

Abstract
The conduct of long‐term conventional randomized clinical trials in rare diseases is very difficult, making evidenced‐based drug development problematic. As a result, real‐world data/evidence are being used more frequently to assess new therapeutic approaches in orphan diseases. In this investigation, inclusion and exclusion criteria from a published trial of maralixibat in Alagille syndrome (ALGS, ITCH NCT02057692) were applied to a prospective longitudinal cohort of children with cholestasis (LOGIC NCT00571272) to derive contextual comparator data for evolving clinical trials of intestinal bile acid transport inhibitors in ALGS. A natural history/clinical care cohort of 59 participants who met adapted inclusion and exclusion criteria of ITCH was identified from 252 LOGIC participants with ALGS with their native liver. Frequency weighting was used to match the age distribution of ITCH and yielded a cohort (Alagille Syndrome Natural History [ALGS NH]) that was very similar to the baseline status of ITCH participants. During a 2‐year prospective follow‐up there was a significant reduction in pruritus in the weighted ALGS NH cohort as assessed by the clinician scratch score (−1.43 [0.28] −1.99, −0.87; mean [SEM] 95% confidence interval). During the same time period, the total bilirubin, albumin, and alanine aminotransferase levels were unchanged, whereas platelet count dropped significantly (−65.2 [16.2] −98.3, −32.1). Weighted survival with native liver was 91% at 2 years in the ALGS NH. These investigations provide valuable real‐world data that can serve as contextual comparators to current clinical trials, especially those without control populations, and highlight the value and importance of funded multicenter, prospective, natural history studies.
Prospective multi‐center longitudinal databases, which may require significant funding, provide critical biomedical information. It is possible to adapt entry criteria for completed and on‐going clinical trials to the participants in these registries, thereby generating a similar natural history cohort. Investment in prospective databases affords unique and invaluable real world data, serving as important comparators in the assessment of the safety and efficacy of novel agents investigated in rare diseases as demonstrated by this study in Alagille syndrome.

INTRODUCTION
Despite focused interest and investment of significant resources, there remains an unmet need in developing proven therapeutic approaches to clinical issues related to rare diseases.[ 1 , 2 , 3 ] Conventional randomized controlled trials are often not feasible due to a variety of issues, most notably the lack of statistical power due to inadequate numbers of potential study participants. Complex ethical issues arise in development of clinical trials in light of the lack of evidenced‐based approaches to therapy for rare diseases. Conventional clinical trial design often necessitates inclusion of a placebo arm, which may not be tenable for individuals and families burdened with poorly understood disorders and debilitating symptoms. This is particularly problematic for severe potentially life‐threatening diseases, in which the studied intervention requires a prolonged course of therapy to demonstrate efficacy.
Alagille syndrome (ALGS) is a rare systemic genetic disorder with a complex clinical phenotype that is not readily predicted on the basis of genotype or family history.[ 4 ] One of the major and debilitating manifestations of ALGS is pruritus related to cholestasis, which has a pronounced impact on sleep and quality of life for patients and their families.[ 5 ] Until very recently, available pharmacologic therapies had limited efficacy in ameliorating itch, leading to the need for invasive surgical interventions including surgical interruption of the enterohepatic circulation (SIEHC) and/or liver transplantation.[ 6 ] In this context there has been ongoing work seeking to identify pharmacologic therapies for pruritus in ALGS. One candidate approach is the use of inhibitors of intestinal bile acid reclamation (ileal bile acid transport inhibitors [IBATi], also known as apical sodium dependent bile acid transport inhibitors, ASBTi) as a surrogate for SIEHC.[ 7 , 8 ] Two of these agents have recently been approved by the US Food and Drug Administration (FDA) for the treatment of cholestatic pruritus, odevixibat for progressive familial intrahepatic cholestasis, and maralixibat for ALGS. The time course of maximal response to SIEHC is not well delineated and may be weeks, months, or years. Coincident with this response to SIEHC is a potential for slow improvement in pruritus in ALGS as part of its “natural history.”[ 9 ]
In the context of this complex clinical problem, investigational trials of IBATi as a potential treatment for pruritus in ALGS have been undertaken.[ 10 ] Short‐term randomized placebo controlled trials have been completed and have demonstrated potential therapeutic efficacy.[ 11 ] An alternative study design has used a randomized placebo‐controlled temporary withdrawal of therapy as a marker of efficacy (ICONIC NCT02160782).[ 12 ] These studies are complicated by significant potential placebo and nocebo effects, which are common in trials of agents directed at the treatment of pruritus.[ 13 ] There is a critical need to determine the long‐term potential efficacy of these agents, but prolonged treatment with placebo is likely not to be acceptable to patients and their families, investigators, clinicians, and potentially regulatory agencies.
The current investigation sought to determine the feasibility of assessing the outcome of a natural history cohort derived from a prospective longitudinal clinical database study focused in part on ALGS that could potentially serve as a control arm for a single‐arm clinical trial. The Childhood Liver Disease Research Network (ChiLDReN) is a multicenter consortium based primarily in the United States, which is funded by the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK). The Longitudinal Study of Genetic Causes of Intrahepatic Cholestasis (LOGIC‐NCT00571272) has been conducted by ChiLDReN since 2007 and has enrolled over 300 participants with ALGS.[ 9 ] The current study sought to use the entry criteria for one of the IBATi ALGS clinical trials (ITCH NCT02057692[ 11 ]) to identify a natural history cohort that might be used as a control population for existing clinical trials for ALGS. This approach addresses the FDA’s Real World Evidence Program (https://www.fda.gov/science‐research/science‐and‐research‐special‐topics/real‐world‐evidence).[ 14 , 15 ]
EXPERIMENTAL PROCEDURES
The inclusion and exclusion criteria for ITCH were adapted to information available in the prospective longitudinal database, LOGIC (Table 1).[ 9 , 11 ] LOGIC participants with ALGS were enrolled between November 1, 2007, and December 31, 2019. Eligibility for ITCH involved an investigational measure of caregiver‐reported pruritus manifestations, ItchRO, and was dependent on an average ItchRO score of ≥ 2 assessed daily during a 2‐week screening period.[ 11 , 16 ] This measure was not available in routine clinical practice, and as such the clinician scratch scale (CSS) developed by Whitington was used in its place.[ 17 ] Although CSS was an outcome measure in ITCH, it must be acknowledged that CSS and ItchRO do not appear to correlate.[ 18 ] The composition of the cohort for this analysis was finalized before the performance of any outcome assessments for the cohort. In addition to applying the inclusion and exclusion criteria of ITCH, eligibility for this study necessitated the potential for at least one follow‐up visit after enrollment in LOGIC, which served as the baseline. Per the LOGIC protocol, follow‐up was scheduled on an annual basis with collection of clinical and laboratory data derived as part of routine clinical care. PedsQL was obtained in participants as logistically possible as a part of the LOGIC research protocol. Participants remained eligible for analysis if their only follow‐up after the baseline visit was for liver transplant or death, as these were sentinel events collected in LOGIC. Participants who were otherwise lost to follow‐up after the baseline enrollment visit with no intervening follow‐up visits were excluded from the analysis. Before calculating changes at 1‐year and 2‐year follow‐up, clinically relevant changes in key parameters were defined based on investigator consensus (Table S1).
