

Abstract
Patients with acute‐on‐chronic liver failure (ACLF) have a high probability of developing systemic inflammation and sepsis due to immune dysregulation. Fifty‐nine patients with ACLF (12 without and 19 with systemic inflammation, and 28 with sepsis) were serially monitored for clinical and immunological changes at baseline, 6 hours, 24 hours, day 3, and day 7 following hospitalization. Ten healthy controls were also included. At all time points, soluble plasma factors and monocyte functions were studied. Patients with ACLF and systemic inflammation showed higher interleukin (IL)–6, vascular endothelial growth factor‐a, monocyte chemoattractant protein 1, and macrophage inflammatory protein 1β than patients with no systemic inflammation. Patients with ACLF with sepsis had raised (p < 0.001) levels of IL‐1Ra, IL‐18, and triggering receptor expressed on myeloid cells 1 (TREM1) compared to patients with ACLF‐systemic inflammation. Five of the 19 (26.3%) patients with systemic inflammation developed sepsis within 48–72 hours with a rapid rise in plasma levels of IL‐1Ra (1203–35,000 pg/ml), IL‐18 (48–114 pg/ml), and TREM1 (1273–4865 pg/ml). Monocytes of patients with ACLF with systemic inflammation and sepsis showed reduced human leukocyte antigen–DR but increased programmed death ligand 1 (PD‐L1) and T‐cell immunoglobulin and mucin domain‐containing protein 3 (TIM3) (p < 0.04) expression with increased ETosis by monocytes at baseline and until day 7. Conclusion: High and rising levels of plasma IL‐1Ra, IL‐18, TREM1 soluble factors, and increased suppressive monocytes (PDL1+ve, TIM3+ve) at baseline can stratify patients with ACLF at high risk of developing sepsis within 48–72 hours of hospitalization.
ACLF is a liver disease with high short‐term mortality. It is associated with immune dysfunction and the development of sepsis. There are few markers of early detection of sepsis. We propose that elevated levels of some biomarkers (IL‐1Ra, IL‐18, and TREM‐1) can be used to identify patients with ACLF who are at risk of developing sepsis. Additionally, the immune cells and monocytes from these patients have suppressive (high suppressive markers, PDL1 and TIM3) and are exhausted (low HLA‐DR) increasing susceptibility to infections.

INTRODUCTION
Acute‐on‐chronic liver failure (ACLF) is a distinct syndrome of liver failure in a patient with chronic liver disease presenting with jaundice, coagulopathy, and ascites and/or hepatic encephalopathy, developing an acute hepatic insult and associated with high 28‐day mortality.[ 1 , 2 , 3 ] Most patients with ACLF with severe alcoholic hepatitis develop systemic inflammation with multi‐organ failure. Chronic ethanol abuse and liver disease lead to intestinal dysbiosis, increased intestinal permeability, and translocation of bacteria.
The systemic inflammation results from a cytokine storm in response to acute hepatocellular injury, which may subsequently lead to immunoparesis and development of sepsis.[ 4 ] Patients with ACLF with systemic inflammation have a high chance of developing sepsis, leading to multiple organ failure and high short‐term mortality.[ 2 ]
White blood cell (WBC) count, C‐reactive protein (CRP), and procalcitonin (PCT) levels are currently being used for the diagnosis and monitoring of infections.[ 5 ] Unfortunately, there are few circulating bio‐markers of sepsis in the setting of systemic inflammation in patients with ACLF. Such tests can help early stratification of patients requiring treatment in the intensive care units. CRP is an acute phase protein that is synthesized and secreted in the liver and belongs to the late‐onset infection index, as it is increased about 24–48 hours after bacterial infection.[ 5 , 6 ] PCT is a prohormone of calcitonin that is produced in the neuroendocrine medullary C‐cells of the thyroid gland in healthy subjects.[ 7 ] Bacterial infection induces a ubiquitous increase in the expression of CALC‐I gene and a constitutive release of PCT from all parenchymal tissues and differentiated cell types throughout the body.[ 8 ] Although PCT and CRP are considered the first markers of inflammation, either alone or in combination they reliably predictor 28‐day mortality in severe sepsis and septic shock.[ 9 , 10 ] Additionally, high baseline international normalized ratio, leukocyte counts, serum creatinine, and low hemoglobin levels are documented to be associated with poor outcome in patients with ACLF[ 11 ] and significantly increased pre‐sepsin associated in conditions of sepsis with proportional rise with severity of sepsis.[ 12 ]
Sepsis develops due to impaired immunity, although innate immune cells act as first line of defense and start the process of pathogen recognition by pathogen‐associated molecular patterns and pattern recognition receptors and activation by damage‐associated molecular patterns or danger‐associated molecular patterns.[ 13 ] The immune status of patient abruptly changes in ACLF condition and is not constant during the hospital stay. The phagocytic activity of monocytes and neutrophils has been shown to be defective in patients with ACLF, and studies showed that using a pharmacological inhibitor of glutamine synthetase,[ 14 ] glucocorticoids, and granulocyte colony–stimulating factor[ 15 ] restored phagocytic and inflammatory capacity of monocytes in ACLF.[ 15 ] However, another study revealed that monocytes from patients with severe alcoholic hepatitis had normal phagocytosis but were defective in mitochondrial oxidative burst due to reduced expression of gp91phox subunit of nicotinamide adenine dinucleotide phosphate (reduced form) oxidase.[ 16 ]
Immune paralysis is characterized by increased soluble CD163, MER proto‐oncogene, tyrosine kinase (MERTK),[ 15 ] and down‐regulation of human leukocyte antigen (HLA)–DR expression on monocyte.[ 17 ] It is common in ACLF and is significantly associated with the severity of organ failure, the risk of sepsis, and high mortality. The intrinsic immune paralysis mechanism of ACLF showed a decrease in pro‐inflammatory immune cells, and an increase in inhibitory immune cells and anti‐inflammatory substances in the microenvironment.[ 15 ]
Activated innate immune cells secrete both pro‐inflammatory and anti‐inflammatory cytokines such as tumor necrosis factor‐α (TNF‐α), interleukin (IL)–1β, IL‐6, monocyte chemoattractant protein 1 (MCP‐1), and their imbalance drives ACLF condition to ACLF systemic inflammation, resulting in monocyte and neutrophil dysfunction.[ 18 , 19 , 20 ] Recently, it has been reported that, like neutrophils, monocytes also control pathogens by releasing extracellular traps (ETs) known as “etosis.” Similar to neutrophils, during inflammation, monocytes release ETs, which are dependent on the oxidative burst capacity of the cell.[ 21 ]
We aimed to identify the soluble factors as early markers of development of systemic inflammation and progression to sepsis, and compared these with available biomarkers like CRP and PCT and possible cause for immune paralysis in patient with sepsis. We also studied the phenotypic and functional changes in monocytes in different categories of patients with ACLF from the time of hospitalization up to 7 days of their hospital stay or death, whichever was earlier. We additionally evaluated the predictive roles of different soluble factors and monocyte changes as early bio‐markers of development of systemic inflammation and sepsis in patients with ACLF.
PATIENTS AND METHODS
Patient recruitment
150 patients with ACLF were screened, 108 patients were included in the study, and 42 patients were excluded (due to superadded viral infections, cardiopulmonary disease, chronic kidney disease, co‐existing hepatocellular carcinoma, with a history of recent plasma exchange, or nonavailability of consent). Of the 108 patients, however, serial blood samples at admission and at all follow‐up time points (6 hours, 24 hours, day 3, and day 7) could be obtained only in 59 patients, and these were finally included in the longitudinal study cohort (Figure S1).
Patients with ACLF between 18 and 65 years of age were included. The diagnosis of ACLF was based on the Asian Pacific Association for the Study of the Liver (APASL) definition.[ 22 ] Systemic inflammation was considered by the occurrence of at least two of the following criteria: (1) fever > 38.0°C or hypothermia < 36.0°C, (2) tachycardia > 90 beats/minute, (3) tachypnea > 20 breaths/min, and (4) leukocytosis > 12 × 109/l or leucopoenia < 4 × 109/l (or immature forms).[ 23 ] The patients were considered to be having sepsis if they had any known or suspected infection at baseline, which was later confirmed by positive bacteriological culture report in addition to fulfilling the systemic inflammation criteria.[ 23 ] Patients with ACLF without systemic inflammation and sepsis served as disease control for the other groups.
