

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common form of liver disease and is often the precursor for more serious liver conditions such as nonalcoholic steatohepatitis and cirrhosis. Although the gut microbiome has been implicated in the development of NAFLD, the strong association of obesity with NAFLD and its effect on microbiome structure has made interpreting study outcomes difficult. In the present study, we examined the taxonomic and functional differences between the microbiomes of youth with obesity and with and without NAFLD. Shotgun metagenome sequencing was performed to profile the microbiomes of 36 subjects, half of whom were diagnosed with NAFLD using abdominal magnetic resonance imaging. Beta diversity analysis showed community‐wide differences between the groups (p = 0.002). Specific taxonomic differences included increased relative abundances of the species Fusicatenibacter saccharivorans (p = 0.042), Romboutsia ilealis (p = 0.046), and Actinomyces sp. ICM47 (p = 0.0009), and a decrease of Bacteroides thetaiotamicron (p = 0.0002), in the NAFLD group as compared with the non‐NAFLD group. At the phylum level, Bacteroidetes (p < 0.0001) was decreased in the NAFLD group. Functionally, branched‐chain amino acid (p = 0.01343) and aromatic amino acid (p = 0.01343) synthesis pathways had increased relative abundances in the NAFLD group along with numerous energy use pathways, including pyruvate fermentation to acetate (p = 0.01318). Conclusion: Community‐wide differences were noted based on NAFLD status, and individual bacterial species along with specific metabolic pathways were identified as potential drivers of these differences. The results of the present study support the idea that the NAFLD phenotype displays a differentiated microbial and functional signature from the obesity phenotype.
NAFLD is a common precursor to more severe liver diseases and has increased in frequency in younger individuals. We found that the microbial communities of youth with obesity and with and without NAFLD differ in both taxonomic and functional aspects.

INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common hepatic complication of obesity in children and adolescents, with an estimated 34% of obese youths affected by this disease.[ 1 , 2 , 3 ] Moreover, data based on histological specimens have shown that inflammation and fibrosis may occur earlier in life in patients who develop NAFLD during childhood,[ 4 ] making them more susceptible to develop liver failure at a younger age.[ 5 ] Although NAFLD is a common complication of obesity in youth, the reason why some patients are susceptible to the disease while others never develop it remains unclear. Nutritional and genetic factors certainly play a key role in conveying susceptibility to NAFLD,[ 6 , 7 , 8 ] but other factors may drive individuals to develop this condition. In particular, it has been suggested that the composition of the gut microbial community may play a pivotal role in this regard.[ 9 , 10 ]
Several studies have shown associations between intrahepatic fat content and gut microbiota, including decreased microbial diversity in severe fatty liver disease and an enrichment of specific taxa, including the bacterial phyla Proteobacteria, Firmicutes and Bacteroidetes, and the genera Escherichia, Veillonella, Faecalibacterium, Eubacterium, Bacteroides, and Oscillospira.[ 10 , 11 , 12 , 13 , 14 , 15 ] The results of seminal studies performed in twin adults with NAFLD demonstrated that within the spectrum of NAFLD, a metagenomic signature can not only differentiate between subjects with differing degrees of fibrosis,[ 12 ] but also between those with and without cirrhosis.[ 16 ]
Based on these and other findings, we previously assessed whether differences exist between the gut microbiomes of youths with and without NAFLD.[ 17 ] We showed that youths with NAFLD have a different metagenomic profile than those without NAFLD.[ 17 ] However, our initial analysis was limited by the use of sequence data from the V4 region of the 16S rRNA gene, which typically allows for only family‐level and genus‐level identification and no direct assessment of the physiological potential of the microbiome.
In the present study, we followed up on our previous findings by shotgun sequencing and analyzing the metagenomes of obese youths with and without NAFLD. Within this group of subjects, we observed community‐wide differences in metagenome composition and identified specific species associated with these differences. We also identified metabolic gene pathways that were increased or decreased in subjects with NAFLD and the species contributing to these differences.
METHODS
Study cohort
Thirty‐eight obese youths (body mass index [BMI] ≥ 95th percentile) were included in the present study. The participants were recruited from the Yale Pediatric Obesity Clinic. Those participants who had known non‐NAFLD hepatic diseases, diabetes, or medication that would interfere with glucose production and liver function were excluded from the study. All participants underwent an oral glucose tolerance test and abdominal fast–magnetic resonance imaging (MRI) to assess abdominal body fat partitioning and intrahepatic fat content as previously described.[ 38 , 39 , 40 ] Hepatic fat fraction (HFF) was assessed by abdominal MRI and used to categorize non‐NAFLD (HFF < 5.5%) and NAFLD (HFF ≥ 5.5%) groups. In addition, fasting blood samples to measure liver function and lipid profiles were obtained. The studies were conducted at the Yale Center for Clinical Investigation at 8 h after a 12‐hour overnight fast. A stool sample from each subject was also collected. Written parental informed consent and written child assent were obtained from all participants. Yale University Human Investigation Committee approved the study.
Statistical analysis
Differences were tested using chi‐square tests for categorical variables, two‐sample t‐tests for normally distributed, continuous variables, and Wilcoxon rank‐sum tests for nonnormally distributed, continuous variables, respectively. Subject characteristic differences were evaluated between the NAFLD and non‐NAFLD groups. Chi‐square testing was applied to the variables of gender (male/female), race (White or Caucasian/Black or African American/Hispanics/Others/Asian), and glucose tolerance (normal/impaired). The distributions of continuous variables were examined by histogram. The following normally distributed variables were examined using two sample t‐tests: visceral fat; BMI, body fat (%); fasting glucose; whole‐body insulin sensitivity index (WBISI); hemoglobin A1C; and total, high‐density lipoprotein, and low‐density lipoprotein cholesterol. Wilcoxon rank‐sum tests were used to test differences for the following variables: hepatic fat fraction (%), subcutaneous, deep subcutaneous, superficial subcutaneous, deep/superficial subcutaneous, age, BMI z‐score, 2‐h glucose, fasting insulin, insulinogenic index, disposition index, triglycerides, alanine aminotransferase (ALT), and aspartate aminotransferase (AST). Subject characteristics analyses were performed using SAS (version 9.4; SAS Institute Inc., Cary, NC, USA).
Shotgun metagenome sample preparation and sequencing
DNA was extracted using a MoBio PowerMag Soil 96‐well kit (MoBio Laboratories, Carlsbad, CA) from 0.25 g of fecal sample. DNA extracts were quantified using the Quant‐iT PicoGreen kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA). Metagenome libraries were produced using a TruSeq PCR‐Free kit (Illumina, San Diego, CA), diluted, and then pooled for loading onto an Illumina HiSeq 2500. The libraries underwent 75 × 75 bp paired‐end sequencing.
Shotgun metagenome preprocessing
Raw paired‐end read data for 36 shotgun metagenomes were downloaded from Basespace (basespace.illumina.com) following demultiplexing. Libraries for two samples failed and were excluded from the study. The reads were then processed using KneadData (version 0.7.4, https://github.com/biobakery/kneaddata) with the default parameters to remove adapter sequences, quality filter and quality trim reads, and remove contaminating host‐associated reads. The median read depth following Quality Control was 46 million reads, and the range was 69 million reads.
