitor (n≥2). C) Calculated CP50 values.
The prodrug 28‐Et exhibited cell penetration ability with a CP50 value in the same range as the control compound, while all the isosteric compounds tested (29–31) showed the same degree of penetration as the parent carboxylic acid (1). That 28‐Et reaches the intracellular matrix more readily than 1 is in agreement with previously published data, showing cytotoxic effect of the prodrug but not the free carboxylic acid‐containing compound (1) against SIRT5‐dependent acute myeloid leukemia (AML) cell lines. [41] Unfortunately, however, all the isosteric compounds that showed high potency in enzymatic assays, exhibited poor cell penetration with CP50 values in the same range as the parent carboxylic acid (1) (Figure 3). We attribute this finding to the presumed negative charge in the side chains of all these analogs that would be expected in medium at neutral pH based on their predicted pK a values (Supporting Table S1). This assumption is further supported by the CP50 values of additional compounds, showing high cell penetration with a simple alkyl side chain and poor cell penetration for additional negatively charged analogs (Supporting Figure S6).
Because the chloroalkane analogs 28–31 showed almost 10‐fold higher CP50 values than the prodrug 28‐Et, and because our SAR study had suggested that an electrostatic interaction between ligand and SIRT5 active site was preferred for high affinity binding, we decided to prepare a compound containing a masked tetrazole functionality. Thus, an analog of compound 24, containing an O‐acyl‐N,O‐isobutyl hemiaminal functionality on the tetrazole moiety (32) was prepared (Figure 4 and Supporting Scheme S16). This functional group was chosen based on previous reports, showing improvement of cell penetration upon masking of tetrazoles using this chemistry.[ 55 , 56 ] Compound 32 would then presumably be cleaved by intracellular esterases to generate a readily hydrolyzed hemiaminal species that would provide the parent tetrazole 24.
Figure 4.
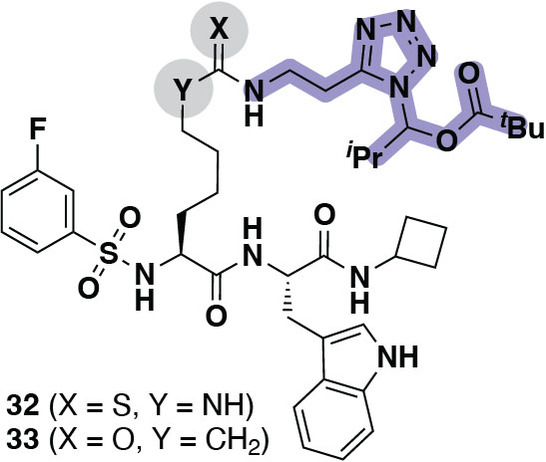
Structures of compounds 32 (masked tetrazole inhibitor) and 33 (masked tetrazole, inverse amide negative control compound).
With compound 32 and its corresponding “inverse amide” version 33 as negative control compound included in our collection, we next turned our attention to investigating the ability of selected compounds to reach the SIRT5 target in cultured cells. First, we chose immunoblot‐based cellular thermal shift assays,[ 57 , 58 ] which we have previously applied for the investigation of engagement of mechanism‐based inhibitors with SIRT1–3 in cells.[ 54 , 59 ] We decided to evaluate the thermal stabilization of SIRT5 in HEK293T cells by determining isothermal dose‐response fingerprinting cellular thermal shift assay (“ITDRF‐CETSA”) curves. [58] The experiments were performed at 52 °C where most of the SIRT5 has denatured according to western blot analysis (Supporting Figure S7A). In the assays, two compounds (16 and 22) showed substantially lower degree of target engagement than the other tested analogs with EC50 values above the highest applied dose (>10 μM) (Figure 5A, B). The parent compound 1 and tetrazole‐containing analog 24 behaved similarly with EC50 values of 0.9 and 1.3 μM, respectively (Figure 5B). This agrees well with the findings that the two compounds have K i values for inhibition of recombinant SIRT5 in the same range (Figure 2 and Supporting Figure S5) and their chloroalkane‐tagged analogs exhibited similar cell penetrating profiles (Figure 3). The two compounds containing masked acidic functional groups (1‐Et and 32) also showed similar ability to bind to SIRT5 in the cultured cells and to a higher extent than their unmasked counterparts (Figure 5A, B), which agrees well with the data recorded for their chloroalkane‐tagged analogs above. Finally, the negative control compound 33, which does not inhibit SIRT5 substantially in its unmasked form (Supporting Table S1), did not exhibit significant stabilization of SIRT5 in the cells (Figure 5A).