Ten healthy subjects were enrolled in the study as control for patients with ACLF. They had no prior history of any liver‐related disease and were negative for all hepatitis viral markers: immunoglobulin M hepatitis A virus, hepatitis E virus, hepatitis B surface antigen, anti–hepatitis C virus, human immunodeficiency virus, cytomegalovirus, and herpes simplex virus.
According to the institute’s antibiotic policy, patients with ACLF received prophylactic antibiotic at admission that was upgraded if sepsis was suspected or confirmed. Choice of the antibiotics was based on the microbial sensitivity patterns. Baseline sample was collected before administration of antibiotics to the patient (i.e., immediately after admission), and all follow‐up samples (6 hours, 24 hours, day 3, and day 7) were after antibiotic treatment. Baseline clinical and laboratory data as well as follow‐up data on the development of cirrhosis complications, organ failures, and bacterial infections were recorded in the Hospital Information System. These data were used for calculating the Model for End‐Stage Liver disease (MELD) and APASL‐ACLF Research Consortium (AARC) scores.
Blood sampling
A total of 10–12 ml of blood was collected in ethylene diamine tetraacetic acid tubes for the experiments. From part of the blood sample, plasma was separated for various biochemical, serological, and laboratory testing, and stored aliquots at −80°C. The rest of the whole‐blood sample was used for immune phenotyping and isolation of peripheral blood mononuclear cells (PBMCs) and polymorphonuclear leucocytes. Monocytes were then separated from the isolated PBMCs.
Analysis of plasma soluble factors using bead array assay
The concentrations of 40 plasma analytes were determined using plasma in different patient groups by using multiplex procartaplex cytokine bead assay (details in the Supporting Information).
Monocyte phenotyping
Fresh whole blood was used to analyze the CD14 and CD16 expression on monocytes (Figure S2), and further HLA‐DR and toll‐like receptor 4 (TLR4) was measured on monocytes by flow cytometry (details in Supporting Information).
PBMC isolation
PBMCs were isolated from the blood of healthy subjects and patients using Ficoll‐hypaque density gradient centrifugation, and the isolated cells were frozen in a medium (90% fetal bovine serum [FBS] + 10% DMSO) and stored in cryocooler overnight at −80°C and subsequently in liquid nitrogen (detailed protocol in Supporting Information).
In vitro culture of adherent monocytes
Frozen PBMCs were used for isolating adherent monocytes. Cells were thawed and rested overnight before being counted using hemocytometer. A total of 1 × 106 isolated PBMCs were plated in six‐well culture grade plates (Eppendorf, Germany) with complete Roswell Park Memorial Institute 1640 medium (RPMI‐1640) media (22400089; Gibco, Thermo Fisher) inclusive of 10% FBS (10270‐106; Gibco, Thermo Fisher) and 10 mM penicillin streptomycin (A001; Hi‐Media,) for 2 hours (37°C, 5% CO2). After 2 hours, the nonadherent cells were removed and adherent monocytes were carefully washed with complete RPMI‐1640 media and gently removed using vigorous pipetting to detach the adhered cells and immediately used for further experiments. Adherent monocytes were further characterized using CD14 and CD16 markers, and more than 80% of the cells were found to be CD14+CD16+ve.
Monocytes and T cell co‐culture assay
Monocytes by the adherent cell method as described previously and T cells were isolated by magnetic separation using CD4 (480010) and CD8 (480012) specific microbeads from MojoSort kit (BioLegend). A total of 1 × 105 monocytes and 1 × 105 T cells were seeded in anti‐CD3/anti‐CD28‐coated 96‐well flat bottom plates for 1 hours (37°C, 5% CO2). Cells were then stimulated with lipopolysaccharide (LPS; 1 µg/ml; Sigma‐Aldrich) for 2 to 3 hours. After that, the cells were treated with Brefeldin A (2 µg/ml; Cayman Chemical) and collected after 4 hours. Later, cells were collected and stained for monocyte and T‐cell markers (details in Supporting Information).
Oxidative burst activity in monocytes and neutrophils
Oxidative burst activity of monocytes and neutrophils was determined using Celonics Phagoburst kit (10‐0200) (Figure S3). Protocol was followed according to manual instructions (details in Supporting Information).
ETosis by monocytes
The formation of extracellular traps by monocytes (METosis) and neutrophils (NETosis) was analyzed in healthy control and patients. Purity and viability of PMNs were analyzed using CD11b, CD16, and propidium iodide in neutrophils isolated from healthy subjects using polymorphoprep (Figure S4). LPS and IL‐8 stimulation was used as positive control (details in Supporting Information).
Quantitative real‐time polymerase chain reaction analysis
Complementary DNA prepared from total RNA isolated from adherent monocytes (isolated from fresh PBMCs) was used to study the expression of various genes (details in Supporting Information).
Statistical analysis
Statistical analysis was performed using Prism (version 6.01; GraphPad) and SPSS version 23 (IBM Corp Ltd.). Data were compared using either t‐tests for paired analysis or nonparametric Wilcoxon signed test unless otherwise stated and appropriate. The comparison for continuous data is carried using one‐way analysis of variance/Kruskal–Wallis test followed by probability adjustment by the Mann–Whitney U test or by Bonferroni test post hoc comparison as appropriate and it is represented as mean ± SD. The change over period of time was seen using repeated‐measure analysis followed by post hoc analysis by least square deviation method. Data with unequal distribution were used as medians. Moreover, this multinomial logistics regression was also applied along with diagnostic tests (receiver operating characteristic [ROC] curve). The significance was seen at 5%.
RESULTS
Demographic profile of patients
Fifty‐nine patients with ACLF with or without clinical systemic inflammation and sepsis (80% males) with a high MELD were enrolled at the time of admission (Table 1).
TABLE 1.
Baseline characteristics of patients with ACLF at the time of admission
| Variable | HC | ACLF no systemic inflammation | ACLF systemic inflammation | ACLF sepsis | p value | p value |
|---|---|---|---|---|---|---|
| (mean ± SD) | (n = 10) | (n = 12) | (n = 19) | (n = 28) | (between HC and ACLF groups) | (between ACLF groups) |
| Age (years) | 37.9 ± 8.1 | 41.4 ± 9.0 | 42.7 ± 12.4 | 46.5 ± 11.9 | ns | ns |
| Temperature (°F) | 98.4 ± 0.4 | 98.5 ± 0.5 | 98.3 ± 0.2 | 98.5 ± 0.6 | ns | ns |
| TLC (×1000 cells/mm3) | 8.6 ± 1.8 | 9.9 ± 4.6 | 15.7 ± 5.4 | 17.3 ± 14.9 | ns | ns |
