

Abstract
Immature dentate granule cells (imGCs) arising from adult hippocampal neurogenesis contribute to plasticity and unique brain functions in rodents1,2 and they are dysregulated in multiple human neurological disorders3–5. Little is known about molecular characteristics of adult human hippocampal imGCs and even their existence is under debate1,6–8. Here we performed single-nucleus RNA-sequencing (snRNA-seq) aided by a validated machine learning-based analytic approach to identify imGCs and quantify their abundance in the human hippocampus during infant, child, adolescent, adult, and aging stages. We identified common molecular hallmarks of human imGCs across the lifespan and discovered age-dependent transcriptional dynamics unique to human imGCs that suggest changes in cellular functionality, niche interactions, and disease relevance, in contrast to those in mice9. We also found a decreased number of imGCs with altered gene expression in Alzheimer’s disease (AD). Finally, we demonstrated the capacity for neurogenesis in the adult human hippocampus with the presence of rare dentate granule cell fate-specific proliferating neural progenitors and with cultured surgical specimens. Together, our findings suggest the presence of a significant number of imGCs in the adult human hippocampus via low frequency de novo generation and protracted maturation, and our study reveals their molecular properties across the lifespan and in AD.
During adult hippocampal neurogenesis, activated neural stem cells generate proliferating intermediate neural progenitors (IPCs) and neuroblasts, which in turn give rise to post-mitotic immature dentate granule cells (imGCs) that mature over time2 (Fig. 1a). The presence of adult-born dentate granule cells (GCs) in humans was first demonstrated in specimens from patients who previously received nucleotide analogs that birth-dated newborn cells10, which was independently confirmed and further characterized by the radiocarbon dating approach11,12. As accumulating evidence has attributed the function of adult neurogenesis to unique properties of immature neurons that are distinct from mature neurons2,4,13–15, the immature neuron population is an important target for analysis. Recent contradictory reports have provided immunohistological evidence for3,5,7,16–19 and against6,20,21 the existence of immature neurons in the adult human dentate gyrus. These studies largely relied on immunostaining of Doublecortin (DCX), an immature neuron marker that requires intricate histological protocols for post-mortem adult human brain specimens5,22. These controversies highlight a major gap in our knowledge about immature neurons in the human hippocampus with limited markers and call for new approaches for their identification and analysis. A more precise identification of immature neurons could be obtained by considering simultaneous expression of multiple genes, ideally the whole transcriptome, at single-cell resolution. To investigate the existence, abundance, and molecular properties of neurons with immature neuronal characteristics in the human hippocampus, we performed snRNA-seq aided by a machine learning-based analytical approach to examine human imGCs across the lifespan (Fig. 1a).
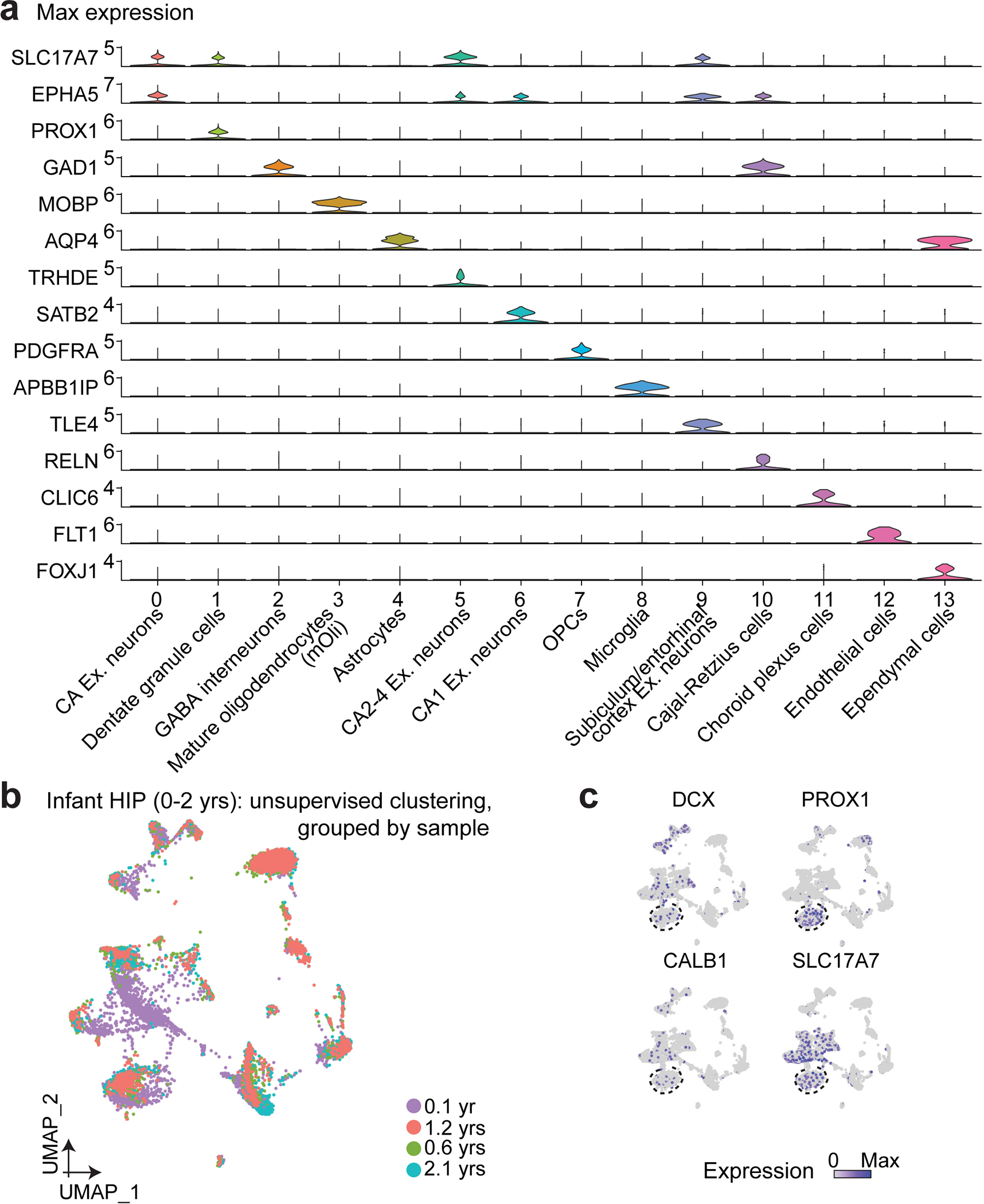
Fig. 1 |. snRNA-seq and immunohistological analyses of imGCs in the human infant hippocampus.
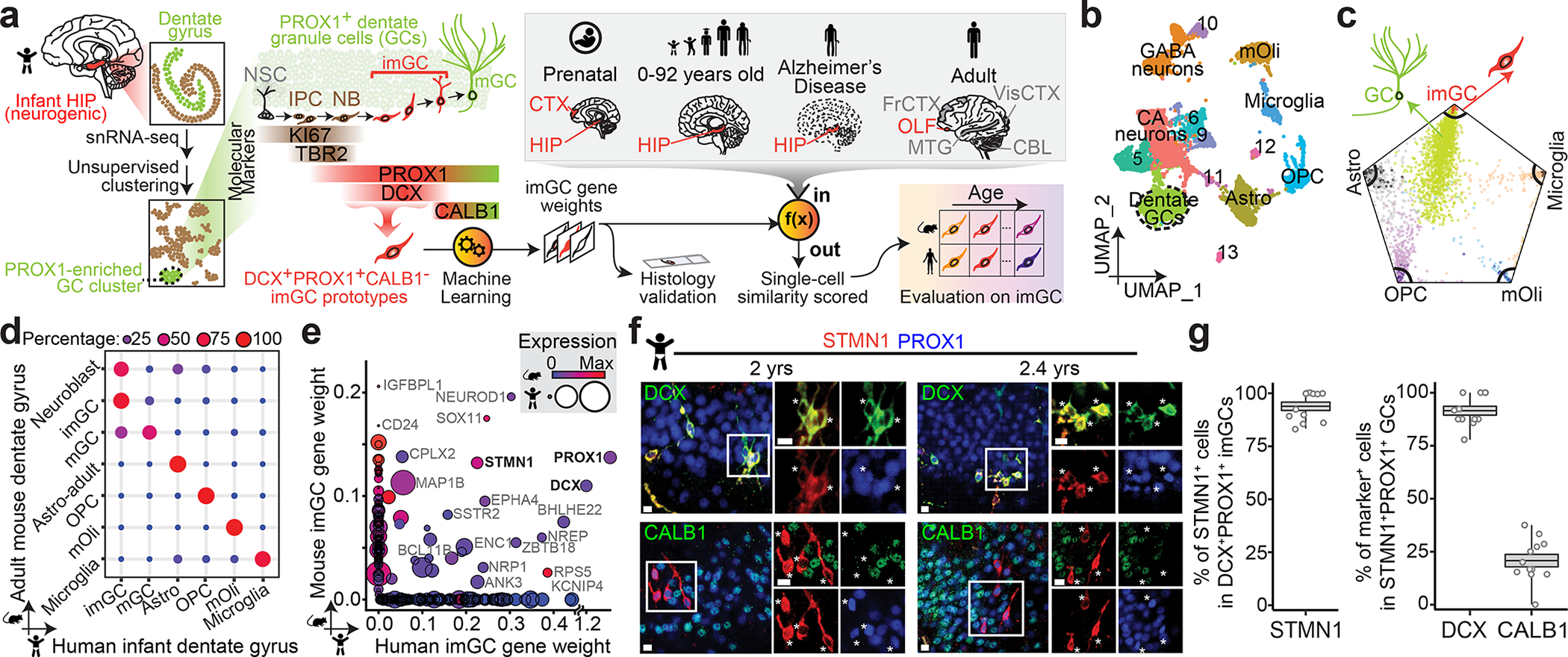
a, A schematic illustration of the experimental design. HIP: hippocampus; NSC: neural stem cell; IPC: intermediate neural progenitor cell; NB: neuroblast; imGC: immature granule cell; mGC: mature granule cell; CTX: cortex; OLF: olfactory epithelium; FrCTX: frontal cortex; VisCTX: visual cortex; CBL: cerebellum; MTG: middle temporal gyrus of cortex. b, UMAP visualization of 15,434 nuclei from four human infant hippocampal specimens, colored by cluster. The GC cluster is highlighted with a dashed line cicle. mOli: mature oligodendrocyte; OPC: oligodendrocyte precursor cell; Astro: astrocyte. c, Wheel plot visualizing scores of each cell to each prototype by the machine learning model. Dots represent individual cells whose distance to each prototype is proportional to the similarity score of that prototype. Each black line indicates a similarity score of 0.85 to each prototypical cell type. d, Transcriptional congruence between the corresponding mouse9 and human cell types measured by a multi-class random forest classifier24,32 trained on different human cell types. Confusion matrix plot indicates the percentage of cells of a given mouse cell cluster (row, based on published annotations9) assigned to a corresponding human cell type (column, classified by the machine learning model). e, Comparison of positive gene weights defining imGCs in humans and mice generated by separate machine learning models. f, g, Sample confocal immunostaining images (f) and quantification (g) of STMN1 enrichment in imGCs in the human infant hippocampus. Scale bars, 10 μm. Asterisks indicate DCX+ or CALB1− among STMN1+PROX1+ cells (f). Dots represent data from individual sections and box values represent mean ± s.e.m. with whiskers for max and min (n = 4 subjects) (g).
snRNA-seq of human infant hippocampi
We first performed snRNA-seq analysis of four infant hippocampus specimens (Supplementary Tables 1, 2), a developmental stage with abundant imGCs16,20. Unsupervised clustering identified fourteen clusters based on their defining markers (Fig. 1b, Extended Data Fig. 1a, b). The imGCs express immature neuronal marker DCX and pan-GC marker Prospero Homeobox 1 (PROX1), whereas Calbindin (CALB1) is expressed in some imGCs, but is more enriched in mature granule cells (mGCs)5,9,23 (Fig. 1a). Within the GC cluster marked by prominent PROX1 enrichment, DCX+ imGCs were intermingled with other cells and could not be separated by finer partitioning (Extended Data Fig. 1c), in contrast to clear clustering of imGCs in the mouse dentate gyrus single-cell mRNA-sequencing (scRNA-seq) dataset9 (Extended Data Figs. 2a, 3). Therefore, the conventional unsupervised method is insufficient to identify the imGC population in human snRNA-seq datasets, similar to previous analyses of the adult human hippocampus24–27.
Identifying imGCs by machine learning
To identify imGCs, we next explored a supervised machine learning approach28. This prototype-based scoring method uses a training set of cell prototypes to extract weighted panels of molecular features de novo, which are then used to quantify the resemblance of each individual cell from query datasets to each prototypical cell type for classification with high fidelity. Applications of supervised models have been shown to be highly effective in differentiating transcriptionally ambiguous cell subtypes in scRNA-seq datasets in multiple systems24,28–32. As a validation of model performance, we first tested this approach to identify immature neurons using scRNA-seq datasets from the mouse hippocampus across ages9 (Supplementary Table 3) for comparison with those from unsupervised clustering, which results in distinct subclusters of neuroblasts, imGCs and mGCs identified via established markers (Extended Data Figs. 2a–c, 3). To mirror our human analysis, we selected high-confidence Dcx+Prox1+Calb1− imGCs from the GC clusters in the P5 mouse hippocampal dataset as a prototype, as well as all major non-neuronal cell-type prototypes (astroglia, oligodendrocyte progenitor cells (OPCs), and microglia) for training (Fig. 1a, Extended Data Fig. 2c; see Methods). The trained model was used to score each cell from the query mouse hippocampal datasets across ages independently of the clustering information (Extended Data Fig. 2d–f, Supplementary Table 4). We identified immature neurons using a conservative, empirical cutoff (p ≥ 0.85) for the similarity score to the mouse imGC prototype and compared our classifications to published clustering annotations9. Model-classified immature neurons were largely within the neuroblast and imGC clusters, with some in the immature CA neuron and GABAergic neuron clusters at P5, yet they resided almost exclusively in the GC lineage in the juvenile and adult mouse hippocampus (Extended Data Fig. 3). Importantly, within the GC lineage, imGCs include the majority of cells within the neuroblast and imGC clusters, fewer in the IPC clusters, and almost none in the mGC clusters (Extended Data Fig. 3c, f, i). Therefore, proof-of-principle analysis using well-defined mouse hippocampal datasets demonstrates the efficacy and selectivity of our machine learning-based approach to reliably identify immature neurons with almost no contamination of mature neurons.