Figure 5.

Cellular target engagement. A) Representative immunoblots for the thermal shift of SIRT5 in HEK293T cells subjected to 2 h treatment with inhibitor at concentrations varying from 2.6 nM to 10 μM and 1‐Et (10 μM) as internal control at 52 °C. B) Fitting of dose–response curves and calculated EC50 values. See the Supporting Information for full immunoblots (n≥2). EC50 values for compounds 16, 22, and 33 were all >10 μM. C) Representative immunoblots for the thermal shift of SIRT3 and SIRT5 in HEK293T cells after 2 h treatment with inhibitor 32 (1 μM) at temperatures ranging from 37–75 °C, comparted to DMSO control. D) Plots of the data and calculated T agg values (n=2; see the Supporting Information for full blots and replicates). Statistical significance of the CETSA shifts were calculated using unpaired t‐test of the T agg values from independent experiments. Adjusted p‐value for 32 against SIRT5: *p<0.05.
Next, we performed full melting experiments with compound 32 against SIRT5 and the other mitochondrial sirtuin, SIRT3, which suggested a high degree of selectivity in cells (Figure 5C, D). Because compound 32, before unmasking, inhibits recombinant SIRT1 quite substantially at 1 μM concentration (Supporting Table S2), we also recorded a full melting curve for this enzyme. This experiment revealed a substantial engagement of SIRT1 in the cells, which we presume arises from incomplete unmasking of the compound during the course of the experiment. This finding suggests that development of more sophisticated masking chemistries could further improve the performance of tetrazole‐containing inhibitors in the future.
We then applied a recently developed assay that reports on SIRT5 enzymatic activity in the mitochondria by evaluating the fluorescence development caused by self‐assembly of a dye‐labeled peptide upon desuccinylation by SIRT5. [60] In this assay, we saw inhibition of SIRT5 activity for co‐treatment of HeLa cells with the labeled peptide substrate and compounds 1‐Et, 24, or 32 dosed at 10 μM concentration (Supporting Figure S8). The relatively high concentration of the inhibitors applied is likely the reason why we observe an effect of compound 24 as well as the masked compounds (1‐Et and 32), which exhibited superior levels of cell penetration and target engagement. However, we do not consider this assay sensitive enough to evaluate compound efficacies. Rather, we find it useful to investigate whether target engagement results in inhibition of the enzymatic activity in living cells as well. Therefore, we decided not to perform dose‐response experiments using this assay.
Finally, we evaluated a selection of compounds for their ability to decrease the viability of SIRT5‐dependent SKM‐1 AML cells versus non‐malignant, immortalized HEK293T cells in culture. None of the acidic compounds 1, 16, and 24 caused substantial decrease in viability of any of the two cell lines at concentrations up to 100 μM (Figure 6). The masked compounds, on the other hand, exhibited GI50 values against SKM‐1 cells of 21 μM (1‐Et) and 9 μM (32), respectively. Inhibition of the growth of two additional SIRT5‐dependent AML cell lines, OCI‐AML2 and MOLM‐13, by 1‐Et and 32 was then tested, which also showed more potent effect of the masked tetrazole (32) than 1‐Et. Moreover, the effect on HEK293T cells was minimal for 32 with <35 % inhibition at 100 μM, indicating cytoselective toxicity towards SIRT5‐selective cells to a higher extent than for compound 1‐Et (Figure 6). To ensure that the effect of compound 32 was not due to toxicity of the cleaved masking group inside the cells, we also tested negative control compound 33, which did not provide any correlation to suggest toxicity issues of the masking group. On the other hand, this compound did affect the viability of the SKM‐1 and HEK293T cells, suggesting cytotoxicity through a different mechanism than inhibition of SIRT5.
