

Abstract
Beckwith‐Wiedemann Syndrome (BWS) is the most common human overgrowth disorder caused by structural and epigenetic changes to chromosome 11p15. Patients with BWS are predisposed to developing hepatoblastoma (HB). To better understand the mechanism of HB oncogenesis in this cancer predisposition background, we performed the first multi‐dimensional study of HB samples collected from patients diagnosed with BWS. This multi‐omic investigation of seven BWS HB and five matched nontumor BWS liver samples from 7 unique patients included examination of whole exome sequences, messenger RNA/microRNA expression, and methylation levels to elucidate the genomic, transcriptomic, and epigenomic landscape of BWS‐associated HB. We compared the transcriptional profiles of the BWS samples, both HB and nontumor, to that of control livers. Genes differentially expressed across BWS tissues were identified as BWS HB predisposition factors; this gene group included cell cycle regulators, chromatin organizers, and WNT, mitogen‐activated protein kinase (MAPK), and phosphoinositide 3‐kinase (PI3K)/AKT members. We also compared transcriptional changes associated with non‐syndromic HB carrying BWS‐like 11p15 alterations compared to those without, as well as to BWS HB. Through this analysis, we identified factors specific to 11p15‐altered HB oncogenesis, termed the BWS oncogenesis network. We propose that 11p15 alterations drive HB oncogenesis by initially dysregulating cell‐cycle regulators and chromatin organizers, including histone deacetylase 1 (HDAC1), ATP‐dependent helicase X, and F‐Box and WD repeat domain containing 7. Furthermore, we found oncogenic factors such as dickkopf WNT signaling pathway inhibitor 1 and 4, WNT16, forkhead box O3 (FOXO3), and MAPK10 are differentially expressed in 11p15‐altered HB in both the BWS and non‐syndromic backgrounds. These genes warrant further investigation as diagnostic or therapeutic targets.
Beckwith‐Wiedemann Syndrome (BWS) is one of the most common human overgrowth and hepatoblastoma (HB) predisposition disorders. Using a mutli‐omic approach, we identified BWS HB predisposition factors by their differential expression in BWS HB and non‐tumor tissue compared to control, as well as BWS oncogenesis factors by their differential expression in BWS HB compared to non‐syndromic or sporadic tumors. We propose that BWS alterations drive HB oncogenesis by initially dysregulating cell cycle regulators and chromatin organizers, then contribute to the dysregulation of known cancer drivers in the WNT, MAPK, and PI3K/AKT pathways.

INTRODUCTION
Hepatoblastoma (HB) is a rare embryonal tumor comprising 1% of pediatric cancer cases[ 1 ]; it is also the most common malignant liver tumor diagnosed in children under 5 years of age.[ 2 ] The most studied driver gene of HB is the WNT pathway member ß‐catenin (CTNNB1); over 60% of HB carry mutations in CTNNB1, specifically in the exon 3 region.[ 3 ] These sequence alterations stabilize the ß‐catenin protein in the nucleus, where it acts as a transcription factor; its targets include the proto‐oncogene MYC, which has been implicated in liver neoplasm malignancy.[ 4 ]
Another locus that contributes to HB development is human chromosome 11p15. Nearly one‐third of all HB samples investigated for structural genomic alterations demonstrated loss of heterozygosity (LOH) at or including 11p15.[ 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 ] Often, these 11p15 LOH are categorized as copy‐neutral and result from the duplication of DNA content from one parental allele with loss of commensurate DNA content from the other allele, or uniparental disomy (UPD).[ 5 , 6 , 8 , 10 , 11 , 12 ] In several studies, parental DNA was available and contained informative variants, allowing researchers to conclude that the maternal allele was lost and the paternal allele was duplicated, thereby resulting in paternal UPD of 11p15 (pUPD11).[ 5 , 10 ] One study also noted an increased risk of HB relapse in patients whose tumors carry this 11p15 LOH.[ 7 ]
Genetic and epigenetic alterations affecting the 11p15 region also cause one of the most common HB predisposition syndromes, Beckwith‐Wiedemann Syndrome (BWS; OMIM 130650). BWS can also be classified as a genomic imprinting disorder, which is caused by disruption of monoallelic parent‐specific gene expression. Within the 11p15 region, there are two independent domains subject to this parent‐specific gene expression: H19/IGF2:IG‐DMR is the imprinting control region (ICR) for Imprinting Center 1 (IC1), and KNCQ1OT1:TSS‐DMR is the ICR for Imprinting Center 2 (IC2) (Figure S1). Within IC1, gain of methylation (GOM) at the ICR causes an increase of the paternally expressed insulin‐like growth factor 2 (IGF2) and its regulatory microRNA (miRNA) miR‐483 as well as a decrease of the maternally expressed noncoding RNA H19 and its regulatory miRNA miR‐675 (Figure S1A). Within IC2, loss of methylation (LOM) at the ICR causes a decrease in the maternally expressed potassium voltage‐gated channel subfamily Q member 1 (KCNQ1) and cyclin‐dependent kinase inhibitor 1C (CDKN1C) as well as an increase in the noncoding antisense RNA KCNQ1OT1 (Figure S1B).
Combinations of these alterations in the 11p15 region cause a range of fetal and neonatal overgrowth characteristics including hepatomegaly and development of embryonal tumors.[ 13 ] BWS occurs in about 1 of 10,000 live births.[ 13 ] The relative risk of HB development in the BWS population was estimated at 2280‐fold compared with age‐matched controls.[ 14 ] Of the 29 HB tumors reported in previously published BWS cohort studies, 20 occurred in patients with pUPD11, five in patients with IC2 LOM, one in a patient with IC1 GOM, and three in patients with other subtypes or who were not molecularly diagnosed.[ 15 , 16 , 17 , 18 , 19 , 20 , 21 ] Due to this increased cancer risk, all patients with BWS are screened by liver ultrasound and serum α‐fetoprotein (AFP) every 3 months from BWS diagnosis until four years of age.[ 22 ]
BWS HB neoplasms may be different from those that arise sporadically in the nonsyndromic population. A meta‐analysis of BWS HB cohorts observed that no individuals developed HB after 30 months of age.[ 23 ] The estimated median age of HB diagnosis is 6 months of age in the BWS population compared with 16 months in the nonsyndromic population.[ 23 , 24 ] Additionally, the genetics driving BWS HB may differ from that of nonsyndromic HB. A previous study of two HB investigated the prevalence of CTNNB1 nuclear localization in BWS HB; CTNNB1 was observed in the nucleus of both BWS tumors, but not normal liver tissue.[ 25 ] Additionally, a CTNNB1 exon 3 mutation common to sporadic HB was only identified in one of the tumors.[ 25 ] These limited data in BWS HB suggest differential HB oncogenic drivers in this patient population.
Aside from this one study focused on two BWS HB tumors,[ 25 ] studies have not investigated the molecular nature of HB in this syndromic background. A recent paper by Nagae et al. noted that their HB cohort included five tumors derived from patients with BWS but did not analyze these samples independently.[ 26 ] Other cohorts reporting on the molecular nature of HB do not note whether any samples were collected from patients with BWS.[ 8 , 27 , 28 , 29 , 30 , 31 ] In this study, we analyzed HB and matched nontumor liver samples from patients with congenital BWS. We use a multi‐omics approach to better understand the molecular contributors to BWS HB predisposition. In addition, we compared our BWS tumor data with that of a nonsyndromic HB cohort to identify BWS HB oncogenesis factors. We believe these genes represent potential targets for future therapeutic development and may inform future personalized cancer screening approaches in cancer predisposition syndromes like BWS.
METHODS
Patients and samples
Samples and clinical information were collected through the BWS Registry, which is a previously established institutional review board protocol (IRB 13–010658) at the Children’s Hospital of Philadelphia (CHOP). Briefly, consent was obtained from all patients and/or legal guardians to collect longitudinal clinical information and samples that became available through clinical care.