TABLE 1.
Major inclusion/exclusion criteria for ITCH relative to LOGIC ALGS approach
| ITCH (from Hep Comm. 2018;2:1184–98) | ALGS LOGIC natural history cohort |
| Inclusion criteria | |
| Diagnosis of ALGS | LOGIC definition of ALGS |
| Age 12 months–18 years inclusive | Age 12 months–18 years inclusive |
| Cholestasis | Cholestasis |
| Total serum bile acid > 3× ULN for age or | Total serum bile acids > 50 μM or |
| Direct bilirubin > 1 mg/dl or | Direct bilirubin or conjugated bilirubin > 1 mg/dl or |
| Fat soluble vitamin deficiency or | Not applied or |
| GGT > 3× ULN for age or | GGT > 150 IU/L or |
| Intractable pruritus based on liver disease | CSS score of 3 or 4 |
| Pruritus—average daily ItchRO (observed) ≥ 2.0 | CSS score ≥ 2.0 |
| Exclusion criteria | |
| Chronic diarrhea requiring intravenous fluids | Not applied |
| Surgical interruption of the enterohepatic circulation | From LOGIC history for surgery |
| Liver transplant | From LOGIC history for surgery |
| ALT > 15× ULN | ALT > 600 insulin resistance/L at baseline |
| Decompensated cirrhosis | Decompensated cirrhosis |
| INR > 1.5 after vitamin K | Not applied—problem ascertaining vitamin K given |
| Albumin < 3.0 gr/dl | Albumin < 3.0 gr/dl |
| History of or presence of significant ascites | History of clinically evident ascites |
| History of variceal hemorrhage | History of gastrointestinal hemorrhage |
| History of encephalopathy | Not applied—not available |
| History of other liver disease | Reviewed historical date—excluded participants with prior Kasai hepatoportoenterostomy and diagnosis of biliary atresia |
| History of disease that could interfere with absorption of, e.g., drugs and bile acids, from intestine | Not applied—data unavailable but unlikely |
| Unable to complete study | Listed for liver transplant |
| Concomitant meds—cholestyramine or other resin, sodium phenylbutyrate, investigational agent trial | Applied as known |
| Nonadherence concern | Not applied |
Abbreviations: GGT, gamma‐glutamyltransferase; INR, international normalized ratio.
The resulting cohort was different from the ITCH cohort with regard to some baseline characteristics, especially in terms of a disproportionate number of participants <2 years of age (Table 2) compared with the ITCH cohort. Because individual‐level data of the baseline ITCH cohort were not available, it was not possible to use the commonly used method, propensity score inverse probability weighting, to adjust for difference between the two cohorts. Therefore, a weighting approach was used to calibrate the age distribution of the ALGS natural history cohort to the age distribution of the ITCH/IMAGINEII cohort. Unweighted and weighted means, medians, or proportions of baseline characteristics in the Alagille Syndrome Natural History (ALGS NH) cohort were calculated. To compare the unweighted and weighted summary statistics of the ALGS NH cohort with the ITCH/IMAGINEII cohort, standardized mean difference for all continuous variables at baseline were calculated. A sensitivity analysis was performed using the last value carried forward for participants who died or underwent liver transplant. Chi‐square test or Fisher’s exact test were used for comparing unweighted distribution of categorical variables in the ALGS NH cohort to their distribution in the ITCH/IMAGINEII cohort. Wald chi‐square tests were used to compare weighted distributions of categorical variables in the ALGS NH cohort to their distribution in the ITCH/IMAGINEII cohort. Taylor series method was used to calculate SD, SEM, and 95% confidence intervals for weighted means and medians, and Kaplan–Meier estimates for transplant‐free survival over time.
TABLE 2.