Figure 6.
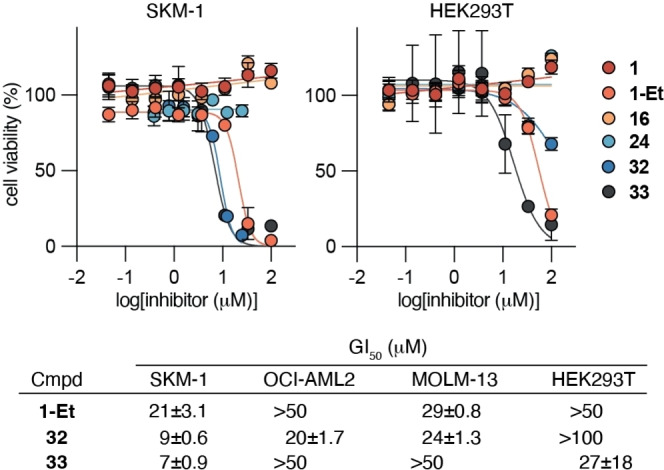
Cell viability upon treatment with 1, 1‐Et, 16, 24, 32, and 33. Dose–response curves from cell viability assays of the selected compounds against cultured SKM‐1 (SIRT5‐dependent acute myeloid leukemia) cells and HEK293T (immortalized, non‐malignant human embryonic kidney) cells as well as GI50 values of 1‐Et, 32, and 33 against OCI‐AML2, MOLM‐13 (SIRT5‐dependent acute myeloid leukemia) cells. All values represent data from three individual assays performed in duplicate (see the Supporting Information Figure S9 for additional dose–response curves).
In summary, we have investigated the prospects of substituting the carboxylic acid moiety, which provides both potency and selectivity to mechanism‐based inhibitors of SIRT5, by isosteric functional groups. In the initial SAR study, we prepared 26 different compounds with a variety of substituents that were envisioned to interact with the Tyr102–Arg105 motif in the active site of SIRT5 and identified the tetrazole (24) and the 1,2,4‐oxadiazol‐5(4H)‐one (16) as potent alternatives to the parent carboxylate (1). Because the compounds were potent enough to reach stoichiometry with respect to the amount of enzyme applied in the assay, we performed a kinetic enzyme inhibition experiment, using continuous assays to provide progression curves for the inhibition over time. Analysis of these data revealed that these potent compounds inhibit SIRT5 through slow, tight‐binding kinetics with estimated K i values ranging from ≈0.5–7 nM. Encouraged by the discovery of compounds containing isosteres that potently bind to the enzyme, we next evaluated the ability of a subset of compounds to reach the intracellular matrix of HeLa cells in culture. The so‐called CAPA procedure was applied to compare selected isosteres by measuring the ability of chloroalkane‐modified versions to enter engineered HeLa cells that express the HaloTag enzyme. The resulting data showed a substantially lower cell penetration of the carboxylate‐containing compound 28 and its most potent heterocyclic isosteres (29–31) than the chloroalkane analog of the ester prodrug (28‐Et). This result led us to design and synthesize a masked version of the tetrazole‐containing compound (32), which was included in the following compound evaluations. Isothermal dose‐response cellular target engagement experiments showed substantially lower EC50 values for the masked compounds compared to their unmasked counterparts [1‐Et vs 1 (3.6‐fold) and 32 vs. 24 (8.7‐fold)]. Both compounds 1‐Et and 32 were also shown to inhibit SIRT5 in HeLa cells by using an assay that produce in‐cell fluorescence in response to SIRT5 activity in the mitochondria. Finally, the two masked compounds were shown to harbor cytoselective toxicity towards SIRT5‐dependent AML cells (SKM‐1) over HEK293T cells in culture, while none of the unmasked compounds tested were able to affect the cell viability in either cell line at the highest concentration applied.
