

Abstract
Laminin-alpha2-related muscular dystrophy (LAMA2-MD) is a genetic condition due to reduced LAMA2, a protein found throughout the nervous system. Late-onset LAMA2-MD may present with proximal muscle weakness, joint contractures, neuropathy, epilepsy and/or cardiorespiratory issues, and is less common than the neonatal form. We describe a novel phenotype of LAMA2-MD with progressive myelopathy and spinal cord abnormalities.
A woman was referred for evaluation of multiple sclerosis (MS) with progressive gait difficulty and abnormal neuroimaging showing white matter abnormalities in the brain and spinal cord. Ancillary testing was not consistent with primary neuroinflammatory disorders, systemic autoimmunity or infection. Metabolic workup revealed low cyanocobalamin. Genetic testing identified two LAMA2-MD variants.
Genetic disorders can mimic treatable neurological conditions. Chronic progressive course, involvement of the peripheral and central nervous systems, and confluent white matter abnormalities should be investigated with molecular testing that includes LAMA2 sequencing to ensure proper diagnosis and management.
Keywords: multiple sclerosis, muscle disease, neuro genetics, neuroimaging, neuromuscular disease
Background
Given the increased prevalence of genetic testing within the field of neurology,1 provider awareness of single gene diseases mimicking treatable neurological diseases, such as multiple sclerosis (MS), is imperative.
Laminin-alpha2 (LAMA2) is found in the alpha2 subunit of laminin-211, a merosin protein2 found in the basement membrane of the peripheral and central nervous system.3 4 When laminin-alpha2 is absent or deficient, laminin-alpha2-related muscular dystrophy (LAMA2-MD) may ensue. LAMA2-MD is responsible for up to 30% of congenital muscular dystrophies and most commonly presents in neonates as hypotonia with failure to thrive (MDC1A variant).5 Late-onset LAMA2-MD is typically less severe, and despite initially preserved ambulation, may cause proximal muscle weakness, joint contractures, demyelinating neuropathy, epilepsy and/or cardiorespiratory issues.2–7
Characteristic brain abnormalities on MRI are common in early and late-onset LAMA2-MD, including diffuse T2 periventricular and confluent subcortical white matter hyperintensities.5 6 Although the laminin-alpha2 protein is found in the spinal cord of mice and humans,3 there does not appear to be reported cases of clinical myelopathy or radiological involvement of the spinal cord. The current report herein describes a novel presentation of LAMA2-MD with progressive myelopathy, originally referred to clinic with possible MS.
Case presentation
A white woman in her late 40s presented for evaluation of progressive gait difficulty associated with an abnormal MRI brain and spine suspicious for progressive MS.
In her 20s, the patient developed vertigo, followed by paresthesia of the extremities without weakness. The symptoms spontaneously resolved after 4 months. Migraines developed in her early 30s, new paresthesia of the hands by her mid-30s, and subjective cognitive decline by her mid-40s. Beginning 2 years prior to her presentation, she developed facial paresthesia, generalised weakness (right greater than left) and imbalance. The weakness slowly progressed over time along with progressive gait difficulty. Review of systems was significant for insomnia, depression and palpitations. Medical and surgical history consisted of bigeminy status post atrioventricular node ablation, 33 pack-year history of smoking, surgery for a high arch foot. Relevant immediate family history consisted of cataracts. Relevant remote family history included amyotrophic lateral sclerosis and stroke after bypass surgery.
General physical examination revealed bilateral high arched feet. Neurological examination showed rightward horizontal nystagmus, hypometric saccades, reduced bulk throughout with normal tone and mild symmetric pyramidal weakness in shoulder abduction, finger extension, hip flexion and ankle dorsiflexion. She had reduced vibration at both halluces with absent and reduced proprioception in the right and left great toes, respectively. Patellar and brachioradialis reflexes were absent, others were normal. Gait revealed exaggerated pelvic girdle distribution of weight, difficulty with tandem walking and inability to walk on heels and toes. Given extraocular movement abnormalities, upper motor neuron pattern of weakness and relative hyporeflexia, there was concern for a multifocal condition indicating both upper and lower motor neuron involvement.