We next implemented the same strategy for the human infant hippocampal dataset by training a new scoring model using high-confidence human infant imGC prototype cells (DCX+PROX1+CALB1− cells from the GC cluster) and prototypes from all major non-neuronal cell types (astrocytes, OPCs, mature oligodendrocytes (mOli), and microglia) at a 99% accuracy rate (Fig. 1a, Extended Data Fig. 4a). Consistent with their immature nature, the positive gene weights for human imGCs are enriched for gene ontology (GO) terms related to nervous system development, neurogenesis, and synaptogenesis, and are closely connected to DCX in the gene network (Extended Data Fig. 4b–d, Supplementary Tables 4 and 5). Immature neurons in the infant human hippocampus were then identified by the trained model using the same conservative similarity score cutoff as for mouse (p ≥ 0.85) (Fig. 1c). As a validation for cell identities, we compared corresponding cell types at the whole transcriptome level between our model-classified human cell types and published cluster annotations in the adult mouse dataset9 using an independent random forest classifier24,32 and found high transcriptomic congruence (Fig. 1d). For example, the identified human imGCs displayed much higher resemblance to mouse neuroblasts and imGCs than to mGCs (Fig. 1d). Human mGCs, defined as cells from the GC cluster with lower similarity scores (p < 0.85) to the imGC prototype, displayed high resemblance to mouse mGCs, but not to neuroblasts or imGCs (Fig. 1d). Despite this general conservation, orthologous positive gene weights generated separately by machine learning models for human and mouse imGCs showed substantial species differences (Fig. 1e, Extended Data Fig. 4e).
To confirm the enriched expression of top weighted genes in imGCs, we screened candidates based on antibody availability and focused on STMN1 (Fig. 1e), a tubulin-depolymerizing protein33, for immunohistology. In independent infant human dentate gyrus specimens (Supplementary Table 1), 93.8% of DCX+PROX1+ imGCs were STMN1+, whereas 91.4% and 20.7% of STMN1+PROX1+ cells were DCX+ and CALB1+, respectively (Fig. 1f, g). Similar results were obtained in the adult mouse dentate gyrus (Extended Data Fig. 4f, g).
Human imGC abundance across lifespan
We then applied our trained model to assess each cell in query scRNA-seq/snRNA-seq datasets from human brain specimens of various developmental stages and regions (Supplementary Tables 2, 3). We first examined published datasets of the prenatal human hippocampus34 and prefrontal cortex35, both containing abundant immature neurons. Indeed, we found a large number of cells with high similarity scores (p ≥ 0.85), including, most prominently, GCs, some CA neurons and GABAergic neurons in the hippocampus, and some neurons in the cortex (Fig. 2a), suggesting that, as in mice (Extended Data Fig. 3a–c), our approach selects for cells with immature neuronal features, but not exclusively for imGCs. We next performed snRNA-seq on human postnatal hippocampal specimens across ages, with four to five subjects each for child, adolescent, adult, and aging stages (Supplementary Tables 1, 2). We integrated the published prenatal34 and all of our postnatal hippocampal datasets using canonical correlation analysis36 (CCA; Supplementary Fig. 1) and classified fifteen cell clusters into four broad classes on Uniform Manifold Approximation and Projection (UMAP) plots of six age groups (Fig. 2a, listed in bold text). Cells with high similarity (p ≥ 0.85) to the human imGC prototype were identified in every hippocampal specimen across all ages, most of which are clustered together in UMAP plots, suggesting their transcriptomic proximity (Fig. 2a). The identified immature neurons resided almost exclusively in the GC cluster in postnatal datasets (Fig. 2a, Extended Data Fig. 5). We also applied the same model to several published datasets from various postnatal human brain regions (Supplementary Table 3). We identified immature neurons in three published adult human hippocampus datasets24,25,27 (Fig. 2a). Adult human olfactory epithelium exhibits continuous neurogenesis; we also identified immature neurons, which matched the published annotations based on unsupervised clustering37 (Fig. 2a). In contrast, almost no immature neurons were identified from datasets of the adult human frontal cortex, cerebellum, visual cortex38, middle temporal gyrus of the cortex39, and prefrontal cortex24 (Fig. 2a, Extended Data Fig. 5). Together, these results demonstrate the sensitivity and specificity of our approach to identify human immature neurons in various brain scRNA-seq/snRNA-seq datasets.
Fig. 2 |. snRNA-seq analysis of human imGCs across ages.
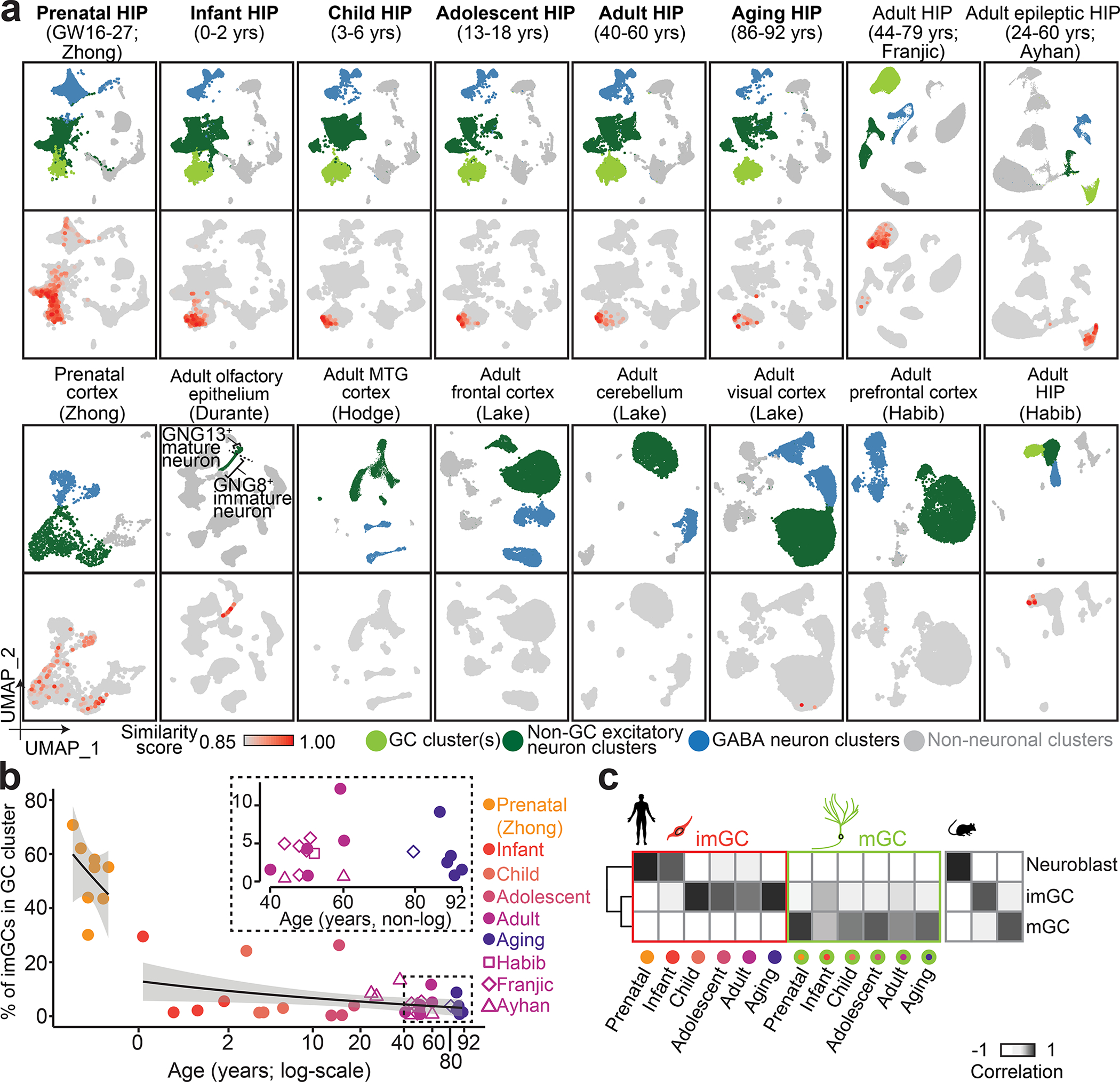
a, UMAP plots showing scRNA-seq/snRNA-seq datasets of human brain specimens colored by four broad cell classes (top rows) and by similarity score to prototypical imGCs (bottom rows). Datasets named in bold were integrated and are shown in aggregate for each age group (with 4 or 5 subjects for each age). GW: gestational week; yrs: years. b, Quantification of proportions of imGCs (with similarity scores p ≥ 0.85) among all GCs in each human hippocampal specimen across ages. Prenatal and postnatal data points are fitted separately with generalized linear model fitting (two black lines) with 95% confidence interval (grey shades). Datasets of 40 to 92 years-old specimens are highlighted in the inset. c, Pearson correlation of gene expression of the corresponding mouse9 and human imGCs and mGCs.
Following imGC identification, quantification of all hippocampal datasets showed that the average percentages of imGCs among all cells in the GC cluster in each age group range from 51.8% in the prenatal stage, 9.4% in infancy, to 3.1–7.5% from four years old and beyond (Fig. 2b), which are very similar to those previously reported based on DCX immunohistology5,21.
As a validation for the imGC identity in our datasets, Pearson correlation analysis showed that the identified human imGCs resembled mouse neuroblasts and imGCs, but not mGCs9, whereas human mGCs resembled mouse mGCs, but not neuroblasts or imGCs (Fig. 2c). Immunohistological analysis using independent post-mortem human dentate gyrus specimens across ages (Supplementary Table 1) showed that over 70% of DCX+PROX1+ imGCs were STMN1+, whereas only 18–39% of STMN1+PROX1+ neurons were CALB1+ (Extended Data Fig. 6a–d). We confirmed the neuronal identity of STMN1+PROX1+ cells with additional markers (Extended Data Fig. 6e, f).
imGC molecular profiles across lifespan
Capturing human imGCs by snRNA-seq across the lifespan enables a systematic analysis of their immature neuronal signature and transcriptomic landscape. To account for batch bias prior to quantitative gene expression comparison, we aligned published prenatal34 and our postnatal human hippocampal datasets across ages using scVI40 in addition to CCA36 (Supplementary Fig. 2). To identify the common molecular signature of human imGCs irrespective of age, we compared imGCs to their mGC counterparts at different ages and found a preferential enrichment of genes in imGCs of all ages related to nervous system development (e.g., NEUROD1, BHLHE22), ion transport (e.g., FXYD7, KCNQ5), and neuron projection development (e.g., SEMA6D, NR2F1) (Fig. 3a, b, Supplementary Fig. 3, Supplementary Table 6). Among these genes, only 15.5% overlapped with orthologous genes enriched in mouse imGCs across ages9 (e.g., FXYD7), indicating substantial interspecies differences (Fig. 3c–e, Supplementary Table 7).
Fig. 3 |. Common and divergent molecular features of imGCs across lifespan and between humans and mice.
