

Abstract
Iron overload disorders represent a variety of conditions that lead to increased total body iron stores and resultant end‐organ damage. An elevated ferritin and transferrin‐iron saturation can be commonly encountered in the evaluation of elevated liver enzymes. Confirmatory homeostatic iron regulator (HFE) genetic testing for C282Y and H63D, mutations most encountered in hereditary hemochromatosis, should be pursued in evaluation of hyperferritinemia. Magnetic resonance imaging with quantitative assessment of iron content or liver biopsy (especially if liver disease is a cause of iron overload) should be used as appropriate. A secondary cause for iron overload should be considered if HFE genetic testing is negative for the C282Y homozygous or C282Y/H63D compound heterozygous mutations. Differential diagnosis of secondary iron overload includes hematologic disorders, iatrogenic causes, or chronic liver diseases. More common hematologic disorders include thalassemia syndromes, myelodysplastic syndrome, myelofibrosis, sideroblastic anemias, sickle cell disease, or pyruvate kinase deficiency. If iron overload has been excluded, evaluation for causes of hyperferritinemia should be pursued. Causes of hyperferritinemia include chronic liver disease, malignancy, infections, kidney failure, and rheumatic conditions, such as adult‐onset Still's disease or hemophagocytic lymphohistiocytosis. In this review, we describe the diagnostic testing of patients with suspected hereditary hemochromatosis, the evaluation of patients with elevated serum ferritin levels, and signs of secondary overload and treatment options for those with secondary iron overload.
This review provides diagnostic evaluation of patients with elevated ferritin and signs of secondary iron overload. It also describes the clinical manifestations and therapeutic options in secondary iron overload.

INTRODUCTION
Iron overload disorders represent a variety of conditions that lead to systemic iron overload (IO) and organ damage. Primary IO is from hereditary hemochromatosis (HH), while secondary IO can result from iatrogenic iron administration, hematologic conditions that lead to ineffective erythropoiesis (IE) or repeated packed red blood cell (PRBC) transfusions, and from liver disease. In this review, we describe the clinical manifestations of secondary IO disorders and a diagnostic approach to IO and treatment options for these disorders.
IRON METABOLISM
Dietary ferric iron (Fe3+) is reduced to ferrous form (Fe2+) through a brush border protein called duodenal cytochrome B in enterocytes and transported through the apical brush border through divalent metal iron transporter (DMT1).[ 1 , 2 , 3 ] Ferroportin, the iron efflux transporter, then transports ferrous iron across the basolateral membrane of enterocytes, following which iron enters into the circulation bound to transferrin.[ 2 , 4 , 5 ] Cells take up the transferrin‐bound iron through transferrin receptor 1‐mediated endocytosis.[ 6 ] Otherwise, iron will remain in the enterocyte and be lost with enterocyte villi sloughing a few days later.[ 2 , 4 ]
Total body iron is approximately 3–4 g, while 1–2 mg are absorbed from the gastrointestinal tract and approximately 20 mg are recycled from hemoglobin after senescent erythrocytes are phagocytosed by reticuloendothelial macrophages.[ 7 , 8 ] There are no effective physiological mechanisms to remove iron, and only approximately 1–2 mg of iron are lost from gastrointestinal mucosal sloughing of cells.[ 7 ] Each unit of transfused blood provides 200–250 mg, and thus it is inevitable that repeated blood transfusions will lead to IO.
Hepcidin is a peptide synthesized by the liver and a key regulator of iron homeostasis.[ 9 ] Hepcidin binds to ferroportin and suppresses ferroportin‐mediated dietary iron absorption and iron transport.[ 5 ] It also promotes ferroportin ubiquination, internalization, and degradation.[ 5 , 10 ] Systemic conditions can lead to hepcidin overexpression and subsequently anemia of chronic disease.[ 9 ]
DISORDERS LEADING TO SECONDARY IO
Thalassemia syndromes
Thalassemia is the most common cause of secondary IO from IE, while myelodysplastic syndromes (MDSs) and sideroblastic anemias (SAs) represent other common causes.[ 11 , 12 ] Thalassemias are inherited genetic disorders caused by mutations in the α‐globin or β‐globin genes. The most severe forms of α‐thalassemia are hemoglobin H (HbH) disease, with mutations/deletions affecting three out of four α‐globin genes, causing chronic hemolysis due to unstable β‐globin tetramers of HbH; and hemoglobin Barts' hydrops fetalis, in which the absence of all four α‐globin genes causes severe anemia in utero. β‐thalassemia phenotypes include β‐thalassemia minor (or trait, with clinically silent anemia without IO), β‐thalassemia intermedia (TI; with mild to moderate anemia), and β‐thalassemia major (TM; with severe anemia and early transfusion dependence).[ 13 ] The combination of thalassemia mutations with the structural variant hemoglobin E (HbE), common in Southeast Asia, causes HbE/β‐thalassemias.
Thalassemias can be transfusion dependent (TDT), encompassing TM and severe HbE/β‐thalassemia, and nontransfusion dependent (NTDT), including TI, mild to moderate HbE/β‐thalassemia, and HbH disease.[ 14 ] Patients with TDT have a higher propensity to develop cardiac siderosis in addition to hepatic and endocrine involvement, whereas patients with NTDT typically have more hepatic involvement.[ 11 ] There is a geographic variation in terms of prevalence of cardiac siderosis, even among TDT (>25% in Southeast Asia versus 15%–20% in Europe/Middle East).[ 14 ]
IO in patients with NTDT is secondary to both transfusions during their lifetime and IE with hepcidin suppression, which leads to increased intestinal absorption and increased iron circulation.[ 14 ] In animal models of β‐thalassemia, bone marrow and spleen erythroid precursors make erythroferrone, which inhibits hepcidin production.[ 14 ] Growth differentiation factor 15 (GDF‐15) is produced by late erythroblasts, and apoptosis of those precursors in IE generates circulating levels that are most elevated in TDT,[ 15 , 16 ] less so in TI, and normal in HbH disease.[ 17 ] This supports GDF‐15 as a marker of IE, even if its role in suppressing hepcidin production has not been confirmed in patients.[ 14 ]
The incidence of hepatocellular carcinoma (HCC) (estimated annual incidence of 2%[ 18 ]) is increasing in patients with thalassemia.[ 19 ] These patients were previously dying at a younger age from iron‐related cardiac disease, but longer life expectancy with effective iron chelation therapy (ICT) thus accounts for a rise in HCC morbidity.[ 19 ] Patients with TI are more affected than patients with TM because they are living longer and have a combination of spontaneous IO and chronic viral hepatitis from blood transfusions.[ 19 ] In a study of 57 patients with thalassemia, 3.5% developed HCC and incidence was higher in patients with TI than those with TM (p = 0.032).[ 20 ] In this cohort, 33% of those patients with HCC had only liver siderosis and advanced fibrosis secondary to delayed introduction of ICT.[ 20 ] However, hepatitis C virus (HCV) antibody is positive in up to 85% of patients with thalassemia and 50%–57% can be viremic.[ 20 , 21 ] In the era of direct antiviral agents, a single‐center study also reported an increased cumulative incidence of HCC among patients with thalassemia from 1.47% to 3.36% despite 50% of patients with HCV eradication,[ 22 ] underscoring the role of aging and IO in the pathogenesis of HCC. Iron can generate reactive oxygen species (ROS) and toxic byproducts that lead to DNA mutations in repair and tumor suppressor genes.[ 19 ] Most guidelines suggest patients with HCC and thalassemia should be treated similarly to patients without thalassemia, with case reports showing good outcomes in liver transplantation.[ 19 , 21 ] In a series of nine patients with thalassemia with HCC, survival was 69 months in a transplant recipient versus a median of 25 months in those without a transplant.[ 23 ] Good outcomes were further reported in an additional five patients with HCC and thalassemia who underwent transplantation, and only two died after the transplant from causes not related to thalassemia.[ 24 , 25 ] In the largest series to date of 62 patients with HCC and thalassemia who were treated with a variety of therapies, including liver transplantation (n = 3), the average survival time from diagnosis to death was only 11.5 months.[ 24 ] However, long‐term outcomes in patients with HCC and thalassemia are currently lacking.[ 19 ]
MDSs