| Respiratory rate | 14.4 ± 1.3 | 21.1 ± 0.9 | 19.6 ± 1.7 | 20.5 ± 2.7 | a , b , c | ns |
| Pulse (beats/min) | 72.5 ± 2.8 | 82.4 ± 9.4 | 93.8 ± 16 | 95.1 ± 14 | b , c | e |
| Bicarbonate | 26 ± 2.4 | 21.3 ± 2.8 | 21.7 ± 3.9 | 19.9 ± 2.1 | a , b , c | ns |
| Hemoglobin (g/dl) | 15.3 ± 1.1 | 9.8 ± 1.7 | 9.4 ± 1.3 | 10 ± 1.9 | a , b , c | ns |
| Hematocrit | 47.7 ± 2.3 | 28.0 ± 5.9 | 25.9 ± 5.4 | 28.3 ± 5.1 | a , b , c | ns |
| N (%) | 62.7 ± 5 | 71.8 ± 14.2 | 71.7 ± 11.3 | 74.3 ± 12.1 | ns | ns |
| L (%) | 29.1 ± 6.6 | 20.6 ± 9.2 | 14.7 ± 9.1 | 12.5 ± 9.1 | b , c | ns |
| M (%) | 3.3 ± 2.2 | 11.5 ± 6.7 | 9.7 ± 1.7 | 10 ± 4.6 | a , b , c | ns |
| E (%) | 2 ± 1.1 | 2.4 ± 4 | 1.2 ± 1 | 1.2 ± 1 | ns | ns |
| NLR | 2.2 ± 0.5 | 5.1 ± 4 | 6.9 ± 6.5 | 10.8 ± 9.5 | c | ns |
| MLR | 0.1 ± 0.08 | 0.8 ± 0.2 | 1.1 ± 0.4 | 1.2 ± 0.5 | a , b , c | e |
| Platelets (×1000 cells/ml) | 291.1 ± 104.3 | 82.6 ± 53.6 | 145.5 ± 63.9 | 128.6 ± 93 | a , b , c | ns |
| Uric acid (mg/dl) | 5.4 ± 1.4 | 3.9 ± 1.5 | 3.6 ± 1 | 4.3 ± 3 | ns | ns |
| K (meq/dl) | 4.1 ± 0.7 | 3.8 ± 0.6 | 3.6 ± 0.6 | 5.3 ± 5.4 | ns | ns |
| Direct bilirubin (mg/dl) | 0.2 ± 0.07 | 9.5 ± 4.9 | 14 ± 6.4 | 14 ± 5.9 | a , b , c | ns |
| Indirect bilirubin (mg/dl) | 0.8 ± 0.3 | 7.1 ± 3.2 | 8.3 ± 2.6 | 11 ± 5.2 | a , b , c | e |
| AST (IU/L) | 30 ± 13.6 | 144.2 ± 58.4 | 152.8 ± 78.8 | 160.4 ± 98.2 | a , b , c | ns |
| ALT (IU/L) | 36.4 ± 13.8 | 52.4 ± 28 | 78 ± 67 | 68.1 ± 60.2 | ns | ns |
| Serum ALP | 75.8 ± 42.3 | 100.8 ± 27.4 | 79.8 ± 27.7 | 173.3 ± 262.5 | ns | ns |
| Gamma‐glutamyltransferase | 15.1 ± 801 | 70.6 ± 49.4 | 116.9 ± 216.1 | 89.2 ± 69.1 | ns | ns |
| T. protein (mg/dl) | 6.9 ± 1 | 6.9 ± 0.9 | 5.7 ± 1 | 6.2 ± 0.8 | b | d |
| Albumin (g/dl) | 3.8 ± 0.8 | 2.5 ± 0.3 | 2.2 ± 0.5 | 2.1 ± 0.6 | a , b , c | ns |
| Globulin | 3.1 ± 0.7 | 4.4 ± 0.9 | 3.8 ± 0.6 | 10.3 ± 30.7 | ns | ns |
| Creatinine (mg/dl) | 1 ± 0.2 | 0.6 ± 0.4 | 0.8 ± 0.5 | 1.5 ± 1.2 | ns | e |
| Total bilirubin (mg/dl) | 1 ± 0.3 | 15.9 ± 8.2 | 24 ± 11.8 | 23.8 ± 9.9 | a , b , c | ns |
| INR | 0.7 ± 0.3 | 2.2 ± 0.5 | 2.8 ± 1.2 | 2.4 ± 0.9 | a , b , c | ns |
| Na (meq/l) | 138.5 ± 6.3 | 129.4 ± 2.6 | 131.5 ± 5.5 | 128.4 ± 5.2 | a , b , c | ns |
| MELD | 0 ± 0 | 24.6 ± 2.3 | 29 ± 3.3 | 30.3 ± 6.4 | a , b , c | e |
| MELD‐Na | 0 ± 0 | 29 ± 0.4 | 29.6 ± 3.1 | 31.9 ± 5.4 | a , b , c | ns |
Analysis was done using Bonferroni post hoc test, and p values < 0.05 were considered significant.
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; INR, international normalized ratio; ns, not significant.
HC versus ACLF no systemic inflammation.
HC versus ACLF systemic inflammation.
HC versus ACLF sepsis.
ACLF no systemic inflammation versus ACLF systemic inflammation.
ACLF no systemic inflammation versus sepsis.
Most patients (94%) had severe alcoholic hepatitis as a cause of acute deterioration, while the remaining had drug‐induced liver injury. Table 1 indicates that patients with ACLF with sepsis have increased total leucocyte count (TLC), pulse, neutrophil to lymphocyte ratio (NLR), uric acid, indirect bilirubin, and creatinine as markers of systemic inflammation compared with no systemic inflammation. MELD was also significantly increased in patients with sepsis compared to patients with no systemic inflammation as indicators of severity. Potassium and indirect bilirubin were also significantly increased in patients with sepsis (Figure 1; Table 1).
FIGURE 1.
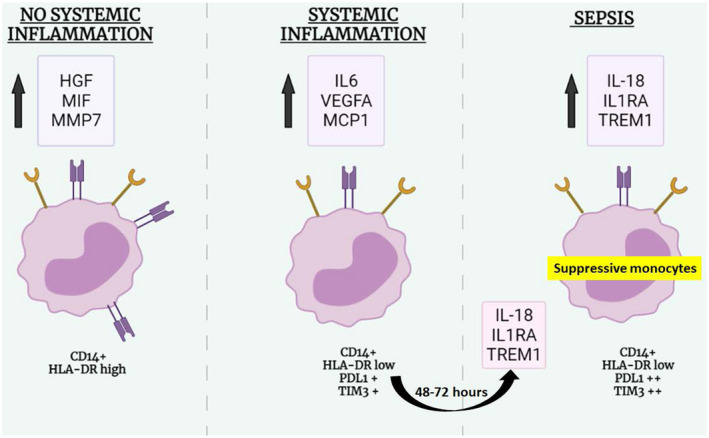
Graphical abstract. Abbreviations: HGF, hepatocyte growth factor; HLA‐DR, human leukocyte antigen–DR; IL, interleukin; MCP1, monocyte chemoattractant protein 1; MIF, migration inhibitory factor; MMP7, matrix metalloproteinase 7; PD‐L1, programmed death ligand 1; TREM1, triggering receptor expressed on myeloid cells 1; VEGFa, vascular endothelial growth factor alpha
Patients with systemic inflammation and sepsis had decreased lymphocyte count and increased neutrophil count compared to those without, which resulted in altered NLR, which was significantly increased in sepsis compared to those with no systemic inflammation (Figure 2A). There was no significant change in differential monocyte count as well as monocyte to lymphocyte ratio in patients with ACLF with or without clinical systemic inflammation and sepsis (Figure 2A).
FIGURE 2.
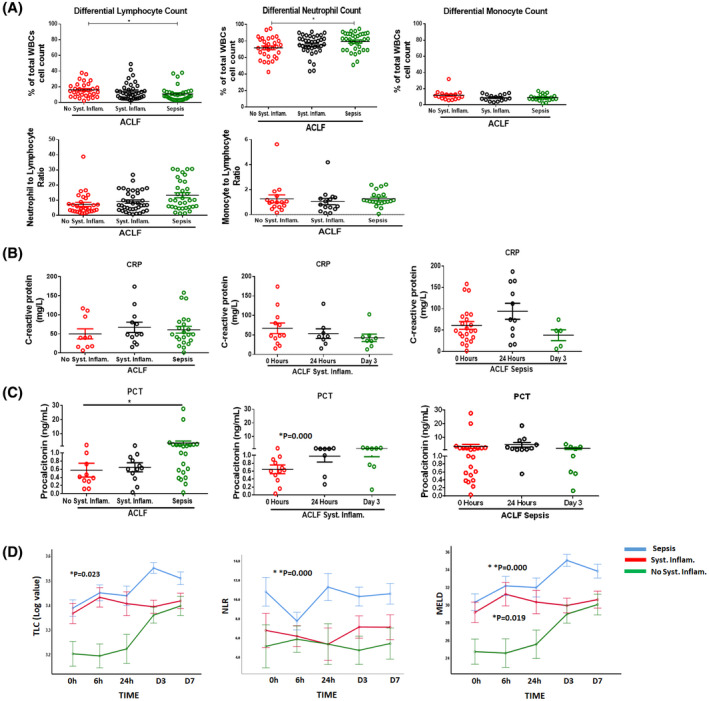
(A) Dot plot graphs showing differential lymphocyte, neutrophil, and monocyte count (as percentage of total white blood cell [WBC] count) in patients with acute‐on‐chronic liver failure (ACLF) (no systemic inflammation, systemic inflammation, and sepsis) at the time of admission. Dot plot graphs of neutrophil to lymphocyte ratio (NLR) and monocyte to lymphocyte ratio (MLR). (B) Dot plot graphs showing C‐reactive protein (in mg/l) in patients with ACLF (no systemic inflammation, systemic inflammation, and sepsis) at the time of admission and at different time points (0 hour, 6 hours, 24 hours, day 3, and day 7). (C) Dot plot graphs showing procalcitonin (in ng/ml) in patients with ACLF (no systemic inflammation, systemic inflammation, and sepsis) at the time of admission and at different time points (0 hour, 6 hours, 24 hours, day 3, and day 7). (D) Line plots showing total leucocyte count (TLC), neutrophil to lymphocyte ratio (NLR), and Model for End‐Stage Liver Disease (MELD) in patients with ACLF systemic inflammation and sepsis at 0 hour, 6 hours, 24 hours, day 3, and day 7. Each data point in graphs (A)–(C) represents an individual sample, and horizontal line represents the mean value. One‐way analysis of variance (ANOVA) and Kruskal‐Wallis test were performed for nonparametric data, respectively, and p < 0.05 was considered significant. Abbreviations: CRP, C‐reactive protein; PCT, procalcitonin
Markers of sepsis
Although CRP was increased in patients with ACLF, there was no significant difference in those without (median and range: 39.7, 6.7–117) or with systemic inflammation (48.4, 15.5–174) or with sepsis (48.8, 1.7–158) (Figure 2B). PCT was significantly increased from no systemic inflammation (median and range: 0.4, 0.12–1.9) compared with the sepsis group (median range: 0.9, 0.03–27.6) (p = 0.04) at the time of admission (Figure 2C). PCT also increased at 24 hours and day 3 in patients with systemic inflammation, but the change was not significant (Figure 2C). Patients were followed until day 7 of their hospital stay, and repeated‐measure analysis revealed (Figure 2D) that TLC and MELD were significantly increased in the groups with systemic inflammation and sepsis at the time of admission and remained elevated.