During surgical procedures for HB management, liver samples were collected from CHOP patients, then snap‐frozen in liquid nitrogen and stored at −80°C. Control liver samples were collected by CHOP Pathology and Laboratory Medicine and were derived from autopsy specimens with appropriate consents provided. Basic and limited clinical information about these samples was provided through an honest‐broker based on their protocol. These samples were selected from individuals who did not have clinical or molecular BWS, nor was the cause of death the result of liver‐associated complications. We analyzed all BWS HB tumor samples available to us through this methodology, which was seven primary HBs collected from 7 patients affected by molecularly confirmed BWS. Five had matched nontumor liver samples available (Table 1).
TABLE 1.
Clinical features of HB cases
| Sex | BWS subtype in blood | BWS subtype in tumor | BWS subtype in NT | Hepato‐megaly | Age at tumor diagnosis | Months off therapy | Tumor histology | Genomic analysis b | Transcriptome analysis | |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 (HB1) | M | pUPD11 | pUPD11 | N/A | No | 7 months | 168 | Fetal | E, M | mRNA, miRNA |
| Patient 2 (HB2/NT2) | M | IC2 LOM | Aberrant IC1/IC2 methylation | IC2 LOM | No | 1 month | 67 | Embryonal with scattered mesenchymal foci | HB: E, M | HB: mRNA, miRNA |
| NT: E, M | NT: N/A | |||||||||
| Patient 3 (HB3/NT3) | F | pUPD11 | IC1 GOM (by pUPD) | IC1 GOM (by pUPD) | Yes | 1 month | 44 | Mixed fetal and embryonal | HB: E, M | HB: mRNA, miRNA |
| NT: E, M | NT: mRNA | |||||||||
| Patient4 (HB4) | F | pUPD11 | pUPD11 with normal IC2 | N/A | No | 4 months | 182 | Mixed fetal and embryonal | E, M | mRNA, miRNA |
| Patient5 (HB5/NT5) | F | pUPD11 | pUPD11 | pUPD11 | No | 4 months | 27 | Mixed fetal and embryonal | HB: E, M | HB: mRNA |
| NT: E, M | NT: mRNA | |||||||||
| Patient6 (HB6/NT6) | F | IC2 LOM | Aberrant IC1/IC2 methylation | No 11p15 change | Yes | 4 months | 20 | Mixed fetal and embryonal | HB: E, M | HB: mRNA |
| NT: E, M | NT: mRNA | |||||||||
| Patient7 (HB7/NT7) | M | pUPD11 | pUPD11 with normal IC1 | pUPD11 | Yes | 13 months | 21 | Mixed fetal and embryonal | HB: E, M | HB: mRNA |
| NT: E, M | NT: N/A | |||||||||
| Control1 | F | N/A | N/A | N/A | No | 27 weeks a | N/A | N/A | N/A | mRNA |
| Control2 | F | N/A | N/A | N/A | No | 20 days a | N/A | N/A | E, M | mRNA, miRNA |
| Control3 | M | N/A | N/A | N/A | No | 10 days a | N/A | N/A | E | mRNA, miRNA |
| Control4 | M | N/A | N/A | N/A | No | 3 months a | N/A | N/A | E, M | mRNA |
| Control5 | F | N/A | N/A | N/A | No | 4 weeks a | N/A | N/A | E, M | N/A |
Abbreviations: F, female; LOM, loss of methylation; M, male; miRNA, microRNA; N/A not applicable; pUPD11, paternal UPD (uniparental disomy) of 11p15.
Age at which control tissue was collected.
“E” denotes whole exome sequencing; “M” denotes methylation array.
Whole genome sequencing and methylation array
Genomic DNA was isolated using the AllPrep DNA/RNA Micro Kit (QIAGEN). Whole genome sequencing (WES) libraries were prepared by the CHOP Center for Applied Genomics (CAG) using the Twist Bioscience Human Core Exome kit with 50 ng of input DNA on controls 1, 2, and 3, as well as all BWS HB and matched nontumor samples presented in this study. DNA from controls 2, 4, and 5 was of suitable quality and concentration for methylation analysis alongside all BWS HB and matched nontumor samples presented in this study (Table 1). The DNA was bisulfite‐converted using the EZ DNA Methylation Kit (Zymo Research) for methylation analysis. The CAG performed Infinium MethylationEPIC array (Illumina) runs.
RNA sequencing
Total RNA was isolated from frozen tumor samples using the AllPrep DNA/RNA Micro Kit (QIAGEN). Large and small RNA fractions were isolated using the Monarch RNA Cleanup kit (NEB). Standard and small RNA‐sequencing (RNA‐seq) was performed by GENEWIZ using the Tru‐Seq RNA Library Prep kit (Illumina) including polyA selection and Tru‐Seq Small RNA Library Prep kit (Illumina), respectively. Large RNA was available from all BWS HB samples as well as BWS nontumor samples 3, 5, and 6. Small RNA at concentrations and qualities suitable for sequencing could only be isolated from BWS HB samples and controls 2 and 3; other controls and BWS nontumor samples are not included in this analysis (Table 1).
Additional details of library preparation and analysis are provided in the Supporting Data and have been deposited in the Database of Genotypes and Phenotypes (dbGAP) of the National Center for Biotechnology Information (United States National Library of Medicine, Bethesda, MD) under accession number phs002614.v1.p1. Nonsyndromic data were retrieved from Gene Expression Omnibus GSE132219 accession.
RESULTS
Overview of the BWS HB cohort
Although HB affects more male than female children,[ 32 ] our cohort represents 43% males and 57% females (Table 1). Hepatomegaly, or enlargement of the liver, is a suggestive feature of BWS[ 13 ] and was previously observed in one‐third of patients with BWS and HB.[ 24 ] In our cohort, 3 of 7 (43%) patients with BWS and HB were also noted to have hepatomegaly (Table 1). The median age of HB diagnosis for these children was 4 months of age (Table 1), in line with the previously reported early development of HB in the BWS population compared with that of the general population.[ 23 ] All patients had their HB detected through increased AFP and/or a liver mass detected as part of the routine BWS tumor surveillance program.[ 22 ] Most of the tumors presented with a mixed fetal and embryonal histologic subtype (Table 1). In the present study, all patients who were affected by BWS HB were alive at last follow‐up, with survival measured between 20 months to 15 years off therapy (Table 1).
Imprinting of 11p15 in BWS HB
All patients were diagnosed with BWS due to IC2 LOM or pUPD11 in blood before HB development (Table 1). Initially, we wanted to confirm these BWS subtypes within the liver samples. We determined 11p15 methylation status based on the results of the methylation array and determined whether UPD was likely present in the sample cell population based on whether single nucleotide polymorphisms (SNPs) were observed in an even proportion of WES reads.
Surprisingly, several liver samples indicated a different BWS subtype compared with that initially identified in blood. For patients 2 and 6, IC2 LOM was indicated in blood, whereas HB presented as IC1 GOM and IC2 LOM without UPD (Table 1 and Figure S2A,B). For patient 3, pUPD11 was observed in blood, but the HB and nontumor samples presented with only IC1 GOM (Table 1 and Figure S2A). Interestingly, we confirmed that IC1 is likely affected by UPD, but not IC2 (Figure S2B), suggesting a shorter extent of UPD across 11p15 in liver samples compared with blood. Finally, for patients 4 and 7, pUPD11 was observed in blood; however, the respective methylation and SNP results in HB and nontumor conflictingly indicated that only IC1 or IC2 aberrant methylation was present along with UPD across the entirety of 11p15 (Table 1 and Figure S2A,B). As such, it is likely that these samples carry pUPD11 but may also have had subsequent methylation changes in a population of cells that transformed. Of note, the confidence interval for methylation at IC2 was wider than that of IC1, which suggests that application of the methylation array at this locus may be less reliable (Figure S2A). In summary, we classified HB1, HB4, HB5, and HB7 as pUPD11; HB3 as IC1 GOM; and HB2 and HB6 as aberrant IC1/IC2 methylation (Table 1 and Figure S2A,B).