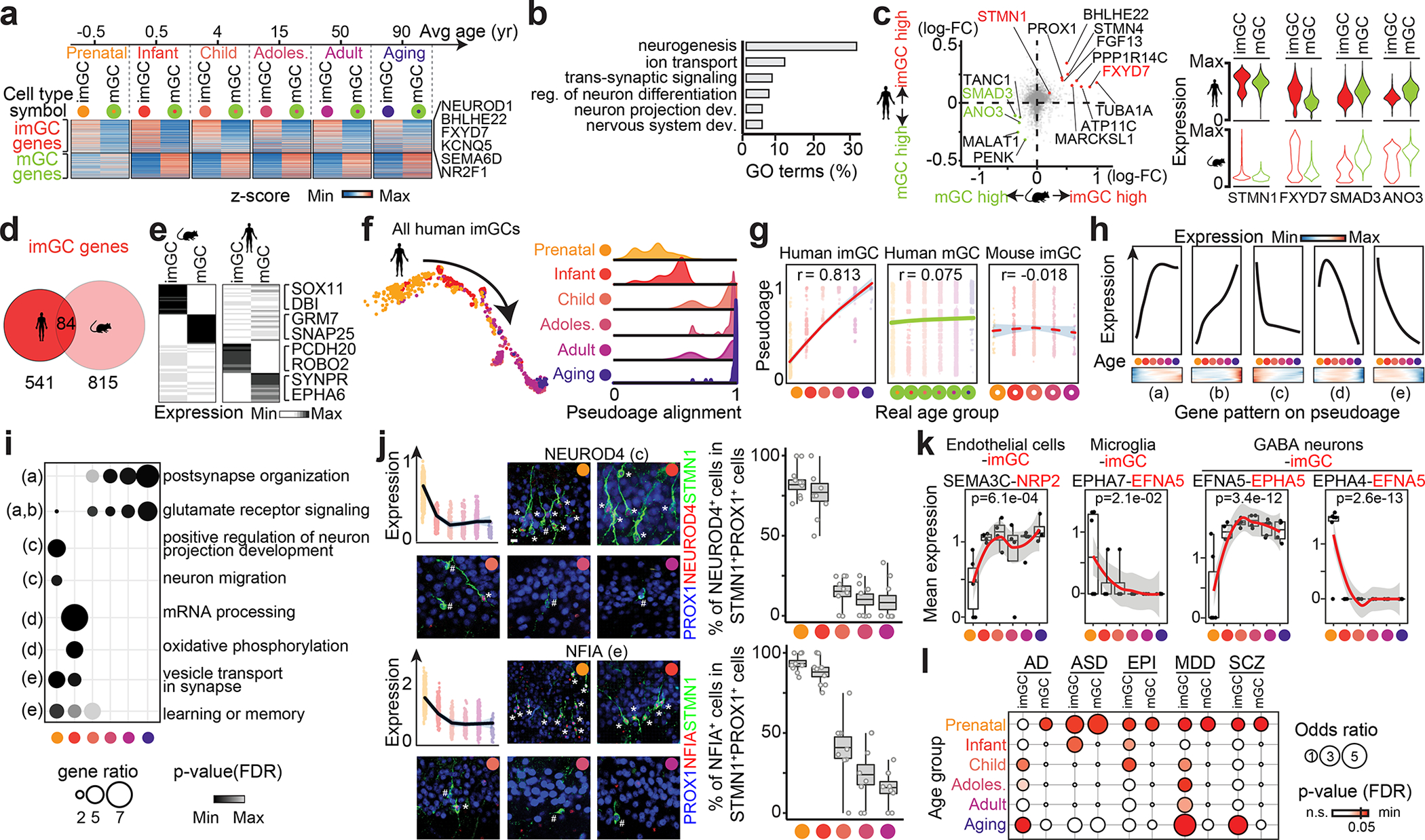
a, Common enriched genes in human imGCs or mGCs across ages (two-sided Wilcoxon rank-sum test, false-discovery rate (FDR)-adjust p-value < 0.05). b, Top GO term groups for common human imGC-enriched genes. reg.: regulation; dev.: development. c. Scatter plot (left) of log2 fold-change (FC) between imGCs and mGCs and violin plots (right) of exemplary genes. d, Venn diagram of imGC-enriched genes in humans and mice. e, Unique features of imGC- and mGC-enriched genes in humans and mice. f, g, Pseudo-age alignment of human imGCs colored by age group, in scatter (left) or density (right) plots (f) and summarized (g). Dots representing imGCs in each age group are fitted with loess fitting (lines) with 95% confidence interval (grey shades) with Pearson’s r for correlations of pseudo- and real-age groups (g). h, i, Distinct patterns of age-dependent gene expression in human imGCs (h, likelihood ratio test, Benjamini-Hochberg-adjusted p, q < 0.01) and representative GO terms (i, one-sided Fisher’s exact test, p(FDR) < 0.05). j, Sample confocal immunostaining images and quantification of two exemplary genes displaying age-dependent expression in human imGCs. Scale bar, 10 μm. Dot plots showing gene expression values similar as in g. Box plots similar as in Fig. 1g (n = 3 subjects per group). k, Exemplary ligand-receptor pairs of imGCs interacting with neighboring cell types (using CellPhoneDB42) with age-dependent gene expression changes (two-sided Moran’s I test, p (Bonferroni) < 0.05; n = 28 specimens). Dots represent mean expression of the ligand-receptor pair for the cell type pair in each specimen with fitting similar as in g. Box plots represent median ± quantiles with whiskers for max and min. l, Enrichment patterns of brain disorder risk gene expression in human imGCs and mGCs across the lifespan (one-sided Fisher’s exact test; p(FDR) < 0.05). EPI: epilepsy; MDD: major depressive disorder; SCZ: schizophrenia.
To deconvolve potential age-dependent molecular changes, we aligned all human imGCs on a pseudo-age trajectory using Monocle41. We observed a striking temporal transcriptomic shift correlated with the specimen age (Pearson’s r = 0.813) (Fig. 3f, g). This correlation was unique to imGCs, but not mGCs, and observed only in humans, but not in mice during the time window examined9 (Fig. 3g, Extended Data Fig. 7). A gene co-variation kinetics analysis encapsulated five distinct age-dependent patterns, including a continuous up-regulation of glutamatergic receptor signaling pathways and down-regulation of neuronal migration- and projection morphogenesis-related genes (Fig. 3h, i, Supplementary Fig. 4, Supplementary Table 8). We confirmed age-dependent expression of two exemplary genes, NEUROD4 and NFIA, in human imGCs, but consistent expression in mouse imGCs across ages using immunohistology (Fig. 3j, Extended Data Fig. 8). Transcriptomic mapping of different cell types in the hippocampus together allows probing cell-cell interactions based on cognate ligand-receptor expression. An imGC-centric analysis using CellPhoneDB42 revealed age-dependent interactions between imGCs and their neighboring cell types in the dentate gyrus (Fig. 3k, Supplementary Fig. 5). To explore the potential contribution of imGCs to different brain disorders, we performed disease risk gene enrichment analysis for AD, autistic spectrum disorders (ASD), epilepsy, major depressive disorder, and schizophrenia, and revealed their selective expression in imGCs at specific ages, many of which coincide with critical periods of the suspected disorder etiology, such as the aging stage for AD and early developmental stages for ASD (Fig. 3l, Supplementary Fig. 6).
Dysregulated imGCs in AD
To directly examine how neurological disorders may impact imGCs, we performed snRNA-seq of hippocampal specimens from eight AD patients and eight matched controls integrated36 together with five controls in the original aging group (Fig. 4a, Supplementary Tables 1, 2). We identified imGCs in all these specimens almost exclusively in GC clusters and quantification showed that the percentage of imGCs among all GCs was two-fold lower in AD compared to that in controls, whereas the percentage of GCs among all cells sequenced per sample was similar (Fig. 4b, c). Our finding is similar to previously reported immunohistological quantifications of DCX+ imGCs among NeuN+ GCs regarding the proportion of imGCs and the level of decrease in AD5,18.
Fig. 4 |. Reduced number and altered gene expression of imGCs in AD patients.
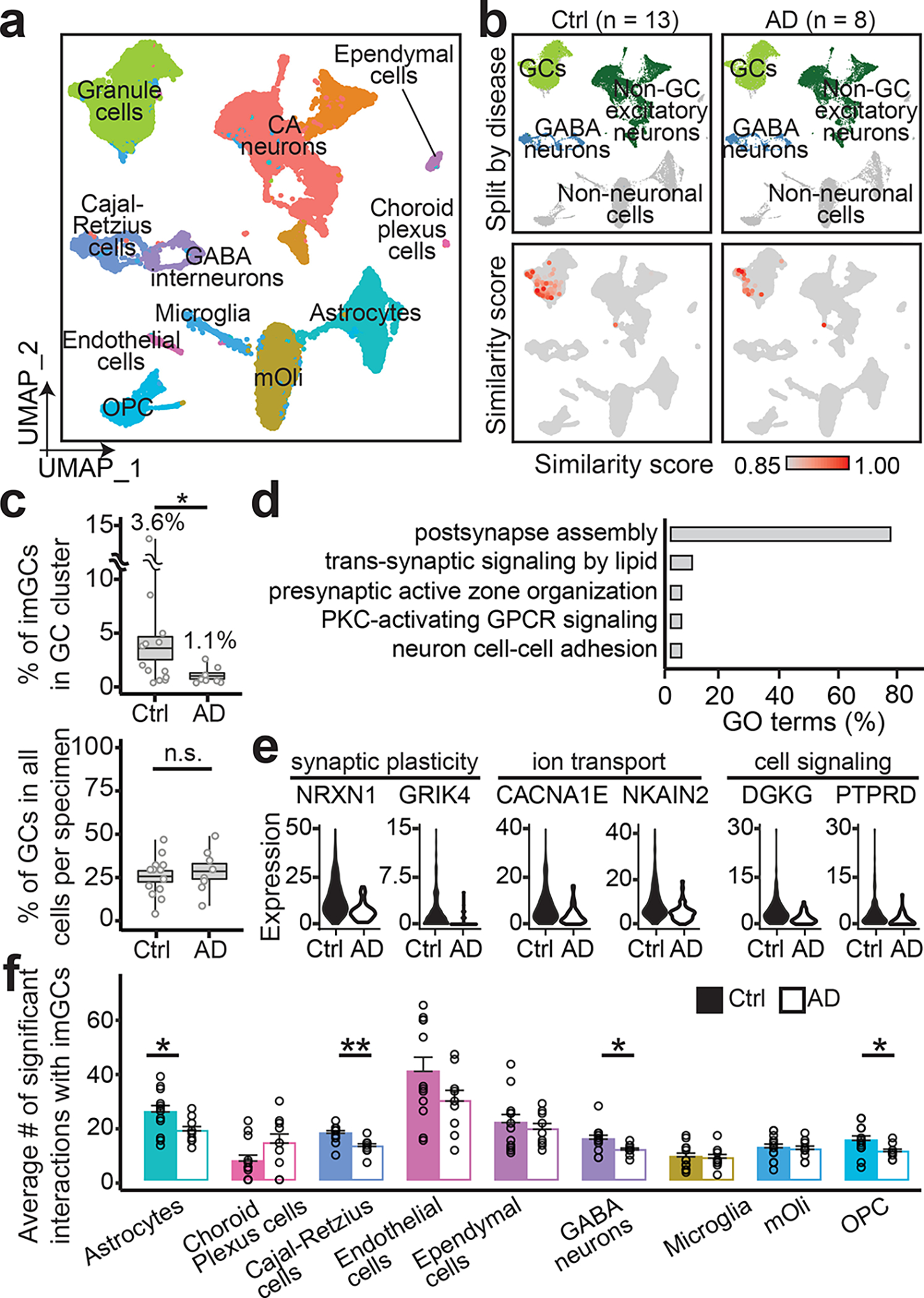
a, b, UMAP plots of the integrated dataset of AD patients and controls (Ctrl) colored by cluster (a) and broad cell class (top row) and similarity score to prototypical imGCs (bottom row) (b). c, Quantifications of proportions of imGCs among GCs (top) and GCs among total cells obtained per specimen (bottom). Each dot represents data from one specimen and boxes represent mean ± s.e.m. with whiskers for max and min (n = 8 and 13 subjects for AD and control, respectively; *: p = 0.0197; n.s., not significant; one-tailed Mann-Whitney test). d, e, GO terms (d) and examples (e) of genes downregulated in imGCs in AD. f, Quantifications of numbers of significant ligand-receptor pairs of imGCs interacting with neighboring cell types (using CellPhoneDB42). Each dot represents data from one specimen. Values represent mean + s.e.m. (n = 8 and 13 subjects from AD and controls, respectively; *p < 0.05; **p < 0.005; p-value of significant pairs from left to right: 0.013, 0.001, 0.017, and 0.012; one-tailed Mann-Whitney test).
Quantitative analysis identified 14 downregulated genes in imGCs in AD, which are mostly associated with synaptic plasticity and signaling (e.g., NRXN1) (Fig. 4d, e, Supplementary Table 9). An imGC-centric cell-cell interaction analysis42 revealed significantly decreased interactions of imGCs with astrocytes, OPCs, GABAergic interneurons, and Cajal-Retzius cells in AD, indicating aberrant niche interactions (Fig. 4f).
Postnatal human hippocampal neurogenesis
Our transcriptomic analysis could not differentiate whether immature neurons in the adult human brain were born later in life versus born earlier and remaining in the immature state8,43. In an attempt to examine the capacity for neurogenesis, we observed GC fate-specific KI67+PROX1+ proliferating neural progenitors and TBR2+PROX1+ IPCs2 in the human hippocampus across ages (Fig. 5a, b). However, the numbers of these precursor cells were very low in the adult human hippocampus (Fig. 5c, d), indicating de novo generation of imGCs at low frequencies.
Fig. 5 |. Capacity for neurogenesis in the postnatal human hippocampus across ages.

a-d, Sample confocal immunostaining images (a, b) and quantifications (c, d) of PROX1+ neuronal progenitors in the human dentate gyrus across ages. Scale bars, 10 μm. Asterisks indicate MKI67+ or TBR2+ among PROX1+ GCs (a, b). Each dot represents the sum value of quantification of multiple sections from one specimen (n = 10 specimens) (c, d). e-g, A slice culture system demonstrating capacity for neurogenesis in the adult human dentate gyrus. Shown are a schematic illustration of the experimental procedure (e), a sample image of a well containing three slices (f; scale bar, 1 cm), and sample confocal staining images (g) of EdU-incorporated newborn imGCs expressing different markers in the postnatal human dentate gyrus. Scale bars, 100 μm (low-magnification images) and 10 μm (insets 3, 4) (g).
Because currently there is almost no practical means to birthdate newborn neurons in humans in vivo, we developed an ex vivo culture method to directly examine the capacity for neurogenesis qualitatively in the postnatal human hippocampus. Fresh surgically resected human hippocampi from patients diagnosed with epilepsy were sliced and cultured in growth factor-free, chemically defined medium44 in the presence of EdU to label dividing cells (Fig. 5e, f, Extended Data Fig. 9a–c). We observed EdU+PROX1+ newborn GCs in 8 out of 10 specimens from patients ranging from 2 to 61 years old at 1–2 weeks in culture (Fig. 5g, Supplementary Table 1), and these cells were S100B−19 and CALB1− (Extended Data Fig. 9d). Over 80% of EdU+PROX1+ cells were DCX+ or STMN1+, and 88.6% of DCX+EdU+ cells were STMN1+ (Fig. 5g, Extended Data Fig. 9e). We also observed EdU+TBR2+PROX1+ IPCs (Fig. 5g). These results indicate the capacity for the adult human dentate gyrus to generate new GCs and validate the enrichment of STMN1 in imGCs.
Discussion
Rather than relying on a few pre-selected marker genes3,5,17–21,45, our study highlights the advantage of the snRNA-seq analysis to precisely define a cell subtype in silico based on its whole transcriptome. Our study reveals dynamic molecular properties of imGCs compared to mGCs in the human hippocampus across the lifespan. As expected, imGCs predominantly express transcripts related to immature neuronal hallmarks, such as development, neurogenesis, and plasticity. The significant differences observed in the molecular landscapes between imGCs and mGCs support the notion of unique contributions of human immature neurons to brain functions. We also mined the datasets for novel candidate genes enriched in human immature neurons, such as STMN1. Interestingly, we found differential transcriptional programs in human imGCs across ages, as well as other properties that diverged from imGCs in mice9, highlighting interspecies variance. Furthermore, we observed a decreased number of imGCs in AD and identified altered gene expression and reduced interactions with niche cells. The mechanisms underlying such age- and disease-related changes in human imGCs remain to be determined.