Taxonomic profiling and comparison
MetaPhlAn (version 3.0)[ 41 ] was used to taxonomically profile the gut microbial communities to the species level using the latest marker gene ChocoPhlAn database (release 2019.01). To investigate potential differences between the viromes of the two study groups, a virus profiling parameter was also included. Individual sample taxonomic profiles were merged using the merge_metaphlan_tables.py utility script, resulting in a single table with relative abundances provided for each taxon. Absolute counts for use in downstream analyses were recovered by multiplying the total read count for each sample by the relative abundances resulting from the merge table script. Species‐level counts were retrieved from this table and reformatted for import into R (version 4.1.0).[ 42 ]
An Operational taxonomic unit table, taxonomy table, and metadata table were imported into RStudio (version 1.4.1717) and read into the package phyloseq (version 1.36.0).[ 43 ] Alpha and beta diversity calculations were performed using the packages phyloseq and microViz (version 0.7.7).[ 44 ] For ecological distance metrics, samples were rarefied to 8,893,658 million reads (the lowest read count for a sample) before calculating distances and ordination. For compositional biplots, a centered log‐ratio transformation was performed as recommended before the generation of principal component analysis (PCA) biplots.
Differential abundance testing was performed using the ANCOM statistical framework[ 45 , 46 ] implemented by the package ANCOM‐BC.[ 47 ] Taxa were aggregated at the species, genus, family, order, class, and phylum levels using the microbiome package (version 1.14)[ 48 ] before performing the ANCOM test on each level. Taxa had to appear in a minimum of 25% of samples to be included in ANCOM‐BC analyses.
Functional profiling and comparison
HUMAnN (version 3.0)[ 49 ] was used to functionally profile the microbial communities. Paired‐end sequence files were first concatenated before running HUMAnN. The full ChocoPhlAn pangenome database (release 2019.01) was used for functional pathway abundance and coverage determination, whereas the UniRef90 database (release 2021.03)[ 50 , 51 ] was used for gene family abundance determination. The output pathway and gene family abundance files for each sample were normalized to relative abundances, and the resulting files were joined.
Enriched pathways and gene families were identified using the R package MaAsLiN2 (version 1.6.0).[ 52 ] Pathways and gene families achieving a corrected p‐value of 0.05 or less were classified as significantly increased within one of the two patient groups. Identified pathways and gene families were then plotted using the bar plot utility script in HUMAnN.
RESULTS
Population characteristics stratified by NAFLD status
The clinical and demographic characteristics of 36 study participants (18 NAFLD and 18 non‐NAFLD) were evaluated for differences with respect to NAFLD status (Table 1). Most of the subject characteristics were not significantly different between the two groups. Subjects with NAFLD had a higher BMI, BMI z‐score, fasting insulin level, alanine transaminase (ALT) level, and visceral and hepatic fat fractions, but a lower WBISI as compared to subjects without NAFLD. These all attained statistical significance (p < 0.05). Although not statistically significant, subjects with NAFLD had elevated fasting blood glucose, body fat percentage, total cholesterol, triglycerides, and AST as compared to the subjects without NAFLD.
TABLE 1.
Characteristics of study population by NAFLD status (n = 36)
| NAFLD status | p | ||
|---|---|---|---|
| non‐NAFLD (n = 18) | NAFLD (n = 18) | ||
| Clinical features | |||
| Age (years) a | 12.22 ± 3.02 | 12.54 ± 2.42 | 0.8628 |
| Gender (M/F) | 8/10 (44.44%/55.56%) | 11/7 (61.11%/38.89%) | 0.3166 |
| Race c | 6/4/6/1/1 (33.33%/22.22%/33.33%/5.56%/5.56%) | 6/2/9/0/1 (33.33%/11.11%/50%/0%/5.56%) | 0.6868 |
| Glucose tolerance (NGT/IGT) b | 15/3 | 10/5 | 0.2660 |
| BMI (kg/m2) b | 30.26 ± 7.04 | 35.21 ± 6.82 | 0.0386 |
| BMI z‐score b | 2.09 ± 0.48 | 2.46 ± 0.28 | 0.0204 |
| Body fat (%) b | 41.34 ± 10.23 | 46.58 ± 7.86 | 0.1100 |
| Glucose metabolism | |||
| Fasting glucose (mg/dl) b | 89.75 ± 5.37 | 94.53 ± 8.31 | 0.0549 |
| Fasting insulin (uU/ml) b | 21.69 ± 5.58 | 50.53 ± 30.40 | 0.0017 |
| 2‐h glucose (mg/dl) b | 118.11 ± 25.35 | 130.80 ± 33.50 | 0.2249 |
| Hemoglobin A1C (%) b | 5.51 ± 0.18 | 5.56 ± 0.30 | 0.2346 |
| WBISI b | 2.25 ± 0.61 | 1.24 ± 0.81 | 0.0003 |
| IGI b | 4.25 ± 3.43 | 6.42 ± 4.38 | 0.0738 |
| DI b | 8.24 ± 4.47 | 8.03 ± 8.32 | 0.2985 |
| Lipid profile | |||
| Total cholesterol (mg/dl) b | 143.72 ± 28.39 | 152.21 ± 24.65 | 0.3816 |
| HDL cholesterol (mg/dL) b | 43.94 ± 8.21 | 44.29 ± 10.41 | 0.9180 |
| LDL cholesterol (mg/dl) b | 84.28 ± 27.19 | 85.64 ± 19.65 | 0.8754 |
| Triglycerides (mg/dl) b | 77.17 ± 28.24 | 111.14 ± 68.63 | 0.2950 |
| Liver function | |||
| Alanine transaminase (U/l) b | 18.33 ± 7.27 | 40.14 ± 33.80 | 0.0463 |
| Aspartate transaminase (U/l) b | 20.83 ± 4.99 | 32.06 ± 26.93 | 0.2313 |
| Body fat composition | |||
| Visceral (cm2) | 52.61 ± 24.86 | 77.24 ± 24.03 | 0.0047 |
| Deep subcutaneous (cm2) | 184.98 ± 202.32 | 171.37 ± 58.03 | 0.1977 |
| Subcutaneous (cm2) b | 467.91 ± 215.92 | 560.69 ± 234.84 | 0.1798 |
| Superficial subcutaneous (cm2) | 161.93 ± 90.37 | 155.98 ± 76.29 | 0.9125 |
| Deep/superficial subcutaneous | 1.14 ± 0.59 | 1.20 ± 0.30 | 0.1419 |
| Hepatic fat fraction (%) | 1.23 ± 1.74 | 20.86 ± 11.36 | < 0.0001 |
Chi‐square tests were used for categorical variables; two‐sample t‐tests were used for normally distributed continuous variables; and Wilcoxon rank test was used for nonnormally distributed continuous variables.