IO in MDS can occur either from repeated PRBC transfusions or from IE. The most commonly used prognostic models for MDS are the International Prognostic Scoring System (IPSS) and revised IPSS (IPSS‐R), which are based on bone marrow blast percentage, cytopenias, and karyotype.[ 26 ] The median survival for IPSS is 5.7, 3.5, 1.2, and 0.4 years for low, intermediate‐1, intermediate‐2, and high risk, respectively,[ 8 ] and 8.8, 5.3, 3.0, 1.6, and 0.8 years for IPSS‐R very low, low, intermediate, high, or very high risk, respectively.[ 27 ] Patients with MDS manifesting primarily with anemia in the absence of other cytopenias, blast percentage increase, or cytogenetic abnormalities will be classified into either IPSS low or IPSS‐R very low/low risk or lower risk MDS (LRMDS). PRBC transfusion is the primary therapy for LRMDS, with approximately one third being transfusion dependent (TD) while two thirds of high‐risk MDS (HRMDS) are TD.[ 26 ] Patients with TD LRMDS have elevated hepcidin but with levels that are significantly lower than healthy controls,[ 28 ] which suggests increased iron absorption and release into an extracellular pool.[ 29 ] In a European registry, TD‐MDS and ringed sideroblast subtype had the highest amount of IO and the worst survival.[ 30 ]
Secondary IO from other hematologic disorders
Most patients with sickle cell disease (SCD) do not require regular PRBC transfusions but are at risk of IO if they do.[ 31 ] Compared to thalassemia, endocrine and cardiac IO are rare in SCD because less of the transfused iron distributes extrahepatically.[ 32 ] Cardiac siderosis in SCD is associated with high transfusion burden, top‐up transfusions instead of red cell exchanges, and noncompliance with ICT.[ 33 ] In thalassemia, transfused PRBCs are mostly destroyed by macrophages, whereas in SCD there is more significant intravascular hemolysis that provides for potential iron elimination through biliary or urinary iron in hemosiderin, heme, and hemoglobin.[ 32 ] In SCD, the bone marrow production of factors, such as GDF‐15 or erythroferrone, is less marked than in thalassemia, with less hepcidin suppression.[ 16 , 32 ]
Thus, ICT in SCD aims to improve hepatic complications. Advanced hepatic fibrosis can be seen in one third of patients with SCD and be associated with liver iron concentration (LIC) >9 mg Fe/g liver dry weight (dw).[ 34 ] Cirrhosis can occur in 11% of patients with SCD.[ 32 ] Ferritin levels correlate well with magnetic resonance imaging (MRI) LIC but not necessarily with cardiac IO.[ 35 ] One study showed that the number of transfusions per year correlated better to LIC than total lifetime transfusions.[ 36 ] None of the patients who had received <10 PRBC units/year had significant IO.[ 36 ]
Other hematologic disorders that can lead to IO include primary myelofibrosis (PMF), Diamond‐Blackfan anemia (DBA), SA, aplastic anemia (AA), and pyruvate kinase deficiency (PKD). PMF is a myeloproliferative neoplasm in which clonal myeloproliferation with atypical megakaryocytic hyperplasia leads to progressive bone marrow reticulin fibrosis; it is clinically characterized by extramedullary hematopoiesis, splenomegaly, and progressive cytopenias.[ 37 ] Approximately 40% of patients with PMF are anemic at diagnosis, but 60% can have progression with frequent transfusion requirements.[ 37 ] IO is not well characterized in PMF, and most studies are extrapolated from MDS cohorts.[ 37 ] No prospective or large retrospective studies have demonstrated the benefit of ICT.[ 37 ] Ferritin >500 μg/L is associated with worse survival, and a very small study showed patients with ICT‐treated PMF had decreased transfusion requirements and better overall survival.[ 37 , 38 ]
AA and SA can both be hereditary due to genetic mutations but can also be acquired.[ 39 , 40 ] In AA, IO is typically from transfusions.[ 39 ] AA is defined by pancytopenia (hemoglobin <10 g/dL, neutrophils <1.5 × 109/L, and platelets <50 × 109/L) and hypocellular bone marrow with absent signs of marrow fibrosis or dysplasia.[ 39 ] Acquired AA can be secondary to viral infections (viral hepatitis, human immunodeficiency virus [HIV], Epstein‐Barr virus, human herpesvirus 6, and other herpesvirus) or exposure to toxic chemicals (e.g., benzene) or medications (e.g., antiseizure agents, nonsteroidal anti‐inflammatory drugs, temozolomide, immune checkpoint inhibitors) but is most frequently autoimmune and can respond to immunosuppression.[ 39 , 41 , 42 ] ICT can improve ferritin, decrease hepatic and cardiac iron, and improve left ventricular ejection fraction (LVEF) and hemoglobin.[ 43 ] SA is characterized by bone marrow findings of ring sideroblasts,[ 44 ] which are erythroid precursors that have ineffective iron utilization leading to nonheme iron deposition in mitochondria.[ 44 ] Common causes of SA are lead toxicity, use of antibiotics, hormone therapy, copper‐chelating agents, isoniazid, and chemotherapy.[ 44 ] X‐linked SA (XLSA) most commonly results from mutations of delta‐aminolevulinic acid synthase 2 (ALAS2) in the heme synthesis pathway, but it can arise from glutaredoxin‐5 (GLRX5) or solute carrier family 25 member 38 (SLC25A38) mutations, and mutations cause XLSA/ataxia syndrome.[ 44 ] Severe SA leads to ineffective erythropoiesis and systemic IO that is similar to what is found in thalassemia.[ 44 ] XLSA/ataxia syndrome caused by adenosine triphosphatase binding cassette B7 (ABCB7) mutations has not been reported to be associated with systemic IO.[ 44 ]
DBA is a rare congenital disorder caused by ribosomal protein mutations that lead to macrocytic anemia, with over 80% of patients presenting within 6 months of age.[ 45 ] Patients with DBA are at increased risk of MDS, acute myeloid leukemia (AML), osteosarcoma, and colon malignancy.[ 45 ] A study found 54% of patients with DBA had significant hepatic IO (LIC ≥15 mg Fe/g liver dw),[ 46 ] with higher LIC than matched patients with TDT. Patients also had hypothyroidism, hypoparathyroidism, diabetes, and subclinical cardiac disease,[ 46 ] and 23% suffered from iron‐related complications that occurred on average 13 years after diagnosis.[ 46 ] Acute‐on‐chronic liver failure has been reported in DBA because of severe massive IO[ 47 ] with a ferritin level of 21,000 μg/L, and autopsy showed advanced hepatic fibrosis and zone 3 hepatocyte necrosis.[ 47 ]
PKD is a rare autosomal recessive hemolytic anemia caused by mutations in the pyruvate kinase L/R (PKLR) gene encoding the glycolytic enzyme pyruvate kinase. In the PKD Natural History Study, IO was found to affect 38%–82% of patients, as defined by ferritin >1000 μg/L or an MRI T2* with LIC >3 mg/g.[ 48 ] IO is caused by transfusions and increased iron absorption associated with elevated GDF‐15 and hepcidin suppression.[ 49 ] Cirrhosis was found in 8/240 patients,[ 50 ] and severe liver failure with the use of liver transplant has been reported.[ 51 ]
IO IN CHRONIC LIVER DISEASE
IO can also be observed in chronic liver diseases (CLDs). Dysmetabolic IO syndrome (DIOS) can be found in up to one third of patients with nonalcoholic fatty liver disease (NAFLD) and is characterized by metabolic syndrome, high serum ferritin (SF) and transferrin‐iron saturation (TS) levels, but normal serum iron levels.[ 52 ] Increased total body iron stores are associated with decreased pancreatic insulin secretion and increased peripheral and hepatic insulin resistance.[ 53 ] In nonalcoholic steatohepatitis (NASH) animal models, mice that developed IO after being fed dietary iron and had increased expression of NLR family pyrin domain containing 3 (Nlrp3; a component of inflammasome) and inflammatory cytokines (tumor necrosis factor alpha [TNF‐α] and interleukin‐6 [IL‐6]), suggestive that IO can lead to hepatic oxidative stress, inflammasome activation, up‐regulation of inflammatory and immune mediators, and hepatocellular ballooning.[ 54 ] In humans, the pattern of hepatic IO has been associated with NAFLD severity.[ 55 ] Stainable iron in DIOS associated with NASH may be observed in a hepatocellular pattern alone, reticuloendothelial system (RES) alone, or a mixed pattern and RES alone pattern is associated with more severe NASH and fibrosis.[ 55 ] A study from the National Institute of Diabetes and Digestive and Kidney Diseases NASH‐Clinical Research Network found 34.5% had stainable iron; an RES alone pattern of IO was associated with advanced fibrosis (p = 0.049), portal inflammation (p = 0.002), hepatocellular ballooning (p = 0.0006), and NASH (p = 0.0007) compared to those with hepatocellular iron alone or even a mixed pattern.[ 55 ] RES iron may contribute to NASH because of increased oxidative stress, hepatocyte apoptosis, and necrosis when compared to those with a hepatocellular pattern of iron alone.[ 56 ]
The pathogenesis of DIOS remains unknown, but in contrast to homeostatic iron regulator (HFE) HH, patients with NAFLD have increased hepcidin, and DIOS may regulate the NAFLD to NASH transition (Figure 1).[ 53 ] Patients with NALFD with hepatic IO had increased hepcidin expression (3.1‐fold, p = 0.04) and lower expression of inflammatory cytokines compared to patients with NAFLD without IO, suggesting that hepcidin may mitigate inflammation.[ 57 ] Iron export and regulation may be impaired in DIOS. In one study, oral iron challenge led to appropriate hepcidin increases but did not regulate increases in TS.[ 58 ] In another study, patients with NAFLD with IO had decreased hepatic hemojuvelin and ferroportin‐1 expression, leading to hepatic iron retention.[ 59 ] Lastly, patients with NASH can have increased duodenal iron absorption through up‐regulated DMT1 despite elevated hepcidin.[ 60 ]
FIGURE 1.