As the similarity score cut-off for human imGCs was determined based on validation in the mouse dataset, we could have under- or over-estimated the numbers of human imGCs in our study. While we could not determine when these imGCs were born, the presence of GC fate-specific proliferating progenitors in the adult human hippocampus revealed by immunohistology indicates that at least some of imGCs are born in adulthood. Future larger-scale snRNA-seq analysis may capture these rare proliferating neural progenitors for molecular analyses. Our slice culture birth-dating study directly demonstrates the capacity for the adult human hippocampus to generate new neurons. Compared to development, newborn GCs in adult rodents46 exhibit immature characteristics for a prolonged period, which lasts longer in aging mice47,48, and over 10 times longer in adult primates49,50. Our results support a model that new neurons are continuously generated at low frequencies, but exhibit protracted neuronal maturation and are maintained in an immature state for a long period of time, leading to an accumulation of a significant number of neurons with immature neuronal characteristics at any given time in the adult human hippocampus (Extended Data Fig. 10). The function of adult neurogenesis primarily arises from the unique properties of immature neurons2,4,13–15, rather than proliferating neural progenitors per se. Our study reveals the molecular landscape of human imGCs across the lifespan and provides new resources and methods that will facilitate future investigations into their functions and disease relevance.
Methods
Tissue specimens
De-identified human tissue specimens were collected and processed under protocols approved by the Institutional Review Boards of the University of Pennsylvania and the Children’s Hospital of Philadelphia. A total of 62 human post-mortem hippocampal specimens between the ages of gestational week (GW) 20 to 92 years old, including 54 specimens from subjects free from neurological disorders and 8 specimens from AD patients, were used for snRNA-seq and immunohistological analyses (Supplementary Table 1). Specimens were collected from tissue banks at the Children’s Hospital of Philadelphia, the Johns Hopkins University Pathology Archive, the Lieber Institute for Brain Development, and the NIH NeuroBioBank at the following repositories: University of Pittsburgh Brain Tissue Donation Program, the University of Maryland Brain and Tissue Bank, the University of Miami Brain Endowment Bank, the Harvard Brain Tissue Resource Center, the Human Brain and Spinal Fluid Resource Center at the VA West Los Angeles Healthcare Center, and the Mount Sinai School of Medicine. All embryonic tissues were from diagnostic autopsies. As post-mortem interval (PMI) could affect results of snRNA-seq51 and immunohistology analysis (e.g., of DCX52), we tried to collect specimens with as short PMIs as possible (listed in Supplementary Table 1). In addition, fresh surgically resected human hippocampal tissue from 10 patients between the ages of 2 to 61 years old were used for ex vivo slice culture, collected from the Children’s Hospital of Philadelphia and the Hospital of the University of Pennsylvania (Supplementary Table 1). Informed consent for each specimen was obtained by its corresponding institution prior to tissue collection.
For mouse immunohistological analysis, P14, P60 and 1.4 years old, wild-type, male and female C57BL/6 mice were used. No obvious sex phenotype was observed in any of the experiments. Animals were housed in a 12-hour dark/ light cycle with food and water ad libitum. Animal procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Single-nucleus RNA sequencing
We used a modified SPLiT-seq approach for nuclei isolation and snRNA-seq53,54. Nuclei isolation from snap-frozen hippocampal tissue was performed as previously described with minor modifications54,55. Briefly, after a visual inspection to include the dentate gyrus by its distinct anatomical structure, tissue was minced with a razor blade and Dounce (Fisher Scientific, 8853000002) homogenized for 5 to 10 strokes using a chilled tissue grinder in 1 mL of chilled homogenization buffer consist of 1 mM DL-Dithiothreitol (DTT, Sigma-Aldrich, D0632), 0.15 mM spermine (Sigma-Aldrich, S4264-1G), 0.5 mM spermidine (Sigma-Aldrich, S0266-1G), EDTA-free protease inhibitor (Roche, 11836170001), 0.3% IGEPAL-630 (Sigma-Aldrich, I8896-50ML), 0.25 M sucrose (Sigma-Aldrich, S5016-500G), 25 mM MgCl2 (ThermoFisher Scientific, AM9530G), and 20 mM Tricine-KOH (Sigma-Aldrich, T5816-100G). Homogenates were filtered through a 40-μm cell strainer (Fisher Scientific, 22-363-547), and mixed with 200 μL Myelin Removal Beads II (Miltenyi Biotec, 130-096-733) for a 15-minute incubation on ice. The mixture was then transferred on top of a sucrose cushion buffer (vol/vol; 0.5 mM MgCl2, 0.5 mM DTT, EDTA-free protease inhibitor, 0.88 M sucrose) at a ratio of 1:1 in the centrifuge tubes, and centrifuged at 2800 g for 10 minutes in a swinging bucket centrifuge at 4 °C. Nuclei were collected as pellets and resuspended in 0.01% Bovine serum albumin (BSA, Sigma-Aldrich, B6917) in chilled phosphate-buffered saline (PBS, Corning, 21-040-CV). Nuclei were spun down for 3 minutes at 500 g at 4 °C before resuspension in 1 mL of chilled PBS-RI containing 0.05 U/μL RNase Inhibitor (Enzymatics, Y924L) in PBS and filtered through a 40-μm cell strainer. For specimens that underwent the SPLiT-seq method, additional processing steps were applied as follows: 3 mL of chilled 1.33% formaldehyde solution was added to the nuclei suspension for fixation for 10 minutes. Next, nuclei were permeabilized with 160 μL of 5% Triton X-100 (Sigma-Aldrich, T9284) in chilled PBS for 3 minutes and spun down at 500 g for 3 minutes at 4 °C. Nuclei were then resuspended in 500 mL chilled PBS-RI before 500 μL of chilled 100 mM Tris-HCl (pH 8) was added. Nuclei were spun down again at 500 g for 3 minutes at 4 °C and resuspended in 300 μL chilled 0.5 X PBS-RI. Finally, nuclei were filtered through a 40-μm strainer again before being counted with a hemocytometer and diluted to one million nuclei per mL.
The majority of the hippocampal specimens was processed using SPLiT-seq as previously described53,54. Briefly, single-nuclei mRNA was tagged in three rounds with barcoded primers (Integrated DNA Technologies) for in-cell ligation using the T4 DNA ligase (New England Biolabs, M0202S). Ligation products were purified with Dynabeads MyOne Streptavidin C1 beads (ThermoFisher Scientific, 65001) and resuspended with Kapa HiFi HotStart Master Mix (KAPA Biosystems, KK2600) for a PCR thermocycling. Next, beads were removed from the PCR products, followed by the addition of EvaGreen dye (Biotium, 31000) for a qPCR thermocycling. The PCR products were then purified using KAPA Pure Beads (KAPA Biosystems, KK8000). One hippocampal specimen was processed using droplet-based snRNA-seq technique56 with modifications57. Briefly, the single-nucleus suspension and the barcoded beads (ChemGenes, MACOSKO-2011-10) were diluted and co-encapsulated using a microfluidic device (μFluidix, Batch #9508). Droplets were broken and reverse transcription was performed to generate cDNA.
Tagmentation was performed with Nextera XT Library Prep Kits (Illumina, FC-131-1024). The tagmented cDNA libraries were further amplified with 12 enrichment PCR cycles using the Illumina Nextera XT i7 primers and the P5-TSO hybrid primer56. After quality control analysis by a Qubit Fluorometer (ThermoFisher Scientific, Q33238) and a Bioanalyzer (Agilent), libraries were sequenced on an Illumina NextSeq 550 instrument using Illumina High Output Kit v2.5 (75-cycle (20024906) for libraries prepared with Drop-seq; 150-cycle (20024907) for libraries prepared with SPLiT-seq). Paired-end sequencing reads were pre-processed using the Drop-seq software (v1.13, http://mccarrolllab.com/dropseq/) with modifications54,56,57. Briefly, each mRNA read was tagged with a barcode and a unique molecular identifier (UMI), trimmed off sequencing adaptors and poly-A sequences, and aligned to the human reference genome assembly (Genome Reference Consortium hg38, Gencode release v28) using Spliced Transcripts Alignment to a Reference (STAR, v2.5.2a)58 using default settings. Both exonic and intronic reads mapped to the predicted strands of annotated genes were retrieved for the cell type classification57. Uniquely mapped reads were grouped by cell barcodes. To generate a digital expression matrix, a list of UMIs in each gene, within each nucleus, was assembled, and UMIs within ED = 1 were merged. The total number of UMIs was counted and reported as the number of transcripts of that gene for a given nucleus. Raw digital expression matrices were generated for each sequencing run (summarized in Supplementary Table 2).
Quality control, cell clustering, and dataset integration
Raw count matrices were loaded into the R (v3.6) package ‘Seurat’ (v3.1.4)36. For each specimen, genes expressed in < 10 nuclei were discarded. Nuclei with < 400 or > 5,000 genes were discarded; nuclei with > 5% UMIs mapped to mitochondrial genes were discarded. For normalization, UMI counts for all nuclei were scaled by library size (total UMI counts), multiplied by 10,000 and transformed to a log scale. Highly variable genes were identified using the function ‘FindVariableFeatures’ in Seurat. The top principal components (PCs), determined by the ‘PCElbowPlot’ function, were selected for dimensionality reduction, clustering, and visualization with Uniform Manifold Approximation and Projection (UMAP) or t-distributed Stochastic Neighbor Embedding (t-SNE). Marker genes for each cluster were identified with a Wilcoxon rank sum test implemented in the ‘FindAllMarkers’ function with the following criteria: adjusted p-value < 0.01 (controlled for false discovery rate (FDR)), log-fold change ≥ 0.5, and genes detected in < 25% of the cells within its corresponding cluster were excluded. In particular, PROX1, the defining marker for excitatory dentate granule cells, was used to determine whether a hippocampal specimen contains cells or nuclei from the dentate gyrus. For the hippocampus, only specimens with a distinct PROX1-enriched excitatory neuronal cluster containing at least 50 cells or nuclei were included. For non-hippocampal specimens, a distinct PROX1-enriched excitatory neuronal cluster precluded further analysis. UMI count matrices from published datasets9,24,25,27,34,35,37–39 were retrieved from the respective repositories and processed independently using the same criteria (summarized in Supplementary Table 3).
To investigate imGCs across the human lifespan under physiological conditions, hippocampal datasets from our postnatal specimens at the infant (0–2 years old, 4 specimens, 15,434 nuclei), child (3–6 years old, 4 specimens, 24,607 nuclei), adolescent (13–18 years old, 4 specimens, 16,310 nuclei), adult (40–60 years old, 5 specimens, 29,832 nuclei), and aging (86–92 years old, 5 specimens, 16,055 nuclei) stages, and from a published prenatal report34 (GW16-GW27) were normalized within each age group to remove sequencing variation (implemented in ‘sctransform’ function in Seurat59) prior to integration using a canonical correlation analysis (CCA) in Seurat36 (Supplementary Fig. 1, Supplementary Tables 1, 2). To investigate imGCs in AD, hippocampal datasets from our AD patient specimens (73–88 years old, 8 specimens, 27,508 nuclei) and matched controls (73–88 years old, 8 specimens, 21,955 nuclei) and the 5 control specimens from the aging group (Supplementary Tables 1, 2) were normalized using the ‘sctransform’ method59 and integrated using CCA36 (Fig. 4a, b). For both integrated datasets, the top 2,000 highly variable genes and the first 30 PCs were used for cell alignment before clustering and UMAP visualization.
Immature neuron signature extraction and prototype-based cell scoring using machine learning
To provide a precise and holistic characterization of human imGCs, we implemented a supervised learning approach to learn comprehensive gene features from imGCs of unambiguous identities (prototypes), which we then used to quantitatively evaluate the similarity of each cell in query (test) brain snRNA-seq datasets. A multinomial machine learning method using a L2-norm regularized logistic regression model (implemented in the ‘LogisticRegression’ function in ‘scikit-learn’60 in Python v3.7) was applied with modifications28. To validate the sensitivity and specificity of this approach, we first recapitulated our analytic paradigm (Fig. 1a) in scRNA-seq datasets from the mouse hippocampus across ages9 by selecting prototypes from the early postnatal dataset (P5) for model training, scoring each cell in the juvenile (P12-35) and the adult (P120-132) datasets, and benchmarking the classifier performance to the published annotations based on unsupervised clustering and known marker expression9 (Extended Data Figs. 2a–c, 3). In the context of the mouse dentate gyrus, despite clear separation by unsupervised clustering, we disregarded the finer partitioning of subtypes in the GC lineage to mimic the scenario within human GCs and selected immature neuron prototypes from all the GC clusters in the mouse P5 dataset. Cell selection criteria described below for the human dataset were strictly followed with one difference, which was the lack of mature oligodendrocytes as a prototypical cell type, as the mouse P5 dataset does not contain a mature oligodendrocyte cluster (Extended Data Fig. 2a).
Given the significant species differences between mice and humans and the potential technical variability in the published datasets, we chose cell-type prototypes from our unsupervised-clustered snRNA-seq dataset of the infant human hippocampi as the training data for the prototype-based scoring model. The prototypes consist of imGCs and all major non-neuronal cell types, including astrocytes (Astro), oligodendrocyte precursor cells (OPC), mature oligodendrocytes (mOli), and microglia. As DCX transcripts are not exclusive to the GC cluster, the imGC prototypes were selected based on consideration of their defining gene expression features, DCX+CALB1−PROX1+, only from the GC cluster, which was included as the only neuronal cell prototype to avoid the potential contamination from non-GC immature neurons in the infant hippocampus. Other cell-type prototypes were selected from their respective clusters by their defining features, including AQP4+ cells from the astrocyte cluster, PDGFRA+ cells from the OPC cluster, MOBP+ cells from the mature oligodendrocyte cluster, and CX3CR1+ cells from the microglia cluster. To further refine the most representative cell populations as prototypes, two negative selection criteria were applied: (1) Cells expressing common markers of the other prototypical cell types were excluded from prototypes. For example, imGC prototypes were expected to exhibit no expression of markers of astrocytes (SLC1A2, AQP4), OPCs (PDGFRA), oligodendrocyte lineage cells (OLIG2, CNP, MBP, MOBP), and microglia (CX3CR1, PTPRC); (2) Cells were excluded from all prototypes if they expressed defining markers of other known cell types in the hippocampal dataset, including GABAergic interneurons (GAD1, GAD2), CA neurons (SATB2), ependymal cells (FOXJ1), endothelial cells (FLT1), and blood cells (HBA1).