Abbreviations: BMI, body mass index; DI, disposition index; HDL cholesterol, high‐density lipoprotein cholesterol; IGI, insulinogenic index, IGT, impaired glucose tolerance; LDL cholesterol, low‐density lipoprotein cholesterol; NGT, normal glucose tolerance; WBISI, whole‐body insulin sensitivity index.
Mean ± SD.
3, 1, 3, 3, 3, 3, 3, 5, 3, 4, 4, 4, 4, 4, 4, 4, 1 missing values.
White or Caucasian/Black or African American/Hispanics/Others/Asian.
Gut microbial community profiles of 36 obese youths
Thirty‐six shotgun metagenomes of the gut microbial communities of 18 subjects with NAFLD and 18 without NAFLD were analyzed in the present study. The results presented in Figure 1 compare the alpha and beta diversity between these groups and profiles the microbial communities at the species and phylum levels. Shannon‐Weiner diversity index values were calculated for all samples and were slightly elevated in the NAFLD group (Figure 1A), although this difference was not significant (p = 0.14, t‐test). Bray‐Curtis dissimilarity values were calculated and plotted using a nonmetric multidimensional scaling ordination to compare overall gut microbial community structure at the species level (Figure 1B). Permutational multivariate analysis of variance (PERMANOVA) testing revealed that the subjects with NAFLD had significantly different clustering from the non‐NAFLD subjects (p adj = 0.001, R2 = 0.1006, PERMANOVA) and showed a significant difference in dispersion (p adj = 0.023, β‐dispersion). These results indicate a significant difference between the centroids of these groups at the species level, and this difference may be due to the tested factor (disease), variability within each group, or a combination of these two factors.[ 18 ] Agglomeration at higher taxonomic levels (genus thru phylum) led to reduced and no longer significant dispersion differences while maintaining the significant clustering differences as measured by PERMANOVA testing (Figure S1).
FIGURE 1.
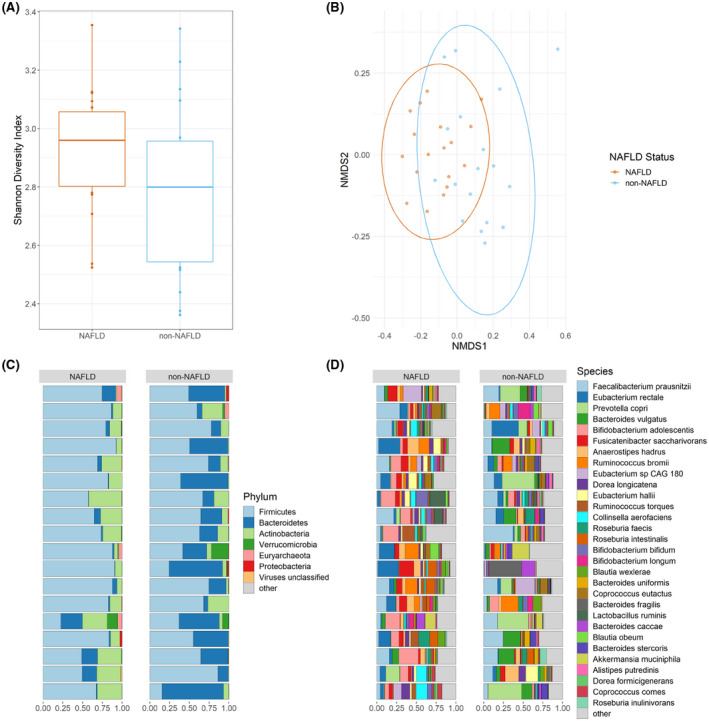
Microbiomes of 36 obese youths. (A) Shannon alpha diversity values are displayed as boxplots with median, interquartile range, and outliers marked. (B) Bray‐Curtis nonmetric multidimensional scaling (NMDS) plot calculated at the species level using rarefied data. Phylum‐level (C) and species‐level (D) compositional bar plots are displayed with each horizontal bar representing an individual microbial community. Nonalcoholic fatty liver disease (NAFLD) status is indicated above each column of bar plots. “Other” indicates all remaining taxa are grouped within this category
Between the NAFLD and non‐NAFLD groups at the phylum level (Figure 1C, Figure S2A), the mean relative abundance of Firmicutes (72.1% vs. 56.1%) and Bacteroidetes (6.7% vs. 33.2%) differed. Actinobacteria (18.8% vs. 7.9%) was the third most prevalent phylum for both groups. Verrucomicrobia (0.9% vs. 1.9%) and Proteobacteria (0.3% vs. 0.5%) were present as minor bacterial phyla along with the archaeal phylum Euryarcheota (0.8% vs. 0.4%) as well as viruses (0.2% vs. 0.1%). At the species level (Figure 1D, Figure S2B), the Firmicutes Faecalibacterium prausnitzii (9.4% vs. 12.7%), Eubacterium rectale (8.6% vs. 4.6%), Fusicatenibacter saccharivorans (6.0% vs. 1.6%), Ruminococcus bromii (3.9% vs. 3.3%), Anaerostipes hadrus (4.1% vs. 3.0%), Eubacterium sp. CAG 180 (2.4% vs. 2.7%), and Dorea longicatena (3.2% vs. 1.7%); the Bacteroidetes Prevotella copri (1.6% vs. 10.0%) and Bacteroides vulgatus (1.4% vs. 7.5%); and the Actinobacteria Bifidobacterium adolescentis (8.5% vs. 2.3%) represented the 10 most abundant species. The next 25 most abundant species belonged to the genera Eubacterium, Ruminococcus, Colinsella, Roseburia, Bifidobacterium, Bacteroides, Lactobacillus, Allistepes, Akkermansia, Dorea, Blautia, Coprococcus, and Parabacteroides.
Taxonomic differences between the NAFLD and non‐NAFLD groups
The differences between the gut microbial communities of individuals with NAFLD and without NAFLD were first compared using PCA biplots. PCA plotting presents a compositional approach in which results are generally more reproducible, and variance in the data is directly accounted for within the plot (as opposed to requiring separate statistical analysis as in principal coordinate plotting).[ 19 ] Figure 2 shows the species‐level and phylum‐level PCA plots, with arrows representing the contribution of individual taxa to each principal coordinate (the length of the arrow represents the strength of the effect). Beneath each PCA biplot is an iris plot showing the taxonomic composition for each sample corresponding to the location on the PCA plot above. The percent of the variation explained in the PCA plot increased from the species level (PC1 12.2%, PC2 10.6%) to the phylum level (PC1 57.1%, PC2 23.0%).
FIGURE 2.