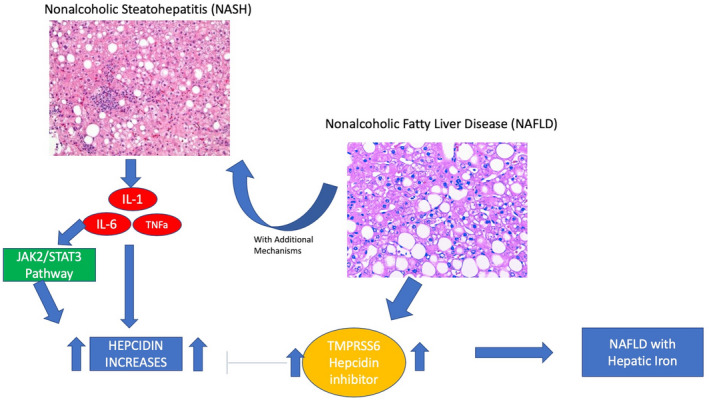
Hepcidin in transition from NAFLD to NASH. IL, interleukin, JAK2, Janus kinase 2; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; STAT3, signal transducer and activator of transcription 3; TMPRSS6, transmembrane serine protease 6; TNFa, tumor necrosis factor alpha.
Hepatic IO is also observed in viral hepatitis (hepatitis B virus [HBV], HCV). Approximately 35%–42% of patients with HCV have stainable iron in the hepatocytes and Kupffer cells.[ 61 , 62 ] Mouse with HCV polyprotein expression have reduced hepatic hepcidin messenger RNA (mRNA) and increased duodenal, hepatic, and spleen ferroportin expression, which leads to increased iron absorption and circulation.[ 63 ] HCV increases ROS, leading to decreased hepcidin expression, and antioxidants have been shown to restore hepcidin function.[ 64 ] While most patients with HCV have mild IO,[ 65 ] IO is associated with more severe HCV disease.[ 62 ] Cirrhosis is observed more frequently in those with IO than those without (17% vs. 3%, p = 0.004).[ 62 ] In HBV, approximately 35% of patients have hepatic iron, and it correlated with ferritin level (odds ratio [OR], 1.2; p = 0.002).[ 66 ] IO is also associated with coinfection with hepatitis delta virus (OR, 4.23; p = 0.003), and patients who are coinfected typically have more advanced fibrosis and higher iron deposition.[ 66 ]
Patients with HCV with HFE mutations (C282Y or H63D) also have more severe disease.[ 67 ] Presence of HFE mutations independently predict risk of having bridging fibrosis or cirrhosis (OR, 18; 95% confidence interval [CI], 1.7–193) after adjusting for disease duration; and HFE mutations correlated with ferritin, transferrin‐iron saturation, and hepatic iron index.[ 68 ] Subsequently, multiple studies show HFE mutations predict a 3‐fold to 6‐fold increased risk of cirrhosis in HCV.[ 67 , 69 , 70 , 71 ] In NAFLD, the data are conflicting in terms of whether HFE mutations are associated with more severe disease. One study showed that patients with NASH with C282Y heterozygosity are at increased risk of having stainable hepatic iron and having bridging fibrosis or cirrhosis (44% vs. 21%, p = 0.05).[ 72 ] However, other studies subsequently showed it did not predict advanced fibrosis or cirrhosis.[ 73 , 74 ]
In alcoholic liver disease (ALD), ethanol metabolism decreases hepcidin mRNA production secondary to oxidative stress and is accompanied by increased DMT1 and ferroportin expression, leading to increased iron circulation and overload.[ 75 , 76 ] Serum ferritin can be elevated in 63% of patients with ALD, and hepatic parenchymal iron and RES iron have been observed in 52% of patients.[ 77 ] Similarly, the data are conflicting in terms of whether HFE mutations increase risk of disease severity. In one study, patients with ALD had a greater probability of carrying HFE mutations as opposed to patients with alcoholism without liver disease (49.5% vs. 31.6%, p = 0.02).[ 78 ] However, another study showed the prevalence of HFE mutations was similar between patients with decompensated alcohol cirrhosis and those with alcoholism without liver disease.[ 79 ] While carrying HFE mutations is also associated with hepatic IO in ALD, it does not predict severity of necroinflammation or liver fibrosis.[ 80 , 81 ] The contribution of hepatic IO to severity of ALD needs further exploration.
CLINICAL MANIFESTATIONS IN SECONDARY IO
Mortality in secondary IO is predominantly from cardiac complications in MDS and thalassemia, but patients also have hepatic and endocrine manifestations. The cardiac events include congestive heart failure (CHF), arrhythmias, and myocardial infarctions.[ 8 ] Patients with anemia and TD‐MDS have increased mortality when compared to those without anemia and those without transfusion dependence.[ 8 , 82 ] Transfusion‐independent chronic anemia in MDS is associated with cardiovascular remodeling, CHF, valvular disease, and arrythmias.[ 8 ] In a retrospective Japanese study of 292 patients with TD‐MDS, AA, and PMF, 24% and 6.7% of deaths were from cardiac and liver failure, respectively.[ 83 ] Similarly, in an Italian cohort of patients with MDS, the most common cause of nonleukemic death was cardiac failure (51%) followed by infections (31%), hemorrhage (8%), and cirrhosis (8%).[ 82 ] In a study using a Medicare database, 73.2% of patients with MDS had a cardiac event during a 3‐year follow‐up and had increased risk of diabetes (40.1% vs. 33.3%), hepatic diseases (0.8% vs. 0.2%), and infections (22.5% vs. 6.1%) (p < 0.01), respectively, when compared to a Medicare population without MDS.[ 84 ]
Although more patients die from cardiac complications in secondary IO, hepatic IO occurs before cardiac siderosis.[ 85 ] In a series of 27 patients with TD‐MDS and myelofibrosis, T2* MRI observed hepatic IO only after a median of 24 units of PRBC transfusion versus 46 units of PRBCs for cardiac IO.[ 85 ] Serum ferritin did not correlate or predict the degree of cardiac or hepatic IO in MDS.[ 85 ]
Patients with TDT present with hepatic and cardiac siderosis along with endocrinopathies depending on how early they start transfusions and ICT. Patients with cardiac MRI T2* <6 milliseconds had a 47% chance of developing CHF in the following year, and cardiac T2* was more accurate in predicting CHF than SF.[ 86 ] In NTDT, clinical manifestations include extramedullary hematopoiesis, thrombotic events, pulmonary hypertension, osteoporosis, hypogonadism, leg ulcers, and liver disease based on abnormal liver enzymes.[ 87 ] In patients with TI, SF ≥800 μg/L predicted a LIC of ≥5 mg Fe/g dw, with a 91.7% positive‐predictive value (PPV) and 53.6% negative‐predictive value (NPV),[ 88 ] while in α‐thalassemia, a lower cutoff ≥600 μg/L predicted similar hepatic iron concentrations.[ 13 ]
In patients with SCD, IO is almost exclusively hepatic and presents later than in TDT.[ 32 ] In a cohort of patients without thalassemia from Latin America with chronic anemias (where most had SCD), hepatic IO was seen in 80% of patients with SCD, 78% with AA, 73% with MDS, 100% with pure red cell aplasia, and 71% with DBA.[ 89 ] Only 19.3% of the patients had cardiac IO in contrast to 36.7% observed in patients with TM.[ 89 , 90 ]
DIAGNOSIS
Patients with elevated liver enzymes or family history of HH should have SF and TS measured, and those with elevated SF (200 μg/L in women and 300 μg/L in men) or TS >45% should have genetic testing for HFE HH (Figure 2).[ 91 ] A normal SF with TS <45% excludes IO with a 97% NPV.[ 92 ] Evaluation for hepatic IO should be pursued either with MRI T2* or consideration of liver biopsy if MRI is unavailable. In cases of secondary IO, quantification of degree of IO can be calculated by lifetime of total PRBC transfused or ferritin can be used as a surrogate measure. However, ferritin can be highly inaccurate, given false elevations in setting of infections, inflammation, and even in malignancy. Liver biopsy is the gold standard for hepatic iron quantification, but because of its risks and invasiveness, MRI T2* is the preferred method for iron quantification. However, if liver biopsy proves to be helpful in diagnosing concomitant CLD, it should be highly considered. MRI T2* is a noninvasive method for hepatic and cardiac iron detection based on how iron affects the protons in water in the tissue of interest.[ 93 ] It leads to signal intensity loss, and iron is quantified by measuring the ratio of signal intensity loss to a reference tissue.[ 93 , 94 , 95 ] An in‐depth review of methods used to quantify tissue iron with MRI is beyond the scope of this review and can be found elsewhere.[ 96 ] Concurrently, in a patient with elevated iron tests with or without evidence of hepatic IO, evaluation for CLD, such as NAFLD, ALD, HCV, and HBV, should be pursued. If there are signs of hepatic IO on MRI, an additional evaluation for secondary causes of hepatic IO should also be considered. Mutations in hemojuvelin, hepcidin, transferrin receptor 2, and ferroportin or non‐HFE hemochromatosis are extremely uncommon causes of IO and should only be pursued if there are no alternative explanations or if there is a family history of IO.[ 97 ] Advanced genetic sequencing can be considered in atypical cases, such as a young patient with cardiac and endocrine involvement.[ 97 ]
FIGURE 2.