The cell scoring model was trained on the log-transformed, max-normalized count matrix of the prototype cells with all genes retained, followed by a gene ranking procedure28,61 to refine for highly variable cell-type specific markers. An optimal regularization parameter of 0.75 for the logistic regression model was chosen by plotting the regularization strength against the classifier accuracy, looking for the most stringent value of regularization with the maximal accuracy rate (~99%). A cross-validation procedure was applied to the training set to estimate the average accuracy of the model (implemented in the ‘LogisticRegressionCV’ function in ‘scikit-learn’) with the following parameters, training set: validation set = 85%:15%, and training set randomly split for 35 iterations (using a stratified k-fold cross-validation approach). The resulting trained model uses a list of positively- and negatively-weighted coefficients to rank genes according to their ability to predict each cell category. Importantly, the trained model relies on a combinatorial gene panel rather than a few arbitrarily picked markers to define the transcriptomic profile of imGCs, which strikes a balance among immature, neuronal, and regional (dentate gyrus) features.
A collection of human scRNA-seq/snRNA-seq datasets from various brain regions and developmental stages were individually prepared using Seurat36 as query (test) datasets (Supplementary Tables 2, 3). For optimal performance of the machine learning-based classification, we ensured that query datasets have similar sequencing characteristics as our training dataset by performing random down-sampling (implemented in the “rbinom” function in R) on query expression matrices with significantly higher average number of genes and reads per cell (> 2,000 genes/cell and > 4,000 reads/cell) to a similar level of depth as our training set (~1,100 genes/cell and ~2,000 reads/cell) prior to quality control and downstream processing on query datasets. This process was repeated 10 times to ensure robustness and consistency. The trained model scored the probabilistic similarity (p) to each prototype of each individual cell from the log-transformed, max-normalized count matrices of the test datasets without prior knowledge of clustering information. The predicted probability, ranging from 0 to 1, was calculated using the ‘softmax’ function (implemented in the ‘predict_proba’ function in ‘scikit-learn’)28. The p(imGC) ≥ 0.85 was empirically set as a conservative cutoff of the similarity score to classify a cell as an immature neuron, which was first validated with the mouse dataset. To ensure the specificity of our method, we compared our scoring model predictions on astrocytes, OPCs, mOlis, and microglia using p ≥ 0.85 as the cutoff of similarity score for cell type classifications to the unsupervised clustering labels in the hippocampal dataset across ages and found that our method classified cells with 94% to 99% specificity. Two types of plots were used for visualization: (1) Each cell was plotted on a ‘wheel plot’ polygon using the ‘polygonalPlot’ function28 to show its similarity to each prototype (Fig. 1c, Extended Data Fig. 2f); (2) A similarity score for each cell to imGC prototypes from the test datasets was projected to its corresponding UMAP or t-SNE plots (Figs. 2a, 4b, Extended Data Fig. 3b, e, h).
Comparison of human and mouse cell type classifications
To independently evaluate the transcriptomic similarity of nuclei or cells in the human and mouse dentate gyrus, a previously described multi-class random forest classifier (using the R package ‘randomForest’ (v4.6.14)24,32) was trained on the human infant hippocampal dataset on six cell types, including imGCs and mGCs (determined by the machine learning model), astrocytes, OPCs, mature oligodendrocytes, and microglia. The number of nuclei from each cluster k used as the training set (Nk) was determined by Nk = min(200, 75% of | nucleik |). A common set of highly variable orthologous genes in both human and mouse datasets (identified using homology tables in Ensembl BioMart62) was used to train the classifier on Nk for 500 decision trees. The remaining nuclei from each of the six cell types were used as the validation set to estimate the accuracy, resulting in an out-of-bag classification error rate of 11.84%. The classifier was then used to map cells of the corresponding cell types from a scRNA-seq dataset of the adult mouse hippocampus9 using the published annotations based on unsupervised clustering (Fig. 1d).
In addition, we performed a Pearson correlation analysis (Fig. 2c) comparing human hippocampal imGCs and mGCs across ages determined by the machine learning model to the mouse neuroblasts, imGCs, and mGCs based on the top orthologous highly variable genes. Dataset A in ref9 was used as the mouse dataset, where imGCs and mGCs were annotated as “Granule-immature” and “Granule-mature”, respectively.
Deep generative model for batch correction
To eliminate sequencing variation within the human hippocampal datasets, we took precautions prior to quantitative gene expression comparison by correcting the data matrix using the single-cell Variational Inference (scVI40, v0.6.8, in Python (v3.7)), a neural network-based deep generative modeling method. Consequently, we obtained a shared, batch-corrected latent space among all the human hippocampal datasets across ages with the following parameters: (1) selecting the 20,000 most variable genes (using the ‘subsample_genes’ function in scVI); (2) training the variational auto-encoder model (VAE) with 90% of cells, holding 10% for validation to monitor overfitting and to measure accuracy (Supplementary Fig. 2a), using 128 hidden layers, and generating 30 latent dimensions (implemented in the ‘UnsupervisedTrainer(model = VAE)’ function in scVI); and (3) training the model at a learning rate of 1e-3 and 100 epochs (implemented in the ‘train’ function in scVI). UMAP was applied to the latent dimensions for visualization (Supplementary Fig. 2b). Robust clustering was achieved post-scVI-correction with excellent cluster correspondence to the results from the canonical correlation analysis by Seurat36, a state-of-the-art cell alignment tool, indicating effective batch correction (Supplementary Fig. 2c). We measured the efficacy of data matrix correction by comparing the expression of the housekeeping “stably expressed genes”39,63 across all age groups, benchmarking the efficacy of the ‘SC Transform’ correction method59 (implemented in Seurat) (Supplementary Fig. 2d). Prior to the differential gene expression comparisons, mouse datasets9 across ages were processed separately with scVI for consistency using the same parameters.
Differentially expressed gene analyses
To investigate differentially expressed genes (DEGs) in imGCs and mGCs across ages under physiological conditions, analysis was performed on the scVI-processed datasets in humans and in mice separately using a two-sided Wilcoxon rank sum test (implemented in the “FindMarkers” function in Seurat). For the human DEG analysis, all human imGCs and mGCs across ages were included. Separately for the mouse DEG analysis, all cells in the mouse “Granule-immature” (imGC) and “Granule-mature” (mGC) populations from Dataset A in ref9 were included. Only DEGs with orthologs in humans (identified using homology tables in Ensembl BioMart62) were included for further analyses. To compare gene expression in imGCs between AD and controls, DEG analysis was performed on the integrated dataset (using the ‘RNA slot’ of the Seurat object) (Fig. 4a, b) using a two-sided Wilcoxon rank sum test (implemented in “FindMarkers”). All imGCs in the AD and control groups were included. For all analyses, “max.cells.per.ident” in the “FindMarkers” function was determined by the cell number of the group with fewer cells for a fair statistical comparison. Genes with an FDR-adjusted p-value < 0.05 and fold change (log scale, absolute value) > 0.1 were considered significantly differentially expressed.
Pseudotime analysis
The R package ‘Monocle’41 (v2.8) was applied to construct single-cell/single-nuclei pseudo-temporal trajectories of human imGCs, human mGCs, mouse imGCs, and mouse mGCs across ages. The scVI-processed data matrices of the four cell types were individually imported into the Monocle pipeline. The highly variable genes within each cell type across ages, identified heuristically using the ‘vst’ method (implemented in the ‘FindVariableFeatures’ function in Seurat), were used to sort cells into a pseudotime order. The ‘DDRTree’ method was used to reduce dimension (implemented in the ‘reduceDimension’ function in Monocle). The minimum spanning tree on cells was plotted for visualization (implemented in the ‘plot_cell_trajectory’ function in Monocle).
Gene expression patterns were grouped after aligning cells on a pseudoage trajectory (Fig. 3h, Supplementary Fig. 4a). Significant genes were determined using a likelihood ratio test (implemented in the ‘differentialGeneTest’ function in Monocle), with Benjamini-Hochberg-adjusted p-value < 0.01 and q-value < 0.01.
Gene ontology, disease risk gene enrichment, and functional protein association analyses
Gene ontology (GO) networks of biological processes were built with the ClueGO (v.2.5.5) plug-in64 in Cytoscape65 (v3.7.2) with the following settings: ‘GO Biological process (January 9, 2020)’ was selected; running the default one-sided hypergeometric test, only pathways with FDR-adjusted p-value < 0.05 were displayed; ‘GO fusion’ option was enabled. Genes identified in the machine learning model or from differential expression analyses were selected as the significantly regulated genes and used as input. For groups with more than 200 significantly regulated genes, a minimum of 7 genes per cluster were used; and for all other groups, a minimum of 3 genes per cluster were used. In addition, the ‘compareCluster’ function (implemented in the R package ‘clusterProfiler’66) was applied to obtain representative GO term enrichment patterns (Fig. 3i) with the following parameters, fun = ‘enrichGO’, ont = ‘BP’, minGSSize = 3, pAdjustMethod = ‘fdr’, FDR-adjusted p-value < 0.05, and q-value < 0.05.
To map risk genes for brain disorders, we calculated the DEGs of imGCs and mGCs at each age using the ‘one_vs_all_degenes’ function (implementing the Bayes’ method) in scVI40 using the following parameters, mode = ‘vanilla’, min_cells = 1, n_samples = 10000. DEGs with natural log Bayes Factor ≥ 1.1 were considered significant. We then analyzed the enrichment of the significant DEGs in each category with disease annotations collected from the Phenopedia database67 (accessed on March 25, 2021) by calculating odds ratios and the enrichment p-values. P-values were determined by a one-tailed Fisher’s exact test (implemented in the ‘fisher.test’ function in R) and corrected by controlling for the FDR for multiple comparisons.
Functional protein association network analysis was performed using the stringApp (v1.5.1) plug-in68 in Cytoscape65 with default settings (Extended Data Fig. 4d). First-degree neighbors representing high-confidence connections were calculated with the following parameter: score = 0.35.
Cell-to-cell interaction analysis
We applied the CellPhone DB42 tool (v2.1.4) with its default settings to infer potential ligand-receptor interactions between imGCs and their neighboring cell types in the dentate gyrus in each sample, including astrocytes, OPC, mOli, microglia, choroid plexus cells, ependymal cells, endothelial cells, GABAergic interneurons, and Cajal-Retzius cells. Other excitatory neurons that were spatially separated from imGCs were excluded from the analysis. To investigate imGC niche interactions across ages, the scVI-processed hippocampal datasets (Supplementary Fig. 2) were used. Gene expression levels ≤ 1.0 were considered negligible and set to 0. To investigate perturbations of AD on imGC niche interactions, the integrated dataset (using the ‘SCT slot’ of the Seurat object) (Fig. 4a, b) were used. For both analyses, the mean expression of each ligand-receptor interaction pair for each cell type pair was calculated. A null distribution was generated by a one-sided random permutation of cell type identities over 1,000 times, followed by computation of the mean of each interaction pair for each iteration. The specificity of each interaction pair was determined by comparing the actual mean expression level against the null distribution. Statistically significant ligand-receptor interaction pairs, called using a threshold of p-value < 0.05, were used to quantify the number of interaction pairs for each cell type pair across ages (Supplementary Fig. 5) or in AD analysis (Fig. 4f). To determine age-related changes of human imGCs across the lifespan, we further assessed expression patterns of each significant ligand-receptor pair across ages using a Moran’s I test69. A specific cell-type interaction pair with a Bonferroni-adjusted p-value < 0.05 was considered age-dependent (exemplary gene pairs shown in Fig. 3k).
Human dentate gyrus ex vivo slice culture analysis
Fresh surgically resected hippocampal tissue was placed in ice-cold sterile cutting solution and taken immediately for vibratome-slicing. The cutting solution, an artificial cerebro-spinal fluid (aCSF) sucrose-based solution, containing 210.3 mM sucrose, 3 mM KCl (Sigma-Aldrich, P9333, CAS 7447-40-7), 1.3 mM MgCl2·6H2O (Sigma-Aldrich, 442615-M, CAS 7791-18-6), 2 mM CaCl2·2H2O (Sigma-Aldrich, C3306, CAS 10035-04-8), 26 mM NaHCO3 (Sigma-Aldrich, S5761, CAS 144-55-8), 1.25 mM NaH2PO4 (Sigma-Aldrich, S3139, CAS 7558-80-7), and 20 mM D-(+)-glucose (Sigma-Aldrich, G6152, CAS 50-99-7), was pre-saturated with carbonated oxygen (95% O2/5% CO2)70. Tissue specimens were first visually inspected to ensure inclusion of the dentate gyrus by its distinct anatomical structure. Slicing was performed within a laminar flow biosafety cabinet in continually oxygenated (95% O2/5% CO2) cutting solution, using a Leica VT 1200S vibratome at 0.1 mm/second speed, 1.2 mm vibration amplitude, and with 300 μm thickness interval54. 1.5-mL-per-well pre-warmed (37 °C) EdU+ BrainPhys medium44,71, containing BrainPhys Neuronal Medium (StemCell Technologies, 05790), 2% SM1 Neuronal Supplement (StemCell Technologies, 05711), 1% N2 Supplement (ThermoFisher Scientific, 17502048), 1% antibiotic-antimycotic (ThermoFisher Scientific, 15240062), and 1 μM EdU (Thermo Fisher Scientific, A10044), was added between a sterile Millicell tissue culture plate well insert (Millipore, PICM03050) and a well of a 6-well plate. Hippocampal slices were transferred and sparsely distributed onto the Millicell well inserts for better access to medium and oxygen during culture. Slices were cultured within a 37 °C, 5% CO2, 90% humidity sterile incubator, with half of the medium in each well replenished with fresh medium every two days. To prepare for whole-mount immunohistological analysis, slices were fixed with 4% paraformaldehyde (wt/vol; in 0.1M phosphate buffer (PB), pH 7.4) for 4–6 hours depending on the tissue size, followed by overnight cryoprotection with 30% sucrose (wt/vol). EdU incorporation was detected using Click-iT EdU Alexa Fluor 647 kit (ThermoFisher Scientific, C10340) prior to primary antibody incubation as previously described72. There are limitations for this approach, such as the use of pathological specimens from patients with epilepsy (Supplementary Table 1), and axonal and neuronal injuries from slicing. We characterized the cellular composition, viability, and oxidative stress state73 of slice cultures and compared them to those of human post-mortem specimens by immunohistology (Extended Data Fig. 9a–c). Importantly, to avoid the contribution from FGF-2 on neural progenitor reprogramming74, we cultured slices in the absence of any exogenous growth factors (EGF or FGF-2) and for a short period of cell culture time to assess the intrinsic capacity for postnatal human neurogenesis.