Comparison of baseline parameters of ITCH participants and LOGIC ALGS natural history cohort
| ITCH (All participants) | LOGIC ALGS natural history cohort (unweighted) | LOGIC ALGS natural history cohort (weighted) | |||
| N | 37 | 59 | 59 | ||
| Summary statistics, mean (SD) [n], median | Summary statistics | SMD% (p value) | Summary statistics | SMD% (p value a ) | |
| Age, years | 6.8 (4.5) [37], 6.0 | 4.1 (3.2) [59], 2.5 | 69% (<0.001) | 6.2 (3.1) [59], 6.5 | 16% (0.32) |
| Age | (<0.001) | (1.00) | |||
| <2 years | 2 (5%) | 26 (44%) | 3.2 (5%) | ||
| 2–4 years | 12 (32%) | 13 (22%) | 19.1 (32%) | ||
| 5–7 years | 11 (30%) | 10 (17%) | 17.5 (30%) | ||
| 8–18 years | 12 (33%) | 10 (17%) | 19.1 (32%) | ||
| Gender | (1.00) | (0.63) | |||
| Female | 16 (43%) | 25 (42%) | 22.2 (38%) | ||
| Male | 21 (57%) | 33 (56%) | 36.7 (62%) | ||
| Not reported | 1 (2%) | 0.1 (0.2%) b | |||
| Race | (0.053) | (0.16) | |||
| White | 29 (78%) | 30 (51%) | 33.7 (57%) | ||
| Black | 5 (14%) | 13 (22%) | 9.3 (16%) | ||
| Non‐White, Non‐Black | 2 (5%) | 9 (15%) | 11.1 (19%) | ||
| Not reported | 1 (3%) | 7 (12%) | 4.9 (8%) | ||
| Ethnicity | (0.63) | (0.91) | |||
| Hispanic | 7 (18.9%) | 9 (15%) | 11.7 (20%) | ||
| Non‐Hispanic | 30 (81%) | 48 (81%) | 47.0 (80%) | ||
| Not reported | 2 (3%) | 0.2 (0.4%) b | |||
| ItchRO | 2.9 (0.60) [37], 2.9 | ||||
| CSS, range 0–4 | 3.0 (1.1) [37], 3.0 | 2,9 (0.7) [59], 3.0 | 11% (0.47) | 2.8 (0.7) [59], 2.4 | 22% (0.16) |
| PesQL parent total score, range 0–100 | 65 (20.1) [35], 67 | 78.8 (12.86) [21], 84.7 | −82% (<0.001) | 79.0 (9.7) [21], 83.1 | −89% (<0.001) |
| Total bilirubin (mg/dl) [3] | 5.3 (6.18) [37], 2.1 | 4.9 (5.42) [57], 2.3 | 7% (0.67) | 3.7 (5.2) [57], 1.4 | 28% (0.07 |
| Cholesterol (mg/dl) | 405.7 (313.6) [37], 320 | 522.4 (342.6) [24], 454 | −36% (0.06) | 408.1 (151.8) [24], 301.4 | −1% (0.94) |
| Serum bile acid (umol/L) | 216.3 (203.3) [37], 155.5 | 174.2 (9.0) [14], 179.2 | 27% (0.14) | 147.5 (29.9) [14], 160.0 | 47% (0.007) |
| ALT (IU/L) | 158.7 (86.5) [37], 137 | 196.7 (99.5) [57], 187 | −41% (0.007) | 184.7 (109.2) [57], 163.9 | −26% (0.07) |
| GGT (IU/L) | 494.9 (379.8) [37], 329 | 542.1 (460.4) [54], 388 | −11% (0.45) | 495.6 (439.6) [54], 292 | 0% (0.99) |
| Albumin (g/dl) | 4.5 (0.4) [37], 4.6 | 4.1 (0.4) [57], 4.1 | 95% (<0.001) | 4.2 (0.4) [57], 4.1 | 76% (<0.001) |
| Platelets (103/mm3) | 287.0 (109.5) [37], 258 | 308.0 (110.4) [51], 308 | −19% (0.214) | 289.7 (105.9) [51], 263.7 | −3% (0.87) |
| Height z score | −1.6 (1.2) [37], −1.6 | −2.3 (1.3) [58], −2.3 | 59% (<0.001) | −1.9 (1.0) [58], −2.0 | 26% (0.09) |
| Weight z score | −1.3 (1.1) [37], −1.3 | −1.7 (0.9) [58], −1.7 | 39% (0.01) | −1.5 (0.9) [58], −1.4 | 17% (0.26) |
Abbreviation: SMD, standardized mean difference.
Compare weighted LOGIC ALGS natural history cohort to the ITCH/IMAGINE II participants. Continuous variables use t‐tests, categorical variables use Wald Chi‐square tests.
Excluded from weighted Wald Chi‐square test due to empty cell issue.
RESULTS
Cohort derivation
Between November 1, 2007, and December 31, 2019, a total of 1261 participants were enrolled in LOGIC. Of these, 1009 were excluded: 856 had a diagnosis other than ALGS; 89 were enrolled after liver transplant; 3 were enrolled as siblings without liver disease (for genetic investigations); 27 did not meet the age criteria for the study; and 34 had no follow‐up information. The inclusion and exclusion criteria were then applied to the remaining 252 participants in LOGIC who had ALGS. The criteria to define cholestasis, as shown in Figure 1, were applied, resulting in 53 participants excluded for absent evidence of this definition of cholestasis. Exclusion criteria were then applied to the remaining 199 participants, leading to the identification of 59 LOGIC participants with ALGS who met the ITCH‐derived inclusion and exclusion criteria.
FIGURE 1.
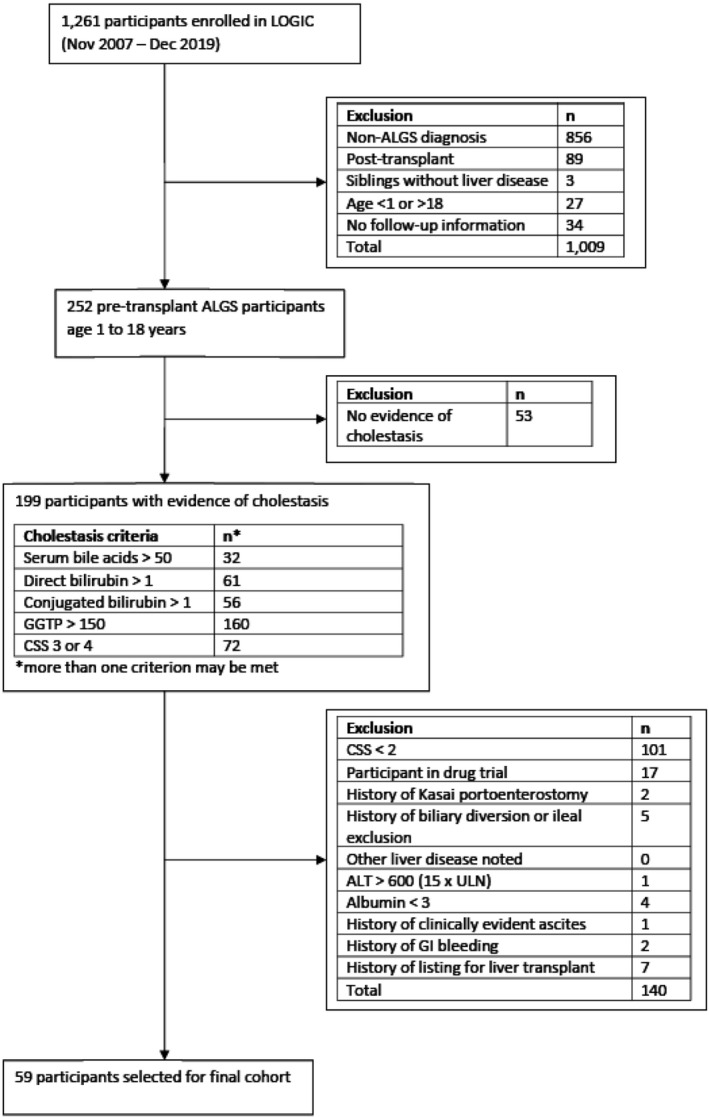
Application of inclusion and exclusion criteria to the LOGIC cohort. From 252 of the 1261 LOGIC participants who had a diagnosis of Alagille syndrome (ALGS), 59 participants could be identified who met the applicable inclusion and exclusion criteria of ITCH. Abbreviations: ALT, alanine aminotransferase; CSS, clinician scratch scale; GGTP, gamma‐glutamyl transpeptidase; GI, gastrointestinal; ULN, upper limit of normal