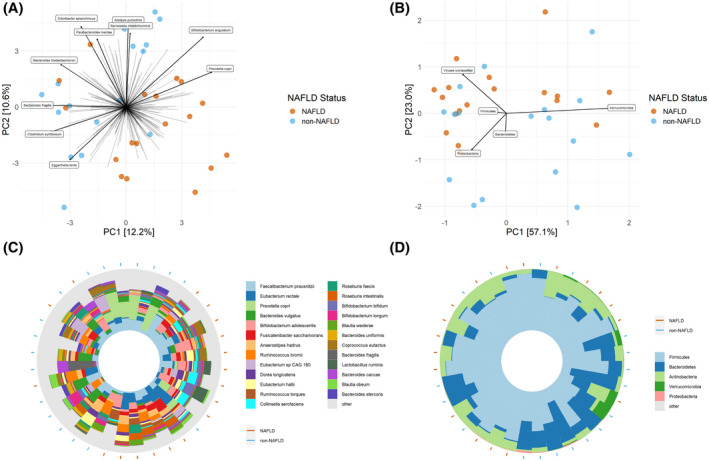
Principal component analysis (PCA) biplots with taxa vectors and iris plots. (A) Species‐level PCA biplot with arrows indicating the abundance gradient for a particular species. The top 10 taxa variable contributors to the principal coordinates (PCs) are shown. (B) Phylum‐level PCA biplot with arrows indicating the abundance gradient for a particular phylum. The top 5 taxa variable contributors to the PCs are shown. (C) Species‐level iris plot showing the taxonomic composition of each sample with its position corresponding to its position in the associated biplot (above). The top 25 species are shown, and all remaining species are grouped into the “other” category. (D) Phylum‐level iris plot showing the taxonomic composition of each sample with its position corresponding to its position in the associated biplot (above). Orange circles on the periphery of the iris plots indicate individuals with NAFLD; blue circles indicate individuals without NAFLD individuals. Genera had to appear in 25% of samples to be included
Species with a large effect size in the quadrant containing most of the non‐NAFLD samples included Alistepes putredinis, Odoribacter splanchnicus, Barnesiella intestinihominis, Parabacteroides merdae, Bacteroides thetaiotamicron, and Bacteroides fragilis (Figure 2A,C). None of the top 10 species shown were dramatically enriched in the individuals with NAFLD. At the phylum level, Bacteroidetes was enriched in the area of the samples from the non‐NAFLD group, while Actinobacteria and viruses were enriched in the area of the samples from the NAFLD group (Figure 2B,D). Proteobacteria and Verrucomicrobia were enriched in a subset of both individuals with and without NAFLD, whereas Firmicutes does not have a clear correlation with either group.
The biplots suggested that specific taxa may correlate with the disease state of the subjects, and differential abundance testing was then used to probe this in a statistically meaningful way. Analysis of composition of microbiomes with bias correction (ANCOM‐BC) was used to determine whether any taxa differed significantly in the microbiomes from subjects with and without NAFLD. All taxonomic levels from species to phylum were tested, and the results are presented in Table 2. The phylum Bacteroidetes, class Bacteroidia, order Bacteroidales, family Bacteroidaceae, and genus Bacteroides were all significantly decreased in the NAFLD group. Three species were shown to have elevated relative abundances in the NAFLD group, including Fusicatenibacter saccharivorans, Romboutsia ilealis, and Actinomyces sp. ICM47. The B. thetaiotamicron species was shown to be decreased in the NAFLD group as compared with the non‐NAFLD group.
TABLE 2.
ANCOM‐BC results
| Species a | Coefficient (beta) b | SEM | Test statistic (W) | p | Adjusted p‐value (q) |
|---|---|---|---|---|---|
| Actinomyces sp. ICM47 | 4.42267 | 0.98863 | 4.47353 | 7.69375E‐06 | 9.23250E‐04 |
| Fusicatenibacter saccharivorans | 1.98506 | 0.55559 | 3.57288 | 3.53079E‐04 | 4.16633E‐02 |
| Romboutsia ilealis | 4.16014 | 1.17304 | 3.54645 | 3.90464E‐04 | 4.56842E‐02 |
| Bacteroides thetaiotaomicron | −9.03164 | 1.88506 | −4.79116 | 1.65800E‐06 | 1.98000E‐04 |
| Genus a | |||||
| Bacteroides | −3.02484 | 0.53084 | −5.69821 | 1.21073E‐08 | 8.47514E‐07 |
| Flavonifractor | −3.20868 | 0.80108 | −4.00544 | 6.19029E‐05 | 4.27130E‐03 |
| Family a | |||||
| Bacteroidaceae | −2.85427 | 0.58112 | −4.91166 | 9.03083E‐07 | 3.07048E‐05 |
| Odoribacteraceae | −5.52603 | 1.38351 | −3.99422 | 6.49073E‐05 | 2.14194E‐03 |
| Order a | |||||
| Bacteroidales | −2.67369 | 0.47544 | −5.62363 | 1.86985E‐08 | 3.55271E‐07 |
| Class a | |||||
| Bacteroidia | −2.78369 | 0.44122 | −6.30911 | 2.80639E‐10 | 3.92895E‐09 |
| Tissierellia | −3.77561 | 1.30288 | −2.89790 | 3.75667E‐03 | 4.88367E‐02 |
| Phylum a | |||||
| Bacteroidetes | −2.84922 | 0.41898 | −6.80041 | 1.04323E‐11 | 6.25940E‐11 |
Only taxa appearing in 25% of samples were included in each taxonomic level’s analysis.
Positive coefficients indicate taxa elevated in the NAFLD group, and negative coefficients indicate reduced in the NAFLD group.
The differentially abundant species identified using ANCOM‐BC were also plotted in Figure S3 as relative abundances in box plots. Certain species, including Bacteroides thetaiotaomicron and Fusicatenibacter saccharivorans, make up a larger portion of the average species consortium (> 0.1%). Other species, including Actinomyces sp. ICM47 and Romboutsia ilealis, amount to a smaller portion (< 0.1%) of the gut microbial community. The significant results from all other taxonomic levels are plotted in Figures S4–S7.
Functional differences between the NAFLD and non‐NAFLD groups
In addition to taxonomic differences, functional differences between the groups were also investigated. Following the functional classification of reads, their abundances were compared, and the top 50 statistically significant results are reported in Table 3. Of these pathways, 29 had an increased relative abundance in the NAFLD group, while 21 were decreased, among which many biosynthetic pathways were identified, including multiple amino acid synthesis pathways. Multiple lysine synthesis pathways had increased relative abundances along with methionine, isoleucine, ornithine, threonine, serine, tryptophan, arginine, aspartate, and glycine pathways. Superpathways for branched‐chain amino acid (BCAA) and aromatic amino acid (AAA) synthesis along with peptidoglycan synthesis were also observed at higher relative abundances in the NAFLD group. Another subset of pathways was decreased in the NAFLD group and included the ornithine (a distinct pathway from the aforementioned one), glutamine, glutamate, and isoleucine (a distinct pathway from the aforementioned one) synthesis pathways. Degradation, use, and energy generation pathways were also identified. Pyruvate fermentation to acetate and lactate, methanogenesis from acetate, lactose and galactose degradation, glycerol degradation, stachyose degradation, guanosine degradation, glycolysis, and the pentose phosphate pathway were increased in individuals with NAFLD. Two histidine degradation pathways as well as the urea and tricarboxylic acid cycles were decreased in the individuals with NAFLD. All significant results are presented in Table S1.
TABLE 3.