Investigations
Prior workup at an outside facility was most significant for abnormal MRI and elevated protein in the cerebrospinal fluid (CSF). MRI brain exhibited confluent periventricular and subcortical white matter abnormalities without enhancement (figure 1A–C). Spinal cord MRI consisted of confluent posterior T2 hyperintensities of the cervicomedullary junction to C6, anterior C3-5, dorsal-central T4-T9, and central T10-T11, without enhancement. Routine CSF constituents were unremarkable other than elevated protein (85 mg/dL). Oligoclonal bands were negative.
Figure 1.
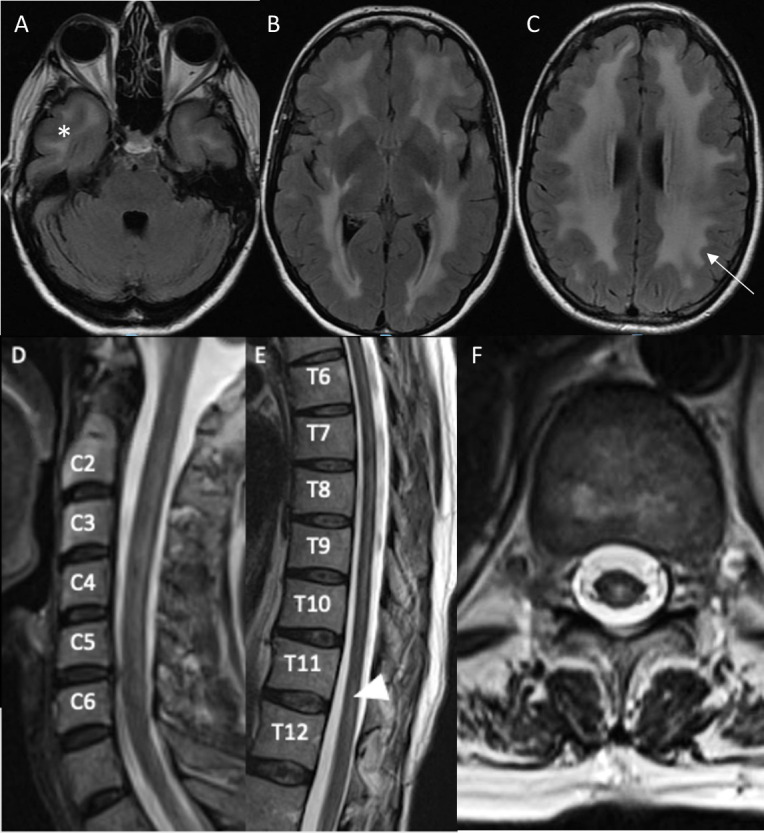
MRI of the brain (panels A–C) 1 year prior to presentation and MRI of the cervical (panel D) and thoracic (panel E–F) spine at time of presentation. Post-gadolinium images are not shown as they were unremarkable. T2-weighted axial fluid-attenuated inversion recovery (FLAIR) images of the brain reveal symmetric confluent white matter hyperintensities in the bilateral anterior temporal lobes (A, asterisk), deep subcortical (B) periventricular and juxtacortical (C) regions of the brain with sparing of most of the occipital lobes and posterior fossa. The U fibres are mostly involved (C, arrow). The sagittal T2-weighted image of the cervical spine reveals a patchy extensive lesion most confluent from C3-5 (D). The sagittal thoracic spinal cord T2-weighted image reveals a longitudinally extensive hyperintense lesion most prominent from T4-T9 (partially seen in F) and a hyperintense lesion at T11-12 (E, F, arrow head) that are central dorsal in location, as corroborated on T2-weighted axial (F) image.