Baseline parameters for the cohort of 59 LOGIC ALGS participants are found in Table 2 and compared with the baseline characteristics of the published cohort of ITCH participants.[ 11 ] The percentage of LOGIC ALGS participants whose baseline visit was at age < 2 years was disproportionately high in the original cohort; frequency weighting was applied to correct for this important discrepancy. In general, the weighted LOGIC ALGS NH cohort was well‐matched to the baseline characteristics of the ITCH cohort. PedsQL parent total score was higher in the weighted ALGS NH cohort. LOGIC participants were eligible to enroll in ITCH, and it is worthwhile noting that 17 LOGIC participants with ALGS were excluded from this cohort due to participation in a drug trial, which was likely ITCH. Serum bile acids and albumin levels were lower in the weighted ALGS NH, although bile acids were measured in only 14 ALGS NH participants, and the difference in mean albumin levels was only 0.3 mg/dl.
Change in key clinical parameters at 1‐year and 2‐year follow‐up
Weighted mean values for key clinical parameters (CSS, total bilirubin [TB], alanine aminotransferase [ALT], platelet count, albumin, weight, and height) at baseline, year 1, and year 2 of follow‐up are given in Table S2. Change from baseline for these parameters is found in Table 3. Similar results were found when the last value measured was carried forward as a sensitivity analysis for the impact of death or transplant in the first 2 years of follow‐up (Table S3). There was a significant reduction in CSS at years 1 and 2 of follow‐up (Figure 2A,B).
TABLE 3.
Change from baseline for key parameters
| Year 1 (baseline; n = 50) | Year 2 (baseline; n = 40) | |||||
|---|---|---|---|---|---|---|
| n | Weighted mean change (SEM) | 95% CI | n | Weighted mean change (SEM) | 95% CI | |
| CSS | 49 | −0.98 (0.24) | (−1.47, −0.49) | 40 | −1.43 (0.28) | (−1.99, −0.87) |
| Total bilirubin (mg/dl) | 44 | 1.20 (0.91) | (−0.64, 3.03) | 36 | 0.02 (0.30) | (−0.58, 0.62) |
| ALT (IU/L) | 44 | −15.8 (13.4) | (−42.8, 11.2) | 36 | 5.7 (14.3) | (−23.4, 34.8) |
| Platelets (×103) | 40 | −19.2 (9.8) | (−39.0, 0.6) | 30 | −65.2 (16.2) | (−98.3, −32.1) |
| Albumin (g/dl) | 43 | 0.09 (0.07) | (−0.04, 0.22) | 35 | 0.09 (0.09) | (−0.09, 0.28) |
| Weight (z score) | 49 | 0.14 (0.08) | (−0.02, 0.29) | 40 | 0.17 (0.13) | (−0.08, 0.43) |
| Height (z score) | 48 | 0.23 (0.07) | (0.08, 0.37) | 39 | 0.28 (0.10) | (0.08, 0.48) |
Note: Bolded numbers represent statistically significant changes.
Abbreviation: CI, confidence interval.
FIGURE 2.
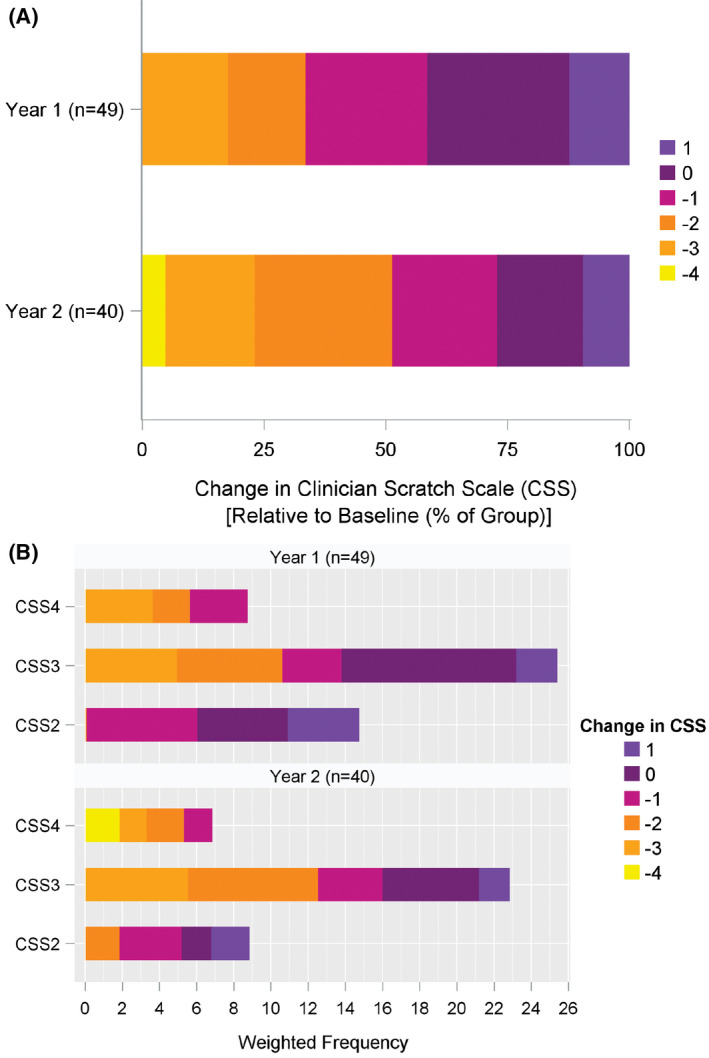
Heatmap of change in CSS from baseline. Colors as noted in the legend indicate the change in CSS. (A) Percentage of the total number of participants with each of the specified changes, shown at year 1 and year 2 of follow‐up (note that −4 is the maximal reduction in CSS that is possible). (B) Absolute number of participants with a given change at year 1 and year 2 relative to the baseline CSS assessment (note that the minimal CSS is 0)
Nine of the 40 participants evaluated at 2 years had complete resolution of their baseline pruritus as assessed by CSS (Figure 2B). Platelet count was reduced at year 2 of follow‐up. Height z score was increased at 1 and 2 years of follow‐up. No changes were observed for TB, ALT, albumin, or weight. The statistically significant changes from baseline for CSS and platelet count were reflected in the percentages of participants with clinically relevant changes in the same parameters (Table S4). By year 2, 73% had a clinically relevant reduction in CSS, and 58% had a clinically relevant reduction in platelet count. One third of the participants had a clinically relevant increase in height and weight z score at 2 years of follow‐up. The percentage of participants receiving various commonly used medications for the treatment of pruritus was stable during the 2 years of follow‐up (Table S5).