We also confirmed that the changes to the 11p15 ICRs influenced their imprinted genes as expected through RNA‐seq analysis. Within IC1, H19 and miR‐675 were both down‐regulated in BWS samples compared with controls, as expected, in samples involving IC1 GOM (Figures S1A,B and S2C). IGF2 was up‐regulated in the BWS nontumor group, but IGF2 and miR‐483 were expressed at a range closer to that of the controls in BWS HB, possibly as result of transcriptome changes associated with the oncogenic process (Figure S2C). Within IC2, CDKN1C was down‐regulated in BWS HB and nontumor groups, as expected, in samples involving IC2 LOM (Figures S1A,C and S2C). The KCNQ1 and KCNQ1OT1 expression patterns were similar to those of H19 and IGF2, respectively (Figure S2C).
Genomic changes associated with BWS HB
Aside from the previously discussed CTNNB1 mutations and 11p15 alterations, several other somatic structural and smaller‐scale genomic alterations have been observed recurrently in HB. These include gains of chromosomal content from 1q, 2q, 6, 8, 12, 17, and 20; losses of 4q have also been noted.[ 3 , 6 , 12 , 29 ] To determine whether these changes work cooperatively with 11p15 in BWS HB development, we examined the WES data for copy number alterations (CNAs). We found that HB6 carried a gain of 1q21‐q44 encompassing nearly the entire arm (Figure 1A). Five other samples presented with 1p36.3 gains or losses, leading to its classification as a recurrent region of alteration (Figure 1A). A recurrent CNA was also identified at 2q35 in five BWS HB samples (Figure 1A). We noted additional recurrent gains and losses at 10q11.2, 12q21.31‐q23.1, and across chromosomes 15, 16, and 17 (Figure 1A).
FIGURE 1.
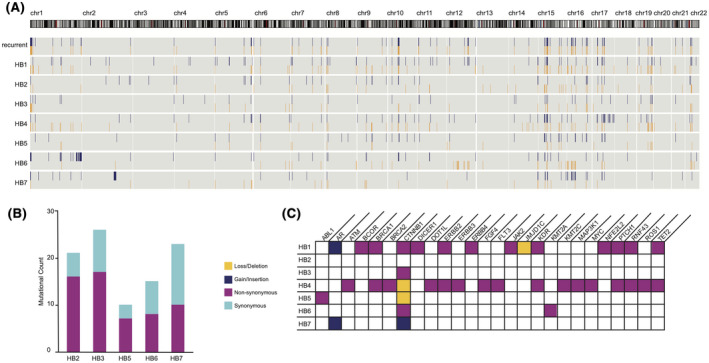
Genomic features of Beckwith‐Wiedemann Syndrome (BWS) hepatoblastoma (HB) indicate genome stability and involvement of few cancer drivers. (A) Copy number alterations (CNAs) identified in BWS HB samples with recurrent alterations identified in at least two samples. Gains are shown in blue; losses are shown in orange. (B) Tumor mutational burden (TMB) in HB samples with a matched nontumor (NT). Synonymous mutations are shown in light blue; non‐synonymous mutations are shown in green. (C) Mutations that map to known cancer‐driving genes. Insertions are shown in blue, deletions in orange, and non‐synonymous substitutions in green
In line with the previously reported HB CNA rate,[ 29 ] we identified just over 200 CNAs identified in tumors without a matched nontumor (HB1 and HB4) and just over 100 CNAs identified in the five HB normalized against their nontumor counterparts (Figure 1A). This result suggests that the BWS HB samples did not have a greater level of genome instability than previously studied sporadic HB by a CNA metric. To confirm, we calculated the tumor mutational burden (TMB). Overall, pediatric cancers are estimated to carry fewer than 25 nonsilent mutations per tumor, with HB carrying the lowest average number of mutations.[ 34 , 35 ] As evidenced in our CNA analysis, samples without matched nontumor samples have an overestimated mutational load. For that reason, HB1 and HB4 were not included in this aspect of the analysis. For the five samples with a matched nontumor sample, we found an average of 19 ± 6.4 coding mutations overall, with an average of 11.6 ± 4.6 non‐synonymous mutations (Figure 1B and Table S1).
While the CNA and TMB estimates indicate that BWS HB is similarly stable to nonsyndromic HB, we wanted to better understand how these mutations might contribute to oncogenesis. During the clinical evaluation of tumors, the Children’s Hospital of Philadelphia uses a gene panel to assess mutations in 238 genes known to contribute to solid tumor development (listed in the Supporting Information). We used these genes to interrogate our WES‐identified CNAs and SNPs to determine whether these common cancer drivers were altered in our BWS HB cohort. We observed a limited number of mutations overlapping with these known cancer‐causing genes in our BWS HB, with the greatest number of mutations identified in HB1 and HB4 likely due to their lack of matched nontumor (Figure 1C). CTNNB1 exon 3 was the only region of recurrent alteration identified in 6 of 7 (86%) samples (Figure 1C): Three of 7 (43%) were non‐synonymous substitutions; 1 of 7 (14%) was an in‐frame insertion; and 2 of 7 (29%) were in‐frame deletions.
BWS HB predisposition transcriptome network
We next investigated transcriptome changes associated with BWS HB. Specifically, we wanted to identify the factors that work alongside CTNNB1 in 11p15‐dysregulated HB. To achieve this, we performed Gene Set Enrichment Analysis (GSEA) in a pairwise manner. While few pathways reached statistical significance, several demonstrated stronger expression correlations based on groups (i.e., KEGG_CELL_CYCLE, SMAD_PROTEIN_SIGNAL_TRANSDUCTION, JNK_CASCADE, and MAPK_CASCADE in BWS HB compared with BWS nontumor as well as JNK_CASCADE and JAK_STAT_SIGNALING in BWS nontumor compared with controls) (Figure 2A and Table S2). As BWS is an imprinting disorder caused by aberrant DNA methylation and chromatin organization, we also tested gene sets related to these terms. Of these, CHROMATIN_REMODELING, HISTONE_METHYLATION, as well as genomic imprinting factors were enriched in BWS tissue (Figure 2A and Table S2).
FIGURE 2.
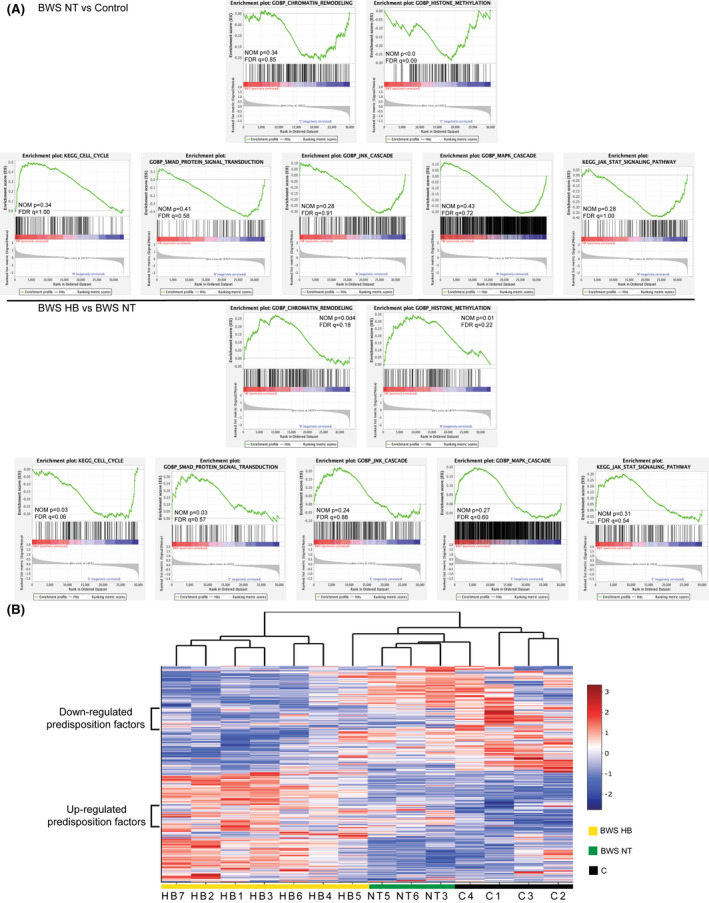
Transcriptome features of BWS liver and HB. (A,C) Preranked Gene Set Enrichment Analysis (GSEA) plots of enriched signaling pathways comparing expression of BWS HB with BWS NT liver (A) or of BWS NT with controls (C). False discovery rate (FDR) q value and nominal (NOM) p values are displayed. (B) Heatmap of genes differentially expressed between BWS HB, BWS NT, and controls identified through GSEA. Genes with similar expression levels between all BWS samples compared with controls are highlighted by the brackets on the left
To visualize differentially expressed genes from these signaling pathways, we developed a heatmap of the 640 genes with the strongest correlation (Figure 2B). Notably, a subset of 139 genes was similarly expressed between BWS HB and BWS nontumor, but up‐regulated or down‐regulated comparing BWS and controls (Figure 2B). We highlight these genes in brackets in Figure 2B and propose these genes as BWS HB predisposition factors, as their dysregulation is not restricted to the cancerous tissue.