The constellation of upper and lower motor neuron involvement on examination, imaging findings and non-specific CSF was atypical for most neuroimmunological disorders. Further extensive workup was pursued. Autoimmune, infectious, paraneoplastic and neuroinflammatory evaluations were unremarkable. Serum investigation revealed the following; cyanocobalamin (B12) deficiency (151 pg/mL), vitamin D deficiency (23 ng/mL), elevated lactate (3.6 mmol/L), elevated glycine and alanine, elevated alanine to lysine ratio and increased total cholesterol (257 mg/dL). Erythrocyte sedimentation rate, C-reactive protein, alkaline phosphatase and aspartate transaminase were mildly elevated (table 1). Electromyogram and nerve conduction study (EMG/NCS) revealed a length dependent, sensory greater than motor, demyelinating polyneuropathy. Genetic testing for cerebral autosomal recessive and dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL and CADASIL) gene testing was negative. Optical coherence tomography (OCT) revealed evidence of subclinical optic neuropathy bilaterally. Repeat brain MRI exhibited confluent periventricular and subcortical white matter abnormalities without contrast enhancement. Spinal cord MRI (figure 1D–F) revealed confluent posterior T2 hyperintensities of the cervicomedullary junction to C6, anterior C3-5, dorsal-central T4-T9 and central T10-T11. Relative to prior MRIs (9 years before), there was mild progression of the number and extent of white matter hyperintensities in both the brain and spinal cord. Pertinent results are summarised in table 1.
Table 1.
Abnormal results of workup performed at Johns Hopkins Hospital
| Category | Tests with abnormal results (reference range) |
| Routine | Alkaline phosphatase: 183 (30–120 U/L) Aspartate transaminase: 38 (0–31 U/L) |
| Nutritional | Vitamin B12: 151 (232–1245 pg/mL) Vitamin D: 23 (30–100 ng/mL) |
| Metabolic | Total cholesterol: 257 mg/dL (0–200 mg/dL) Lactic acid: 3.6 mmol/L (0.5–2.0 mmol/L) Amino acid chromatography: Increased glycine, increased alanine, increased alanine to lysine |
| Immunologic/inflammatory | ESR: 26 (4–25 mm/hour) CRP: 1.2 (<0.5 g/dL) Total complement CH50: >60 U/mL (31–60 U/mL) IgG2: 154 mg/dL (242–700 mg/dL) IgG4: 3.4 mg/dL (3.9–86.4 mg/dL) SPEP: Gamma region decreased |
| Infectious | JCV Antibody Index: 1.89 (<0.20 negative, >0.40 positive) |
| Genetic | WES: compound heterozygosity for two variants of LAMA2-MD (c.526 T>C, p.C176R and c.443G>, p.R148Q) |
| Ancillary tests | OCT: Evidence of bilateral optic neuropathy EMG/NCS: Demyelinating length dependent polyneuropathy |
| Imaging | MRI brain, cervical, thoracic spine with and without contrast (figure 1) |
Abnormal results of diagnostic workup that was completed at Johns Hopkins Hospital. Laboratory values for normal results are not listed but included the following: Routine tests: Complete blood count, comprehensive metabolic panel. Nutritional labs: Copper, ceruloplasmin, zinc vitamins (B1, B6, B12, D, E), folate, homocysteine, methylmalonic acid. Metabolic: Thyroid stimulating hormone, thyroxine, haemoglobin A1C, lactic acid, pyruvic acid, free fatty acids, essential fatty acids, very long chain fatty acids, arylsulfatase, alpha galactosidase triglycerides, amino acid chromatography, lipid panel. Immunologic/inflammatory: Antibodies: antinuclear (ANA), antineutrophil cytoplasmic (ANCA), perinuclear antibody, cytoplasmic, double-stranded DNA, ribonucleotide protein, smith, Ro 52, Ro 60, La, scleroderma (Scl-70), cyclic citrullinated peptide 3, thyroid peroxidase, histone, glutamic acid decarboxylase-65, Beta2-Glycoprotein (IgG, IgM, IgA), cardiolipin (APL/MPL/GPL), NMO, MOG, Mayo Clinic paraneoplastic panel. Other: erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), rheumatoid factor, DRVVT, total complement CH50, C3, C4, ACE, immunoglobulins A, M, G (including subclasses), serum protein electrophoresis (SPEP), kappa and lambda free light chains. Infectious: Treponema pallidum CL antibody, Burgdorferi antibody, Whipples PCR, varicella zoster virus IgG, hepatitis C antibody, hepatitis B surface antigen, hepatitis B core antibody, total hepatitis B surface antibody, T Spot tuberculosis, HIV 1 and 2, JC virus antibody index. Genetic: Notch 3/CADASIL, HTRA1/CARASIL, GLA gene test for Fabry’s, mitochondrial DNA Screen, whole exome sequencing (WES).