Immunostaining and confocal microscopy
Brain tissue sections were pre-treated and immunohistology was performed following a published protocol22 with modifications for optimal antigen retrieval. Briefly, brain tissue blocks were fixed with 4% paraformaldehyde (PFA) at 4 °C for 24–48 hours, and cryoprotected with 30% sucrose (wt/vol). 40-μm-thick sections were cut on a frozen sliding microtome (Leica, SM2010R) as previously described75. A small proportion of the brain specimens was prepared as formalin-fixed, paraffin-embedded (FFPE) sections. Prior to further pre-treatment, 10 μm FFPE sections were deparaffinized in 4 times xylene (Fisher Scientific, X5-1), 4 times 100% ethanol, and 4 times 95% ethanol, each for 5 minutes. Tissue sections were incubated with fresh-made 0.5% NaBH4 (Sigma-Aldrich, 213462; in 0.1 N phosphate buffer) for 30 minutes and washed 4 times with PBS, each for 5 minutes. The sections then underwent antigen retrieval prior to antibody application by being incubated in 1X target retrieval solution (DAKO) at 95 °C for 12.5 minutes, followed by a 15-minute cooling to room temperature. Antibodies were diluted in Tris buffered saline (TBS) with 0.1% Triton X-100, 5% (vol/vol) donkey serum (Millipore, S30), and sodium azide (Sigma, S2002, 1:100). Sections were incubated with primary antibodies at 4 °C for five days. The following primary antibodies were applied: Atf4 (CREB-2, rabbit, Abcam, ab28830, 1:250), Calbindin (rabbit, Abcam, ab49899, 1:250), Calbindin (rabbit, SWANT, D-28k, CB38, 1:500), cleaved Caspase 3 (rabbit, Cell Signaling Technology, 9661s, 1:250), Doublecortin (rabbit, Cell Signaling Technology, 4604s, 1:500), Doublecortin (goat, Santa Cruz Biotechnology, sc8066, 1:250, only works against mouse tissue), Iba1 (rabbit, WAKO, 019-19741, 1:500), Math3 (Neurod4, mouse, Santa Cruz Biotechnology, sc393724, 1:100), Mki67 (mouse, BD Biosciences, 550609, 1:500), NeuN (mouse, Millipore, MAB377X, 1:500), Neurod1 (mouse, Abcam, ab60704, 1:250), Nfia (mouse, CDI Laboratories, 1.2C6, 1:500), Olig2 (goat, R&D Systems, AF2418, 1:500), Op18 (Stmn1, mouse, Santa Cruz Biotechnology, sc48362, 1:250, only works against human tissue), Prox1 (rabbit, Abcam, ab101851, ab11941, 1:500), Prox1 (goat, R&D Systems, AF2727, 1:500), S100b (rabbit, Sigma, s2644, 1:500), Stmn1 (goat, GeneTex, GTX89411, 1:500, only works against mouse tissue), Stmn1 (rabbit, Abcam, ab24445, 1:500), and Tbr2 (Eomes, rabbit, Abcam, ab216870, 1:250). The Cyanine (Cy)-conjugated secondary antibodies raised in donkey (Jackson ImmunoResearch; 1:300), including Cy2 anti-goat (705-225-147), mouse (715-225-151), rabbit (711-225-152), Cy3 anti-goat (705-165-147), mouse (715-165-151), rabbit (711-165-152), Cy5 anti-goat (705-175-147), mouse (715-175-151), and rabbit (711-175-152), were incubated at room temperature for 2 hours along with DAPI (Roche, 10236276001, 1:1000). After washing with TBS, sections were incubated with 1X TrueBlack (Biotium, 23007; diluted 1:20 in 70% ethanol) for 1 minute to block the autofluorescent lipofuscin and blood components. After washing with PBS, stained sections were mounted and imaged as Z-stacks on a Zeiss LSM 800 confocal microscope (Carl Zeiss) using a 20X or a 40X objective with Zen 2 software (Carl Zeiss).
Image processing and data analysis
All confocal images were blindly acquired among different specimens under the same laser power and gain, and analyzed as Z-stacked images using Imaris 9.0 software (BitPlane) as previously described76,77. The Spots module in Imaris was used to digitize cell-nucleus locations in 3D space and to code cell type classifications according to distinct morphological and molecular markers. A minimum of three randomly chosen areas of equal dimensions within each dentate gyrus tissue were quantitated. The sum of quantifications of these areas per section was considered as one data point. In Fig. 5c, d, due to the sparsity of marker positive cells, quantification of all sections from one patient specimen were summed as one data point. No statistical methods were used to predetermine sample size.
To quantify total numbers of PROX1+ GCs (Fig. 5c, d) or DAPI+ cells (Extended Data Fig. 9b), semi-automated nuclear staining quantification was performed using Fiji-ImageJ78. DAPI and PROX1 channels of confocal image files (.czi) were converted to “.tiff” format and imported to Image J. The DAPI channel was utilized to manually select the sub-granular zone and GC layer as a region of interest (ROI) in each image using the ‘polygon’ tool. The resulting cropped image was utilized to generate individual ROIs for each nucleus in an image by background subtraction with a rolling ball radius of 50, auto-thresholding with the default algorithm, despeckling, nuclear segmentation using the ‘watershed’ function, and finally ROI generation via the ‘Analyze Particles’ function with a minimal size of 5 and circularity 0.2 to 1.0. For quantification of number of PROX1+ GCs (Fig. 5c, d), the corresponding PROX1 channel image for each file was then opened and each nucleus was background subtracted with rolling ball radius of 50. Mean intensities of each nucleus within the previously determined ROIs were measured, and results were inspected in the R software with attention to the overall intensity distributions. Thresholds for assigning marker positivity were determined manually by measuring the mean intensity of nuclei with the minimal signal that would have been determined to be marker positive by traditional manual counting. This process was repeated three times for each image file and results were averaged to ensure consistency and reproducibility.
Quantification and statistical analysis
The studies were blinded during data collection and quantification. Data in figure panels reflect several independent experiments performed on different days. No data were excluded. An estimate of variation within each group of data is indicated using standard error of the mean (s.e.m.). All data are shown as mean ± s.e.m. All statistical analyses are indicated in the text or Figure Legends, performed with the R language for Statistical Computing (v3.6; https://www.r-project.org/).
Extended Data
Extended Data Fig. 1 |. Characteristics of the snRNA-seq dataset of the infant human hippocampus.
a, Expression patterns of marker genes used to determine cluster identities. Ex.: excitatory; OPC: oligodendrocyte precursor cells. b, Uniform Manifold Approximation and Projection (UMAP) visualization of all cells from the four infant hippocampi (0–2 years) colored by specimen. HIP: hippocampus; yrs: years. c, UMAP plots of nuclei from four human infant hippocampal specimens by marker gene expression. The dentate granule cell cluster is highlighted with a dashed line circle.
Extended Data Fig. 2 |. Machine learning model trained with the mouse early postnatal hippocampal scRNA-seq dataset.
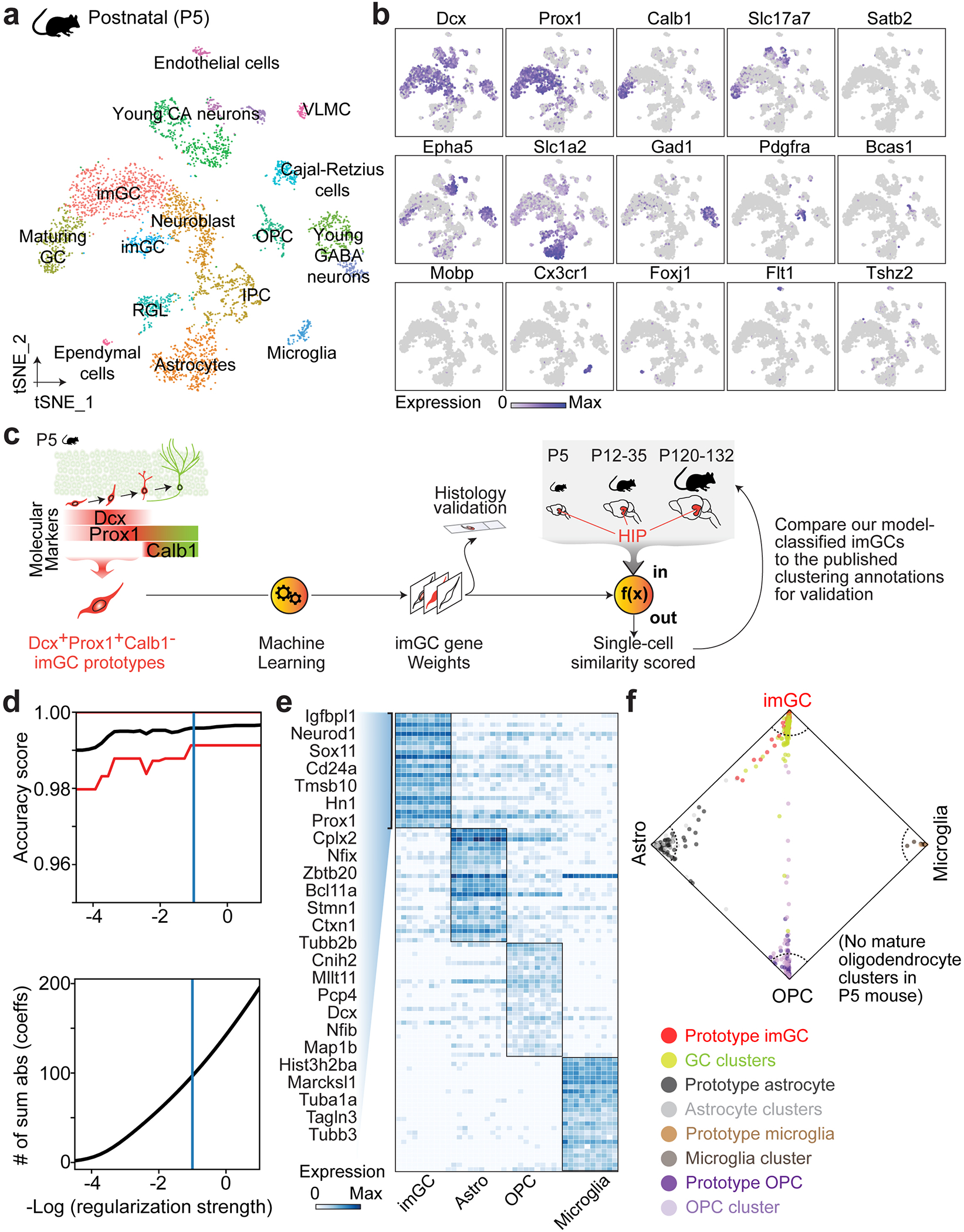
a, b, Unsupervised clustering and t-distributed Stochastic Neighbor Embedding (t-SNE) visualization of all cells from the mouse postnatal (P5) hippocampus9 colored by cluster (a) and marker gene expression (b). imGC: immature dentate granule cell; GC: dentate granule cell; IPC: intermediate progenitor cell; OPC: oligodendrocyte precursor cell. RGL: radial glia-like cell; VLMC: vascular and leptomeningeal cell. c, A schematic illustration of the machine learning-aided analysis using the mouse hippocampal scRNA-seq datasets9, mirroring our analysis pipeline in human studies (Fig. 1a). In brief, Dcx+Calb1−Prox1+ imGCs in the P5 mouse dentate gyrus were selected as prototypes to train a scoring model to comprehensively learn their gene features. The trained model containing an aggregate of weighted features (“gene weights”) was then used to quantitatively evaluate the similarity of each cell to the imGC prototype in query (test) datasets of the early postnatal (P5; self-scoring), the juvenile (P12-35) and the adult (P120-132) hippocampus9. To assess the efficacy of our method, we classified cells with high similarity scores to the imGC prototype as imGCs and compared our model classifications to the published annotations based on unsupervised clustering9 (Shown in Extended Data Fig. 3). d, Measuring performance of the machine learning model. Line plot showing the accuracy score of the machine learning classifier varying with decreasing regularization strength as estimated by cross-validation. Red line shows 95% confidence interval on the estimation of the accuracy score. #Sum abs (coeffs): sum of the absolute value of regression coefficients. e, Heatmap showing expression of top-weighted genes in top-scoring cells of each prototype determined by the machine learning model. Genes listed are the top 25 weights defining mouse imGCs. f, Wheel plot visualizing the scores of each cell to each prototype. Dots represent individual cells whose distance to each prototype is proportional to the score of that prototype. Red and lime green dots represent the prototypical imGCs and all other GCs, respectively. Dotted line indicates a similarity score of 0.85 to each prototypical cell type. Note that unlike in the human system (Fig. 1c), no mature oligodendrocyte (mOli) cluster was present in the P5 mouse hippocampus.
Extended Data Fig. 3 |. Validation of prototype-based scoring of mouse imGCs across ages by the trained machine learning model with published annotations based on unsupervised clustering.