To understand how these 139 genes work together to influence BWS HB predisposition, we generated a protein–protein interaction (PPI) network (Figure 3A, Figure S3A). The top 10% of interactions were made by the following 10 hub genes: up‐regulated MYC, tumor growth factor beta R2 (TGFBR2), histone deacetylase 1 (HDAC1), mitogen‐activated protein kinase (MAPK8), and interleukin‐2 (IL‐2), and down‐regulated HRAS, endothelial growth factor, IL‐6, SHC adaptor protein 1, and WNT1 (Figure 3A,B). There were an additional 18 BWS HB predisposition network genes significantly differentially expressed between the BWS HB/nontumor versus control samples (Figure S3B). Of these, eight function in chromatin organization and/or cell‐cycle regulation (growth factor independent 1B transcriptional repressor (GFI1B), RNA polymerase‐associated protein CTR9 homolog, ubiquitin specific peptidase 8, ATP‐dependent helicase X (ATRX), lysine methyltransferase 2E (KMT2E), RTF1, F‐Box and WD repeat domain containing 7 (FBXW7), and AT‐Rich interaction domain 4A (ARID4A)) and three are associated with MAPK and/or PI3K/AKT signaling (erythropoietin, coiled‐coil domain containing 88A, and histone deacetylase 3) (Figure S3). In fact, of the GSEA pathways initially enriched in our analysis, chromatin organization and cell‐cycle regulation were the two most represented processes in this network (Figure 3C and Figure S3A). Overall, 77% of the predisposition factors belong to these pathways and/or insulin response, WNT, MAPK, and PI3K/AKT signaling pathways, even though the overall WNT and PI3K/AKT signaling pathways were not statistically significant (Figure 3C and Figure S3A).
FIGURE 3.
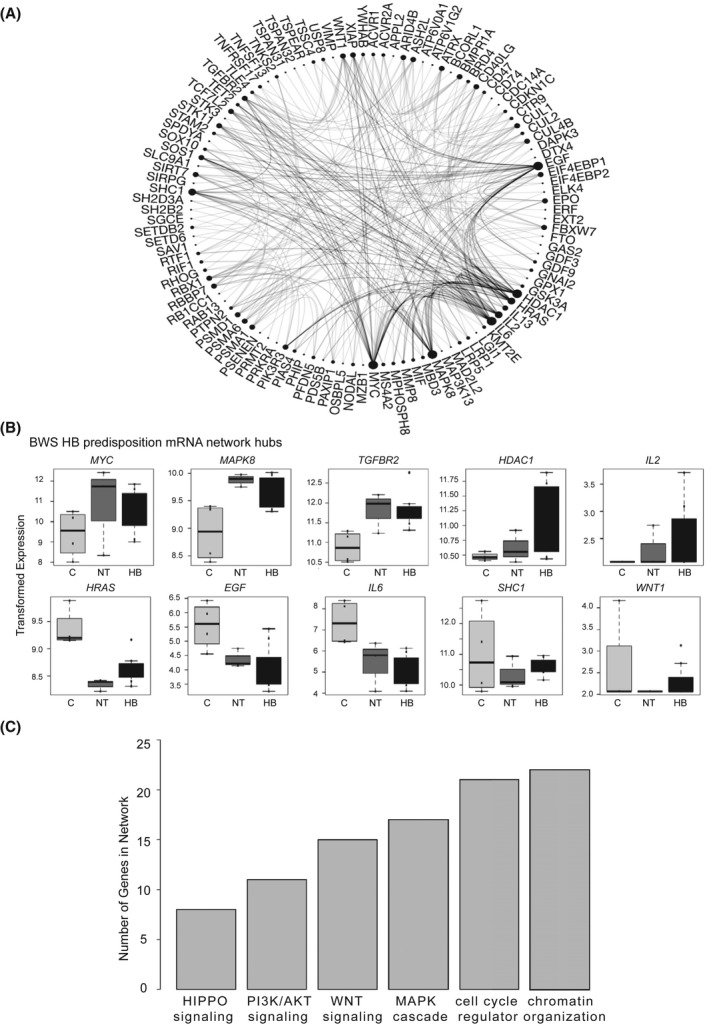
Features of expression factors comprising the BWS HB predisposition network. (A) Protein–protein interaction (PPI) of the 139 genes with similar expression between BWS HB and NT with differential expression in controls. Hub genes are indicated by the proportionally large number of interactions. (B) Expression of hub genes in control (C), BWS NT, and BWS HB samples. Variance stabilizing–transformed read counts detected by RNA‐sequencing (RNA‐seq) are shown. (C) Pathways represented in the predisposition PPI network based on the number of genes attributed to the biological process
BWS HB oncogenesis transcriptome network
Previously Carrillo‐Reixach et al. presented a multi‐omic analysis of sporadic HB.[ 28 ] We wanted to use these data to explore how BWS HB development might be similar or distinct from that of nonsyndromic HB. First, we needed to confirm that none of the samples were derived from patients with an 11p15 alteration indicative of congenital BWS. Comparing nontumor samples from this independent cohort with the controls generated in our cohort, we observed IC1 GOM in NT12, NT2, NT33, and NT7, but none of the samples exhibited robust IC2 LOM (Figure S4A). Although it is possible that these samples were derived from individuals with BWS, it is unlikely that all 4 of these patients had congenital BWS before HB development based on established (epi)genotype–phenotype correlations.
Previously, Carrillo‐Reixach et al. evaluated their cohort for 11p15 LOH; 41% of the HB exhibited this structural alteration including tumors T12, T2, T33, and T7.[ 28 ] We confirmed aberrant methylation in these samples as well as seven additional samples; Nineteen of 28 (68%) of the nonsyndromic tumors presented with BWS‐like 11p15 alterations (Figure S4B). The samples with normal 11p15 methylation and no pUPD11 were T4, T5, T6, T9, T14, T15, T16, T17, and T18 (Figure S4B). To directly compare the BWS HB samples with these nonsyndromic HB, we performed a batch correction as described in the Supporting Information. After correction, we performed GSEA to compare HB‐carrying 11p15 alterations, including those derived from patients with BWS, to HB with normal 11p15 methylation and structure. Here, KEGG_CELL_CYCLE, CHROMATIN_REMODELING, RAS_SIGNAL_TRANSDUCTION, MAPK_CASCADE, JNK_CASCADE, and JAK_STAT_SIGNALING were significantly differentially expressed between the groups (Figure 4A and Table S2).
FIGURE 4.