Differential diagnosis
Given the potential relapsing-remitting symptoms over the years with more recent progression, brain and spinal cord abnormalities, and subclinical optic neuropathy, a neuroinflammatory condition, such as secondary progressive MS, was considered. However, confluent brain and longitudinally extensive spinal cord findings are not consistent with MS in which lesions are more typically discrete and multifocal. MS lesions can accrue and appear more confluent overtime mimicking longitudinally extensive lesions more often observed in neuromyelitis optica and myelin oligodendrocyte glycoprotein associated disease. However, the clinical course and negative results for relevant associated antibody tests made these differentials unlikely. In addition, the CSF profile was non-specific and less supportive of a neuroinflammatory condition.
Additionally, the presence of peripheral neuropathy and metabolic abnormalities on testing suggested a more diffuse process. Vitamin B12 deficiency was the most remarkable abnormality found and may be associated with subacute combined degeneration of the spinal cord (demyelination of lateral corticospinal tracts and posterior columns,8 9 peripheral neuropathy and optic neuropathy,10 among other neurological abnormalities). However, this would less likely explain the anterior and central cord findings on MRI. Another consideration, given the diffuse nature, was a paraneoplastic process. Although the decade-long duration of symptoms without declaration of a malignancy and negative paraneoplastic antibody panel made this extremely unlikely. Additional autoimmune and/or infectious disorders were also deemed highly unlikely based on history and testing.
The confluent, diffuse nature of white matter involvement in the brain alone, and in combination with a chronic progressive course raised suspicion for a leukodystrophy or mitochondrial disorder. Migraines, memory changes, white matter abnormalities suggested CADASIL or CARASIL. However, the lack of lacunar infarcts on MRI and negative genetic testing made these unlikely. Mitochondrial DNA sequencing was also negative (despite minimally abnormal metabolic labs). Ultimately, whole exome sequencing (WES) was pursued and provided the diagnosis.
Treatment
Although the pattern of spinal cord affliction would be highly atypical of vitamin B12 deficiency, a partial contribution from vitamin B12 deficiency cannot be excluded and supplementation was recommended. Also advised was continued follow-up with primary care, cardiology, neuropsychiatry, rehabilitation and genetics.
Outcome and follow-up
WES identified compound heterozygosity variants (c.526T>C, p.C176R and c.443G>A, p.R148Q) of uncertain significance associated with LAMA2. Similar to the classically described phenotype of late-onset LAMA2-MD, this patient had a demyelinating neuropathy, characteristic white matter abnormalities of the brain, muscle weakness and a cardiac conduction abnormality. Although muscle biopsy and immunohistochemistry may provide additional supportive information, the abnormality may not be profound enough to be detected.4 5
Our conclusion is that the spinal cord abnormalities and resultant progressive myelopathy are most likely related to the underlying late-onset LAMA2-MD. Although not previously described, the LAMA2 protein is known to be present within the spinal cord of mice and humans and thus its presence and association with a progressive myelopathy is possible. She has remained neurologically stable and continues to follow with multispecialty providers for supportive care and monitoring.
Discussion
This case report suggests a novel presentation of late-onset LAMA2-MD consisting of progressive myelopathy with associated white matter abnormalities on spinal MRI. Given the historical paucity of spinal cord involvement with LAMA2-MD, the possibility of two conditions was considered. After extensive workup, a concomitant B12 deficiency was thought to be a possible contributor to optic and peripheral neuropathy and myelopathy but could not fully explain the entire presentation nor MRI findings. Therefore, our final conclusion was that this patient’s presentation, and associated genotype, represents a novel presentation of late-onset LAMA2-MD.