Sentinel events
The occurrence of sentinel events was tracked during the available follow‐up for the ALGS NH cohort of 59 participants. The duration of observed follow‐up for this cohort ranged from 4 months to 12 years with an average of 5 years. During that time period, 3 participants developed clinically evident ascites (at ages 3, 12, and 23 years); none had variceal hemorrhage; and 3 underwent surgical interruption of the enterohepatic circulation (1 had partial biliary diversion, 1 had ileal exclusion, and 1 underwent both procedures; partial biliary diversion followed 1.5 years later by ileal exclusion). SIEHC took place between the baseline and the year‐1 visit in the 3 cases in which surgery was undertaken. After SIEHC and within 2 years of baseline, there was no change in CSS in 1 and reductions of CSS by 1 and 2 in the other 2. Three children died with their native liver during follow‐up at an average age of 3.5 years. Thirteen children underwent liver transplant at a mean age of 5.3 years. Of those who died or underwent liver transplant during follow‐up, 9 were under 2 years of age; 6 were 2 to 3 years old; and only 1 was 3 years or older at enrollment. Survival with native liver is shown in Figure 3A. Given the preponderance of young children who did not survive with their native liver, a weighted survival curve is shown in Figure 3B and reveals 91% survival with native liver 2 years from baseline.
FIGURE 3.
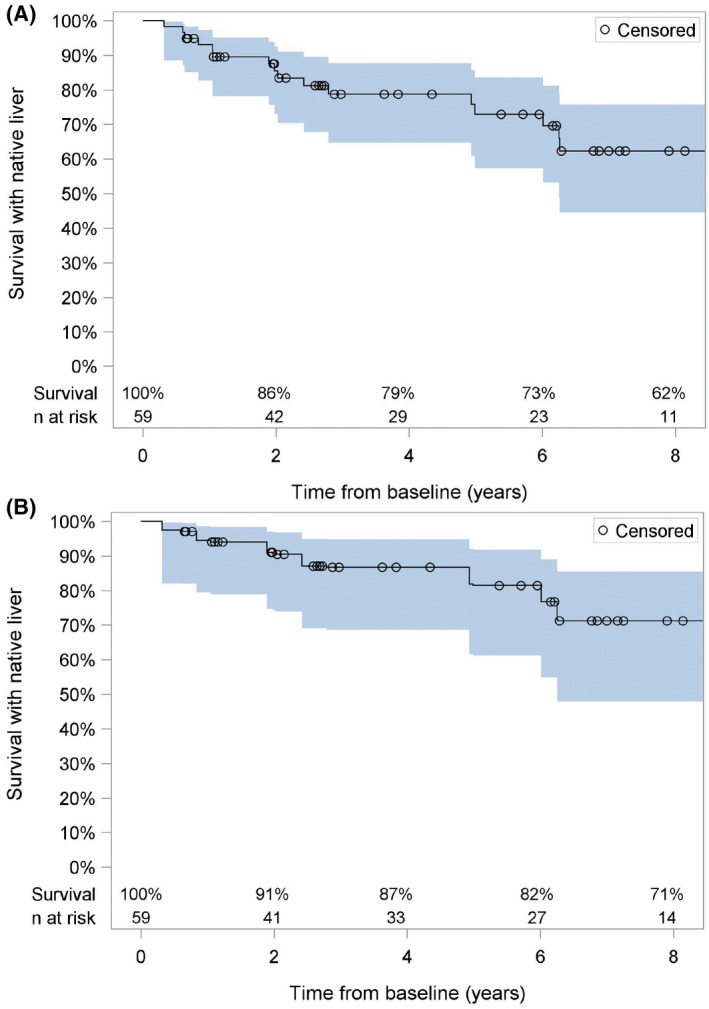
Survival with native liver. Kaplan–Meier survival curves are shown relative to time from baseline. (A) Unweighted analysis of all 59 natural history cohort participants. (B) Weighted analysis of natural history participants. The gray‐shaded area represents the 95% confidence interval for the measurement
DISCUSSION
These investigations demonstrate the feasibility and potential utility of using funded, multicenter, prospective, clinical registries as contextual comparators for clinical trials in rare diseases. The approach meets the mandate of the CURES Act to accelerate medical intervention by using Real World Data and Evidence in the context of guidance from the FDA. In this circumstance, the cohort was relatively contemporaneous to the model clinical trial in question,[ 11 ] thereby enhancing its utility. In addition, the application of inclusion/exclusion criteria increased the relevance of the data set. This clinical care/natural history cohort was derived from a large‐scale NIDDK‐funded effort that was conducted over a period of greater than 10 years and encompassed investigations in more than 15 academic centers with clinical expertise in pediatric hepatology. Access to data from 5‐fold more participants than were selected as a comparator was required. This kind of effort almost certainly necessitates significant funding. The information found within the LOGIC database was generally robust enough for a fairly accurate application of the inclusion and exclusion criteria of ITCH. The definition of ALGS was well established, and the collaborative development of ITCH by ChiLDReN and Lumena/Shire/Takeda/Mirum through a Cooperative Research and Development Agreement enhanced the concordance of the ALGS definition. The ITCH protocol specified a number of parameters that were collected as part of the operation of the clinical trial. The LOGIC protocol specified collection of clinical information that was obtained primarily as part of clinical care. This resulted in some discrepancies, the most significant of which is the baseline criteria for pruritus, which is addressed subsequently. Enrollment in ITCH necessitated an assessment by the local investigator that the potential participant would be able to complete the 13‐week trial. This was difficult to apply to the LOGIC database, and listing for liver transplant was used as a surrogate for advanced liver disease that might preclude completion of the clinical trial. The other factor that could not be accurately assessed within LOGIC was the severity of cardiac disease, which can be significant in ALGS.[ 19 ] A number of historical clinical issues were queried in ITCH, which were not recorded in the LOGIC database, including as examples, chronic diarrhea requiring intravenous fluids, history of encephalopathy, and concerns for nonadherence. Serum bile acid levels and fat‐soluble vitamin sufficiency were used in ITCH entry criteria, but were sporadically available in the clinical care of LOGIC participants.