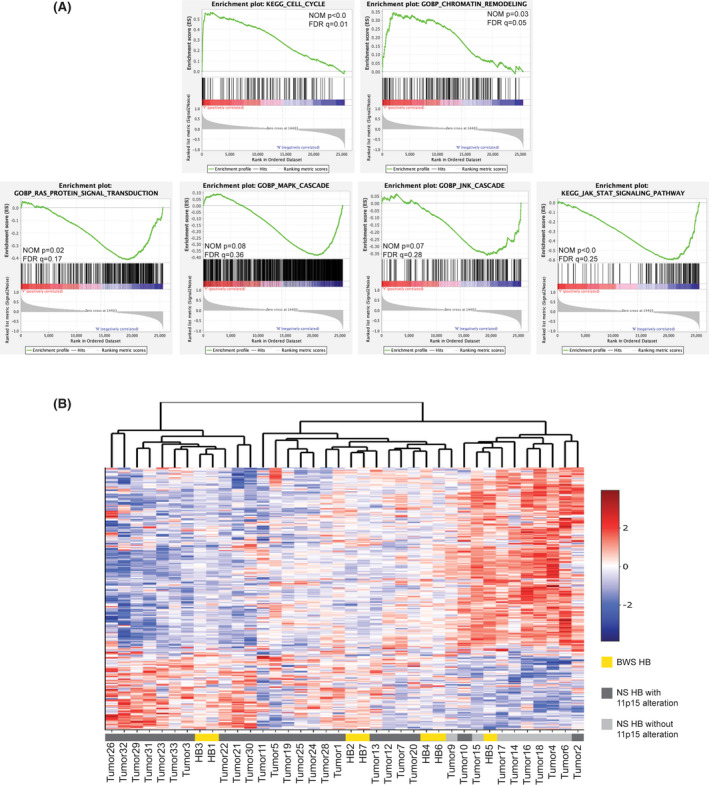
Transcriptome features of 11p15‐altered HB. (A) Preranked GSEA plots of enriched signaling pathways comparing expression of BWS HB and nonsyndromic (NS) HB with 11p15 alterations versus nonsyndromic HB without 11p15 changes. FDR q value and NOM p values are displayed. (B) Heatmap of genes differentially expressed between HB groups stratified by 11p15 status identified through GSEA
Again, we visualized the top genes with the strongest expression correlation. These 303 genes cluster the HB without 11p15 alterations together on the right, while most of the nonsyndromic HB carrying 11p15 changes cluster with the BWS HB on the left (Figure 4B). The BWS HB formed three pairs that were not based on 11p15 status (i.e., whether aberrant methylation or pUPD11) (Figure 4B, Figure S2A,B, and Table 1). As the BWS HB did not form a single, distinct cluster, they are likely transcriptionally similar to those arising in a nonsyndromic background with spontaneous 11p15 changes (Figure 4B).
Of the 303 differentially expressed genes, 54 genes overlap with the analysis presented in Figure 2A (Figure 5A). To better understand the relationship between these genes identified by multiple comparisons, we generated a BWS HB oncogenesis PPI network (Figure 5B and Figure S5A). Supporting the similarity between BWS and nonsyndromic HB in the broader expression profile (Figure 4B), we identified the following hub genes: 11p15‐down‐regulated janus kinase 2 (JAK2), forkhead box O3 (FOXO3), and proto‐oncogene KIT, 11p15‐up‐regulated proto‐oncogene SRC, and similarly expressed among all HB is cyclin dependent kinase inhibitor 2A (CDKN2A) (Figure 5B,C). There were an additional 10 genes significantly differentially expressed between HB stratified by 11p15 status (Figure S5B). While several cell‐cycle regulator and chromatin organization factors are represented in this network, a larger number of genes represent WNT, MAPK, PI3K/AKT, and JAK–signal transducer and activator of transcription (STAT) signaling factors (Figure 5C and Figure S5A).
FIGURE 5.
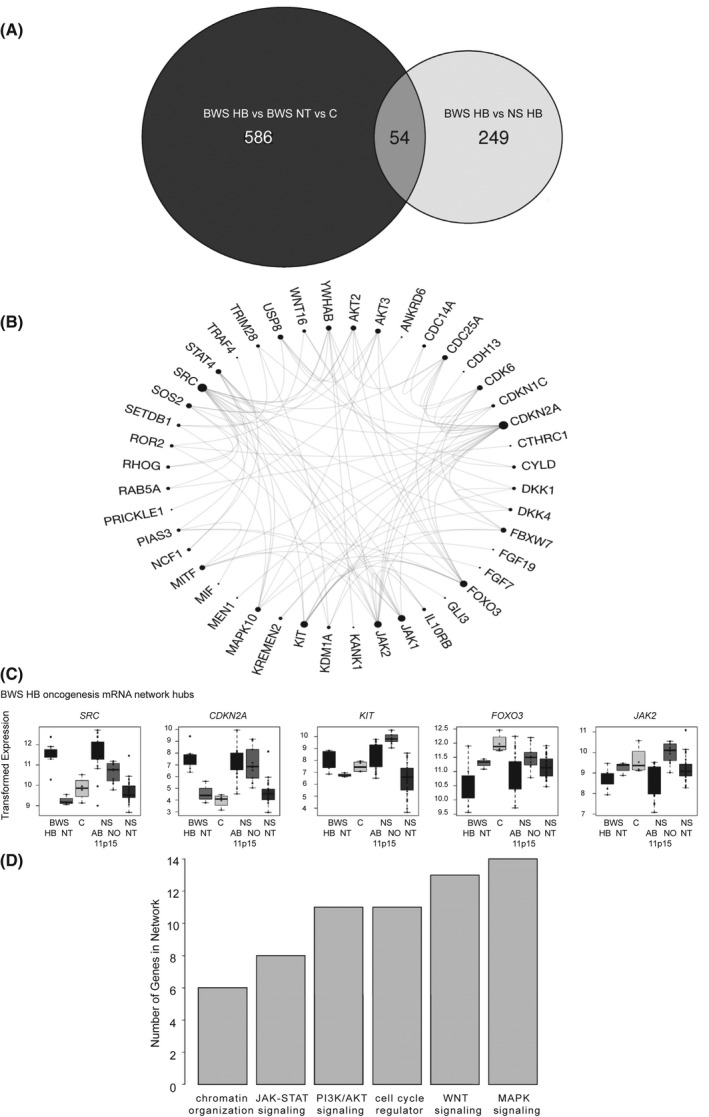
Features of expression factors comprising the BWS HB oncogenesis network. (A) Venn diagram comparing the genes identified in the pairwise comparison among BWS HB, BWS NT, and control (C) comparison (black), and in the BWS HB versus nonsyndromic (NS) HB comparison (white). (B) PPI of the 54 genes central to the Venn diagram. Hub genes are indicated by the proportionally large number of interactions. (C) Expression of hub genes in control, BWS NT and HB, and NS with normal (NO) or aberrant (AB) 11p15 methylation samples. Variance‐stabilizing transformed read counts detected by RNA‐seq are shown. (D) Pathways represented in the oncogenesis PPI network based on the number of genes attributed to the biological process. mRNA, messenger RNA
Although these results suggest how BWS HB oncogenesis might be distinct from HB without 11p15 imprint disruption, we also wanted to determine how similar the BWS HB transcriptome might be compared with other HB. To this end, we tested whether gene signatures proposed to stratify HB in previous publications were enriched in BWS HB (Figure S6A–E). Aside from the cell population and epigenetic machinery stratification proposed by Rivas et al.,[ 36 ] the signatures were enriched in BWS HB samples (Figure S6A–E and Table S2).
miRNA regulation of BWS HB networks
Previously, miRNA dysregulation has been shown to contribute to HB oncogenesis, such as through WNT signaling and MYC regulation.[ 37 ] As with the messenger RNA analysis, we wanted to determine whether previously proposed HB miRNA signatures were conserved in BWS HB. None were well‐correlated with BWS HB miRNA expression patterns (Figure S6F–H and Table S2). To elucidate miRNAs that might play a more prominent role in BWS HB, we identified those miRNAs predicted to bind BWS HB predisposition and oncogenesis factors.