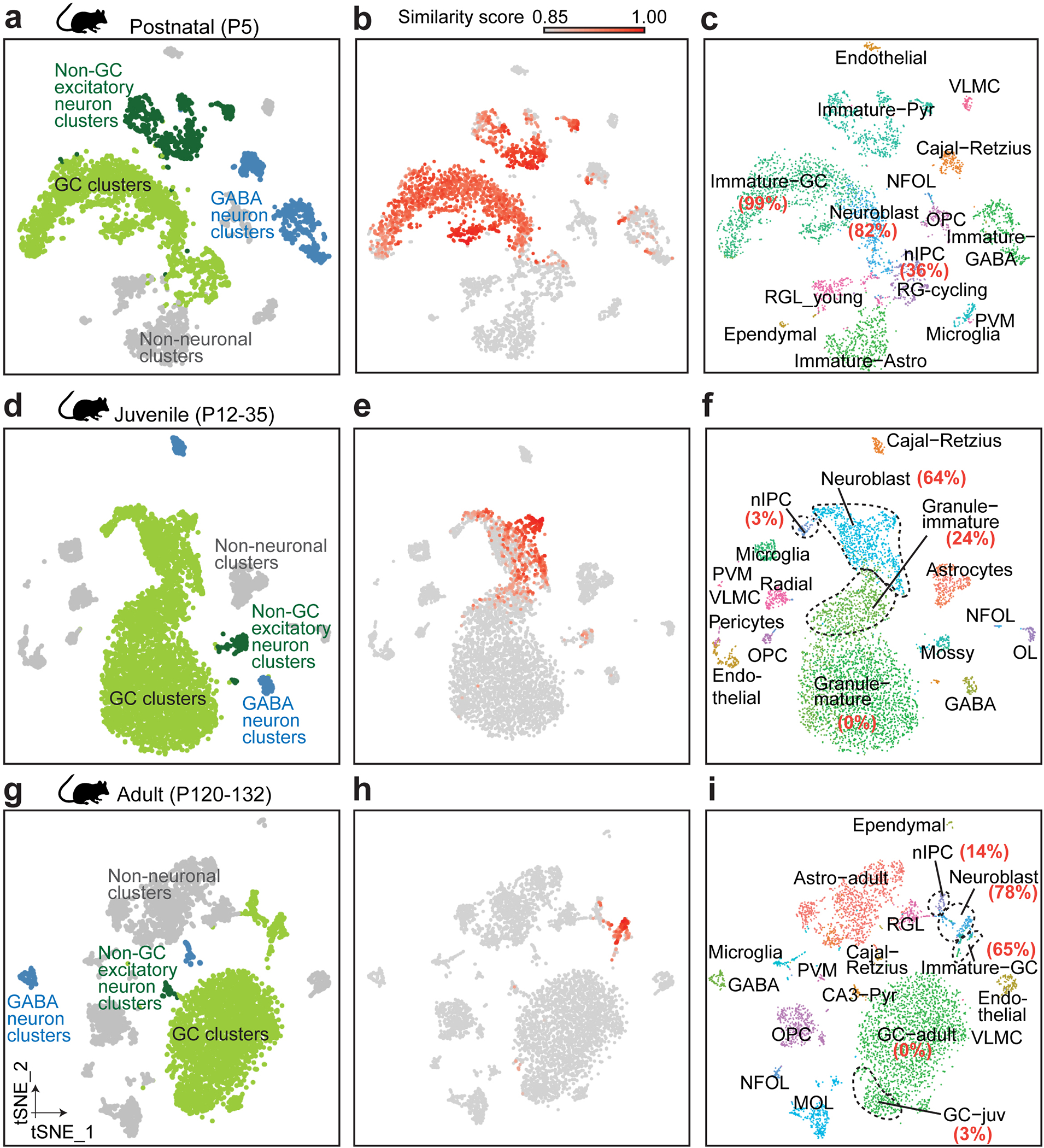
a, b, d, e, g, h, t-SNE visualization of previously published mouse hippocampal datasets9 at postnatal (a), juvenile (d), and adult (g) stages, colored by four broad cell classes and by similarity score to prototypical imGCs (b, e, h). c, f, i, Benchmarking cells with high similarity scores (p ≥ 0.85) with the published annotations9. Percentage of cells in the GC lineage clusters (based on published annotations9) that are selected as imGCs by our trained machine learning model are indicated in red, bold text.
Extended Data Fig. 4 |. Machine learning model performance and feature extraction of gene weights defining human imGCs.
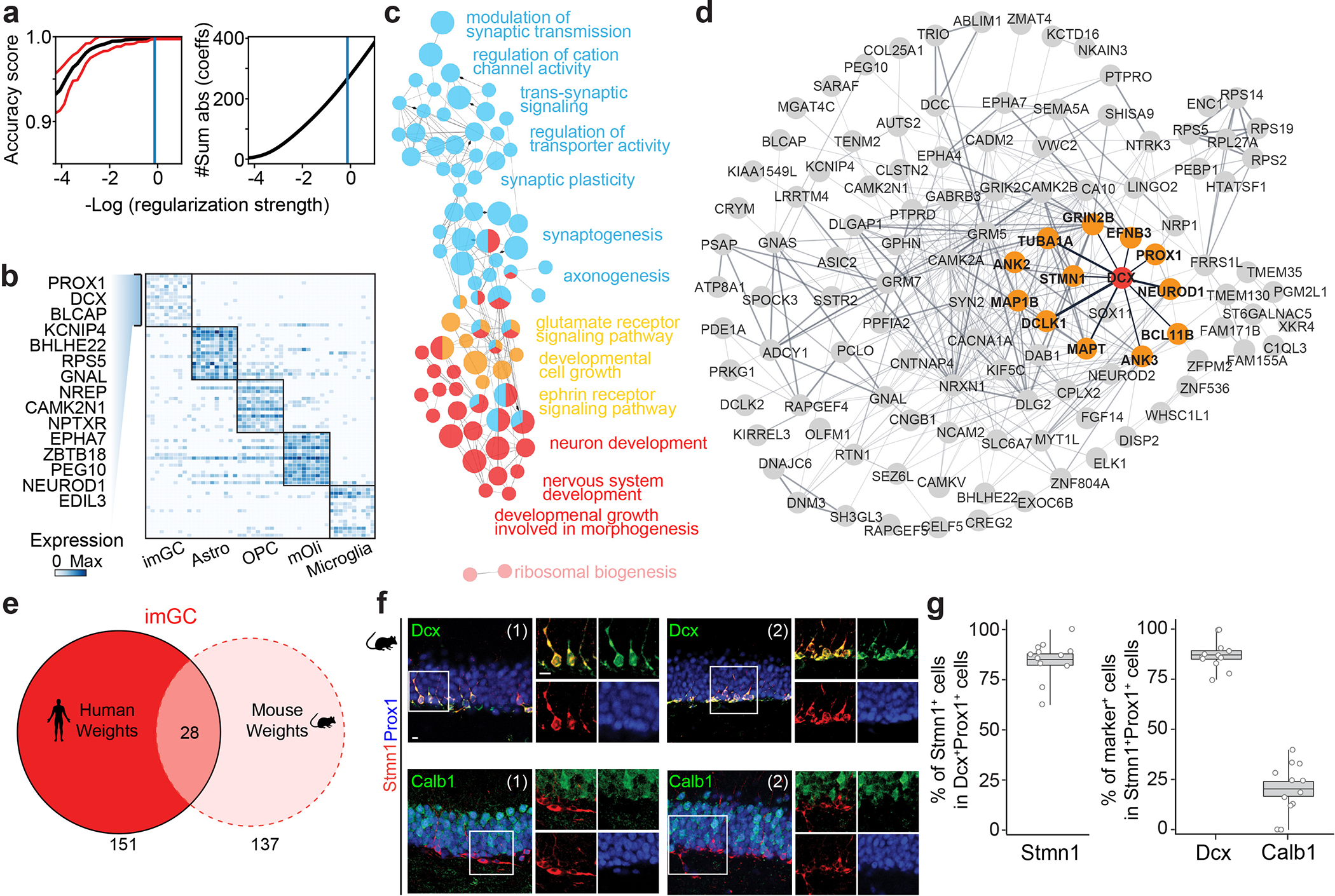
a, Efficacy of the machine-learning approach. Line plot showing the accuracy score of the machine learning model varying with decreasing regularization strength as estimated by cross-validation. Red line shows 95% confidence interval on the estimation of the accuracy score. b, Heatmap showing expression of top gene weights in top-scoring cells of each prototype determined by the machine learning model. Genes listed are the top 15 weights defining human imGCs. c, Gene ontology (GO) network of biological processes of the positive gene weights defining human imGCs, colored by functionally related ontology group. Only significantly enriched nodes are displayed (one-sided hypergeometric test, false-discovery rate-adjust p value (FDR) < 0.05). The node size represents the term enrichment significance. Examples of the most significant terms per group are shown. See also Supplementary Table 5 for the list of GO terms. d, Functional protein-protein association network68 of the positive gene weights defining human imGCs, highlighting the first-degree neighbors (high-confidence connections) in orange related to DCX. e, Overlap of the positive gene weights defining imGCs in humans and in mice that were generated by separate machine learning models. See Supplementary Table 4 for the lists of genes. f, g, Immunohistological analysis showing Stmn1 enrichment in immature neurons in the adult mouse dentate gyrus. Shown are sample confocal images (f) and quantification (g) of Stmn1 expression in imGCs in the adult mouse hippocampus. Individual dots represent value of quantification for different sections (f). Scale bars, 10 μm. Box plots similar as in Fig. 1g (n = 4 mice) (g).
Extended Data Fig. 5 |. Specificity of the machine learning approach for identification of human immature neurons.
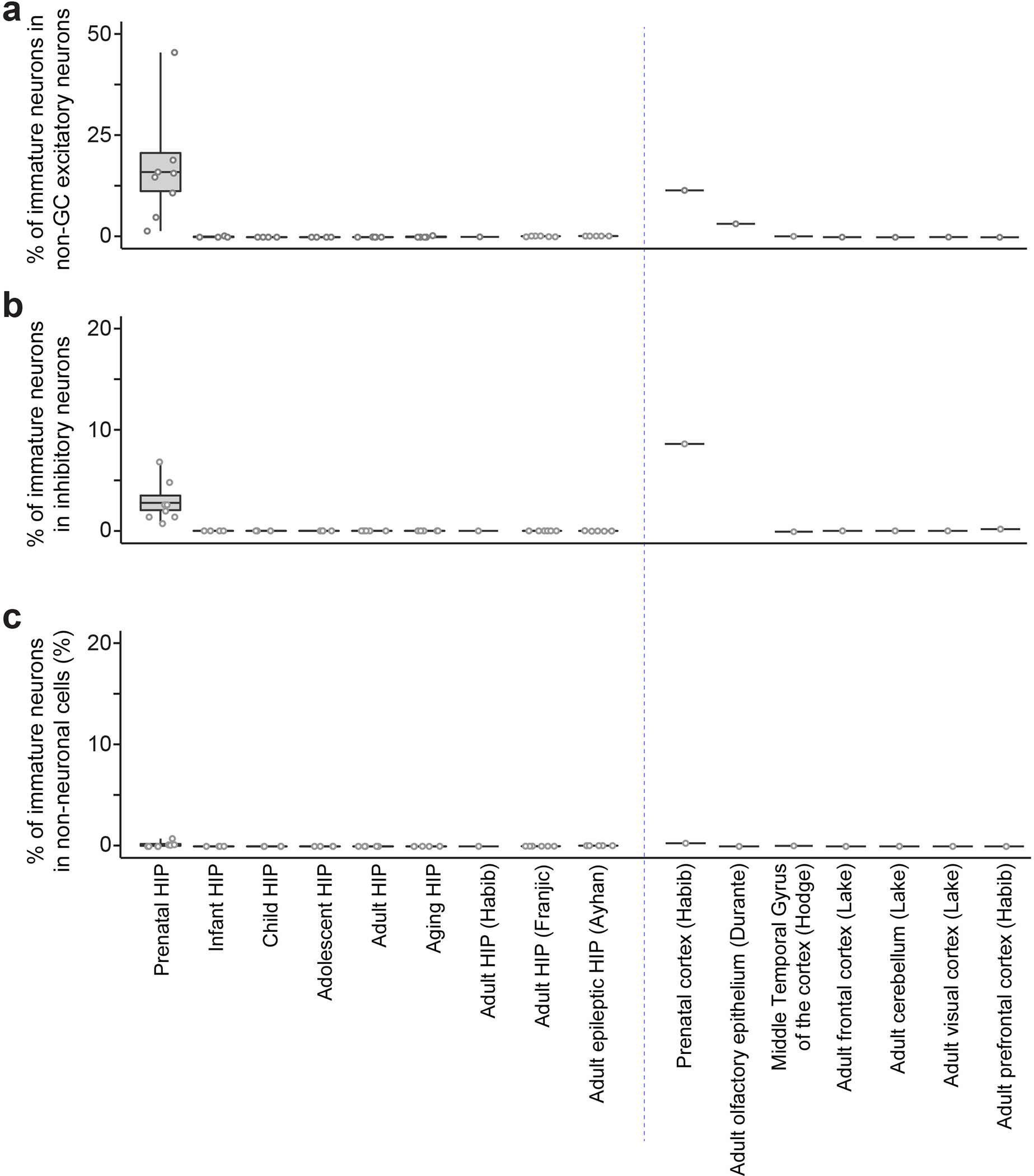
The fractions of cells with high similarity scores (p ≥ 0.85) among non-GC excitatory neuron (a), GABA interneuron (b), and non-neuronal cell (c) clusters in various scRNA-seq/snRNA-seq datasets of the human brains. Box plots represent mean ± s.e.m. with whiskers for max and min. See Supplementary Tables 1, 2, 3 for the specimens used in ours and all published datasets.
Extended Data Fig. 6 |. Immunohistological analysis of STMN1 enrichment in human imGCs across the lifespan.