Conclusion
Our SAR demonstrated that size, orientation, hydrogen bonding properties, and charge of the bioisosteres were important for retaining affinity by interaction with the Tyr102–Arg105 motif in the active site of SIRT5. The most promising chemotypes in our series of inhibitors had pK a values in the same range as the parent carboxylate, which appeared to hamper cell penetration and, in turn, needed to be further masked to provide an inhibitor with activity in cells. We therefore designed a masked tetrazole‐containing compound, which exhibited selective cytotoxic effect and enhanced potency against SIRT5‐dependent AML cell lines compared to a previously developed ester prodrug of a carboxylic acid. Thus, these data provide insight into future inhibitor design principles and expand the functionalities that can be used to inhibit SIRT5. Further, we expect the developed inhibitors to enable more detailed investigation of the biology of SIRT5 and help illuminate its potential as a target for pharmacological intervention in cancer treatment. It is our hope that the insight gained from this study will inspire the exploration of new avenues in the pursuit of drug discovery efforts targeting sirtuin enzymes.
Supporting Information
Supporting Schemes, Figures, and Tables, general methods and materials, biochemical methods, supporting references, as well as copies 1H, 13C, and 19F NMR spectra (PDF).
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank Dr. Carlos Moreno‐Yruela for helpful discussion regarding fitting of kinetic data. Dr. Mette I. Rosenbaum and Dr. Laura Cesa are acknowledged for establishing the CAPA assay platform and Professor Joshua Kritzer for providing the HaloTag‐expressing HeLa cell line. We gratefully acknowledge financial support from the Hørslev Foundation, the Carlsberg Foundation (2013‐01‐0333, CF15‐011, and CF18‐0442; C.A.O.), the Novo Nordisk Foundation (NF17OC0029464; C.A.O.), and the Independent Research Fund Denmark‐Medical Sciences (0134‐00435B; C.A.O.). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement number CoG‐725172 “SIRFUNCT”; C.A.O.).
N. Rajabi, T. N. Hansen, A. L. Nielsen, H. T. Nguyen, M. Bæk, J. E. Bolding, O. Ø. Bahlke, S. E. G. Petersen, C. R. O. Bartling, K. Strømgaard, C. A. Olsen, Angew. Chem. Int. Ed. 2022, 61, e202115805; Angew. Chem. 2022, 134, e202115805.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2021‐29rtf).
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Michan S., Sinclair D., Biochem. J. 2007, 404, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Houtkooper R. H., Pirinen E., Auwerx J., Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bheda P., Jing H., Wolberger C., Lin H., Annu. Rev. Biochem. 2016, 85, 405–429. [DOI] [PubMed] [Google Scholar]
- 4. Sabari B. R., Zhang D., Allis C. D., Zhao Y., Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rajabi N., Galleano I., Madsen A. S., Olsen C. A., Progress in Molecular Biology and Translational Science, Academic Press, New York, 2018, pp. 25–69. [DOI] [PubMed] [Google Scholar]
- 6. Wang M., Lin H., Annu. Rev. Biochem. 2021, 90, 245–285. [DOI] [PubMed] [Google Scholar]