Patients with sepsis showed increasing trend of MELD and TLC at follow‐up time points, but patients with systemic inflammation had almost the same MELD and TLC as at the time of admission (Figure 2D). Patients with no systemic inflammation showed increased TLC and MELD at day 3 and day 7 (Figure 2D). In addition, patients with ACLF without any clinical systemic inflammation also showed significant (p < 0.05) deterioration from the time of admission to day 7 in respiratory rate, hemoglobin, hematocrit, neutrophils, and lymphocyte counts (Table S3). However, patients with ACLF systemic inflammation showed changes in MELD‐Na within 6 hours immediately after admission (Table S4). Respiratory rate was also increased at day 1 compared to time of admission, and this change was significant at day 3 and day 7 from the 6‐h time point. Similarly, uric acid was increased at day 1 compared to 0 hours and 6 hours, which was stabilized later at day 3 and day 7 compared to day 1. Patients with ACLF sepsis also showed a significant decline in hemoglobin and hematocrit in the first 6 hours of hospital admission (Table S5).
Soluble components at the time of admission and follow‐up
Distinct pattern of soluble factors in ACLF with and without systemic inflammation
Along with deranged clinical parameters associated with patients with ACLF, the cytokine profile of all three groups was distinctly different at the time of admission and in follow‐up time points. As compared to healthy subjects, patients with ACLF with no clinical systemic inflammation showed markedly increased levels of hepatocyte growth factor (HGF), migration inhibitory factor (MIF), and matrix metalloproteinase 7 (MMP7) (p = 0.01, 0.001, and 0.0006, respectively) (Figure 3A). These soluble factors had sustained expression at all follow‐up time points, as depicted in line graphs (Figure 3B). The ROC curve of HGF also showed a significant change in area under the receiver operating characteristic (AUROC) with high sensitivity and specificity (0.963; p = 0.000) (Figure 3C).
FIGURE 3.
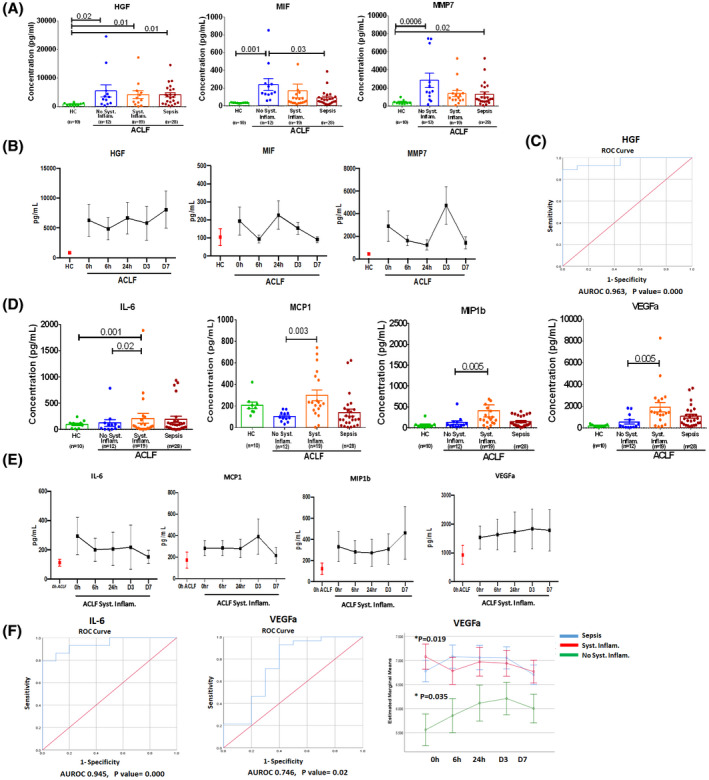
Distinct pattern of soluble factors in patients with ACLF. (A) Dot plot graph represents concentrations of HGF, MIF, and MMP7 in plasma (pg/ml) in healthy controls (HC) and patients with ACLF (no systemic inflammation, systemic inflammation, and sepsis) at the time of admission. (B) Line plot represents the mean concentrations of HGF, MIF, and MMP7 in patients with ACLF but no systemic inflammation at 0 hour, 6 hours, 24 hours, day 3, and day 7. Red dot in graph represents mean concentration of analytes in HC to compare with patients with ACLF without systemic inflammation. (C) Receiver operating characteristic (ROC) curve for HGF values obtained from all patients with ACLF: area under the curve (AUC) = 0.963 (95% confidence interval [CI] = 0.735 to 1.0; p < 0.000). (D) Dot plot graph represents concentrations of IL‐6, monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein 1 beta (MIP‐1b), vascular endothelial growth factor alpha (VEGFa) (pg/ml) in plasma of HC and patients with ACLF (no systemic inflammation, systemic inflammation, and sepsis) at the time of admission. (E) Line plot represents the mean concentrations of IL‐6, MCP1, VEGFa, and MIP‐1β in patients with ACLF systemic inflammation at 0 hour, 6 hours, 24 hours, day 3, and day 7. Red dot in graph represents mean concentration of soluble factor in patients with ACLF with no systemic inflammation. (F) ROC curve for IL‐6 and VEGFa; values obtained from all patients with ACLF systemic inflammation: AUC = 0.945 (95% CI = 0.89–1.0; p < 0.000) for IL‐6 and 0.746 (95% CI = 0.75–1.0; p < 0.02) for VEGFa. Line plot represents the mean concentrations of VEGFa in patients with ACLF systemic inflammation and sepsis at 0 hour, 6 hours, 24 hours, day 3, and day 7. Each data point in (A) and (D) represents an individual sample, and horizontal line represents the mean value. One‐way ANOVA and Kruskal‐Wallis test were performed for nonparametric data, respectively, and p < 0.05 was considered significant
We observed that patients with ACLF with systemic inflammation had significantly higher levels of IL‐6, monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein 1 beta (MIP‐1β), and vascular endothelial growth factor alpha (VEGFa) (p = 0.02, 0.003, 0.005, and 0.005, respectively) at the time of admission compared to ACLF without systemic inflammation (Figure 3D). These factors were significantly and exclusively increased in these patients and continued until day 7 but not in the other groups (Figure 3E). Increase in VEGFa and IL‐6 predicted systemic inflammation condition with AUROC of 0.945 with p = 0.000, and 0.746 with p = 0.02 (Figure 3F). In addition, the line diagram in Figure 3F showed that both patients with systemic inflammation and patients with sepsis had significantly increased VEGFa at baseline and in follow‐up time points. Furthermore, we observed that at the time of admission, few plasma soluble factors like IL‐17a, IL‐12p40, granulocyte‐macrophage colony‐stimulating factor (GMCSF), IL‐1Ra, transforming growth factor β, angiopoietin, and eotaxin were drastically decreased in patients with ACLF with or without clinical systemic inflammation compared with healthy subjects (Table 2). Logistic regression model negatively predicted IL‐12p40 and IL‐17a for ACLF and ACLF systemic inflammation with ROC (0.988 and 0.926 with p = 0.000; Figure S5).
TABLE 2.