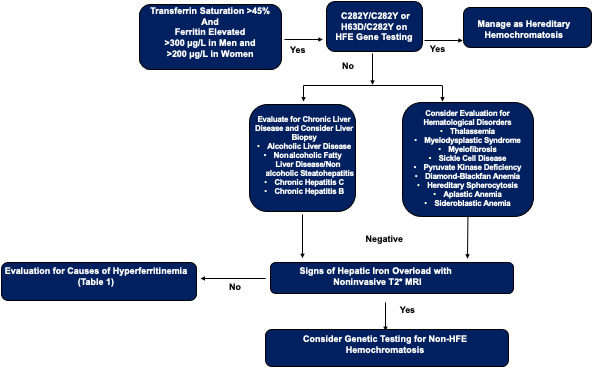
Evaluation of elevated ferritin and transferrin saturation. HFE, homeostatic iron regulator; MRI, magnetic resonance imaging.
EVALUATION OF HYPERFERRITINEMIA
In patients with elevated SF who are negative for common HFE mutations and without IO, evaluation for reactive hyperferritinemia should be considered. Ferritin is an acute‐phase reactant and is elevated in inflammatory conditions, including rheumatic, hematologic, and infectious diseases and ALD, NAFLD, and viral hepatitis.[ 91 ] In ineffective erythropoiesis, ferritin increases as a result of macrophages phagocytosing senescent red blood cells and directly from IO in liver tissue.[ 98 ]
Serum ferritin levels >1000 μg/L should prompt evaluation for diagnoses listed in Table 1.[ 91 , 98 , 99 ] In a US study, the most common cause of high SF levels was malignancy (24%) followed by IO syndromes (22%) and hepatocellular disease (17%).[ 100 ] However, in younger patients (<50 years old), the top three causes were IO (30%), hepatocellular disease (18%), and infectious causes (17%).[ 100 ] In a Japanese study, the most common cause was non‐HIV infection (44.8%) followed by solid tumor (26.3%), liver dysfunction (20.3%), and renal failure (20.3%).[ 101 ] Thus, in patients with elevated SF and lack of IO, malignancy and liver disease should be considered. With SF >10,000 μg/L, conditions, such as adult‐onset Still's disease (AOSD) and hemophagocytic lymphohistiocytosis (HLH), may be suspected.[ 91 ] In both US and Japanese studies, mean ferritin was highest for inflammatory diseases, with average ferritin often >10,000 μg/L in patients with AOSD, systemic juvenile idiopathic arthritis, and HLH.[ 100 , 101 ] In AOSD, high ferritin is thought to be cytokine driven.[ 102 ] Ferritin has various isoforms, with H‐ferritin found predominantly in heart and pancreas whereas L‐ferritin is found in liver, spleen, and the histiocyte–macrophage system.[ 102 ] Inflammatory mediators, such as IL‐1α, interferon‐gamma, and TNF‐α induce H‐ferritin expression.[ 102 ] Similarly, the cytokine storm in HLH has been implicated in H‐ferritin production through nuclear factor‐kappa B signaling by TNF‐α.[ 103 ] Hereditary hyperferritinemia‐cataract syndrome is a rare disorder caused by an autosomal‐dominant mutation in the iron‐responsive element of the L‐ferritin gene, which leads to deposition of L‐ferritin in various fluids and tissues.[ 99 ] Patients present with elevated SF (603–3432 μg/L) and normal TS; a personal or family history of early onset bilateral cataracts may be useful to identify such patients who are not at risk of systemic IO and its complications.[ 99 ]
TABLE 1.
Etiologies of hyperferritinemia
Ferritin >1000 μg/L
|
Ferritin >10,000 μg/L
|
Abbreviations: HAMP, hepcidin antimicrobial peptide; HFE, homeostatic iron regulator; HIV, human immunodeficiency virus; HJV, hemojuvelin.
Source: Adapted from Beaton and Adams[ 91 ].
TREATMENT AND MANAGEMENT
The mainstay of treatment for HFE HH is induction and subsequent maintenance phlebotomy, with target SF between 50 μg/L and 100 μg/L.[ 97 ] Compound heterozygotes (H63D/C282Y) and H63D homozygotes have a lower risk of developing significant IO, but advanced fibrosis has been reported in patients with concomitant liver disease (NAFLD) and with other risk factors, such as significant alcohol use and type 2 diabetes.[ 97 ] Thus, the decision to pursue phlebotomy should be individualized in those patients and based on risk factors.[ 97 ] Patients with HH should have family members tested for ferritin, TS, and HFE genetic testing. Genetic testing is particularly important to characterize the risk of liver disease as even C282Y and H63D carriers may have significant risk factor of advanced fibrosis or CLD with concomitant alcohol use.[ 97 ] Further management of HFE HH is reviewed in detail in another review.[ 97 ] We do not recommend phlebotomy for isolated hyperferritinemia without further evaluation.
In secondary IO, ICT is the primary therapy as patients have anemia and are intolerant of phlebotomy. ICT has been recommended for patients with SF >1000 μg/L or LIC of 7 mg Fe/g dw in TD‐SCD and other transfusion‐dependent chronic anemias.[ 32 , 39 ] National Institutes of Health guidelines recommend ICT in patients with >120 cm3 of PRBC/kg of transfusions. 32 In TM, ICT is commonly started after 2–3 years of regular PRBC transfusions, >10 PRBC transfusions, or if SF is ≥1000 μg/L.[ 104 ] There are three iron chelators currently approved for clinical use: (1) deferoxamine (DFO), (2) deferiprone (DFP), (3) deferasirox (DFX) (Table 2).[ 104 ] DFO is the oldest drug and administered intravenously or subcutaneously, while DFP and DFX are taken orally. Patients with TDT are initially treated with a single agent, and effectiveness of ICT can be followed with serial iron quantification on MRI.[ 104 ] Ferritin correlates well to LIC in thalassemia but is only used to guide ICT if MRI iron quantification is unavailable. Because early mortality in TDT is predominantly cardiac, aggressive ICT is indicated for cardiac siderosis.[ 104 ] Cardiac MRI T2* of 10–20 milliseconds indicates mild to moderate involvement, while <10 milliseconds indicates severe myocardial overload.[ 104 ]
TABLE 2.
Various types of iron chelation therapya
| Drug | Deferoxamine | Deferiprone | Deferasirox |
|---|---|---|---|
| Route of administration | Subcutaneous or infusion | Oral tablet or suspension | Oral tablet for suspension |
| Dose | 20–40 mg/kg/day over 8–24 hours, 5 days/week | 75–100 mg/kg/day over three divided doses | 20 mg/kg/day and up to 40 mg/kg/day |
| Half‐life | 20–30 minutes | 3–4 hours | 8–16 hours |
| Excretion | Fecal/urinary | Urinary | Fecal |
| Adverse events | Local skin reactions | Gastrointestinal | Gastrointestinal |
| Allergic reactions | Agranulocytosis/neutropenia (weekly CBC recommended) | Rash | |
| Growth retardation and bone abnormalities | Arthralgias | Increased creatinine (one third of patients but rarely significantly abnormal) | |
| Auditory | Elevated liver enzymes | Proteinuria | |
| Ophthalmologic | Elevated liver enzymes (rare fulminant hepatic failure, LFTs every 2 weeks for first month followed by monthly recommended) | ||
| Pulmonary and neurologic (high doses) | Auditory | ||
| Ophthalmologic |
Abbreviations: CBC, complete blood count; LFT, liver function test.