As noted previously, one of the key issues in the application of the ITCH entry criteria to the LOGIC cohort was the assessment of pruritus severity. Quantitative assessment of pruritus remains an elusive goal in hepatology.[ 18 ] In the ITCH trial, a novel tool, ItchRO, was developed to quantify caregiver and/or patient‐reported features of pruritus that were most relevant as determined by qualitative interviews and formal instrument development.[ 16 ] Key questions in ItchRO relate to observed scratching, skin manifestations of scratching, and sleep disturbance. This instrument is not in use in routine clinical care and was not available within LOGIC. ItchRO was used not only as an entry criterion but as a major endpoint of the randomized placebo‐ controlled trial of maralixibat.[ 11 ] The entry criteria for ITCH relied on twice daily assessment of pruritus, as measured by ItchRO for 2 weeks. The CSS, a very different assessment of pruritus that is used in clinical care of children with cholestasis, was collected in LOGIC.[ 17 ] This assessment of pruritus relies on clinical observation of scratching behavior and/or skin manifestations of scratching during a clinical encounter. CSS was collected in ITCH. Clearly these are different assessments of pruritus, and it is not entirely unexpected that the two measures do not correlate with each other.[ 18 ] In ITCH, both ItchRO and CSS were reduced in participants who received maralixibat compared with those who received placebo. As such, CSS was felt to be a reasonable, although not equivalent, parameter for this analysis.
The comparator cohort from LOGIC had many essential similarities at baseline compared with the ITCH cohort. A major and critical difference, however, was the age distribution, with a very disproportionate number of young participants in the LOGIC cohort. This was felt to be a crucial difference that could impact on outcomes. Very young children with ALGS and cholestasis are more predisposed to worse outcomes and a need for early transplantation, particularly for intractable pruritus.[ 20 ] In light of this potential confounder, a weighting method was used to match age distribution at enrollment between the ITCH and LOGIC cohorts. The decision to use this weighting method was arrived at before any outcome measures were assessed. The weighted ALGS NH cohort was very similar to the ITCH cohort.
Notable differences, however, were serum bile acid and albumin levels along with PedsQL. Although statistically significant, the clinical significance of the differences in bile acids and albumin are unclear. Bile acids are not routinely measured in the clinical practice of many, and as such, only 14 LOGIC participants had this important parameter assessed. Serum albumin was lower in the weighted cohort, which could be interpreted as being consistent with more severe disease, although the values do not really support the conclusion. The findings in PedsQL are potentially important, although this important assessment was only conducted in 21 of the participants in the weighted cohort. PedsQL was the parental total score and was significantly lower in the ITCH cohort. Parental perception of poor quality of life may have been an incentive to enroll in ITCH, and as such may have led to the observed discrepancy.
The data derived from this kind of analysis can be quite useful to put the results of clinical trials into context, but should not be used for direct comparisons with distinct clinical trials. The outcomes focused on in this investigation were ones that were commonly assessed in routine clinical practice and ones that were amenable to an empiric a priori assignation of clinical relevance. One of the major findings was that pruritus—as measured by CSS—improved significantly in the 2 years of clinical follow‐up in LOGIC. There was not an easily identifiable change in clinical practice that would explain this change. It has been surmised by many that pruritus improves over time in ALGS. One could also hypothesize a potential bias of clinicians to support the value of their clinical practice by identifying an improvement in one of the major manifestations of the disease that they are treating in their patients. As CSS is clinician‐scored, this potential bias could have affected the change over time in this parameter. An additional important parameter that changed over time was platelet count, which fell significantly after 2 years of follow‐up. This is not unexpected, as progressive fibrosis and portal hypertension in cholestatic liver disease could lead to increased hypersplenism and a commensurate reduction in platelet count. While statistically significant, the increase in height z score in this study is of uncertain clinical significance. The contextual meaning of these findings may be apparent from review of experiences with the use of maralixibat over the long term in ALGS. In the combined experiences of several recent trials (IMAGO [NCT 01903460]/ITCH [NCT 02057692]/IMAGINE [NCT 02047318]/IMAGINE II [NCT 02117713] and the ICONIC trial [NCT 02160782]), there was also the observation of a significant improvement of pruritus over 1 to 2 years of treatment with maralixibat.[ 21 ] Platelet count was also observed to decrease in these clinical trials. One notable difference was the increase in ALT observed in these clinical trials, which was not seen in this natural history cohort.