- 7. Feldman J. L., Baeza J., Denu J. M., J. Biol. Chem. 2013, 288, 31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Madsen A. S., Andersen C., Daoud M., Anderson K. A., Laursen J. S., Chakladar S., Huynh F. K., Colaço A. R., Backos D. S., Fristrup P., Hirschey M. D., Olsen C. A., J. Biol. Chem. 2016, 291, 7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klein M. A., Liu C., Kuznetsov V. I., Feltenberger J. B., Tang W., Denu J. M., J. Biol. Chem. 2020, 295, 1385–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin L., Wei W., Jiang Y., Peng H., Cai J., Mao C., Dai H., Choy W., Bemis J. E., Jirousek M. R., Milne J. C., Westphal C. H., Perni R. B., J. Biol. Chem. 2009, 284, 24394–24405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang H., Khan S., Wang Y., Charron G., He B., Sebastian C., Du J., Kim R., Ge E., Mostoslavsky R., Hang H. C., Hao Q., Lin H., Nature 2013, 496, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bin Teng Y., Jing H., Aramsangtienchai P., He B., Khan S., Hu J., Lin H., Hao Q., Sci. Rep. 2014, 5, 8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davenport A. M., Huber F. M., Hoelz A., J. Mol. Biol. 2014, 426, 526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaffney D. O., Jennings E. Q., Anderson C. C., Marentette J. O., Shi T., Schou Oxvig A. M., Streeter M. D., Johannsen M., Spiegel D. A., Chapman E., Roede J. R., Galligan J. J., Cell Chem. Biol. 2020, 27, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moreno-Yruela C., Zhang D., Wei W., Bæk M., Liu W., Gao J., Danková D., Nielsen A. L., Bolding J. E., Yang L., Jameson S. T., Wong J., Olsen C. A., Zhao Y., Sci. Adv. 2022, 8, eabi6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anderson K. A., Huynh F. K., Fisher-Wellman K., Stuart J. D., Peterson B. S., Douros J. D., Wagner G. R., Thompson J. W., Madsen A. S., Green M. F., Sivley R. M., Ilkayeva O. R., Stevens R. D., Backos D. S., Capra J. A., Olsen C. A., Campbell J. E., Muoio D. M., Grimsrud P. A., Hirschey M. D., Cell Metab. 2017, 25, 838–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pannek M., Simic Z., Fuszard M., Meleshin M., Rotili D., Mai A., Schutkowski M., Steegborn C., Nat. Commun. 2017, 8, 1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mathias R. A. A., Greco T. M. M., Oberstein A., Budayeva H. G. G., Chakrabarti R., Rowland E. A. A., Kang Y., Shenk T., Cristea I. M. M., Cell 2014, 159, 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tong Z., Wang M., Wang Y., Kim D. D., Grenier J. K., Cao J., Sadhukhan S., Hao Q., Lin H., ACS Chem. Biol. 2017, 12, 300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang M., Li Z., Zhang C., Lu X., Tu B., Cao Z., Li Y., Chen Y., Jiang L., Wang H., Wang L., Wang J., Liu B., Xu X., Wang H., Zhu W. G., Sci. Adv. 2019, 5, eaav1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bao X., Liu Z., Zhang W., Gladysz K., Fung Y. M. E., Tian G., Xiong Y., Wong J. W. H., Yuen K. W. Y., Li X. D., Mol. Cell 2019, 76, 660–675. [DOI] [PubMed] [Google Scholar]
- 22. Tong Z., Wang Y., Zhang X., Kim D. D., Sadhukhan S., Hao Q., Lin H., ACS Chem. Biol. 2016, 11, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., Yang K., Zwaans B. M. M., Tishkoff D., Ho L., Lombard D., He T. C., Dai J., Verdin E., Ye Y., Zhao Y., Mol. Cell. Proteomics 2011, 10, M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., Lin H., Science 2011, 334, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G. R., Green M. F., Madsen A. S., Schmiesing J., Peterson B. S., Xu G., Ilkayeva O. R., Muehlbauer M. J., Braulke T., Mühlhausen C., Backos D. S., Olsen C. A., McGuire P. J., Pletcher S. D., Lombard D. B., Hirschey M. D., Zhao Y., Cell Metab. 2014, 19, 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park J., Chen Y., Tishkoff D. X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B. M. M., Skinner M. E., Lombard D. B., Zhao Y., Mol. Cell 2013, 50, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rardin M. J., He W., Nishida Y., Newman J. C., Carrico C., Danielson S. R., Guo A., Gut P., Sahu A. K., Li B., Uppala R., Fitch M., Riiff T., Zhu L., Zhou J., Mulhern D., Stevens R. D., Ilkayeva O. R., Newgard C. B., Jacobson M. P., Hellerstein M., Goetzman E. S., Gibson B. W., Verdin E., Cell Metab. 2013, 18, 920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishida Y., Rardin M. J., Carrico C., He W., Sahu A. K., Gut P., Najjar R., Fitch M., Hellerstein M., Gibson B. W., Verdin E., Mol. Cell 2015, 59, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin Z. F., Xu H. B., Wang J. Y., Lin Q., Ruan Z., Liu F. B., Jin W., Huang H. H., Chen X., Biochem. Biophys. Res. Commun. 2013, 441, 191–195. [DOI] [PubMed] [Google Scholar]