For the BWS HB predisposition and oncogenesis networks, 89 and 169 predicted that regulatory miRNAs were differentially expressed, respectively. Several miRNAs were predicted to co‐regulate hub and significant factors to form three predisposition networks and four oncogenesis networks (Figure S7A,B). Of these miRNAs, 19 stem‐loop structures (miR‐22, miR‐30b, miR‐33b, miR‐92a‐1, miR‐100, miR‐139, miR‐203a, miR‐205, miR‐449b, miR‐450a‐2, miR‐490, miR‐579, miR‐676, miR‐3622a, and miR‐4728) were ranked with high annotation confidence by miRBase. miR‐30b‐3p and miR‐3622a‐5p were predicted components of both the predisposition and oncogenic networks. miR‐30b‐3p was up‐regulated and may contribute to the down‐regulation of 14 BWS HB predisposition factors as well as BWS HB oncogenesis hub genes JAK2 and FOXO3 (Figure S7A–C). miR‐3622a‐5p was down‐regulated, which may contribute to the up‐regulation of hub genes MAPK8, HDAC1, and CDKN2A (Figure S7A–C). Also of note, miR‐139 and miR‐490 were predicted to co‐regulate the previously mentioned dickkopf WNT signaling pathway inhibitor 1 and 4 (DKK1/4), WNT16, and SRC members of the BWS HB oncogenesis network (Figure S7B and Figure 5B).
Epigenetic regulation of BWS HB networks
Another mechanism of gene regulation that plays a role in HB development is DNA methylation. At a global level, the HB methylome exhibits hypomethylation compared with nontumor livers; more specifically, while CpG islands become hypermethylated, the rest of the genome loses methylation.[ 28 ] Global hypomethylation was observed in BWS HB compared with both BWS nontumor and controls, but was not statistically significant (Figure 6A). For further classification of the BWS HB methylome, we looked at regions designated as CpG islands, shores, shelves, and open sea. While significantly differentially methylated probes at CpG islands were hypermethylated between BWS HB and nontumor, hypomethylation of significantly differentially methylated probe mapping to shores, shelves, and open sea was observed (Figure 6B; Tables S3 and S4).
FIGURE 6.
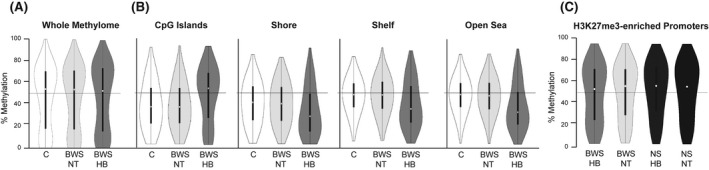
Methylome features of BWS HB. (A) Violin plot of the global methylation of BWS HB compared with NT and controls (one‐way analysis of variance: df = 2, F = 2.67, p value = 0.07). (B) Violin plots of the methylation levels at CpG islands, as well as the shores, shelves, and at open sea. (C) Violin plots of the methylation levels of genes known to have H3K27me3 at the promoter in BWS HB and NT compared with NS HB and NT. White circle represents the average methylation level
When we compared BWS HB methylation with that of nonsyndromic HB without 11p15 alterations, GSEA revealed that genes with histone H3 trimethylation of lysine 27 (H3K27me3) promoter‐enrichment also displayed statistically significant hypomethylation (Figure 6C and Table S5). At these probe sites, methylation levels in the matched nontumor samples from both BWS and nonsyndromic backgrounds were more similar to those of nonsyndromic HB (Figure 6C). However, these H3K27me3‐promoter genes did not match those identified in the BWS HB predisposition or oncogenesis networks (Table S5). In fact, only four probes with differential methylation mapped to genes from these networks, and each mapped to a different gene (Tables S3 and S4). As such, it is unlikely that changes to the BWS HB methylome, aside from those at 11p15, drive BWS HB.
DISCUSSION
It has been established that changes to human chromosome 11p15 are associated with HB development[ 5 , 6 , 7 , 8 , 10 , 11 , 12 ]; however, a mechanistic link to the oncogenic process has yet to be elucidated. In this study, we present a multi‐omic profile specific to HB occurring in the BWS background and the molecular changes associated with 11p15 alterations in liver oncogenesis. While our cohort is modestly sized compared with others describing the molecular nature of sporadic HB, this report represents an important step toward understanding the effect of 11p15 on growth regulation.
Data in this report point to BWS‐associated 11p15 alterations acting in conjunction with cell cycle and chromatin regulators as well as MYC and its targets to create an environment of liver overgrowth (Figures 2 and 3). Up‐regulation of MYC is a “hallmark” of general cancer development.[ 38 ] CTNNB1, a commonly mutated driver of HB, is known to act as a transcription factor to increase levels of MYC; however, the 11p15‐imprinted KCNQ1 has been observed to regulate both factors.[ 39 ] Decreased levels of KCNQ1 may therefore contribute to increased MYC in BWS liver before altered CTNNB1 activity (Figure S2C and Figure 3B). Fukuzawa et al. previously noted that nuclear localization of CTNNB1 was specific to BWS HB and was not observed in BWS nontumor samples.[ 25 ] As such, CTNNB1 was suggested to be a late‐acting factor, rather than a key cause of hepatomegaly and BWS HB predisposition.[ 25 ] Absent of DNA methylation changes, we propose that chromatin regulators, such as SET domain containing 6, protein lysine methyltransferase, HDAC1, GFI1B, KMT2E, and ATRX, along with cell‐cycle restrictors such as FBXW7, ARID4A, cullin 1/2/3/4b, and 11p15‐imprinted CDKN1C, function with MYC‐targeted transcription in normal, nontumor BWS liver to create the landscape for liver overgrowth and cancer development (Figure 3 and Figure S3B).
Our previous study of BWS hepatocytes differentiated from patient‐derived induced pluripotent stem cells indicated that 11p15 alterations and insulin signaling dysregulation were associated with differential expression of MYC and FOXO1.[ 40 ] Our current investigation of primary BWS HB demonstrated loss of expression of FOXO3 (Figure 5C). Generally, this family of transcription factors induces cell‐cycle arrest and, subsequently, apoptosis.[ 41 ] Conflictingly, both high and low levels of FOXO3 have been associated with liver oncogenesis; in contrast, MAPK10 is frequently down‐regulated in liver cancer and therefore thought to function as a tumor suppressor.[ 42 , 43 , 44 ] As MAPK10 is a predicted binding target of FOXO3 and was also down‐regulated in BWS HB tissue, we posit that the loss of these factors cooperatively restricts cell death in BWS HB development (Figure 5 and Figure S5B).
In addition to these BWS HB data, we present an investigation of sporadic HB stratified by 11p15 status. Previously, studies by Sumazin et al. and Sekiguchi et al. identified WNT‐antagonists DKK1/4 as part of their HB signatures.[ 30 , 31 ] However, we demonstrate that HB with 11p15 alterations largely drive this trend (Figure 5C and Figure S5B). We also observed that overexpression of the DKK1/4 membrane receptor kringle containing transmembrane protein 2, as well as WNT16, was specific to 11p15‐altered HB both in BWS and nonsyndromic backgrounds (Figures 4B and 5B; Figure S5B). It has previously been noted that the function of WNT antagonists is abrogated in HB carrying a CTNNB1 mutation.[ 45 ] As such, WNT16 may independently promote cell division.[ 46 ] DKK4 has also been shown to promote liver cell proliferation in a high glucose environment[ 47 ]; as such, early dysregulation of 11p15‐imprinted IGF2, as previously proposed by Honda et al.,[ 48 ] may contribute to this pro‐proliferative process (Figures S1 and S2C).