Neurogenetics is becoming increasingly accessible and relevant within the field of neurology.1 As such, more variants are being recognised that have uncertain clinical significance. It is imperative to combine molecular findings with the clinical picture to determine the pathological significance of variants identified. In LAMA2-MD, there are significant white matter abnormalities that occur in almost all affected individuals.5 6 This may prompt referral to a subspecialist, such as a neuroimmunologist. However, providers who see patients with neurological complaints (regardless of specialty) should remain vigilant regarding single gene diseases that can clinically or radiologically mimic treatable neurological conditions. Extensive workup may be necessary to help guide the differential and/or rule out concomitant diagnoses. History, examination and ancillary testing are imperative to ensure proper diagnosis. If diagnostic uncertainty remains after thorough evaluation, genetic testing and/or clinical genetics referral may be necessary to ensure proper counselling, diagnosis, management and monitoring. Recognising and diagnosing neurological single gene diseases are particularly important when considering the prospect of future treatments that may become available.5 11 12
Learning points.
A chronic progressive course, involvement of the peripheral and central nervous systems, and confluent white matter abnormalities should raise suspicion for a genetic aetiology.
Although rare, genetic disorders can mimic multiple sclerosis (MS) and other common neurological conditions, in both clinical presentation and imaging.
Genetics assessment may be necessary to ensure proper counselling, diagnosis, management and monitoring.
The landscape for treatment options for genetic conditions is evolving, highlighting the importance of their recognition.
Footnotes
Contributors: All authors contributed to patient care. SS conceived the plan for the case report. JK wrote the manuscript and prepared figures and tables with support and editing from NdMS and SS.
Funding: This study was funded by the National Human Genome Research Institute/National Heart, Lung, and Blood Institute (grant number: NHGRI/NHLBI UM1 HG006542); National Multiple Sclerosis Society (grant number: RG-1606-08768 and RG-2001-36011).
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Obtained.
References
- 1.Jayadev S, Smith CO, Bird TD. Neurogenetics. Neurol Clin Pract 2011;1:41–8. 10.1212/CPJ.0b013e31823c0f5f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarkozy A, Foley AR, Zambon AA, et al. Lama2-Related dystrophies: clinical phenotypes, disease biomarkers, and clinical trial readiness. Front Mol Neurosci 2020;13:123. 10.3389/fnmol.2020.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arreguin AJ, Colognato H. Brain dysfunction in Lama2-related congenital muscular dystrophy: lessons from human case reports and mouse models. Front Mol Neurosci 2020;13:118. 10.3389/fnmol.2020.00118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen Q, Lim KRQ, Yokota T. Current understanding and treatment of cardiac and skeletal muscle pathology in laminin-α2 chain-deficient congenital muscular dystrophy]]>. TACG 2019;12:113–30. 10.2147/TACG.S187481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliveira J, Gruber A, Cardoso M, et al. LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum Mutat 2018;39:1314–37. 10.1002/humu.23599 [DOI] [PubMed] [Google Scholar]
- 6.Oliveira J, Freixo JP, Santos M, et al. Lama2 muscular dystrophy. muscular dystrophy;25. [Google Scholar]
- 7.Natera-de Benito D, Muchart J, Itzep D, et al. Epilepsy in Lama2-related muscular dystrophy: an electro-clinico-radiological characterization. Epilepsia 2020;61:971–83. 10.1111/epi.16493 [DOI] [PubMed] [Google Scholar]
- 8.Ravina B, Loevner LA, Bank W. Mr findings in subacute combined degeneration of the spinal cord. American Journal of Roentgenology 2000;174:863–5. 10.2214/ajr.174.3.1740863 [DOI] [PubMed] [Google Scholar]
- 9.Yamada K, Shrier DA, Tanaka H, et al. A case of subacute combined degeneration: MRI findings. Neuroradiology 1998;40:398–400. 10.1007/s002340050610 [DOI] [PubMed] [Google Scholar]
- 10.Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients 2013;5:4521–39. 10.3390/nu5114521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durbeej M. Chapter Two - Laminin-α2 Chain-Deficient Congenital Muscular Dystrophy: Pathophysiology and Development of Treatment. In: Miner JH, ed. urrent Topics in Membranes [Internet. vol. 76. Academic Press, 2015: 31–60. https://www.sciencedirect.com/science/article/pii/S1063582315000563 [DOI] [PubMed] [Google Scholar]
- 12.Wood AJ, Currie PD. Analysing regenerative potential in zebrafish models of congenital muscular dystrophy. Int J Biochem Cell Biol 2014;56:30–7. 10.1016/j.biocel.2014.10.021 [DOI] [PubMed] [Google Scholar]