Source: Adapted from Poggiali et al.[ 104 ]
In patients with TDT, a combination of DFO and DFP has been shown to be superior in improving cardiac iron, LVEF, and SF levels[ 105 ] than DFO alone.[ 104 , 106 ] ICT with DFO or DFX has been shown to also reverse hepatic fibrosis (by two stages) or maintains stability of fibrosis (−1, 0, +1) in 82.6% of patients with TDT, independent of LIC or HCV positivity.[ 107 ]
In NTDT, transfusion therapy reduces the risk of pulmonary hypertension, extramedullary hematopoiesis, heart failure, cholelithiasis, thrombotic events, and leg ulcers.[ 87 ] However, transfusions are associated with increased risk of hypogonadism, hypothyroidism, and osteoporosis.[ 87 ] The largest data for ICT in NTDT come from the Assessment of Exjade in Nontransfusion‐Dependent Thalassemia (THALASSA) study, which enrolled 166 patients (22 with α‐thalassemia) ≥18 years old with signs of hepatic IO (LIC ≥5 mg Fe/g dw) and SF >300 μg/L.[ 88 ] Patients received either DFX (5 mg/kg or 10 mg/kg) or placebo for 1 year with crossover to DFX for a 1‐year extension.[ 88 ] Ferritin correlated with LIC, and Taher et al.[ 88 ] provided recommendations for continuation, escalation, and interruption of ICT based on ferritin levels when MRI was unavailable. With SF in the range of 1700‐2000 μg/L, there was 100% PPV in predicting LIC >7 mg Fe/g dw. Thus, ferritin ≥2000 μg/L has been proposed for dose escalation in NTDT.[ 88 ] Ferritin of 300 μg/L could be used as a threshold to interrupt chelation therapy in the absence of MRI evaluation because of a PPV of 86% for LIC >3 mg Fe/g dw.[ 88 ] In patients with α‐thalassemia, a lower cutoff of ≥600 μg/L predicted LIC >7 mg Fe/g dw, with area under the receiver operating characteristic curve of 0.76.[ 13 ] Therefore, it is recommended that patients with thalassemia be evaluated and followed serially with MRI for hepatic and cardiac IO improvement and ferritin alone only if MRI iron quantification is unavailable.
ICT in TD‐MDS has been controversial, with initial studies showing patients with ICT to have better survival than patients without ICT.[ 8 ] However, most studies were retrospective or not randomized.[ 8 ] A prospective study showed ICT improved overall survival (median 5.2 vs. 2.1 years in chelated vs. nonchelated) in patients with low‐risk TD‐MDS.[ 108 ] Subsequently, the MDS Event Free Survival With Iron Chelation Therapy Study (TELESTO), the only prospective, placebo‐controlled, randomized trial, showed ICT to prolong event‐free survival by almost 1 year (3.9 vs. 3.0 years for chelated vs. nonchelated with a hazard ratio [HR] of 0.64 (95% CI, 0.42–0.96]).[ 109 ] In that study, 225 patients with IPSS low to intermediate‐1 risk TD‐MDS were randomized to either DFX or placebo, and the primary endpoint was a composite of cardiac events, liver dysfunction, transformation to AML, or death.[ 109 ] The 3‐year probability of event‐free survival was higher (61.5% vs. 47.3) in treated patients compared to untreated patients.[ 109 ] However, there were no statistically significant differences in hematologic improvement, hypothyroidism, glucose intolerance, or infections.[ 109 ] This study was limited by the lack of long‐term follow‐up data, small number of primary endpoint events, relative younger age of participants in Western countries, and imbalances in baseline characteristics.
DFX has improved LIC, hemoglobin and platelets, and liver enzymes.[ 110 , 111 ] In another MDS‐specific study, ICT improved hematologic parameters and decreased the risk of AML progression (34% of the chelated group vs. 17% of the nonchelated group, p = 0.087).[ 112 , 113 ] Despite the suggested benefits from ICT, most MDS trials evaluate only DFX, and the discontinuation rate secondary to adverse effects is high (48%–75%).[ 109 , 112 , 114 , 115 ] In one study, 46% discontinued its use because of side effects and sepsis was the most common cause of death.[ 114 ] Thus, the decision to pursue ICT in MDS should be weighed against risks and typically managed by an expert hematologist. Currently, most guidelines suggest starting ICT for patients with TD‐MDS who have had >20 units of PRBCs or have SF 1000–2500 μg/L, with a goal to reduce ferritin to <1000 μg/L.[ 8 ]
Similarly, DFX has also been shown to improve ferritin and survival and reduce risk of leukemic transformation in primary myelofibrosis.[ 116 , 117 ] In SA, iron chelators are recommended after at least 10 blood transfusions or if SA is >1000 μg/L, but in patients whose anemia can be normalized with pyridoxine, phlebotomy can be considered for IO.[ 44 ]
With regards to phlebotomy and NAFLD/DIOS, phlebotomy was originally shown to be effective in improving insulin resistance,[ 118 ] but subsequent randomized control trials (RCTs) have led to opposite results. In one study in patients with SF ≥250 μg/L and NAFLD activity score >1, the iron‐depleted group had improved liver enzymes, more histologic improvement, and reduction in hepatic steatosis at the 2‐year follow‐up than the nondepleted group.[ 119 ] However, an RCT of 74 subjects showed no difference in liver enzymes, hepatic steatosis, or insulin resistance.[ 120 ] Phlebotomy is not currently recommended for DIOS and NAFLD.
CONCLUSIONS
In patients with elevated iron tests but HFE genetic testing negative for C282Y or H63D, evaluation for CLD is indicated and hematologic disorders, such as thalassemia or MDS, should be considered if the patient is anemic. Suspicion of IO should be pursued either with noninvasive T2* MRI or liver biopsy in such patients. If there are no signs of organ IO, evaluation for other causes of hyperferritinemia should be considered. For patients with secondary IO, ICT should be implemented and preferably monitored with MRI.
CONFLICTS OF INTEREST
Kleber Fertrin received honoraria from, consults for, is on the speakers' bureau of, and received grants from Agios Pharmaceuticals; he consults for and received honoraria and research support from Sanofi Genzyme. Kris Kowdley received honoraria and research support from Gilead, HighTide, Mirum, Pliant, Genfit, CymaBay, GSK, Viking, Pfizer, Intercept, NGM Bio, 89Bio, Celgene, BMS, Corcept, Metacrine, Hanmi, Terns, Madrigal, Enanta, and PTG; he is a consultant for Gilead, Intercept, HighTide, Mirum, Genfit, CymaBay; Inipharm, Madrigal, and NGMBio and serves on the speakers' bureau for AbbVie, Gilead, and Intercept. The other authors have nothing to report.
Hsu CC, Senussi NH, Fertrin KY, Kowdley KV. Iron overload disorders. Hepatol Commun. 2022;6:1842–1854. 10.1002/hep4.2012
REFERENCES
- 1. Krishnamurthy P, Xie T, Schuetz JD. The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Ther. 2007;114(3):345–58. [DOI] [PubMed] [Google Scholar]
- 2. Anderson GJ, Frazer DM, McLaren GD. Iron absorption and metabolism. Curr Opin Gastroenterol. 2009;25(2):129–35. [DOI] [PubMed] [Google Scholar]
- 3. Riedel HD, Remus AJ, Fitscher BA, Stremmel W. Characterization and partial purification of a ferrireductase from human duodenal microvillus membranes. Biochem J. 1995;309:745–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366(4):348–59. Erratum in: N Engl J Med. 2012;366(8):771. [DOI] [PubMed] [Google Scholar]
- 5. Billesbølle CB, Azumaya CM, Kretsch RC, Powers AS, Gonen S, Schneider S, et al. Structure of hepcidin‐bound ferroportin reveals iron homeostatic mechanisms. Nature. 2020;586(7831):807–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson GJ, Darshan D, Wilkins SJ, Frazer DM. Regulation of systemic iron homeostasis: how the body responds to changes in iron demand. Biometals. 2007;20(3–4):665–74. [DOI] [PubMed] [Google Scholar]
- 7. Johnson‐Wimbley TD, Graham DY. Diagnosis and management of iron deficiency anemia in the 21st century. Therap Adv Gastroenterol. 2011;4(3):177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mitchell M, Gore SD, Zeidan AM. Iron chelation therapy in myelodysplastic syndromes: where do we stand? Expert Rev Hematol. 2013;6(4):397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ganz T. Hepcidin in iron metabolism. Curr Opin Hematol. 2004;11(4):251–4. [DOI] [PubMed] [Google Scholar]
- 10. Rishi G, Subramaniam VN. The liver in regulation of iron homeostasis. Am J Physiol Gastrointest Liver Physiol. 2017;313(3):G157–65. [DOI] [PubMed] [Google Scholar]
- 11. Taher AT, Cappellini MD. How I manage medical complications of β‐thalassemia in adults. Blood. 2018;132(17):1781–91. [DOI] [PubMed] [Google Scholar]