This study also provides evidence of the combination of genomic, transcriptomic, and epigenomic changes that distinguish BWS HB from that of sporadic HB. Regarding the genome, the TMB between BWS HB and other HB was similar, but BWS HB mutations did not recurrently include HB‐common genes axis inhibition protein 2, nuclear factor erythroid 2‐related factor 2, BCL6 corepressor like 1, or 8q gains[ 8 , 28 , 34 ] (Figure 1). Regarding the epigenome, significant differences in methylation have been observed in genes such as Ras association domain family member 1 and suppressor of cytokine signaling 1 (SOCS1), which are correlated with prognostic outcomes of HB.[ 49 ]. We did not observe differential methylation of either gene in BWS HB, nor others commonly implicated in HB (Tables S3 and S4). Regarding the transcriptome, we observed increases of IL‐6 and KIT in 11p15‐altered HB, but not to the same extent as other HB (Figure 5C). The relatively moderate expression in BWS HB of these two genes, which normally increase liver progenitor cell proliferation, may contribute to decreased BWS‐associated malignancy.[ 50 ]
Lastly and importantly, we observed that the BWS subtypes observed in blood did not always match the BWS subtypes observed in liver samples (Table 1 and Figure S2A,B), suggesting that tumor‐risk stratification by blood epigenotype in BWS is not necessarily sufficient. This unexpected result requires further investigation to better understand its frequency and effect. However, while these data presented herein represent several potential targets for future BWS HB surveillance and treatment development, further work and confirmational cohort information are required before clinical care for this patient population can be modified. Additionally, further research into the individual contributions of the 11p15‐imprinted protein‐coding genes, miRNAs, and noncoding RNAs to liver and HB development is essential to understanding the 11p15‐HB relationship. Our multi‐omic profiling provides a map for these future studies.
CONFLICT OF INTEREST
Nothing to report.
AUTHOR CONTRIBUTIONS
Study concept: Jennifer M. Kalish and Natali S. Sobel Naveh. Data analysis: Natali S. Sobel Naveh and Emily M. Traxler. Clinical information: Kelly A. Duffy. Manuscript draft: Natali S. Sobel Naveh, Kelly A. Duffy, and Jennifer M. Kalish. Manuscript editing: Natali S. Sobel Naveh, Kelly A. Duffy, Emily M. Traxler, and Jennifer M. Kalish. Study supervision: Jennifer M. Kalish.
Supporting information
Supinfo
Table S1
Table S2
Table S3
Table S4
Table S5
Supinfo
ACKNOWLEDGMENT
We would first and foremost like to thank the patients and families who are members of the BWS Registry and provided their tissue samples for research purposes. We thank Maria Lemma, Dr. Fernanda Thompson, Dr. Renata Pellegrino Da Silva from the CHOP CAG, and Dr. Xi Chen with GENEWIZ, Inc. for coordinating sequencing. We would also like to thank Dr. Joseph Glessner and Dr. Hakon Hakonarson from the CHOP CAG for providing control exome data for the analysis. Thank you to Dr. Rebecca Linn from the Division of Pathology for coordinating the control liver samples used in this study; we would also like to respectfully acknowledge the individuals from whom these samples were derived.
Sobel Naveh NS, Traxler EM, Duffy KA, Kalish JM. Molecular networks of hepatoblastoma predisposition and oncogenesis in Beckwith‐Wiedemann syndrome. Hepatol Commun. 2022;6:2132–2146. 10.1002/hep4.1972
Funding informationSupported by National Institutes of Health (CA193915), a Damon Runyon Clinical Investigator Award supported by the Damon Runyon Cancer Research Foundation (105–19), Alex’s Lemonade Stand Foundation, and the Lorenzo “Turtle” Sartini Jr. Endowed Chair in Beckwith‐Wiedemann Syndrome Research. These funding sources did not play a role in the study design, the collection, analysis and interpretation of data, the writing of the report, nor in the decision to submit the article for publication.
REFERENCES
- 1. Schnater JM, Kohler SE, Lamers WH, von Schweinitz D, Aronson DC. Where do we stand with hepatoblastoma? A review. Cancer. 2003;98:668–78. [DOI] [PubMed] [Google Scholar]
- 2. Feng J, Polychronidis G, Heger U, Frongia G, Mehrabi A, Hoffmann K. Incidence trends and survival prediction of hepatoblastoma in children: a population‐based study. Cancer Commun (Lond). 2019;39:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta‐catenin gene. Cancer Res. 1999;59:269–73. [PubMed] [Google Scholar]
- 4. Buendia MA, Bourre L, Cairo S. Myc target miRs and liver cancer: small molecules to get Myc sick. Gastroenterology. 2012;142:214–8. [DOI] [PubMed] [Google Scholar]
- 5. Albrecht S, von Schweinitz D, Waha A, Kraus JA, von Deimling A, Pietsch T. Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res. 1994;54:5041–4. [PubMed] [Google Scholar]
- 6. Arai Y, Honda S, Haruta M, Kasai F, Fujiwara Y, Ohshima J, et al. Genome‐wide analysis of allelic imbalances reveals 4q deletions as a poor prognostic factor and MDM4 amplification at 1q32.1 in hepatoblastoma. Genes Chromosomes Cancer. 2010;49:596–609. [DOI] [PubMed] [Google Scholar]
- 7. Chitragar S, Iyer VK, Agarwala S, Gupta SD, Sharma A, Wari MN. Loss of heterozygosity on chromosome 11p15.5 and relapse in hepatoblastomas. Eur J Pediatr Surg. 2011;21:50–3. [DOI] [PubMed] [Google Scholar]
- 8. Eichenmuller M, Trippel F, Kreuder M, Beck A, Schwarzmayr T, Häberle B, et al. The genomic landscape of hepatoblastoma and their progenies with HCC‐like features. J Hepatol. 2014;61:1312–20. [DOI] [PubMed] [Google Scholar]
- 9. Hartmann W, Waha A, Koch A, Goodyer CG, Albrecht S, von Schweinitz D, et al. p57(KIP2) is not mutated in hepatoblastoma but shows increased transcriptional activity in a comparative analysis of the three imprinted genes p57(KIP2), IGF2, and H19. Am J Pathol. 2000;157:1393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montagna M, Menin C, Chieco‐Bianchi L, D’Andrea E. Occasional loss of constitutive heterozygosity at 11p15.5 and imprinting relaxation of the IGFII maternal allele in hepatoblastoma. J Cancer Res Clin Oncol. 1994;120:732–6. [DOI] [PubMed] [Google Scholar]
- 11. Suzuki M, Kato M, Yuyan C, Takita J, Sanada M, Nannya Y, et al. Whole‐genome profiling of chromosomal aberrations in hepatoblastoma using high‐density single‐nucleotide polymorphism genotyping microarrays. Cancer Sci. 2008;99:564–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. von Schweinitz D, Kraus JA, Albrecht S, Koch A, Fuchs J, Pietsch T. Prognostic impact of molecular genetic alterations in hepatoblastoma. Med Pediatr Oncol. 2002;38:104–8. [DOI] [PubMed] [Google Scholar]
- 13. Duffy KA, Cielo CM, Cohen JL, Gonzalez‐Gandolfi CX, Griff JR, Hathaway ER, et al. Characterization of the Beckwith‐Wiedemann spectrum: Diagnosis and management. Am J Med Genet C Semin Med Genet. 2019;181:693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. MR DB, Tucker MA. Risk of cancer during the first four years of life in children from The Beckwith‐Wiedemann Syndrome Registry. J Pediatr. 1998;132:398–400. [DOI] [PubMed] [Google Scholar]
- 15. Coktu S, Spix C, Kaiser M, Beygo J, Kleinle S, Bachmann N, et al. Cancer incidence and spectrum among children with genetically confirmed Beckwith‐Wiedemann spectrum in Germany: a retrospective cohort study. Br J Cancer. 2020;123:619–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luk HM. Clinical and molecular characterization of Beckwith‐Wiedemann syndrome in a Chinese population. J Pediatr Endocrinol Metab. 2017;30:89–95. [DOI] [PubMed] [Google Scholar]