Elevated metabolic pathways
| Pathway | Class | Coefficient a | SEM | Prevalence b | p | q |
|---|---|---|---|---|---|---|
| COA PWY: coenzyme A biosynthesis I: prokaryotic | Biosynthesis | 0.257 | 0.052 | 36 | 0.00002 | 0.01318 |
| GLYCOGENSYNTH PWY: glycogen biosynthesis I: from ADP D Glucose | Biosynthesis | 0.751 | 0.142 | 36 | 0.00001 | 0.01318 |
| PWY 4242: pantothenate and coenzyme A biosynthesis III | Biosynthesis | 0.285 | 0.056 | 36 | 0.00001 | 0.01318 |
| PWY 5100: pyruvate fermentation to acetate and lactate II | Degradation/use | 0.747 | 0.149 | 36 | 0.00002 | 0.01318 |
| PWY 6471: peptidoglycan biosynthesis IV: Enterococcus faecium | Biosynthesis | 1.265 | 0.249 | 36 | 0.00001 | 0.01318 |
| ARO PWY: chorismate biosynthesis I | Biosynthesis | 0.304 | 0.068 | 36 | 0.00009 | 0.01343 |
| BRANCHED CHAIN AA SYN PWY: superpathway of branched chain amino acid biosynthesis | Biosynthesis | 0.268 | 0.063 | 36 | 0.00017 | 0.01343 |
| COMPLETE ARO PWY: superpathway of aromatic amino acid biosynthesis | Biosynthesis | 0.309 | 0.070 | 36 | 0.00009 | 0.01343 |
| HSERMETANA PWY: L methionine biosynthesis III | Biosynthesis | 0.472 | 0.099 | 36 | 0.00003 | 0.01343 |
| LACTOSECAT PWY: lactose and galactose degradation I | Degradation/use | 1.592 | 0.379 | 36 | 0.00018 | 0.01343 |
| PWY 5097: L lysine biosynthesis VI | Biosynthesis | 0.216 | 0.051 | 36 | 0.00017 | 0.01343 |
| PWY 5103: L isoleucine biosynthesis III | Biosynthesis | 0.311 | 0.070 | 36 | 0.00009 | 0.01343 |
| PWY 6270: isoprene biosynthesis I | Biosynthesis | 0.562 | 0.121 | 36 | 0.00005 | 0.01343 |
| PWY 7221: guanosine ribonucleotides de novo biosynthesis | Biosynthesis | 0.237 | 0.051 | 36 | 0.00005 | 0.01343 |
| PWY 724: superpathway of L lysine: L threonine and L methionine biosynthesis II | Biosynthesis | 0.262 | 0.058 | 36 | 0.00006 | 0.01343 |
| PWY 7357: thiamine phosphate formation from pyrithiamine and oxythiamine: yeast | Biosynthesis | 0.419 | 0.093 | 36 | 0.00008 | 0.01343 |
| PWY 7560: methylerythritol phosphate pathway II | Biosynthesis | 0.597 | 0.129 | 36 | 0.00005 | 0.01343 |
| GLUTORN PWY: L ornithine biosynthesis I | Biosynthesis | 0.369 | 0.091 | 36 | 0.00028 | 0.01710 |
| METH ACETATE PWY: methanogenesis from acetate | Energy generation | 1.690 | 0.421 | 36 | 0.00031 | 0.01780 |
| NONOXIPENT PWY: pentose phosphate pathway: non oxidative branch: I | Energy generation | 0.606 | 0.154 | 36 | 0.00038 | 0.02008 |
| PWY 6527: stachyose degradation | Degradation/use | 0.677 | 0.176 | 36 | 0.00051 | 0.02400 |
| P4 PWY: superpathway of L lysine: L threonine and L methionine biosynthesis I | Biosynthesis | 1.269 | 0.331 | 36 | 0.00051 | 0.02401 |
| CALVIN PWY: Calvin Benson Bassham cycle | Biosynthesis | 0.358 | 0.094 | 36 | 0.00057 | 0.02470 |
| PWY 5188: tetrapyrrole biosynthesis I: from glutamate | Biosynthesis | 0.792 | 0.209 | 36 | 0.00058 | 0.02470 |
| PWY 6121: 5 aminoimidazole ribonucleotide biosynthesis I | Biosynthesis | 0.223 | 0.059 | 36 | 0.00058 | 0.02470 |
| DAPLYSINESYN PWY: L lysine biosynthesis I | Biosynthesis | 1.041 | 0.283 | 36 | 0.00080 | 0.02734 |
| PWY 6163: chorismate biosynthesis from 3 dehydroquinate | Biosynthesis | 0.255 | 0.069 | 36 | 0.00078 | 0.02734 |
| PWY 6606: guanosine nucleotides degradation II | Degradation/use | 0.869 | 0.236 | 36 | 0.00080 | 0.02734 |
| SER GLYSYN PWY: superpathway of L serine and glycine biosynthesis I | Biosynthesis | 0.439 | 0.118 | 36 | 0.00074 | 0.02734 |
| ARGININE SYN4 PWY: L ornithine biosynthesis II | Biosynthesis | −3.235 | 0.636 | 32 | 0.00001 | 0.01318 |
| HISDEG PWY: L histidine degradation I | Degradation/use | −1.876 | 0.383 | 36 | 0.00002 | 0.01318 |
| PWY 1269: CMP 3 deoxy D manno octulosonate biosynthesis | Biosynthesis | −1.837 | 0.373 | 36 | 0.00002 | 0.01318 |
| PWY 5973: cis vaccenate biosynthesis | Biosynthesis | −1.440 | 0.288 | 36 | 0.00002 | 0.01318 |
| PWY 7663: gondoate biosynthesis: anaerobic | Biosynthesis | −1.556 | 0.300 | 36 | 0.00001 | 0.01318 |
| CITRULBIO PWY: L citrulline biosynthesis | Biosynthesis | −2.367 | 0.540 | 35 | 0.00011 | 0.01343 |
| POLYISOPRENSYN PWY: polyisoprenoid biosynthesis: E: coli | Biosynthesis | −1.875 | 0.443 | 36 | 0.00017 | 0.01343 |
| PWY 4984: urea cycle | Degradation/use | −2.402 | 0.547 | 35 | 0.00010 | 0.01343 |
| PWY 5030: L histidine degradation III | Degradation/use | −1.743 | 0.381 | 36 | 0.00006 | 0.01343 |
| PWY 5505: L glutamate and L glutamine biosynthesis | Biosynthesis | −2.094 | 0.483 | 35 | 0.00012 | 0.01343 |
| PWY0 845: superpathway of pyridoxal 5: phosphate biosynthesis and salvage | Biosynthesis | −2.621 | 0.560 | 33 | 0.00004 | 0.01343 |
| PYRIDOXSYN PWY: pyridoxal 5: phosphate biosynthesis I | Biosynthesis | −2.892 | 0.602 | 33 | 0.00003 | 0.01343 |
| PWY 6859: all trans farnesol biosynthesis | Biosynthesis | −2.037 | 0.493 | 36 | 0.00022 | 0.01441 |
| PWY 7539: 6 hydroxymethyl dihydropterin diphosphate biosynthesis III: Chlamydia | Biosynthesis | −1.551 | 0.376 | 36 | 0.00023 | 0.01461 |
| PWY 5104: L isoleucine biosynthesis IV | Biosynthesis | −2.218 | 0.546 | 33 | 0.00027 | 0.01663 |
| PWY 6147: 6 hydroxymethyl dihydropterin diphosphate biosynthesis I | Biosynthesis | −1.529 | 0.380 | 36 | 0.00030 | 0.01751 |
| PWY 7332: superpathway of UDP N acetylglucosamine derived O antigen building blocks biosynthesis | Biosynthesis | −2.241 | 0.557 | 17 | 0.00030 | 0.01751 |
| PWY 7392: taxadiene biosynthesis: engineered | Biosynthesis | −1.781 | 0.448 | 36 | 0.00035 | 0.01877 |
| TCA: TCA cycle I: prokaryotic | Energy generation | −0.855 | 0.218 | 36 | 0.00040 | 0.02066 |
| NAD BIOSYNTHESIS II: NAD salvage pathway III: to nicotinamide riboside | Biosynthesis | −2.145 | 0.582 | 33 | 0.00079 | 0.02734 |
| PWY 7282: 4 amino 2 methyl 5 diphosphomethylpyrimidine biosynthesis II | Biosynthesis | −1.428 | 0.388 | 35 | 0.00080 | 0.02734 |
Positive coefficients indicate an elevated pathway in the NAFLD group, and negative coefficients indicate a reduced pathway in the NAFLD group.