- 12. Pippard MJ, Callender ST, Warner GT, Weatherall DJ. Iron absorption and loading in beta‐thalassaemia intermedia. Lancet. 1979;2(8147):819–21. [DOI] [PubMed] [Google Scholar]
- 13. Teawtrakul N, Sirijerachai C, Chansung K, Jetsrisuparb A. The serum ferritin levels and liver iron concentrations in patients with alpha ‐ thalassemia: is there a good correlation? Hematology. 2021;26(1):473–7. [DOI] [PubMed] [Google Scholar]
- 14. Taher AT, Saliba AN. Iron overload in thalassemia: different organs at different rates. Hematology Am Soc Hematol Educ Program. 2017;2017(1):265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13(9):1096–101. [DOI] [PubMed] [Google Scholar]
- 16. Fertrin KY, Lanaro C, Franco‐Penteado CF, de Albuquerque DM, de Mello MRB, et al. Erythropoiesis‐driven regulation of hepcidin in human red cell disorders is better reflected through concentrations of soluble transferrin receptor rather than growth differentiation factor 15. Am J Hematol. 2014;89(4):385–90. [DOI] [PubMed] [Google Scholar]
- 17. Origa R, Cazzola M, Mereu E, Danjou F, Barella S, Giagu N, et al. Differences in the erythropoiesis‐hepcidin‐iron store axis between hemoglobin H disease and β‐thalassemia intermedia. Haematologica. 2015;100(5):e169–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mancuso A, Sciarrino E, Renda MC, Maggio A. A prospective study of hepatocellular carcinoma incidence in thalassemia. Hemoglobin. 2006;30(1):119–24. [DOI] [PubMed] [Google Scholar]
- 19. Moukhadder HM, Halawi R, Cappellini MD, Taher AT. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: a comprehensive review. Cancer. 2017;123(5):751–8. [DOI] [PubMed] [Google Scholar]
- 20. Fragatou S, Tsourveloudis I, Manesis G. Incidence of hepatocellular carcinoma in a thalassemia unit. Hemoglobin. 2010;34(3):221–6. [DOI] [PubMed] [Google Scholar]
- 21. Mangia A, Bellini D, Cillo U, Laghi A, Pelle G, Valori VM, et al. Hepatocellular carcinoma in adult thalassemia patients: an expert opinion based on current evidence. BMC Gastroenterol. 2020;20(1):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ricchi P, Costantini S, Spasiano A, Cinque P, Esposito S, Filosa A. Hepatocellular carcinoma in patients with thalassemia in the post‐DAA era: not a disappearing entity. Ann Hematol. 2021;100(7):1907–10. [DOI] [PubMed] [Google Scholar]
- 23. Restivo Pantalone G, Renda D, Valenza F, D'Amato F, Vitrano A, Cassarà F, et al. Hepatocellular carcinoma in patients with thalassaemia syndromes: clinical characteristics and outcome in a long term single centre experience. Br J Haematol. 2010;150(2):245–7. [DOI] [PubMed] [Google Scholar]
- 24. Borgna‐Pignatti C, Garani MC, Forni GL, Cappellini MD, Cassinerio E, Fidone C, et al. Hepatocellular carcinoma in thalassaemia: an update of the Italian Registry. Br J Haematol. 2014;167(1):121–6. [DOI] [PubMed] [Google Scholar]
- 25. Mancuso A. Hepatocellular carcinoma in thalassemia: a critical review. World J Hepatol. 2010;2(5):171–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carraway HE, Saygin C. Therapy for lower‐risk MDS. Hematology Am Soc Hematol Educ Program. 2020;2020(1):426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia‐Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cui R, Gale RP, Zhu G, Xu Z, Qin T, Zhang Y, et al. Serum iron metabolism and erythropoiesis in patients with myelodysplastic syndrome not receiving RBC transfusions. Leuk Res. 2014;38(5):545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weber S, Parmon A, Kurrle N, Schnütgen F, Serve H. The clinical significance of iron overload and iron metabolism in myelodysplastic syndrome and acute myeloid leukemia. Front Immunol. 2021;11:627662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoeks M, Bagguley T, van Marrewijk C, Smith A, Bowen D, Culligan D, et al. Toxic iron species in lower‐risk myelodysplastic syndrome patients: course of disease and effects on outcome. Leukemia. 2021;35(6):1745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wood JC. Cardiac iron across different transfusion‐dependent diseases. Blood Rev. 2008;22(Suppl 2):S14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Porter J, Garbowski M. Consequences and management of iron overload in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2013;2013:447–56. [DOI] [PubMed] [Google Scholar]
- 33. Tavares AHJ, Benites BD, Fertrin KY. Myocardial iron overload in sickle cell disease: a rare but potentially fatal complication of transfusion. Transfus Med Rev. 2019;33(3):170–5. [DOI] [PubMed] [Google Scholar]
- 34. Olivieri NF. Progression of iron overload in sickle cell disease. Semin Hematol. 2001;38(1 Suppl 1):57–62. [DOI] [PubMed] [Google Scholar]
- 35. Badawy SM, Liem RI, Rigsby CK, Labotka RJ, DeFreitas RA, Thompson AA. Assessing cardiac and liver iron overload in chronically transfused patients with sickle cell disease. Br J Haematol. 2016;175(4):705–13. [DOI] [PubMed] [Google Scholar]
- 36. Inati A, Musallam KM, Wood JC, Sheikh‐Taha M, Daou L, Taher AT. Absence of cardiac siderosis by MRI T2* despite transfusion burden, hepatic and serum iron overload in Lebanese patients with sickle cell disease. Eur J Haematol. 2009;83(6):565–71. [DOI] [PubMed] [Google Scholar]
- 37. Carreau N, Tremblay D, Savona M, Kremyanskaya M, Mascarenhas J. Ironing out the details of iron overload in myelofibrosis: lessons from myelodysplastic syndromes. Blood Rev. 2016;30(5):349–56. [DOI] [PubMed] [Google Scholar]
- 38. Elli EM, Belotti A, Aroldi A, Parma M, Pioltelli P, Pogliani EM. Iron chelation therapy with deferasirox in the management of iron overload in primary myelofibrosis. Mediterr J Hematol Infect Dis. 2014;6(1):e2014042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miano M, Dufour C. The diagnosis and treatment of aplastic anemia: a review. Int J Hematol. 2015;101(6):527–35. [DOI] [PubMed] [Google Scholar]
- 40. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430685/ [Google Scholar]
- 41. Kroll MH, Yee C, Rojas Hernandez CM. Hematologic complications of immune checkpoint inhibitors. Blood. 2021. 10.1182/blood.2020009016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park AK, Waheed A, Forst DA, Al‐Samkari H. Characterization and prognosis of temozolomide‐induced aplastic anemia in patients with central nervous system malignancies. Neuro Oncol. 2021. 10.1093/neuonc/noab240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Du Y, Long Z, Chen M, Han B, Hou B, Feng F. Observational monitoring of patients with aplastic anemia and low/intermediate‐1 risk of myelodysplastic syndromes complicated with iron overload. Acta Haematol. 2017;138(2):119–28. [DOI] [PubMed] [Google Scholar]
- 44. Camaschella C. Hereditary sideroblastic anemias: pathophysiology, diagnosis, and treatment. Semin Hematol. 2009;46(4):371–7. [DOI] [PubMed] [Google Scholar]
- 45. Diaz‐de‐Heredia C, Bresters D, Faulkner L, Yesilipek A, Strahm B, Miano M, et al. Recommendations on hematopoietic stem cell transplantation for patients with Diamond‐Blackfan anemia. On behalf of the Pediatric Diseases and Severe Aplastic Anemia Working Parties of the EBMT. Bone Marrow Transplant. 2021;56(12):2956–63. [DOI] [PubMed] [Google Scholar]
- 46. Roggero S, Quarello P, Vinciguerra T, Longo F, Piga A, Ramenghi U. Severe iron overload in Blackfan‐Diamond anemia: a case–control study. Am J Hematol. 2009;84(11):729–32. [DOI] [PubMed] [Google Scholar]
- 47. Assis‐Mendonça GR, Cunha‐Silva M, Fernandes MF, Torres LD, de Almeida Verissimo MP, MTN O, et al. Massive iron overload and acute‐on‐chronic liver failure in a patient with Diamond‐Blackfan anaemia: a case report. BMC Gastroenterol. 2020;20(1):332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Beers EJ, van Straaten S, Morton DH, Barcellini W, Eber SW, Glader B, et al. Prevalence and management of iron overload in pyruvate kinase deficiency: report from the Pyruvate Kinase Deficiency Natural History Study. Haematologica. 2019;104(2):e51–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Finkenstedt A, Bianchi P, Theurl I, Vogel W, Witcher DR, Wroblewski VJ, et al. Regulation of iron metabolism through GDF15 and hepcidin in pyruvate kinase deficiency. Br J Haematol. 2009;144(5):789–93. [DOI] [PubMed] [Google Scholar]
- 50. Grace RF, Bianchi P, van Beers EJ, Eber SW, Glader B, Yaish HM, et al. Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood. 2018;131(20):2183–92. [DOI] [PubMed] [Google Scholar]