- 17. Bliek J, Gicquel C, Maas S, Gaston V, le Bouc Y, Mannens M. Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith‐Wiedemann syndrome (BWS). J Pediatr. 2004;145:796–9. [DOI] [PubMed] [Google Scholar]
- 18. Sasaki K, Soejima H, Higashimoto K, Yatsuki H, Ohashi H, Yakabe S, et al. Japanese and North American/European patients with Beckwith‐Wiedemann syndrome have different frequencies of some epigenetic and genetic alterations. Eur J Hum Genet. 2007;15:1205–10. [DOI] [PubMed] [Google Scholar]
- 19. Mussa A, Russo S, de Crescenzo A, Freschi A, Calzari L, Maitz S, et al. (Epi)genotype‐phenotype correlations in Beckwith‐Wiedemann syndrome. Eur J Hum Genet. 2016;24:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ibrahim A, Kirby G, Hardy C, Dias RP, Tee L, Lim D, et al. Methylation analysis and diagnostics of Beckwith‐Wiedemann syndrome in 1,000 subjects. Clin Epigenetics. 2014;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weksberg R, Nishikawa J, Caluseriu O, Fei YL, Shuman C, Wei C, et al. Tumor development in the Beckwith‐Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. 2001;10:2989–3000. [DOI] [PubMed] [Google Scholar]
- 22. Kalish JM, Doros L, Helman LJ, Hennekam RC, Kuiper RP, Maas SM, et al. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res. 2017;23:e115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mussa A, Duffy KA, Carli D, Ferrero GB, Kalish JM. Defining an optimal time window to screen for hepatoblastoma in children with Beckwith‐Wiedemann syndrome. Pediatr Blood Cancer. 2019;66:e27492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trobaugh‐Lotrario AD, Venkatramani R, Feusner JH. Hepatoblastoma in children with Beckwith‐Wiedemann syndrome: does it warrant different treatment? J Pediatr Hematol Oncol. 2014;36:369–73. [DOI] [PubMed] [Google Scholar]
- 25. Fukuzawa R, Hata J, Hayashi Y, Ikeda H, Reeve AE. Beckwith‐Wiedemann syndrome‐associated hepatoblastoma: WNT signal activation occurs later in tumorigenesis in patients with 11p15.5 uniparental disomy. Pediatr Dev Pathol. 2003;6:299–306. [DOI] [PubMed] [Google Scholar]
- 26. Nagae G, Yamamoto S, Fujita M, Fujita T, Nonaka A, Umeda T, et al. Genetic and epigenetic basis of hepatoblastoma diversity. Nat Commun. 2021;12:5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cairo S, Armengol C, de Reyniès A, Wei Y, Thomas E, Renard CA, et al. Hepatic stem‐like phenotype and interplay of Wnt/beta‐catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14:471–84. [DOI] [PubMed] [Google Scholar]
- 28. Carrillo‐Reixach J, Torrens L, Simon‐Coma M, Royo L, Domingo‐Sàbat M, Abril‐Fornaguera J, et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol. 2020;73:328–41. [DOI] [PubMed] [Google Scholar]
- 29. Jia D, Dong R, Jing Y, Xu D, Wang Q, Chen L, et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology. 2014;60:1686–96. [DOI] [PubMed] [Google Scholar]
- 30. Sekiguchi M, Seki M, Kawai T, Yoshida K, Yoshida M, Isobe T, et al. Integrated multiomics analysis of hepatoblastoma unravels its heterogeneity and provides novel druggable targets. NPJ Precis Oncol. 2020;4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sumazin P, Chen Y, Treviño LR, Sarabia SF, Hampton OA, Patel K, et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology. 2017;65:104–21. [DOI] [PubMed] [Google Scholar]
- 32. Heck JE, Meyers TJ, Lombardi C, Park AS, Cockburn M, Reynolds P, et al. Case‐control study of birth characteristics and the risk of hepatoblastoma. Cancer Epidemiol. 2013;37:390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yeh YA, Rao PH, Cigna CT, Middlesworth W, Lefkowitch JH, VVVS M. Trisomy 1q, 2, and 20 in a case of hepatoblastoma: possible significance of 2q35‐q37 and 1q12‐q21 rearrangements. Cancer Genet Cytogenet. 2000;123:140–3. [DOI] [PubMed] [Google Scholar]
- 34. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 35. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rivas M, Aguiar T, Fernandes G, Lemes R, Caires‐Júnior L, Goulart E, et al. DNA methylation as a key epigenetic player for hepatoblastoma characterization. Clin Res Hepatol Gastroenterol. 2021;45:101684. [DOI] [PubMed] [Google Scholar]
- 37. Cristobal I, Sanz‐Álvarez M, Luque M, Caramés C, Rojo F, García‐Foncillas J. The role of microRNAs in hepatoblastoma tumors. Cancers (Basel). 2019;11:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4:a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fan H, Zhang M, Liu W. Hypermethylated KCNQ1 acts as a tumor suppressor in hepatocellular carcinoma. Biochem Biophys Res Commun. 2018;503:3100–7. [DOI] [PubMed] [Google Scholar]
- 40. Chang S, Hur SK, Sobel Naveh NS, Thorvaldsen JL, French DL, et al. Derivation and investigation of the first human cell‐based model of Beckwith‐Wiedemann syndrome. Epigenetics. 2021;16:1295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y, Gan B, Liu D, Paik JH. FoxO family members in cancer. Cancer Biol Ther. 2011;12:253–9. [DOI] [PubMed] [Google Scholar]
- 42. Li J, Qin X, Wu R, Wan L, Zhang L, Liu R. Circular RNA circFBXO11 modulates hepatocellular carcinoma progress and oxaliplatin resistance through miR‐605/FOXO3/ABCB1 axis. J Cell Mol Med. 2020;24:5152–61. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43. Lu M, Hartmann D, Braren R, Gupta A, Wang B, Wang Y, et al. Oncogenic Akt‐FOXO3 loop favors tumor‐promoting modes and enhances oxidative damage‐associated hepatocellular carcinogenesis. BMC Cancer. 2019;19:887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tang L, Zhu S, Peng W, Yin X, Tan C, Yang Y. Epigenetic identification of mitogen‐activated protein kinase 10 as a functional tumor suppressor and clinical significance for hepatocellular carcinoma. PeerJ. 2021;9:e10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koch A, Waha A, Hartmann W, Hrychyk A, Schüller U, Waha A, et al. Elevated expression of Wnt antagonists is a common event in hepatoblastomas. Clin Cancer Res. 2005;11:4295–304. [DOI] [PubMed] [Google Scholar]
- 46. Mendoza‐Reinoso V, Beverdam A. Epidermal YAP activity drives canonical WNT16/beta‐catenin signaling to promote keratinocyte proliferation in vitro and in the murine skin. Stem Cell Res. 2018;29:15–23. [DOI] [PubMed] [Google Scholar]
- 47. Chouhan S, Singh S, Athavale D, Ramteke P, Pandey V, Joseph J, et al. Glucose induced activation of canonical Wnt signaling pathway in hepatocellular carcinoma is regulated by DKK4. Sci Rep. 2016;6:27558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Honda S, Arai Y, Haruta M, Sasaki F, Ohira M, Yamaoka H, et al. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br J Cancer. 2008;99:1891–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Honda S, Haruta M, Sugawara W, Sasaki F, Ohira M, Matsunaga T, et al. The methylation status of RASSF1A promoter predicts responsiveness to chemotherapy and eventual cure in hepatoblastoma patients. Int J Cancer. 2008;123:1117–25. [DOI] [PubMed] [Google Scholar]
- 50. Wang W, Shui L, Liu L, Zheng M. C‐Kit, a double‐edged sword in liver regeneration and diseases. Front Genet. 2021;12:598855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo
Table S1
Table S2
Table S3
Table S4
Table S5
Supinfo