- 30. Guedouari H., Daigle T., Scorrano L., Hebert-Chatelain E., Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 169–176. [DOI] [PubMed] [Google Scholar]
- 31. Wang F., Wang K., Xu W., Zhao S., Ye D., Wang Y., Xu Y., Zhou L., Chu Y., Zhang C., Qin X., Yang P., Yu H., Cell Rep. 2017, 19, 2331–2344. [DOI] [PubMed] [Google Scholar]
- 32. Lu W., Zuo Y., Feng Y., Zhang M., Tumor Biol. 2014, 35, 10699–10705. [DOI] [PubMed] [Google Scholar]
- 33. Ye X., Niu X., Gu L., Xu Y., Li Z., Yu Y., Chen Z., Lu S., Oncotarget 2017, 8, 6984–6993.28036303 [Google Scholar]
- 34. Wang Y. Q., Wang H. L., Xu J., Tan J., Fu L. N., Wang J. L., Zou T. H., Sun D. F., Gao Q. Y., Chen Y. X., Fang J. Y., Nat. Commun. 2018, 9, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang L., Xi L., Liu Y., Liu R., Wu Z., Jian Z., Mol. Med. 2018, 17, 342–349. [DOI] [PubMed] [Google Scholar]
- 36. Kumar S., Lombard D. B., Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.L. Xu, X. Che, Y. Wu, N. Song, S. Shi, Sh. O. Wang, C. Li, Lingy. N. Zhang, X. Zhang, Xi. A. Qu, Y. E. Teng, Oncol. Rep. 2018, 39, 2315–2323. [DOI] [PubMed]
- 38. Yang X., Wang Z., Li X., Liu B., Liu M., Liu L., Chen S., Ren M., Wang Y., Yu M., Wang B., Zou J., Zhu W. G., Yin Y., Gu W., Luo J., Cancer Res. 2018, 78, 372–386. [DOI] [PubMed] [Google Scholar]
- 39. Sun X., Wang S., Gai J., Guan J., Li J., Li Y., Zhao J., Zhao C., Fu L., Li Q., Front. Oncol. 2019, 9, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abril Y. L. N., Fernandez I. R., Hong J. Y., Chiang Y. L., Kutateladze D. A., Zhao Q., Yang M., Hu J., Sadhukhan S., Li B., He B., Remick B., Bai J. J., Mullmann J., Wang F., Maymi V., Dhawan R., Auwerx J., Southard T., Cerione R. A., Lin H., Weiss R. S., Oncogene 2021, 40, 1644–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yan D., Franzini A., Pomicter A. D., Halverson B. J., Antelope O., Mason C. C., Ahmann J. M., Senina A. V., Vellore N. A., Jones C. L., Zabriskie M. S., Than H., Xiao M. J., van Scoyk A., Patel A. B., Clair P. M., Heaton W. L., Owen S. C., Andersen J. L., Egbert C. M., Reisz J. A., D'Alessandro A., Cox J. E., Gantz K. C., Redwine H. M., Iyer S. M., Khorashad J. S., Rajabi N., Olsen C. A., O'Hare T., Deininger M. W., Blood Cancer Discovery 2021, 2, 266–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Madsen A. S., Olsen C. A., J. Med. Chem. 2012, 55, 5582–5590. [DOI] [PubMed] [Google Scholar]
- 43. Roessler C., Nowak T., Pannek M., Gertz M., Nguyen G. T. T., Scharfe M., Born I., Sippl W., Steegborn C., Schutkowski M., Angew. Chem. Int. Ed. 2014, 53, 10728–10732; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10904–10908. [Google Scholar]
- 44. Rajabi N., Auth M., Troelsen K. R., Pannek M., Bhatt D. P., Fontenas M., Hirschey M. D., Steegborn C., Madsen A. S., Olsen C. A., Angew. Chem. Int. Ed. 2017, 56, 14836–14841; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15032–15037. [Google Scholar]
- 45. He B., Du J., Lin H., J. Am. Chem. Soc. 2012, 134, 1922–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.“Methods for Treatment of Cancer by Targeting SIRT5”: Lin H., Cerione R., (Cornell University), US 2014213530A1, Jul 31, 2014, n.d., US2014197775.