Plasma concentrations of soluble factors in HC and in patients with ACLF with and without systemic inflammation
| Analyte (pg/ml) | HC (n = 10) | ACLF without systemic inflammation (n = 12) | ACLF with systemic inflammation (n = 19) | p value |
|---|---|---|---|---|
| IL‐12p40 | 16.7 ± 1.7 | 9.8 ± 2.4 | 7.8 ± 0.6 | a 0.000, b 0.000 |
| IL‐17a | 104 ± 10.9 | 23.8 ± 9.9 | 16 ± 3.9 | a 0.000, b 0.000 |
| GMCSF | 447.2 ± 68.6 | 75.9 ± 33.1 | 47.8 ± 21.8 | a 0.000, b 0.000 |
| IL‐1Ra | 3438 ± 746 | 744 ± 238 | 1365 ± 660 | a 0.014, b 0.01 |
| TGF‐β | 742 ± 95 | 307 ± 74 | 348 ± 50 | a 0.000, b 0.000 |
| Angiopoietin | 883 ± 259 | 32.3 ± 5 | 142.9 ± 60 | a 0.07, b 0.06 |
| Eotaxin | 114.9 ± 31.7 | 88 ± 28.2 | 70 ± 19.8 | a 0.05, b 0.06 |
Data presented with mean and SEM.
Abbreviations: GMCSF, granulocyte‐macrophage colony‐stimulating factor; TGF‐β, transforming growth factor beta.
Significance between HC and ACLF without systemic inflammation.
Significance between HC and ACLF with systemic inflammation.
Marked increase of soluble factors in sepsis
At the time of admission, patients with ACLF with sepsis showed significantly raised levels of IL‐1Ra, IL‐18, and TREM1 and had sustained rise in these factors in all follow‐up time points (Figure 4A,B). Repeated‐measure analysis revealed a continuous increase in TREM1, IL‐18, and IL‐1RA in patients with sepsis compared to patients with or without systemic inflammation (Figure 4C).
FIGURE 4.
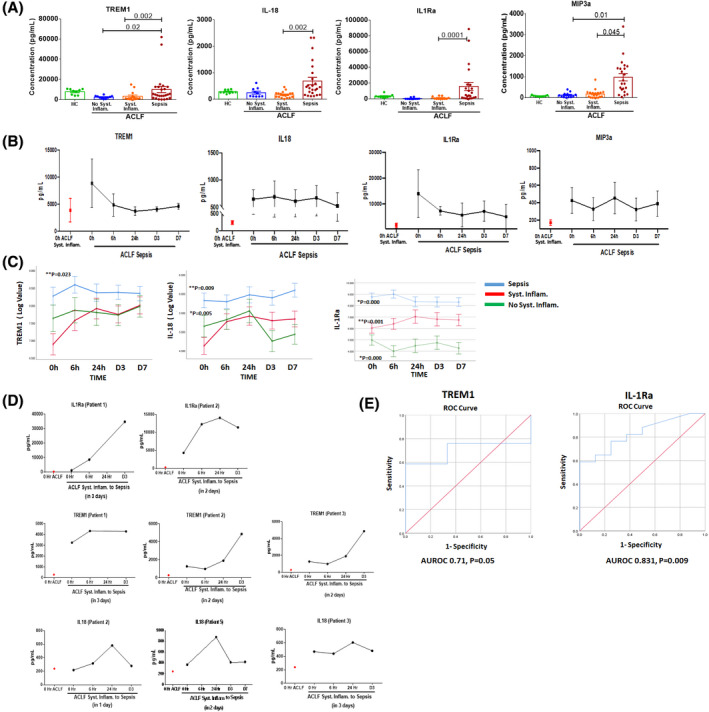
Plasma soluble factors in sepsis. (A) Dot plot graph represents concentrations of TREM1, IL‐18, IL‐1Ra, and MIP‐3a (pg/ml) in HC and patients with ACLF (no systemic inflammation, systemic inflammation, and sepsis) at the time of admission. Each data point in graph represents an individual sample, and horizontal line represents the mean value. One‐way ANOVA and Kruskal‐Wallis test were performed for nonparametric data, respectively, and p < 0.05 was considered significant. (B) Line plot represents the mean concentrations of TREM1, IL‐18, IL‐1Ra, and MIP‐3a in patients with ACLF sepsis at 0 hour, 6 hours, 24 hours, day 3, and day 7. Red dot in graph represents mean concentration of soluble factor in patients with ACLF systemic inflammation for comparison. (C) Line plot graph showing TREM1, IL‐18, and IL1Ra across different time points (0 hour, 6 hours, 24 hours, day 3, and day 7) in patients with ACLF. (D) Line plot graph represents increase in IL‐1Ra, TREM1, and IL‐18 in 5 patients with ACLF systemic inflammation who developed sepsis within 72 hours. (E) ROC curve for TREM1 and IL‐1Ra made using values obtained from all patients with ACLF sepsis. AUC of TREM1 was 0.71 (95% CI = 0.735–1.0; p < 0.04) and IL‐1Ra was 0.831 (95% CI = 0.75–1.0; p < 0.000). (F) Combined ROC curve for TREM1, IL‐18, and IL‐1Ra in patients with ACLF sepsis and table listing the AUC, SEM, and significance of the analytes
Furthermore, we observed that out of 19 patients with ACLF with systemic inflammation, 5 patients started showing raised levels of IL‐1Ra, IL‐18, and TREM1 within 24 hours of admission and developed sepsis within 48–72 hours (Figure 4D). Serum IP10 level was also increased in the first 6 hours during progression from systemic inflammation to sepsis in a few patients, but this was not consistent and showed a fall by 24 hours (Figure S6).
IL‐1Ra and TREM1 were able to predict sepsis at the time of admission with AUROC of 0.831 and 0.71 (Figure 4E). Combined ROC curve shows that area under the curve (AUC) of IL‐18, IL‐1Ra, and TREM1 is 85.6%, 85.4%, and 71.7%, respectively (Figure 4F). Messenger RNA (mRNA) expression analysis in isolated monocytes revealed that along with increased soluble factors in plasma, monocytes from patients with sepsis also had increased expression of IL‐18 and TREM1, and TREM1 downstream signaling molecules DNAX‐activation protein 12 (DAP12) also known as TYRO protein kinase‐binding protein (TYROBP), bruton tyrosine kinas (BTK), and high mobile group box 1 (HMGB1) (Figure S7).
Cellular components
Reduced HLA‐DR but increase in PDL1 and T‐cell immunoglobulin and mucin domain‐containing protein 3 expression in monocytes
Soluble factors in the plasma are representative of immune response but they do not elucidate the immune cell status; therefore, we have dynamically analyzed monocyte functionality. At admission, circulating monocyte numbers was not significantly different in patients compared to healthy controls (Figure 5A), but when we compared percentage and mean fluorescence intensity (MFI) of HLA‐DR expression on monocytes, it was significantly low in patients of ACLF with systemic inflammation and sepsis compared to patients with ACLF without it and controls (Figure 5B).
FIGURE 5.

Reduced HLA‐DR and increased PD‐L1 and TIM3 expression on monocytes. (A) Dot plot graphs represent the percentage of monocytes. (B) Percentage and mean fluorescence intensity (MFI) of HLA‐DR expression on total monocytes in HC and patients with ACLF with or without systemic inflammation and sepsis at the time of admission. (C) Percentage and MFI of HLA‐DR expression on total monocytes in patients with ACLF with or without systemic inflammation and sepsis at follow‐up time points. In patients with ACLF without systemic inflammation, HLA‐DR expression was compared with mean expression in HC; in patients with ACLF with systemic inflammation, HLA‐DR expression was compared to patients with ACLF without systemic inflammation and sepsis with ACLF with systemic inflammation. (D) Dot plot graph shows PD‐L1 and TIM3 expression on monocytes. Each data point represents an individual sample, and horizontal line represents the mean value. (E) Bar graphs represent the percentages of PD‐L1, TIM3, and PD‐L1 with TIM3 expression on unstimulated and stimulated by LPS in HC and the ACLF group (no systemic inflammation, systemic inflammation, and sepsis). Each data point in (A)–(D) represents an individual sample, and horizontal line represents the mean value. One‐way ANOVA and Kruskal‐Wallis test were performed for nonparametric data, respectively, and p < 0.05 was considered significant
During all follow‐up time points, in the patients with systemic inflammation and sepsis, despite standard‐of‐care treatment, there was persistent low MFI of HLA‐DR expression (Figure 5C). There was no significant change in the HLA‐DR expression in the patients with ACLF without systemic inflammation.
With loss of HLA‐DR, circulating monocytes showed gain of programmed death ligand 1 (PD‐L1) and T‐cell immunoglobulin and mucin domain‐containing protein 3 (TIM3) expression as a suppressive phenotype in patients with ACLF systemic inflammation and sepsis (Figure 5D). Furthermore, ex vivo LPS‐stimulated monocytes from patients with ACLF systemic inflammation and sepsis showed significant increase in the single and dual expression of PD‐L1 and TIM3 positivity (Figure 5E).