Ultimately, one strives to identify approaches that will alter the natural history of ALGS and other rare diseases. Often this requires the assessment of endpoints that consist of sentinel events: in the case of liver disease markers of hepatic decompensation, the perceived need for liver transplant, or death. This analysis provides critical information about progression to these important endpoints during clinical practice that avails currently available therapies. In this cohort, the development of sentinel events was relatively infrequent. Interestingly there were no episodes of variceal hemorrhage, and only 3 individuals developed ascites. Many more underwent liver transplantation, which suggests that it may have been for intractable and incapacitating pruritus. Survival with native liver at 2 and 4 years was 91% and 87% in the weighted cohort, respectively. An intervention would need to be extremely efficacious to yield a significant improvement in this survival in the setting of the relatively small numbers of individuals affected by this rare disorder who might be eligible for a conventional randomized clinical trial. It is noteworthy that the FDA approval of odexivibat and maralixibat were for impact on pruritus, not changes in long‐term disease progression.
In summary, the current studies have shown that prospective longitudinal database registries can be used to provide important and unique contextual information regarding interventional clinical trials in rare diseases, especially in those that are single‐arm studies or lack a control population. Application of inclusion and exclusion criteria to a robust registry provides information that is potentially more precise and relevant than broadly constructed real‐world natural history data. The findings from this kind of real‐world evidence are critically important, given the difficulties of conducting long‐term placebo controlled trials in orphan diseases. There are potential limitations on application of entry, inclusion, and exclusion criteria related to the scope of data collected in routine clinical practice relative to a formal clinical trial. With adequate numbers of participants in a registry, relevant matching to important clinical features is feasible. Caution should be exercised in the comparisons of the data derived from these observational databases with interventional trials. Formal statistical comparisons are not warranted. Given the potential high value of this information and the scope and cost of the prospective longitudinal databases, new approaches to funding these natural history studies should be considered, including use of funds from the prescription drug user fee act for prospective studies of rare diseases relevant to new advances from the pharmaceutical industry.
CONFLICT OF INTEREST
SK consults for Albeireo, Mirum, and Intercept. SH received grants from Mirum and consults for Albireo. KL received grants from and consults for Albireo and Mirum. PR received grants from and consults for Albireo, Gilead, Taverse, and Mirum. He received grants from Abbvie, Arrowhead, and Merck. He also consults for Ambys, Audentes, BioMarin, Dicerna, Enconded, Takeda/Vertex, and MediCell. BK consults for and received grants from Mirum and Albireo. She also consults for Audentes. RS consults for Albireo and Mirum.
Supporting information
Table S1–S5
ACKNOWLEDGMENT
Shauna Leighton, Medical Editor with Arbor Research Collaborative for Health, provided editorial assistance on this manuscript.
Shneider BL, Kamath BM, Magee JC, Goodrich NP, Loomes KM, Ye W, for the Childhood Liver Disease Research Network (ChiLDReN) . Use of funded multicenter prospective longitudinal databases to inform clinical trials in rare diseases—Examination of cholestatic liver disease in Alagille syndrome. Hepatol Commun. 2022;6:1910–1921. 10.1002/hep4.1970
Funding informationAnn & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL (supported by National Institute of Diabetes and Digestive and Kidney Diseases [NIDDK] DK62436 and National Center for Advancing Translational Sciences [NCATS] UL1TR001422): Estella Alonso, MD, Lee Bass, MD, Susan Kelly, RN, BSN, Mary Riordan, CCRP, Hector Melin‐Aldana, MD. Cincinnati Children’s Hospital Medical Center, Cincinnati, OH (supported by NIDDK DK62497 and NCATS UL1TR000077): Jorge Bezerra, MD, Kevin Bove, MD, James Heubi, MD, Alexander Miethke, MD, Greg Tiao, MD, Julie Denlinger, BSN, RN, Erin Chapman. Children’s Hospital Colorado, Aurora, CO (supported by NIDDK DK62453 and NCATS UL1TR002535): Ronald Sokol, MD, Amy Feldman, MD, Cara Mack, MD, Michael Narkewicz, MD, Frederick Suchy, MD, Shikha Sundaram, MD, Johan Van Hove, MD, Benigno Garcia, Mikaela Kauma, Kendra Kocher, CCRP, Matthew Steinbeiss, MS, CCRP, Mark Lovell, MD. The Children’s Hospital of Philadelphia, Philadelphia, PA (supported by NIDDK DK62481): Kathleen Loomes, MD, David Piccoli, MD, Elizabeth Rand, MD, Pierre Russo, MD, Nancy Spinner, PhD, Jessi Erlichman, MPH, Samantha Stalford, MPH, Dina Pakstis, Sakya King. Children’s Hospital of Pittsburgh, Pittsburgh, PA (supported by NIDDK DK62466 and NCATS UL1TR000005): Robert Squires, MD, Rakesh Sindhi, MD, Veena Venkat, MD, Kathy Bukauskas, RN, CCRC, Patrick McKiernan, MD, Lori Haberstroh, James Squires, MD, MS. UCSF Children’s Hospital, San Francisco, CA (supported by NIDDK DK62500 and NCATS UL1TR000004): Philip Rosenthal, MD, Laura Bull, PhD, Joanna Curry, Camille Langlois, Grace Kim, MD. Saint Louis University School of Medicine, St. Louis, MO (supported by NIDDK DK62453): Jeffery Teckman, MD, Vikki Kociela, BSN, CCRC, Rosemary Nagy, RDN, MBA, Shraddha Patel, PhD, Jacqueline Cerkoski, BSN. Riley Hospital for Children, Indiana University School of Medicine, Indianapolis, IN (supported by NIDDK DK84536 and NCATS UL1TR001108): Jean P. Molleston, MD, Molly Bozic, MD, Girish Subbarao, MD, Ann Klipsch, RN, Cindy Sawyers, BSRT, Oscar Cummings, MD. Seattle Children’s Hospital, Seattle WA (supported by NIDDK DK84575 and NCATS UL1TR000423): Simon Horslen, MB, ChB, FRCPCH, Karen Murray, MD, Evelyn Hsu, MD, Kara Cooper, CCRC, Melissa Young, CCRC, Laura Finn, MD. The Hospital for Sick Children, Toronto, Ontario, Canada (supported by NIDDK DK103135): Binita Kamath, MD, Vicky Ng, MD, Claudia Quammie, CCRP, Juan Putra, MD, Deepika Sharma, MSc, Aishwarya Parmar, BSc. University of Utah, Salt Lake City, UT (supported by NIDDK DK103140): Stephen Guthery, MD, Kyle Jensen, MD, Ann Rutherford, Amy Lowichik, MD, PhD, Linda Book, MD, Rebecka Meyers, MD, Tyler Hall. Children’s Hospital Los Angeles, Los Angeles, CA (supported by NIDDK DK84538 and NCATS UL1TR000130): Kasper Wang, MD, Sonia Michail, MD, Danny Thomas, MD, Catherine Goodhue, CPNP, Rohit Kohli, MBBS, MS, Larry Wang, MD, PhD, Nisreen Soufi, MD, Daniel Thomas, MD. Children’s Healthcare of Atlanta, Atlanta, GA (supported by NIDDK DK062470 and NCATS UL1TR000454): Saul Karpen, MD, PhD, Nitika Gupta, MD, DCH, DNB, MRCPH, Rene Romero, Jr., MD, Miriam B. Vos, MD, MSPH, Rita Tory, MS, CCRP, John‐Paul Berauer, MD, Carlos Abramowsky, MD, Jeanette McFall, MPH. Texas Children’s Hospital, Houston, TX (supported by NIDDK DK103149): Benjamin Shneider, MD, Sanjiv Harpavat, MD, Paula Hertel, MD, Daniel Leung, MD, Mary Tessier, MD, Deborah Schady, MD, Laurel Cavallo, Diego Olvera, Christina Banks, Cynthia Tsai. King’s College Hospital, London, UK: Richard Thompson, BM, BCh, MRCP, MRCPCH. National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD: Edward Doo, MD, Jay Hoofnagle, MD, Averell Sherker, MD, FRCP, Rebecca Torrance, RN, MSN, Sherry Hall, MS. Scientific Data Coordinating Center, Ann Arbor, MI (supported by NIDDK DK62456): John Magee, MD, Robert Merion, MD, FACS, Cathie Spino, DSc, Wen Ye, PhD.