- 47. Zang W., Hao Y., Wang Z., Zheng W., Bioorg. Med. Chem. Lett. 2015, 25, 3319–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Polletta L., Vernucci E., Carnevale I., Arcangeli T., Rotili D., Palmerio S., Steegborn C., Nowak T., Schutkowski M., Pellegrini L., Sansone L., Villanova L., Runci A., Pucci B., Morgante E., Fini M., Mai A., Russo M. A., Tafani M., Autophagy 2015, 11, 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peraro L., Zou Z., Makwana K. M., Cummings A. E., Ball H. L., Yu H., Lin Y.-S., Levine B., Kritzer J. A., J. Am. Chem. Soc. 2017, 139, 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peraro L., Deprey K. L., Moser M. K., Zou Z., Ball H. L., Levine B., Kritzer J. A., J. Am. Chem. Soc. 2018, 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mientkiewicz K. M., Peraro L., Kritzer J. A., ACS Chem. Biol. 2021, 16, 1184–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deprey K., Batistatou N., Debets M. F., Godfrey J., VanderWall K. B., Miles R. R., Shehaj L., Guo J., Andreucci A., Kandasamy P., Lu G., Shimizu M., Vargeese C., Kritzer J. A., ACS Chem. Biol. 2022, 17, 348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jing Y., Bergholtz S. E., Omole A., Kulkarni R. A., Zengeya T. T., Yoo E., Meier J. L., ChemBioChem 2022, 23, e2021004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nielsen A. L., Rajabi N., Kudo N., Lundø K., Moreno-Yruela C., Bæk M., Fontenas M., Lucidi A., Madsen A. S., Yoshida M., Olsen C. A., RSC Chem. Biol. 2021, 2, 612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ryono D. E., Lloyd J., Poss M. A., Bird J. E., Buote J., Chong S., Dejneka T., Dickinson K. E. J., Gu Z., Mathers P., Moreland S., Morrison R. A., Petrillo E. W., Powell J. R., Schaeffer T., Spitzmiller E. R., White R. E., Bioorg. Med. Chem. Lett. 1994, 4, 201–206. [Google Scholar]
- 56. O'Brien N. J., Amran S., Medan J., Cleary B., Deady L. W., Jennings I. G., Thompson P. E., Abbott B. M., ChemMedChem 2013, 8, 914–918. [DOI] [PubMed] [Google Scholar]
- 57. Molina D. M., Jafari R., Ignatushchenko M., Seki T., Larsson E. A., Dan C., Sreekumar L., Cao Y., Nordlund P., Martinez Molina D., Jafari R., Ignatushchenko M., Seki T., Larsson E. A., Dan C., Sreekumar L., Cao Y., Nordlund P., Science 2013, 341, 84–87. [DOI] [PubMed] [Google Scholar]
- 58. Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundbäck T., Nordlund P., Molina D. M., Nat. Protoc. 2014, 9, 2100–2122. [DOI] [PubMed] [Google Scholar]
- 59. Troelsen K. S., Bæk M., Nielsen A. L., Madsen A. S., Rajabi N., Olsen C. A., RSC Chem. Biol. 2021, 2, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang L., Peltier R., Zhang M., Song D., Huang H., Chen G., Chen Y., Zhou F., Hao Q., Bian L., He M., Wang Z., Hu Y., Sun H., J. Am. Chem. Soc. 2020, 142, 18150–18159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.