Decreased protective and functional ability of monocytes with increased expression of PD‐L1, TIM3, and METosis
Monocyte phenotype may mirror the magnitude of inflammatory immune response in patients but does not assure cellular functionality; therefore, we analyzed monocyte functionality in patients with ACLF.
Monocytes showed decreased oxidative burst activity in all patients. Spontaneous oxidative burst activity of monocytes was severely compromised in all patients, especially in patients with sepsis (Figure 6A). Furthermore, ex vivo stimulated monocytes revealed that either with low (fMLP) or high (E. coli and phorbol 12‐myristate 13‐acetate [PMA]) stimulus, oxidative burst activity of monocytes in sepsis remained low. ACLF systemic inflammation monocytes showed little better oxidative burst activity with E. coli and PMA, but it was still insignificant (Figure 6A). Neutrophils from patients with sepsis revealed decreased oxidative burst activity compared to healthy and no systemic inflammation (Figure S8).
FIGURE 6.

Functionality of monocytes. (A) Dot plot graphs represent the percentage of oxidative burst activity of monocytes, spontaneously (without stimulation), fMLP (low stimulation), E. coli (high stimulation), and phorbol 12‐myristate 13‐acetate (PMA) (high stimulation as positive control) in HC and patients with ACLF. (B) The healthy CD4+ T cells (low programmed cell death‐1 [PD‐1] and TIM3 expression) with suppressive monocytes (high PD‐L1 and high TIM3 expression) cells co‐cultured and stimulated by lipopolysaccharide (LPS) for 2 hours. CD4 T cells expressed high PD‐1 and high TIM3. (C) Bar graphs represent the percentage of CD4 total, CD4 with PD‐1, and CD4 with Tim3 cells on nonstimulated and stimulated with LPS in HC and the ACLF group (no systemic inflammation, systemic inflammation, and sepsis). (D) Bar graphs represent the percentage of CD8 total, CD8 with PD‐1, and CD8 with TIM3 cells on nonstimulated and stimulated with LPS in HC and the ACLF group (no systemic inflammation, systemic inflammation, and sepsis). Each data point in (A) represents an individual sample, and horizontal line represents the mean value. One‐way ANOVA and Kruskal‐Wallis test were performed for nonparametric data, respectively, and p < 0.05 was considered significant
Monocytes from patients with systemic inflammation and sepsis ACLF showed increased PD‐L1 and TIM3 expression, and when these monocytes were co‐cultured ex vivo with T cells from healthy subjects with and without LPS stimulation (Figure 6B), it was observed that T cells showed increased expression of programmed cell death‐1 (PD‐1) and TIM3 after the co‐culture (Figure 6C,D).
Furthermore, we observed increased release of extracellular traps (ETosis) in monocytes of patients with ACLF with systemic inflammation and sepsis, which indicates increased cell death.
In fact, when monocytes of healthy subjects were stimulated with plasma of patients with ACLF with systemic inflammation and sepsis, we observed that healthy monocytes started to generate extracellular traps (METosis) similar to when they are stimulated with PMA and IL‐8‐positive controls (Figure S9). Although we observed increased extracellular traps by neutrophils (NETosis) in patients with systemic inflammation and sepsis compared to no systemic inflammation, the METosis phenomenon was more dominant than NETosis (Figure S9).
When monocytes and natural killer cells encounter a new infection, they undergo epigenetic reprogramming, which confers trained immunity resulting in preparedness for future recurrence. Hence, the monocytes were assessed for trained immunity by analyzing gene expression through real‐time polymerase chain reaction, which showed decreased mRNA levels of hypoxia inducible factor 1 alpha subunit, phosphatase and tensin homolog, and AKT. This is indicative of diminished functionality of monocytes and hence a subdued trained immune response (Figure S10).
DISCUSSION
ACLF results from an acute injury in a patient with a pre‐existing chronic liver disease, resulting in a high 4‐week mortality. Sepsis is the most common trigger for organ failure and results in increased mortality. Only a few biomarkers of sepsis have been identified in patients with ACLF.
The results of this prospective and longitudinal study show dynamic evolution of soluble and cellular factors in patients with ACLF who transitioned from systemic inflammation to sepsis. The results of our study highlight that high and rising levels of three plasma soluble factors (IL‐1Ra, IL‐18, and TREM1) and increased number of suppressive monocytes (PDL1+ve and TIM3+ve) at baseline can clearly identify the patients with ACLF who are at high risk of developing sepsis within 48–72 hours of hospitalization.
Clinically, along with bacterial sample culture positivity, rise in WBC, CRP, PCT, and presepsin are used as systemic inflammatory and sepsis markers. However, bacterial culture assay is time‐consuming, and its delay can result into severe pathogenesis.[ 5 , 6 , 7 , 8 ] CRP, PCT, and presepsin alone are also of limited utility due to low sensitivity and specificity. PCT has caught attention as a specific and early marker for systemic inflammation and sepsis, but it is unreliable due to false positives (i.e., increased in noninfectious conditions[ 9 , 10 ]), and false negatives may remain low in infections.[ 8 ] Furthermore, to evaluate the usefulness of CRP and PCT together, a multicenter study evaluated CRP as more predictive than PCT. However, compared with either CRP or PCT, an increase in both was associated with the greater mortality rate.[ 5 ]
In our study, CRP and PCT levels were found to be raised in patients with ACLF with systemic inflammation and sepsis compared to no systemic inflammation, but this increase was not significant. Furthermore, despite increased CRP and PCT levels, it was difficult to evaluate progression from systemic inflammation to sepsis, as there was no change from day 1 to day 3.
Therefore, an array of plasma soluble factors including cytokines, chemokines, growth factors, and matrix metalloproteinases were used to evaluate progression from systemic inflammation to sepsis.
Elevated levels of pro‐inflammatory cytokines in patients with liver cirrhosis were correlated with changes in microbiome and clinical complications.[ 10 , 11 , 12 , 18 , 19 , 20 ] Cytokines and growth factors play a pivotal role in the regulation of the local immune microenvironment and establishment of systemic inflammation. Therefore, serum biomarkers can be used for identifying the critically ill patients who require quick diagnosis and treatment of sepsis for improved prognosis.
Recently, six proteins including MIP3a, E‐selectin, and HGF were found to be increased in hepatitis B virus–induced ACLF.[ 24 ] Although increased inflammatory response in the initial hours was evidently documented,[ 25 ] dynamic changes in the pattern of inflammatory response in patients with ACLF with or without systemic inflammation and sepsis were not known.
We have observed distinct alteration of cytokines, growth factors, and matrix metalloproteinases in patients with ACLF with or without systemic inflammation and sepsis at the time of admission. Increased levels of HGF, MIF, and MMP7 may have a role in developing ACLF and systemic inflammation in patients with no clinical systemic inflammation, as enhanced MIF results in stimulation of pro‐inflammatory cytokines related to systemic inflammation.[ 26 ] MIF interacts with CD74, chemokine (C‐X‐C motif) receptor 2 (CXCR2), and CXCR4 expressing major histocompatibility complex II (MHC II)–positive macrophages, lymphocytes, dendritic and endothelial cells, and induce production of TNF‐α, interferon gamma (IFN‐γ), IL‐1β, IL‐6, and IL‐8 pro‐inflammatory cytokines.[ 26 , 27 , 28 ] On activation, CXCR4 and CXCR2 also promote the recruitment of lymphocytes and neutrophils.[ 28 ] Neutralization or deletion of MIF confers protection from LPS‐induced septic shock.[ 28 , 29 ] At the same time, rise in matrix metalloproteinases remodels the intestinal layer for increased permeability, as it was evident that MMP7‐deficient mice had reduced LPS‐induced intestinal permeability and bacterial translocation.[ 30 , 31 ]
Although sustained rise in MIF, MMP7, and HGF reflects the systemic inflammation, additionally VEGFa and MCP1 were positively correlated with MELD and AARC score in ACLF systemic inflammation (0.619 [p < 0.032] and 0.827 [p < 0.001]). However, increase in these factors also supports the liver rejuvenation and regeneration.