Prevalence indicates the number of samples in which this particular pathway was detected.
Additionally, the specific contributions of individual species to a particular metabolic pathway were calculated and then compared between groups to identify differentially abundant taxonomic contributions. Bar plots are provided in Figures S9–S14 for a subset consisting of pathways of interest. Eubacterium hallii and Fusicatenibacter saccharivorans were by far the two most common taxa contributing to increased pathways in the NAFLD group. Additionally, Blautia wexlerae, Blautia obeum, Streptococcus salivarius, Ruminococcus torques, Streptococcus parasanguinis, Coprococcus catus, Streptococcus thermophilus, Romboutsia ilealis, Roseburia sp. CAG 471, and Dorea formicigenerans were also increased in relative abundance in certain pathways. Reduced taxa contributing to decreased metabolic pathways in the NAFLD group included Bacteroides vulgatus, Bacteroides thetaiotaomicron, Bacteroides ovatus, Bacteroides stercoris, Parabacteroides distasonis, Bacteroides fragilis, Bacteroides xylanisolvens, Ruthenibacterium lactatiformans, and Parabacteroides merdae. All significant results are presented in Table S1.
DISCUSSION
In the present study, we used shotgun metagenomic sequencing to illuminate a number of taxonomic and functional differences between the gut microbial communities of obese youths with and without NAFLD. Previous studies sequenced relatively short variable regions of the 16S rRNA gene, which limits the taxonomic resolution capacity and lacks important functional information.[ 20 ] The analysis of the shotgun metagenome data at the species level revealed that the composition of the microbiomes differed significantly between the NAFLD and non‐NAFLD groups (Figure 1). The high degree of variability of the human microbiome, especially at the species and genus levels, has been well documented.[ 21 , 22 ] This variability may explain the dispersion visualized in Figure 1B, particularly for the non‐diseased group. The reduction in variance at higher taxonomic levels suggests that a larger number of genera and species vary among the subjects without NAFLD than the subjects with NAFLD.
The taxonomic contributors of the observed difference in beta diversity were analyzed using PCA biplots. Interestingly, a number of species appeared to correlate with the non‐NAFLD group, whereas the NAFLD group lacks obvious ones. This result indicates that a lack of certain microbial taxa and general dysbiosis may contribute to the pathogenesis of NAFLD as opposed to the presence of specific taxa being causative.[ 23 ] The most commonly observed and significant taxonomic difference was the relatively decreased abundance of bacteria belonging to the phylum Bacteroidetes in subjects with NAFLD. Both the PCA biplot and ANCOM differential abundance testing identified this group as strongly decreased in individuals with NAFLD. This was also the finding of an earlier study that we performed on a larger cohort using 16S rRNA gene sequencing and that included the subjects in this study.[ 17 ] A commonly cited overabundance of Proteobacteria[ 24 ] in individuals with NAFLD was not observed in our data set, although a subset of both study groups possessed increased relative abundances of Proteobacteria. A number of species were identified as being differentially abundant, among which B. thetaiotamicron was shown to be significantly decreased in the NAFLD group and was identified as a major driver in PCA (Figure 2). B. thetaiotamicron has been shown to reduce diet‐induced body weight gain and adiposity in mice and was observed at lower abundances in obese human subjects compared with healthy ones.[ 25 ] O. splanchnicus, a species within the family Odoribacteriaceae that was decreased in the NAFLD group, was shown to be associated with decreased incidence of NAFLD,[ 26 ] cystic fibrosis,[ 27 ] and inflammatory bowel disease[ 28 ] in previous studies. This species was identified as a major driver in PCA, and its bacterial family was identified via ANCOM analysis. Fusicatenibacter saccharivorans, the lone species within a relatively newly recognized genus,[ 29 ] comprised a fairly large portion of the NAFLD community. This species has been associated with a diet high in processed foods[ 30 ] and is capable of fermenting a wide variety of saccharides, producing short‐chain fatty acids (SCFAs) as a result.[ 29 ] The species Actinomyces sp. ICM47 is primarily associated with the human oral microbiome,[ 31 ] and its overabundance in individuals with NAFLD could be partly due to increased saliva entering the gastrointestinal tract through more frequent eating or an increased salivary response (although this is only speculation). It is important to note that detecting DNA from specific bacterial species does not establish their living presence within a certain niche. The presence of oral microbial DNA in fecal samples is to be expected but does not necessarily indicate that a species is a gut resident.
Functionally, an even more expansive list of features was shown to be differentially abundant between the two groups. BCAA (Figure S9) and AAA (Figure S10) biosynthetic genes were observed to be at higher relative abundances in the group with NAFLD, and previous studies have reported BCAAs and AAAs to be associated with NAFLD.[ 32 , 33 ] Interestingly, pathways for isoleucine (a BCAA) were shown to be increased in both groups. Isoleucine pathways I (Figure S11) and III (Figure S12) were increased in individuals with NAFLD, while pathway IV (Figure S13) was decreased. Pathways 1 and 3 use 2‐oxobutanoate and glutamate, respectively, whereas pathway 4 uses propanoate. Among the three aforementioned pathways, contributions by Eubacterium, Blautia, and Streptococcus spp. were noted as increased in individuals with NAFLD, while Bacteroides and Parabacteroides spp. were decreased. Although these shotgun data only detect the presence of certain genes and do not provide information on expression levels, it is possible that these pathways are active and lead to an increase in free isoleucine associated with NAFLD.