REFERENCES
- 1. Miller KL, Mueller C, Liu G, Miller Needleman KI, Maynard J. FDA orphan products clinical trial grants: assessment of outcomes and impact on rare disease product development. Orphanet J Rare Dis. 2020;15:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shah KK, Kogut S, Slitt A. Challenges in evaluating safety and efficacy in drug development for rare diseases: a review for pharmacists. J Pharm Pract. 2021;34:472–79. [DOI] [PubMed] [Google Scholar]
- 3. Stockler‐Ipsiroglu S, Potter BK, Yuskiv N, Tingley K, Patterson M, van Karnebeek C. Developments in evidence creation for treatments of inborn errors of metabolism. J Inherit Metab Dis. 2021;44:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mitchell E, Gilbert M, Loomes KM. Alagille syndrome. Clin Liver Dis. 2018;22:625–41. [DOI] [PubMed] [Google Scholar]
- 5. Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N. Systematic review: the epidemiology, natural history, and burden of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2018;67:148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang KS, Tiao G, Bass LM, Hertel PM, Mogul D, Kerkar N, et al. Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology. 2017;65:1645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neimark E, Shneider B. Novel surgical and pharmacological approaches to chronic cholestasis in children: partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2003;36:296–7. [DOI] [PubMed] [Google Scholar]
- 8. Kamath BM, Stein P, Houwen RHJ, Verkade HJ. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020;40:1812–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kamath BM, Ye W, Goodrich NP, Loomes KM, Romero R, Heubi JE, et al. Outcomes of childhood cholestasis in Alagille syndrome: results of a multicenter observational study. Hepatol Commun. 2020;4:387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karpen SJ, Kelly D, Mack C, Stein P. Ileal bile acid transporter inhibition as an anticholestatic therapeutic target in biliary atresia and other cholestatic disorders. Hepatol Int. 2020;14:677–89. [DOI] [PubMed] [Google Scholar]
- 11. Shneider BL, Spino C, Kamath BM, Magee JC, Bass LM, Setchell KD, et al. Placebo‐controlled randomized trial of an intestinal bile salt transport inhibitor for pruritus in Alagille syndrome. Hepatol Commun. 2018;2:1184–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gonzales E, Hardikar W, Stormon M. ICONIC. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomized phase 2 study. Lancet. 2021;398:1581–92. [DOI] [PubMed] [Google Scholar]
- 13. van Laarhoven AI, van der Sman‐Mauriks IM, Donders AR, Pronk MC, van de Kerkhof PC, Evers AW. Placebo effects on itch: a meta‐analysis of clinical trials of patients with dermatological conditions. J Invest Dermatol. 2015;135:1234–43. [DOI] [PubMed] [Google Scholar]
- 14. Schurman B. The framework for FDA’s Real World Evidence Program. Appl Clin Trials. 2019;28:15–7. [Google Scholar]
- 15. Sherman RE, Anderson SA, Dal Pan GJ, Gray GW, Gross T, Hunter NL, et al. Real‐world evidence—what is it and what can it tell us? N Engl J Med. 2016;375:2293–7. [DOI] [PubMed] [Google Scholar]
- 16. Kamath BM, Abetz‐Webb L, Kennedy C, Hepburn B, Gauthier M, Johnson N, et al. Development of a novel tool to assess the impact of itching in pediatric cholestasis. Patient. 2018;11:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology. 1988;95:130–6. [DOI] [PubMed] [Google Scholar]
- 18. Kamath BM, Spino C, McLain R, Magee JC, Fredericks EM, Setchell KD, et al. Unraveling the relationship between itching, scratch scales, and biomarkers in children with Alagille syndrome. Hepatol Commun. 2020;4:1012–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kohut TJ, Gilbert MA, Loomes KM. Alagille syndrome: a focused review on clinical features, genetics, and treatment. Semin Liver Dis. 2021;41:525–37. [DOI] [PubMed] [Google Scholar]
- 20. Kamath BM, Yin W, Miller H, Anand R, Rand EB, Alonso E, et al. Outcomes of liver transplantation in alagille syndrome: the split experience. Liver Transpl. 2012;18:940–8. [DOI] [PubMed] [Google Scholar]
- 21. Shneider BL, Spino C, Kamath BM, Magee JC, Sokol RJ, et al. Impact of long‐term Administration of Maralixibat on children with cholestasis secondary to Alagille syndrome. Hepatol Commun. 2022. [Submitted]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1–S5