Furthermore, MCP1 and VEGFa being inflammatory induces chemotaxis to recruit monocytes and confers them from M1 to M2 during liver injury.[ 19 , 20 , 32 , 33 ] Although, in systemic inflammation, MCP‐1 and VEGF support the monocyte recruitment, these factors were also associated with their tolerant functions,[ 32 , 33 , 34 ] disease severity, and act as an independent predictor of hospital readmission.[ 32 ]
In this study, patients with systemic inflammation moved to sepsis condition within 72 hours; however, it was observed that TREM1 was significantly increased in patients within 12 hours of admission. In liver, TREM1 played a role as master regulator of Kupffer cell activation, which escalates chronic liver inflammatory responses.[ 35 ] Raised TREM1 not only promotes pro‐inflammatory cytokine secretion but also mobilizes neutrophils and monocytes to the site of injury and is involved in liver injury and fibrosis.[ 35 ] Recently, another group also proposed increased TREM1 and pre‐sepsin as potential biomarkers for development of sepsis in patients with ACLF.[ 36 ]
Deletion of TREM1 reduced liver injury, inflammatory cell infiltration, and fibrogenesis, and reconstitution of Trem1‐deficient mice with Trem1 supported the recruitment of inflammatory monocytes and the severity of liver injury. Activation of TREM1 signaling is facilitated through signaling adaptor protein DAP12.[ 37 ] We also observed increased downstream molecules of TREM1 signaling, HMGB1, DAP12, and BTK in monocytes of patients with sepsis, confirming involvement of TREM1 in increased inflammation and promotion from systemic inflammation to sepsis condition.
After brief stimulation, naïve innate immune cells, especially monocytes, undergo a series of epigenetic and metabolic changes. This induces a lasting change in phenotype, so that these cells mount an altered response to secondary unrelated stimuli. It is observed that IL‐18 and IL‐1Ra was drastically increased in patients with sepsis. Although IL‐1 family members modulate trained immunity in monocytes, they also have a strong role in systemic inflammation and matrix biosynthesis inhibition.[ 38 , 39 ] Both IL‐18 as pro‐inflammatory accounted for IFN‐γ production and apoptosis,[ 40 ] but IL‐1Ra as anti‐inflammatory antagonizes IL‐1α, IL‐1β, and IL‐36.[ 41 ] Therefore, antagonists to IL‐1Ra are being considered as the most effective targeting therapy for inflammation.[ 41 ] Recently, it is observed that the IL‐1 family (especially IL‐18 and IL1Ra) not only confers inflammation but also intrinsically modulates monocytes for protective trained immunity.[ 38 ]
In parallel to the systemic inflammation, immunosuppression is evident in patients with ACLF with multi‐organ failure.[ 2 , 42 , 43 , 44 ] Monocytes of patients with ACLF showed immunosuppressive behavior by reduced HLA‐DR, increased MERTK, impaired phagocytic ability, compromised antibacterial response and antigen presentation, and decreased ex vivo cytokine production following LPS stimulation, but elevated IL‐10 secreting cells.[ 4 , 45 , 46 ] Immunosuppressive behavior of monocytes supported their impaired phagocytic capacity, chemotaxis, and resting oxidative burst in “Golden Window” phase, which led to the development of secondary infections, sepsis, multi‐organ failure, and death.
In this study, patients with ACLF systemic inflammation and sepsis had severely decreased HLA‐DR expression with higher PD‐L1 and TIM3 expression at the time of admission and follow‐up time points. Indeed, PDL1+veTIM3+ve suppressive monocyte from patients with sepsis induced T‐cell suppression in healthy subjects. PD‐L1‐expressing monocytes promoted T‐cell apoptosis in tertiary lymphoid organs in patients with sepsis, and PD‐L1 blockade reversed the dysfunctional monocytes and inhibited lymphocytes apoptosis.[ 47 , 48 ]
The suppressive monocytes appeared to be associated with high risk of nosocomial infections, increased morbidity, and mortality.[ 48 ] Earlier, suppressive monocyte functionality was reversed with pharmacological inhibitor, and glutamine synthetase showed biological relevance in reversing the immunosuppressive monocytes in cirrhosis.[ 15 ]
Therefore, we believe that increased PD‐L1 and TIM3 expression not only blunts phagocytic response of monocytes but also does metabolic rewiring to make them tolerant suppressive monocytes. Furthermore, monocytes of patients with ACLF with no clinical systemic inflammation sets into trained protective immunity; however, increased surge of IL‐18 and IL‐1Ra in sepsis condition may revoke the monocyte protective immunity and change to be pro‐inflammatory monocytes.
We conclude that systemic liver inflammation and increase in VEGFa and MCP1 in patients with ACLF with no systemic inflammation drive monocyte recruitment from bone marrow to rescue further deterioration. However, increased TREM1 and IL‐1Ra change protective monocytes into suppressive monocytes with increased PD‐L1 and TIM3 expression. We propose that TREM1, IL‐1Ra, and IL‐18 measurement at baseline and serial follow‐up can predict sepsis early.
CONFLICT OF INTEREST
Nothing to report.
AUTHOR CONTRIBUTIONS
Patient recruitment and characterization: Shiv K. Sarin, Rakhi Maiwall, Rakesh Kumar Jagdish, and Rajan Vijayaraghavan. Study concept: Nirupama Trehanpati and Shiv K. Sarin. Experiments: Pushpa Yadav, Rashi Sehgal, and Sadam Bhat. Data analysis: Pushpa Yadav. Cytokine bead array assay: Rashi Sehgal, MI, Deepanshu Maheshwari, and Anupam Kumar. Manuscript draft: Nirupama Trehanpati. Input, data presentation, and final manuscript: Shiv K. Sarin and Rakhi Maiwall.
ETHICS APPROVAL
The study was approved by the Research and Ethics committee of the Institutional Review Board No. IEC/2017/50/NA02.
PATIENT CONSENT
Written informed consent was obtained from the patients or their first relative if they were unable to give informed consent themselves.
Supporting information
Supplementary Material
ACKNOWLEDGMENT
The authors thank the excellent technical assistance provided by Dileep Kumar and Surinder Kapoor in the study, as well as the healthy controls and patients who consented to take part in this study.
Yadav P, Trehanpati N, Maiwall R, Sehgal R, Singh R, Islam M, et al. Soluble factors and suppressive monocytes can predict early development of sepsis in acute‐on‐chronic liver failure. Hepatol Commun. 2022;6:2105–2120. 10.1002/hep4.1949
Funding information
The Department of Science and Technology (SB/EF/02/2016 dated March 23, 2017) and the Government of India
Contributor Information
Nirupama Trehanpati, Email: trehanpati@gmail.com, Email: trehanpati@ilbs.in.
Shiv K. Sarin, Email: shivsarin@gmail.com, Email: shivsarin@ilbs.in.
DATA AVAILABILITY STATEMENT
The data generated during and/or analyzed during the current study are available with the corresponding authors on reasonable request.