The fermentation of pyruvate to lactate and acetate, a SCFA, is another interesting pathway that was observed at higher relative abundances in the NAFLD group compared with the non‐NAFLD group (Figure S14)—a difference primarily driven by the abundances of Fusicatenibacter saccharivorans and two Streptococcus species. Total SCFA content in the gut has been correlated with obesity status.[ 23 ] The classification of both evaluated groups as obese may indicate that certain bacterial community members further exacerbate SCFA concentration, increasing the chances for more severe obesity and NAFLD progression. The increase or decrease in specific SCFAs (acetate, propionate, and butyrate) may also contribute to disease progression. Butyrate and acetate are the most and least potent anti‐inflammatory SCFAs, respectively.[ 34 ] The gene potential for increased acetate production by F. saccharivorans and a decrease in butyrate production from a decreased abundance of F. prausnitzii (the most abundant species in these metagenomes and a known butyrate producer) (Figure S2B) may contribute to more severe gut inflammation in the NAFLD group.[ 35 ]
This study has some limitations, the main one being the relatively small sample size (n = 36) used, which could have led to a potentially high false discovery rate (FDR). To account for this issue, we made FDR‐related statistical adjustments and used minimum prevalence values to exclude taxa or features only present in a small subset of samples. Another consideration, particularly for the functional data, is that shotgun metagenomic data provide the functional potential of the community present rather than the expression levels of these genes. This distinction is important, as certain genes may be present but not actively transcribed, whereas others may be overexpressed. Although a metatranscriptomic analysis could provide a functional snapshot of the community, this approach has its own set of caveats, as the transcriptomes of fecal microbiome samples are not necessarily representative of the physiology in the colon. The significant differences noted for BMI and body fat percentage between the two groups may also present as a confounding variable. The NAFLD group had consistently higher BMI and percent body fat measurements, indicating more severe obesity, which has been shown to affect microbiome diversity.[ 36 ]
In the present study, we profiled a largely undercharacterized age group within the population,[ 37 ] including thorough clinical phenotyping of the patient groups using MRI and shotgun deep sequencing of the gut microbial communities. In summary, we found that the two groups had significantly different microbial community compositions, and that these differences were at least partially driven by a specific subset of bacterial species and that biosynthetic pathways producing metabolites (BCAAs, AAAs, SCFAs) previously correlated with disease were again identified here. There is great potential for future studies to investigate these identified species as probiotics or drug targets as well as using correlated metabolite production as a biomarker for increased disease risk to inform clinical interventions. The comparison examined was also unique in that only obese youth with and without NAFLD were included, thereby controlling for the effect of obesity on the microbiome. Given NAFLD’s link with type 2 diabetes, cardiovascular disease, and advanced liver disease, as well as its increasing prevalence, NAFLD poses a major concern to public health and begs more effective strategies for prevention and treatment. While gut dysbiosis has been postulated as a potential contributor to the pathogenesis of NAFLD and NASH, more research is needed to elucidate how gut‐liver axis signaling influences hepatic fat accumulation and inflammation and how these mechanisms might be modulated for therapeutic benefit. Future studies that combine clinical, metabolomic, and microbiome data will be extremely important for prospective therapies for NAFLD and its more severe progressions, especially considering children and adolescents in high‐income countries are in the midst of an obesity epidemic.
CONFLICTS OF INTEREST
Nothing to report.
ETHICS APPROVAL
The Yale University Human Investigation committee approved the study.
CONSENT TO PARTICIPATE AND CONSENT FOR PUBLICATION
Written child assent and written parental permission were obtained from all participants involved in the study.
AVAILABILITY OF DATA AND MATERIAL
Data can be found in the NCBI SRA database under project ID PRJNA328258.
CODE AVAILABILITY
Code can be found at https://github.com/joerggraflab/Code‐for‐Testerman‐Li‐2021.
Supporting information
Fig S1‐S14
Table S1
ACKNOWLEDGMENT
The authors are grateful for the clinical and research staff of the Hospital Research Unit, Church Street Research Unit, and YCCI Core Lab at Yale School of Medicine. They also thank Dr. Jeremiah Marden for providing feedback on the manuscript.
Testerman T, Li Z, Galuppo B, Graf J, Santoro N. Insights from shotgun metagenomics into bacterial species and metabolic pathways associated with NAFLD in obese youth. Hepatol Commun. 2022;6:1962–1974. 10.1002/hep4.1944
Funding information
National Institute of Diabetes and Digestive and Kidney Disease (R01 DK113403‐01A1) and the Yale Center for Clinical Investigation
REFERENCES
- 1. Castillo‐Leon E, Cioffi CE, Vos MB. Perspectives on youth‐onset nonalcoholic fatty liver disease. Endocrinol Diabetes Metab. 2020;3:e00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marzuillo P, Giudice EM, Santoro N. Pediatric non‐alcoholic fatty liver disease: new insights and future directions. World J Hepatol. 2014;6:217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. D'Adamo E, Santoro N, Caprio S. Metabolic syndrome in pediatrics: old concepts revised, new concepts discussed. Pediatr Clin North Am. 2011;58:1241–55. [DOI] [PubMed] [Google Scholar]
- 4. Patton HM, Yates K, Unalp‐Arida A, Behling CA, Huang T‐K, Rosenthal P, et al. Association between metabolic syndrome and liver histology among children with nonalcoholic fatty liver disease. Am J Gastroenterol. 2010;105:2093–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non‐alcoholic fatty liver disease in children: a follow‐up study for up to 20 years. Gut. 2009;58:1538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marzuillo P, del Giudice EM, Santoro N. Pediatric fatty liver disease: Role of ethnicity and genetics. World J Gastroenterol. 2014;20:7347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwimmer JB, Ugalde‐Nicalo P, Welsh JA, Angeles JE, Cordero M, Harlow KE, et al. Effect of a low free sugar diet vs usual diet on nonalcoholic fatty liver disease in adolescent boys: a randomized clinical trial. JAMA. 2019;321:256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Name MA, Savoye M, Chick JM, Galuppo BT, Feldstein AE, Pierpont B, et al. A low ω‐6 to ω‐3 PUFA ratio (n‐6:n‐3 PUFA) diet to treat fatty liver disease in obese youth. J Nutr. 2020;159:2314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hrncir T, Hrncirova L, Kverka M, Hromadka R, Machova V, Trckova E, et al. Gut microbiota and NAFLD: pathogenetic mechanisms, microbiota signatures, and therapeutic interventions. Microorganisms. 2021;9:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharpton SR, Schnabl B, Knight R, Loomba R. Current concepts, opportunities, and challenges of gut microbiome‐based personalized medicine in nonalcoholic fatty liver disease. Cell Metab. 2021;33:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S, et al. A universal gut‐microbiome‐derived signature predicts cirrhosis. Cell Metab. 2020;32:878–88.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome‐based metagenomic signature for non‐invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 2017;25:1054–62.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharpton SR, Yong GJM, Terrault NA, Lynch SV. Gut microbial metabolism and nonalcoholic fatty liver disease. Hepatol Commun. 2019;3:29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Del Chierico F, Nobili V, Vernocchi P, Russo A, De Stefanis C, Gnani D, et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta‐omics‐based approach. Hepatology. 2017;65:451–64. [DOI] [PubMed] [Google Scholar]