- 51. Chartier ME, Hart L, Paganelli M, Ahmed N, Bilodeau M, Alvarez F. Successful liver transplants for liver failure associated with pyruvate kinase deficiency. Pediatrics. 2018;141(Suppl 5):S385–9. [DOI] [PubMed] [Google Scholar]
- 52. Milic S, Mikolasevic I, Orlic L, Devcic E, Starcevic‐Cizmarevic N, Stimac D, et al. The role of iron and iron overload in chronic liver disease. Med Sci Monit. 2016;22:2144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Datz C, Felder TK, Niederseer D, Aigner E. Iron homeostasis in the metabolic syndrome. Eur J Clin Invest. 2013;43(2):215–24. [DOI] [PubMed] [Google Scholar]
- 54. Handa P, Morgan‐Stevenson V, Maliken BD, Nelson JE, Washington S, Westerman M, et al. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am J Physiol Gastrointest Liver Physiol. 2016;310(2):G117–27. [DOI] [PubMed] [Google Scholar]
- 55. Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp‐Arida A, et al. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology. 2011;53(2):448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maliken BD, Nelson JE, Klintworth HM, Beauchamp M, Yeh MM, Kowdley KV. Hepatic reticuloendothelial system cell iron deposition is associated with increased apoptosis in nonalcoholic fatty liver disease. Hepatology. 2013;57(5):1806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Handa P, Vemulakonda AL, Maliken BD, Morgan‐Stevenson V, Nelson JE, Dhillon BK, et al. Differences in hepatic expression of iron, inflammation and stress‐related genes in patients with nonalcoholic steatohepatitis. Ann Hepatol. 2017;16(1):77–85. [DOI] [PubMed] [Google Scholar]
- 58. Rametta R, Dongiovanni P, Pelusi S, Francione P, Iuculano F, Borroni V, et al. Hepcidin resistance in dysmetabolic iron overload. Liver Int. 2016;36(10):1540–8. [DOI] [PubMed] [Google Scholar]
- 59. Aigner E, Theurl I, Theurl M, Lederer D, Haufe H, Dietze O, et al. Pathways underlying iron accumulation in human nonalcoholic fatty liver disease. Am J Clin Nutr. 2008;87(5):1374–83. [DOI] [PubMed] [Google Scholar]
- 60. Hoki T, Miyanishi K, Tanaka S, Takada K, Kawano Y, Sakurada A, et al. Increased duodenal iron absorption through up‐regulation of divalent metal transporter 1 from enhancement of iron regulatory protein 1 activity in patients with nonalcoholic steatohepatitis. Hepatology. 2015;62(3):751–61. [DOI] [PubMed] [Google Scholar]
- 61. Di Bisceglie AM, Axiotis CA, Hoofnagle JH, Bacon BR. Measurements of iron status in patients with chronic hepatitis. Gastroenterology. 1992;102(6):2108–13. [DOI] [PubMed] [Google Scholar]
- 62. Hézode C, Cazeneuve C, Coué O, Roudot‐Thoraval F, Lonjon I, Bastie A, et al. Liver iron accumulation in patients with chronic active hepatitis C: prevalence and role of hemochromatosis gene mutations and relationship with hepatic histological lesions. J Hepatol. 1999;31(6):979–84. [DOI] [PubMed] [Google Scholar]
- 63. Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y, et al. Hepatitis C virus‐induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology. 2008;134(1):226–38. [DOI] [PubMed] [Google Scholar]
- 64. Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus‐induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48(5):1420–9. [DOI] [PubMed] [Google Scholar]
- 65. Price L, Kowdley KV. The role of iron in the pathophysiology and treatment of chronic hepatitis C. Can J Gastroenterol. 2009;23(12):822–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sebastiani G, Tempesta D, Alberti A. Hepatic iron overload is common in chronic hepatitis B and is more severe in patients coinfected with hepatitis D virus. J Viral Hepat. 2012;19(2):e170–6. [DOI] [PubMed] [Google Scholar]
- 67. Martinelli AL, Franco RF, Villanova MG, Figueiredo JF, Secaf M, Tavella MH, et al. Are haemochromatosis mutations related to the severity of liver disease in hepatitis C virus infection? Acta Haematol. 2000;102(3):152–6. [DOI] [PubMed] [Google Scholar]
- 68. Tung BY, Emond MJ, Bronner MP, Raaka SD, Cotler SJ, Kowdley KV. Hepatitis C, iron status, and disease severity: relationship with HFE mutations. Gastroenterology. 2003;124(2):318–26. [DOI] [PubMed] [Google Scholar]
- 69. Geier A, Reugels M, Weiskirchen R, Wasmuth HE, Dietrich CG, Siewert E, et al. Common heterozygous hemochromatosis gene mutations are risk factors for inflammation and fibrosis in chronic hepatitis C. Liver Int. 2004;24(4):285–94. [DOI] [PubMed] [Google Scholar]
- 70. Erhardt A, Maschner‐Olberg A, Mellenthin C, Kappert G, Adams O, Donner A, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003;38(3):335–42. [DOI] [PubMed] [Google Scholar]
- 71. Piperno A, Vergani A, Malosio I, Parma L, Fossati L, Ricci A, et al. Hepatic iron overload in patients with chronic viral hepatitis: role of HFE gene mutations. Hepatology. 1998;28(4):1105–9. [DOI] [PubMed] [Google Scholar]
- 72. Nelson JE, Bhattacharya R, Lindor KD, Chalasani N, Raaka S, Heathcote EJ, et al. HFE C282Y mutations are associated with advanced hepatic fibrosis in Caucasians with nonalcoholic steatohepatitis. Hepatology. 2007;46(3):723–9. [DOI] [PubMed] [Google Scholar]
- 73. Bugianesi E, Manzini P, D'Antico S, Vanni E, Longo F, Leone N, et al. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology. 2004;39(1):179–87. [DOI] [PubMed] [Google Scholar]
- 74. Raszeja‐Wyszomirska J, Kurzawski G, Lawniczak M, Miezynska‐Kurtycz J, Lubinski J. Nonalcoholic fatty liver disease and HFE gene mutations: a Polish study. World J Gastroenterol. 2010;16(20):2531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Harrison‐Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, et al. Alcohol metabolism‐mediated oxidative stress down‐regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006;281(32):22974–82. [DOI] [PubMed] [Google Scholar]
- 76. Bridle K, Cheung TK, Murphy T, Walters M, Anderson G, Crawford DG, et al. Hepcidin is down‐regulated in alcoholic liver injury: implications for the pathogenesis of alcoholic liver disease. Alcohol Clin Exp Res. 2006;30(1):106–12. [DOI] [PubMed] [Google Scholar]
- 77. Czaja AJ. Review article: iron disturbances in chronic liver diseases other than haemochromatosis ‐ pathogenic, prognostic, and therapeutic implications. Aliment Pharmacol Ther. 2019;49(6):681–701. [DOI] [PubMed] [Google Scholar]
- 78. Machado MV, Ravasco P, Martins A, Almeida MR, Camilo ME, Cortez‐Pinto H. Iron homeostasis and H63D mutations in alcoholics with and without liver disease. World J Gastroenterol. 2009;15(1):106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gleeson D, Evans S, Bradley M, Jones J, Peck RJ, Dube A, et al. HFE genotypes in decompensated alcoholic liver disease: phenotypic expression and comparison with heavy drinking and with normal controls. Am J Gastroenterol. 2006;101(2):304–10. [DOI] [PubMed] [Google Scholar]
- 80. Costa‐Matos L, Batista P, Monteiro N, Henriques P, Girão F, Carvalho A. Hfe mutations and iron overload in patients with alcoholic liver disease. Arq Gastroenterol. 2013;50(1):35–41. [DOI] [PubMed] [Google Scholar]
- 81. Grove J, Daly AK, Burt AD, Guzail M, James OFW, Bassendine MF, et al. Heterozygotes for HFE mutations have no increased risk of advanced alcoholic liver disease. Gut. 1998;43(2):262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Malcovati L, Porta MG, Pascutto C, Invernizzi R, Boni M, Travaglino E, et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol. 2005;23(30):7594–603. [DOI] [PubMed] [Google Scholar]
- 83. Takatoku M, Uchiyama T, Okamoto S, Kanakura Y, Sawada K, Tomonaga M, et al. Retrospective nationwide survey of Japanese patients with transfusion‐dependent MDS and aplastic anemia highlights the negative impact of iron overload on morbidity/mortality. Eur J Haematol. 2007;78(6):487–94. [DOI] [PubMed] [Google Scholar]
- 84. Goldberg SL, Chen E, Corral M, Guo A, Mody‐Patel N, Pecora AL, et al. Incidence and clinical complications of myelodysplastic syndromes among United States Medicare beneficiaries. J Clin Oncol. 2010;28(17):2847–52. [DOI] [PubMed] [Google Scholar]
- 85. Di Tucci AA, Matta G, Deplano S, Gabbas A, Depau C, Derudas D, et al. Myocardial iron overload assessment by T2* magnetic resonance imaging in adult transfusion dependent patients with acquired anemias. Haematologica. 2008;93(9):1385–8. [DOI] [PubMed] [Google Scholar]
- 86. Kirk P, Roughton M, Porter JB, Walker JM, Tanner MA, Patel J, et al. Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassemia major. Circulation. 2009;120(20):1961–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Taher AT, Musallam KM, Karimi M, el‐Beshlawy A, Belhoul K, Daar S, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–92. [DOI] [PubMed] [Google Scholar]