REFERENCES
- 1. Sarin SK, Choudhury A. Management of acute‐on‐chronic liver failure: an algorithmic approach. Hep Int. 2018;12:402–16. [DOI] [PubMed] [Google Scholar]
- 2. Sarin SK, Choudhury A. Acute‐on‐chronic liver failure: terminology, mechanisms and management. Nat Rev Gastroenterol Hepatol. 2016;13:131–49. [DOI] [PubMed] [Google Scholar]
- 3. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute‐on‐chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144:1426–37. [DOI] [PubMed] [Google Scholar]
- 4. Wasmuth HE, Kunz D, Yagmur E, Timmer‐Stranghöner A, Vidacek D, Siewert E, et al. Patients with acute on chronic liver failure display ‘sepsis‐like’ immune paralysis. J Hepatology. 2005;42:195–201. [DOI] [PubMed] [Google Scholar]
- 5. Ryoo SM, Han KS, Ahn S, Shin TG, Hwang SY, Chung SP, et al. The usefulness of C‐reactive protein and procalcitonin to predict prognosis in septic shock patients: a multicenter prospective registry‐based observational study. Sci Rep. 2019;9:6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Attar BM, Moore CM, George M, Ion‐Nedelcu N, Turbay R, Zachariah A, et al. Procalcitonin, and cytokines document a dynamic inflammatory state in non‐infected cirrhotic patients with ascites. World J Gastroenterol. 2014;20:2374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muller B, White JC, Nylen ES, Snider RH, Becker KL, Habener JF. Ubiquitous expression of the calcitonin‐i gene in multiple tissues in response to sepsis. J Clin Endocrinol Metab. 2001;86:396–404. [DOI] [PubMed] [Google Scholar]
- 8. Aabenhus R, Jensen JU. Procalcitonin‐guided antibiotic treatment of respiratory tract infections in a primary care setting: are we there yet? Prim Care Respir J. 2011;20:360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Christ‐Crain M, Müller B. Procalcitonin in bacterial infections—hype, hope, more or less? Swiss Med Wkly. 2005;135:451–60. [DOI] [PubMed] [Google Scholar]
- 10. Mitaka C. Clinical laboratory differentiation of infectious versus noninfectious systemic inflammatory response syndrome. Clin Chim Acta. 2005;351:17–29. [DOI] [PubMed] [Google Scholar]
- 11. Jindal A, Sharma MK, Sarin SK. Predictors and outcomes of liver failure in severe hepatitis B reactivation. J Clin Exp Hepatol. 2013;3:S51. [Google Scholar]
- 12. Zhao J, Tan Y, Wang LI, Shi YI. Discriminatory ability and prognostic evaluation of presepsin for sepsis‐related acute respiratory distress syndrome. Sci Rep. 2020;10:9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delano M, Ward P. The immune system's role in sepsis progression, resolution, and long‐term outcome. Immunol Rev. 2016;274:330–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Korf H, du Plessis J, van Pelt J, De Groote S, Cassiman D, Verbeke L, et al. Inhibition of glutamine synthetase in monocytes from patients with acute‐on‐chronic liver failure resuscitates their antibacterial and inflammatory capacity. Gut. 2018;68:1872–83. [DOI] [PubMed] [Google Scholar]
- 15. Chen P, Wang Y, Chen C, Guan J, Zhu H, Chen Z. The immunological roles in acute‐on‐chronic liver failure: an update. Hepatobiliary Pancreat Dis Int. 2019;18:403–11. [DOI] [PubMed] [Google Scholar]
- 16. Vergis N, Khamri W, Beale K, Sadiq F, Aletrari MO, Moore C, et al. Defective monocyte oxidative burst predicts infection in alcoholic hepatitis and is associated with reduced expression of NADPH oxidase. Gut. 2016;66:519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin C‐Y, Tsai I‐F, Ho Y‐P, Huang C‐T, Lin Y‐C, Lin C‐J, et al. Endotoxemia contributes to the immune paralysis in patients with cirrhosis. J Hepatol. 2007;46:816–26. [DOI] [PubMed] [Google Scholar]
- 18. Kany S, Vollrath JT, Relja B. Cytokines in Inflammatory Disease. Int J Mol Sci. 2019;20:6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khanam A, Kottilil S. Abnormal innate immunity in acute‐on‐chronic liver failure: immunotargets for therapeutics. Front Immunol. 2020;11:2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Solé C, Solà E, Morales‐Ruiz M, Fernàndez G, Huelin P, Graupera I, et al. Characterization of inflammatory response in acute‐on‐chronic liver failure and relationship with prognosis. Sci Rep. 2016;6:32341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Devin MB, Brenda JC, Michael MC, Jill AL, Elizabeth JK. Extracellular traps and macrophages: new roles for the versatile phagocyte. J Leukoc Biol. 2015;97:1023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sarin SK, Choudhury A, Sharma MK, Maiwall R, Mahtab MA, et al APASL ACLF Research Consortium (AARC) for APASL ACLF working Party. Acute‐on‐chronic liver failure: consensus recommendations of the Asian Pacific association for the study of the liver (APASL): an update. Hepatol Int. 2019;13:353–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choudhury A, Kumar M, Sharma BC, Maiwall R, Pamecha V, Moreau R, et al. Systemic inflammatory response syndrome in acute‐on‐chronic liver failure: relevance of ‘golden window’: a prospective study. J Gastroenterol Hepatol. 2017;32:1989–97. [DOI] [PubMed] [Google Scholar]
- 24. Miller AM, Horiguchi N, Jeong WI, Radaeva S, Gao B. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res. 2011;35:787–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mehta G, Mookerjee R, Sharma V, Jalan R. Systemic inflammation is associated with increased intrahepatic resistance and mortality in alcohol‐related acute‐on‐chronic liver failure. Liver Int. 2014;35:724–34. [DOI] [PubMed] [Google Scholar]
- 26. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–96. [DOI] [PubMed] [Google Scholar]
- 27. Martínez‐Esparza M, Tristán‐Manzano M, Ruiz‐Alcaraz AJ, García‐Peñarrubia P. Inflammatory status in human hepatic cirrhosis. World J Gastroenterol. 2015;21:11522–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Farr L, Ghosh S, Moonah S. Role of MIF cytokine/CD74 receptor pathway in protecting against injury and promoting repair. Front Immun. 2020;11:1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Larson DF, Horak K. Macrophage migration inhibitory factor: controller of systemic inflammation. Crit Care. 2006;10:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nighot P, Al‐Sadi R, Rawat M, Guo S, Watterson DM, Ma T. Matrix metalloproteinase 9‐induced increase in intestinal epithelial tight junction permeability contributes to the severity of experimental DSS colitis. Am J Physiol Gastrointest Liver Physiol. 2015;309:G988–G997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vandenbroucke RE, Vanlaere I, Van Hauwermeiren F, Van Wonterghem E, Wilson C, Libert C. Pro inflammatory effects of matrix metalloproteinase 7 in acute inflammation. Mucosal Immunol. 2014;7:579–88. [DOI] [PubMed] [Google Scholar]
- 32. Queck A, Bode H, Uschner FE, Brol MJ, Graf C, Schulz M, et al. Systemic MCP‐1 levels derive mainly from injured liver and are associated with complications in cirrhosis. Front Immunol. 2020;11:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sekiguchi K, Ito Y, Hattori K, Inoue T, Hosono K, Honda M, et al. VEGF receptor 1‐expressing macrophages recruited from bone marrow enhances angiogenesis in endometrial tissues. Sci Rep. 2019;2019:7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Korbecki J, Kojder K, Siminska D, Bohatyrewicz R, Gutowska I, Chlubek D, Baranowska‐Bosiacka I. CC Chemokines in a tumor: a review of pro‐cancer and anti‐cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int J Mol Sci. 2020;21:8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nathan C, Ding A. TREM‐1: a new regulator of innate immunity in sepsis syndrome. Nat Med. 2001;7:530–2. [DOI] [PubMed] [Google Scholar]
- 36. Chen J, Huang Z‐B, Li H, Zheng X, Chen J‐J, Wang X‐B, et al. Early diagnostic biomarkers of sepsis for patients with acute‐on‐chronic liver failure: a multicenter study. Infect Dis Ther. 2021;10:281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen‐Lefebvre AT, Ajith A, Portik‐Dobos V, Horuzsko DD, Arbab AS, Dzutsev A, et al. The innate immune receptor TREM‐1 promotes liver injury and fibrosis. J Clin Invest. 2018;128:4870–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moorlag SJCFM, Röring RJ, Joosten LAB, Netea MG. The role of the interleukin‐1 family in trained immunity. Immunol Rev. 2018;281:28–39. [DOI] [PubMed] [Google Scholar]
- 39. McEntee CP, Finlay CM, Lavelle EDC. Divergent roles for the IL‐1 family in gastrointestinal homeostasis and inflammation. Front Immunol. 2019;10:1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bossaller L, Chiang PI, Schmidt‐Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, et al. Cutting edge: FAS (CD95) mediates noncanonical IL‐1β and IL‐18 maturation via caspase‐8 in an RIP3‐independent manner. J Immunol. 2012;189:5508–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dinarello CA, Simon A, van der Meer JWM. Treating inflammation by blocking interleukin‐1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arroyo V, Moreau R, Kamath PS, Jalan R, Ginès P, Nevens F, et al. Acute‐on‐chronic liver failure in cirrhosis. Nat Rev Dis Primers. 2016;2:16041. [DOI] [PubMed] [Google Scholar]
- 43. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. [DOI] [PubMed] [Google Scholar]
- 44. Moreau R. The pathogenesis of ACLF: the inflammatory response and immune function. Semin Liver Dis. 2016;36:133–40. [DOI] [PubMed] [Google Scholar]
- 45. Bernsmeier C, Pop OT, Singanayagam A, Triantafyllou E, Patel VC, Weston CJ, et al. Patients with acute‐on‐chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology. 2015;148:603–615.e14. [DOI] [PubMed] [Google Scholar]
- 46. Bretz NP, Ridinger J, Rupp A‐K, Rimbach K, Keller S, Rupp C, et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via toll‐like receptor signaling. J Biol Chem. 2013;288:36691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. PDL1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. 2010;14:R220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xia QI, Wei LI, Zhang Y, Sheng J, Wei WU, Zhang YI. Immune checkpoint receptors tim‐3 and PD‐1 regulate monocyte and T lymphocyte function in septic patients. Mediators Inflamm. 2018;2018:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data generated during and/or analyzed during the current study are available with the corresponding authors on reasonable request.