- 15. Michail S, Lin M, Frey MR, Fanter R, Paliy O, Hilbush B, et al. Altered gut microbial energy and metabolism in children with non‐alcoholic fatty liver disease. FEMS Microbiol Ecol. 2015;91:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caussy C, Tripathi A, Humphrey G, Bassirian S, Singh S, Faulkner C, et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat Commun 2019;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kravetz AM, Testerman T, Galuppo B, Graf J, Pierpont B, Siebel S, et al. Effect of gut microbiota and PNPLA3 rs738409 variant on nonalcoholic fatty liver disease (NAFLD) in obese youth. J Clin Endocrinol Metab. 2020;105:e3575–e3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anderson MJ. Permutational multivariate analysis of variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online. 2017. 10.1002/9781118445112.stat07841 [DOI] [Google Scholar]
- 19. Gloor GB, Reid G. Compositional analysis: a valid approach to analyze microbiome high‐throughput sequencing data. Can J Microbiol. 2016;62:692–703. [DOI] [PubMed] [Google Scholar]
- 20. Del Chierico F, Nobili V, Vernocchi P, Russo A, De Stefanis C, Gnani D, et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta‐omics‐based approach. Hepatology. 2017;65:451–4. [DOI] [PubMed] [Google Scholar]
- 21. Gilbert J, Blaser MJ, Caporaso JG, Jansson J, Lynch SV, Knight Rb. Current understanding of the human microbiome. Nat Med. 2018;24:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Senghor B, Sokhna C, Ruimy R, Lagier JC. Gut microbiota diversity according to dietary habits and geographical provenance. Hum Microbiome J. 2018;7–8:1–9. [Google Scholar]
- 23. Sharpton S, Ajmera V, Loomba R. Emerging role of the gut microbiome in nonalcoholic fatty liver disease: from composition to function. Clin Gastroenterol Hepatol. 2019;17:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lau E, Carvalho D, Freitas P. Gut microbiota: association with NAFLD and metabolic disturbances. BioMed Res Int. 2015;2015:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, et al. Gut microbiome and serum metabolome alterations in obesity and after weight‐loss intervention. Nat Med. 2017;23:859–68. [DOI] [PubMed] [Google Scholar]
- 26. Hiippala K, Barreto G, Burrello C, Diaz‐Basabe A, Suutarinen M, Kainulainen V, et al. Novel Odoribacter splanchnicus strain and its outer membrane vesicles exert immunoregulatory effects in vitro. Front Microbiol. 2020;11:2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Price CE, O’Toole GA. The gut‐lung axis in cystic fibrosis. J Bacteriol. 2021;203:e0031121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jeffery IB, O’toole PW, Hman LO, Claesson MJ, Deane J, Quigley EMM, et al. An irritable bowel syndrome subtype defined by species‐specific alterations in faecal microbiota. Gut. 2012;61:997–1006. [DOI] [PubMed] [Google Scholar]
- 29. Takada T, Kurakawa T, Tsuji H, Nomoto K. Fusicatenibacter saccharivorans gen. nov., sp. nov., isolated from human faeces. Int J syst Evol Microbiol. 2013;63:3691–6. [DOI] [PubMed] [Google Scholar]
- 30. Matsuyama M, Morrison M, Cao K‐AL, Pruilh S, Davies PSW, Wall C, et al. Dietary intake influences gut microbiota development of healthy Australian children from the age of one to two years. Sci Rep. 2019;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. JGI IMG Actinomyces sp . ICM47 [Internet]. 2013. [cited 2021 Oct 11]. Available from: https://img.jgi.doe.gov/cgi‐bin/m/main.cgi?section=TaxonDetail&page=taxonDetail&taxon_oid=2537561777.
- 32. Yamakado M, Tanaka T, Nagao K, Imaizumi A, Komatsu M, Daimon T, et al. Plasma amino acid profile associated with fatty liver disease and co‐occurrence of metabolic risk factors. Sci Rep. 2017;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goffredo M, Santoro N, Tricò D, Giannini C, D’Adamo E, Zhao H, et al. A branched‐chain amino acid‐related metabolic signature characterizes obese adolescents with non‐alcoholic fatty liver disease. Nutrients. 2017;9:642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vinolo MAR, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3:858–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)‐mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, le Chatelier E, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585–8. [DOI] [PubMed] [Google Scholar]
- 37. Derrien M, Alvarez A‐S, de Vos WM. The gut microbiota in the first decade of life. Trends Microbiol. 2019;27:997–1010. [DOI] [PubMed] [Google Scholar]
- 38. Tricò D, Caprio S, Rosaria Umano G, Pierpont B, Nouws J, Galderisi A, et al. Metabolic features of nonalcoholic fatty liver (NAFL) in obese adolescents: findings from a multiethnic cohort. Hepatology. 2018;68:1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Galderisi A, Giannini C, Weiss R, Kim G, Shabanova V, Santoro N, et al. Trajectories of changes in glucose tolerance in a multiethnic cohort of obese youths: an observational prospective analysis. Lancet Child Adolescent Health. 2018;2:726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hershkop K, Besor O, Santoro N, Pierpont B, Caprio S, Weiss R. Adipose insulin resistance in obese adolescents across the spectrum of glucose tolerance. J Clin Endocrinol Metab. 2016;101:2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Beghini F, McIver LJ, Blanco‐Míguez A, Dubois L, Asnicar F, Maharjan S, et al. Integrating taxonomic, functional, and strain‐level profiling of diverse microbial communities with bioBakery 3. eLife. 2021;10:e65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. R Core Team . R: a language and environment for statistical computing. 2017. [Google Scholar]
- 43. McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barnett D. david‐barnett/microViz: microViz 0.7.7. 2021 [cited 2021 Jun 18]. Available from: https://zenodo.org/record/4898887.
- 45. Mandal S, van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microbial Ecol Health Dis. 2015;26:27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kaul A, Mandal S, Davidov O, Peddada SD. Analysis of microbiome data in the presence of excess zeros. Front Microbiol. 2017;8:2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin H, Peddada S, das. Analysis of compositions of microbiomes with bias correction. Nat Communi. 2020;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. microbiome/microbiome: microbiome R package [Internet]. [cited 2021 Jun 18]. Available from: https://github.com/microbiome/microbiome.
- 49. Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, et al. Species‐level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15:962–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suzek BE, Huang H, McGarvey P, Mazumder R, UniRef WuCH. Comprehensive and non‐redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–8. [DOI] [PubMed] [Google Scholar]
- 51. Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Tickle TL, et al. Multivariable association discovery in population‐scale meta‐omics studies 3. bioRxiv. 2021. 10.1101/2021.01.20.427420 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S14
Table S1
Data Availability Statement
Data can be found in the NCBI SRA database under project ID PRJNA328258.