- 88. Taher AT, Porter JB, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, et al. Defining serum ferritin thresholds to predict clinically relevant liver iron concentrations for guiding deferasirox therapy when MRI is unavailable in patients with non‐transfusion‐dependent thalassaemia. Br J Haematol. 2015;168(2):284–90. [DOI] [PubMed] [Google Scholar]
- 89. Cancado R, Watman NP, Lobo C, Chona Z, Manzur F, Traina F, et al. Assessment of liver and cardiac iron overload using MRI in patients with chronic anemias in Latin American countries: results from ASIMILA study. Hematology. 2018;23(9):676–82. [DOI] [PubMed] [Google Scholar]
- 90. Aydinok Y, Porter JB, Piga A, Elalfy M, el‐Beshlawy A, Kilinç Y, et al. Prevalence and distribution of iron overload in patients with transfusion‐dependent anemias differs across geographic regions: results from the CORDELIA study. Eur J Haematol. 2015;95(3):244–53. [DOI] [PubMed] [Google Scholar]
- 91. Beaton MD, Adams PC. Treatment of hyperferritinemia. Ann Hepatol. 2012;11(3):294–300. [PubMed] [Google Scholar]
- 92. Bassett ML, Halliday JW, Ferris RA, Powell LW. Diagnosis of hemochromatosis in young subjects: predictive accuracy of biochemical screening tests. Gastroenterology. 1984;87(3):628–33. [PubMed] [Google Scholar]
- 93. Westphalen AC, Qayyum A, Yeh BM, Merriman RB, Lee JA, Lamba A, et al. Liver fat: effect of hepatic iron deposition on evaluation with opposed‐phase MR imaging. Radiology. 2007;242(2):450–5. [DOI] [PubMed] [Google Scholar]
- 94. Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS, American Association for the Study of Liver Diseases . Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(1):328–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sirlin CB, Reeder SB. Magnetic resonance imaging quantification of liver iron. Magn Reson Imaging Clin N Am. 2010;18(3):359–81, ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Labranche R, Gilbert G, Cerny M, Vu KN, Soulières D, Olivié D, et al. Liver iron quantification with MR imaging: a primer for radiologists. Radiographics. 2018;38(2):392–412. [DOI] [PubMed] [Google Scholar]
- 97. Kowdley KV, Brown KE, Ahn J, Sundaram V. ACG clinical guideline: hereditary hemochromatosis. Am J Gastroenterol. 2019;114(8):1202–18. Erratum in: Am J Gastroenterol. 2019;114(12):1927. [DOI] [PubMed] [Google Scholar]
- 98. Kernan KF, Carcillo JA. Hyperferritinemia and inflammation. Int Immunol. 2017;29(9):401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ferrante M, Geubel AP, Fevery J, Marogy G, Horsmans Y, Nevens F. Hereditary hyperferritinaemia‐cataract syndrome: a challenging diagnosis for the hepatogastroenterologist. Eur J Gastroenterol Hepatol. 2005;17(11):1247–53. [DOI] [PubMed] [Google Scholar]
- 100. Moore C, Ormseth M, Fuchs H. Causes and significance of markedly elevated serum ferritin levels in an academic medical center. J Clin Rheumatol. 2013;19(6):324–8. [DOI] [PubMed] [Google Scholar]
- 101. Senjo H, Higuchi T, Okada S, Takahashi O. Hyperferritinemia: causes and significance in a general hospital. Hematology. 2018;23(10):817–22. [DOI] [PubMed] [Google Scholar]
- 102. Mehta B, Efthimiou P. Ferritin in adult‐onset still's disease: just a useful innocent bystander? Int J Inflam. 2012;2012:298405. Erratum in: Int J Inflam. 2012;2012:254858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM. Role for NF‐kappa B in the regulation of ferritin H by tumor necrosis factor‐alpha. J Biol Chem. 1995;270(25):15285–93. [DOI] [PubMed] [Google Scholar]
- 104. Poggiali E, Cassinerio E, Zanaboni L, Cappellini MD. An update on iron chelation therapy. Blood Transfus. 2012;10(4):411–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tanner MA, Galanello R, Dessi C, Smith GC, Westwood MA, Agus A, et al. A randomized, placebo‐controlled, double‐blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation. 2007;115(14):1876–84. [DOI] [PubMed] [Google Scholar]
- 106. Pantalone GR, Maggio A, Vitrano A, Capra M, Cuccia L, Gagliardotto F, et al. Sequential alternating deferiprone and deferoxamine treatment compared to deferiprone monotherapy: main findings and clinical follow‐up of a large multicenter randomized clinical trial in ‐thalassemia major patients. Hemoglobin. 2011;35(3):206–16. [DOI] [PubMed] [Google Scholar]
- 107. Deugnier Y, Turlin B, Ropert M, Cappellini MD, Porter JB, Giannone V, et al. Improvement in liver pathology of patients with β‐thalassemia treated with deferasirox for at least 3 years. Gastroenterology. 2011;141(4):1202–11, 1211.e1‐3. Erratum in: Gastroenterology. 2012;142(1):186. [DOI] [PubMed] [Google Scholar]
- 108. Leitch HA, Parmar A, Wells RA, Chodirker L, Zhu N, Nevill TJ, et al. Overall survival in lower IPSS risk MDS by receipt of iron chelation therapy, adjusting for patient‐related factors and measuring from time of first red blood cell transfusion dependence: an MDS‐CAN analysis. Br J Haematol. 2017;179(1):83–97. [DOI] [PubMed] [Google Scholar]
- 109. Angelucci E, Greenberg P, Izquierdo M, Garcia‐Manero G. Iron chelation in transfusion‐dependent patients with low‐ to intermediate‐1‐risk myelodysplastic syndromes. Ann Intern Med. 2020;173(7):595–6. [DOI] [PubMed] [Google Scholar]
- 110. Cheong JW, Kim HJ, Lee KH, Yoon SS, Lee JH, Park HS, et al. Deferasirox improves hematologic and hepatic function with effective reduction of serum ferritin and liver iron concentration in transfusional iron overload patients with myelodysplastic syndrome or aplastic anemia. Transfusion. 2014;54(6):1542–51. [DOI] [PubMed] [Google Scholar]
- 111. Ko BS, Chang MC, Chiou TJ, Chang TK, Chen YC, Lin SF, et al. Long‐term safety and efficacy of deferasirox in patients with myelodysplastic syndrome, aplastic anemia and other rare anemia in Taiwan. Hematology. 2019;24(1):247–54. [DOI] [PubMed] [Google Scholar]
- 112. List AF, Baer MR, Steensma DP, Raza A, Esposito J, Martinez‐Lopez N, et al. Deferasirox reduces serum ferritin and labile plasma iron in RBC transfusion‐dependent patients with myelodysplastic syndrome. J Clin Oncol. 2012;30(17):2134–9. [DOI] [PubMed] [Google Scholar]
- 113. Rose C, Brechignac S, Vassilief D, Pascal L, Stamatoullas A, Guerci A, et al. Does iron chelation therapy improve survival in regularly transfused lower risk MDS patients? A multicenter study by the GFM (Groupe Francophone des Myélodysplasies). Leuk Res. 2010;34(7):864–70. [DOI] [PubMed] [Google Scholar]
- 114. Gattermann N, Finelli C, Porta MD, Fenaux P, Ganser A, Guerci‐Bresler A, et al. Deferasirox in iron‐overloaded patients with transfusion‐dependent myelodysplastic syndromes: results from the large 1‐year EPIC study. Leuk Res. 2010;34(9):1143–50. [DOI] [PubMed] [Google Scholar]
- 115. Angelucci E, Santini V, Di Tucci AA, Quaresmini G, Finelli C, Volpe A, et al. Deferasirox for transfusion‐dependent patients with myelodysplastic syndromes: safety, efficacy, and beyond (GIMEMA MDS0306 Trial). Eur J Haematol. 2014;92(6):527–36. [DOI] [PubMed] [Google Scholar]
- 116. Elli EM, Iurlo A, Aroldi A, Caramella M, Malato S, Casartelli E, et al. Deferasirox in the management of iron‐overload in patients with myelofibrosis: a multicentre study from the Rete Ematologica Lombarda (IRON‐M study). Br J Haematol. 2019;186:e123–6. [DOI] [PubMed] [Google Scholar]
- 117. Elalfy MS, Adly AM, Wali Y, Tony S, Samir A, Elhenawy YI. Efficacy and safety of a novel combination of two oral chelators deferasirox/deferiprone over deferoxamine/deferiprone in severely iron overloaded young beta thalassemia major patients. Eur J Haematol. 2015;95(5):411–20. [DOI] [PubMed] [Google Scholar]
- 118. Valenti L, Fracanzani AL, Dongiovanni P, Bugianesi E, Marchesini G, Manzini P, et al. Iron depletion by phlebotomy improves insulin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: evidence from a case–control study. Am J Gastroenterol. 2007;102(6):1251–8. [DOI] [PubMed] [Google Scholar]
- 119. Valenti L, Fracanzani AL, Dongiovanni P, Rovida S, Rametta R, Fatta E, et al. A randomized trial of iron depletion in patients with nonalcoholic fatty liver disease and hyperferritinemia. World J Gastroenterol. 2014;20(11):3002–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Adams LA, Crawford DH, Stuart K, House MJ, St. Pierre TG, Webb M, et al. The impact of phlebotomy in nonalcoholic fatty liver disease: a prospective, randomized, controlled trial. Hepatology. 2015;61(5):1555–64. [DOI] [PubMed] [Google Scholar]