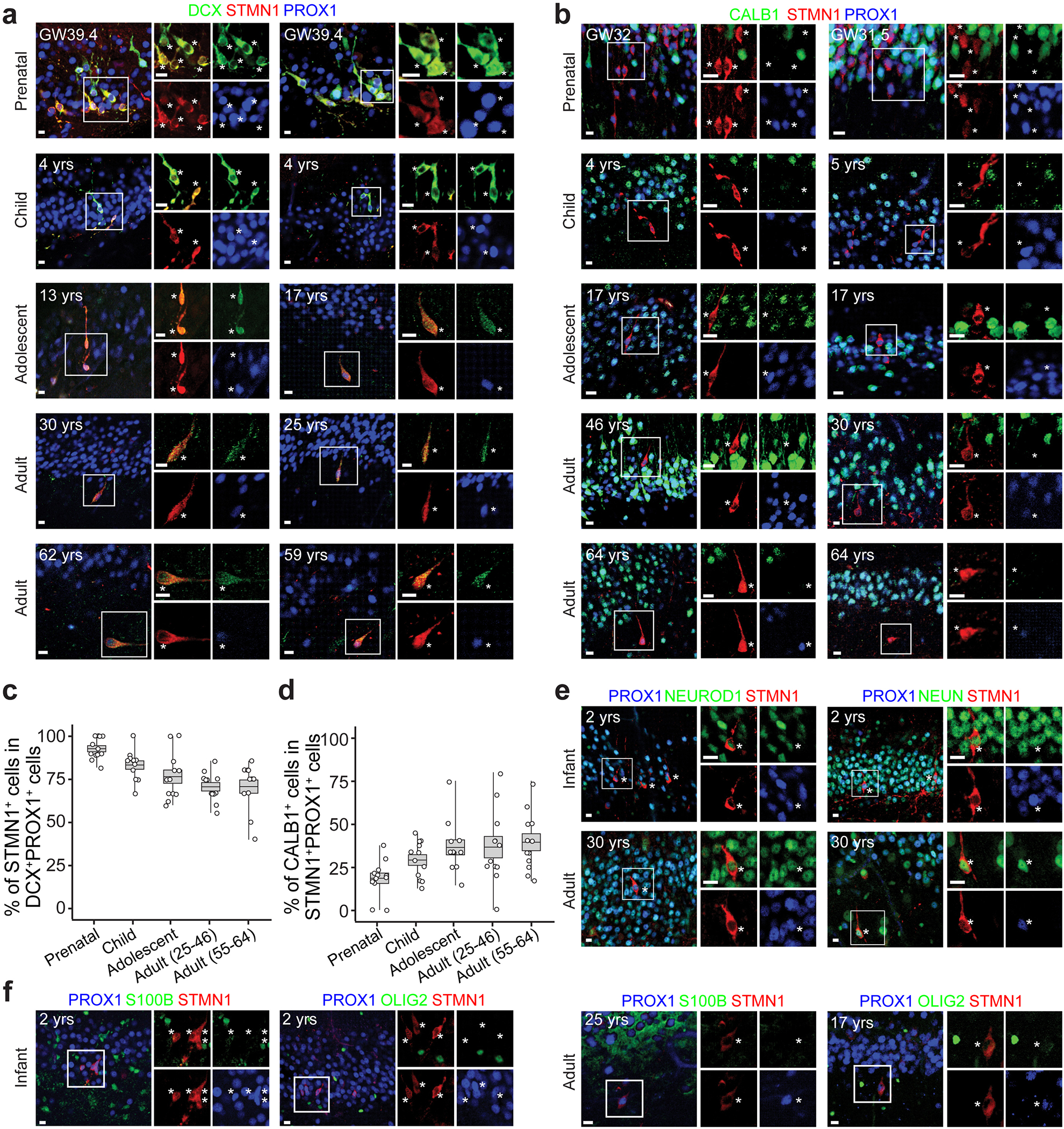
a-d, Sample confocal images (a, b) and quantifications (c, d) of imGCs in the human dentate gyrus across the lifespan. Asterisks indicate DCX+ or CALB1− among STMN1+PROX1+ GCs (a, b). Box plots similar as in Fig. 1g (n = 4 subjects each group) (c, d). The immunohistological signal of STMN1 was noticeably more robust than that of DCX in adult specimens. e, f, Sample confocal images showing NEUROD1+, NEUN+ (e), S100B−, or OLIG2− (f) among STMN1+PROX1+ imGCs in infant or adult human dentate gyrus, confirming their neuronal identity. Asterisks indicate STMN1+PROX1+ imGCs (n = 1 specimen for each immunostaining). All scale bars, 10 μm.
Extended Data Fig. 7 |. Age-dependent transcriptomic dynamics are specific to human imGCs.

a-c, In contrast to human imGCs (Fig. 3f), pseudo-age cell alignment of human mature (a), mouse immature (b), and mouse mature (c) GCs9 shows very little age-related divergence, visualized as scatter plots. Cells were colored by age group. Distribution of cells within each age group on the pseudo-age trajectory is displayed in the density plots (bottom left). See summary plots in Fig. 3g. d, Summary plot comparing pseudo-age alignment (y-axis) of mouse mGCs to real age groups (x-axis), with each mGC of the different age groups plotted as a data point in the background. Data points are fitted with loess fitting (lines) with 95% confidence interval (grey shades). Pearson’s r was measured for correlation of pseudo-age and real-age groups. Mouse datasets9 at prenatal (E16.5), neonatal (P0), or early postnatal (P5) stages do not contain mGC populations.
Extended Data Fig. 8 |. Consistent expression of Neurod4 and Nfia in imGCs of the postnatal mouse hippocampus across ages.
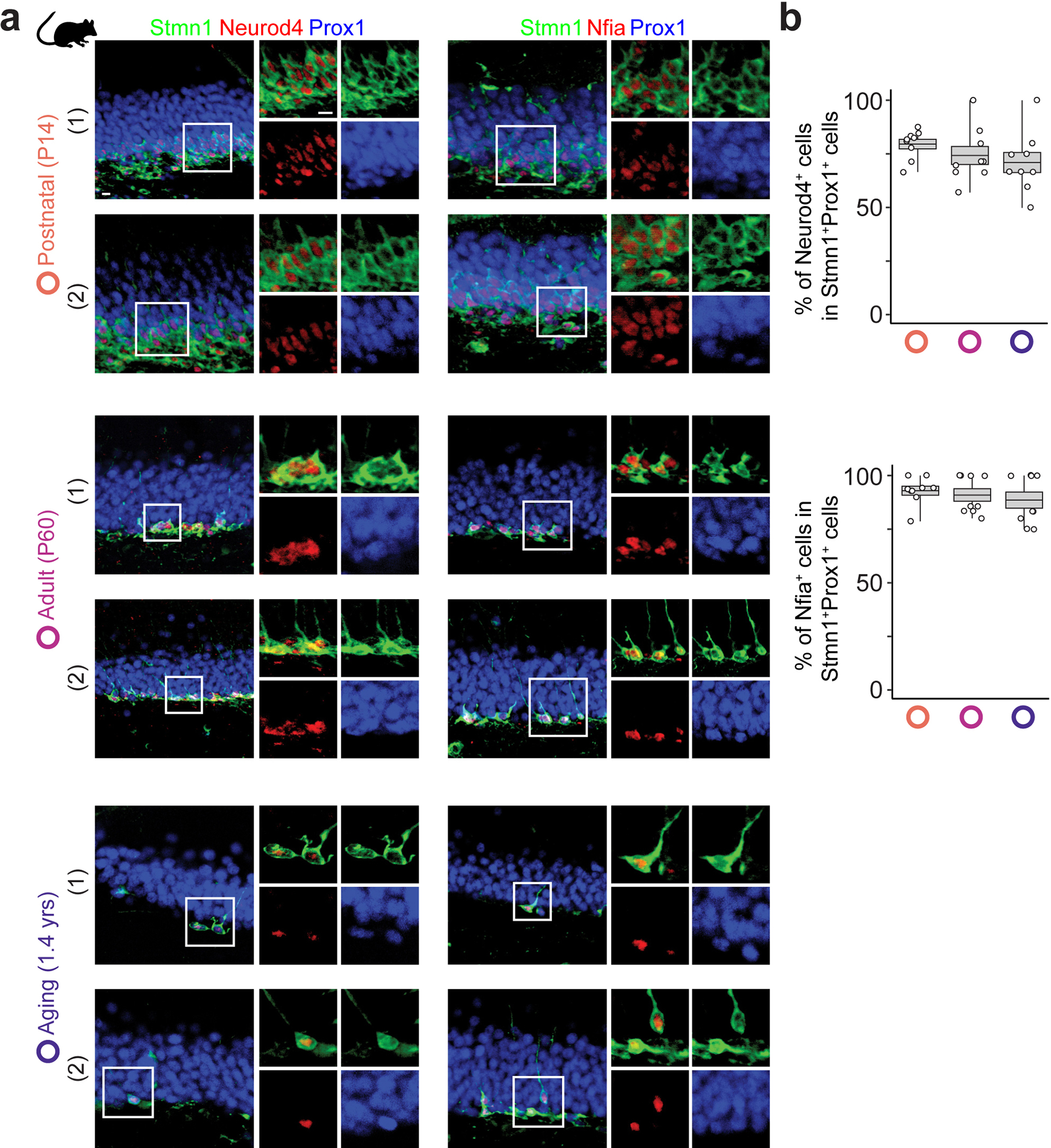
a, b, Sample confocal immunostaining images (a) and quantification (b) of two exemplary genes that display age-dependent expression patterns in human imGCs (Fig. 3j), but consistent expression in mouse imGCs across ages. Scale bar, 10 μm (a). Box plots similar as in Fig. 1g (n = 3 mice per age group) (b).
Extended Data Fig. 9 |. Characterization of the slice culture system.
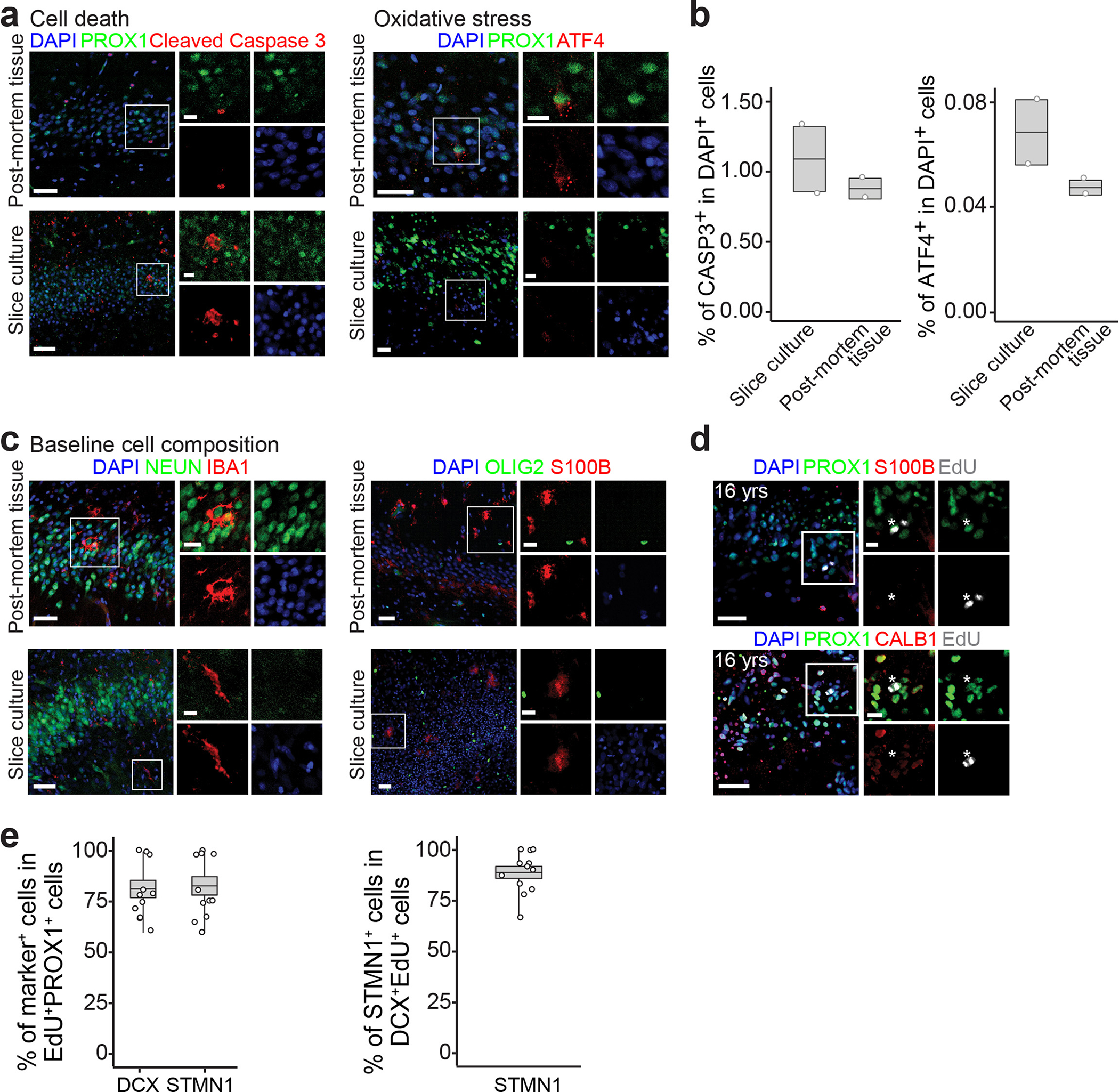
a, b, Sample confocal images (a) and quantification (b) of cell death and oxidative stress level measured in our human hippocampal slice culture in comparison to the post-mortem tissue, using immunohistological analysis of cleaved Caspase 3 and ATF4 (a marker of oxidative stress73), respectively. Dots represent value of quantification for individual sections and boxes represent mean ± s.e.m with whiskers for max and min (n = 2 sections) (b). c, Sample confocal immunostaining images showing baseline cellular composition of slice culture and post-mortem tissue. NEUN+ neurons, IBA1+ microglia, S100B+ astrocytes, and OLIG2+ oligodendrocyte lineage cells were observed. d, Sample confocal images showing EdU-incorporated PROX1+ newborn GCs are absent of the astrocyte marker S100B or the more mature neuron marker CALB1 in slice cultures. Asterisks indicate EdU+PROX1+ GCs. For c, d, n = 1 section for each immunostaining. Scale bars: 50 μm for main panels and 10 μm for insets. e, Quantification of EdU-incorporated newborn imGCs expressing different markers in the postnatal human dentate gyrus. Box plots similar as in Fig. 1g (n = 4 subjects).
Extended Data Fig. 10 |. Protracted neuronal maturation leads to accumulation of immature neurons in the presence of low frequency of de novo new neuron generation.
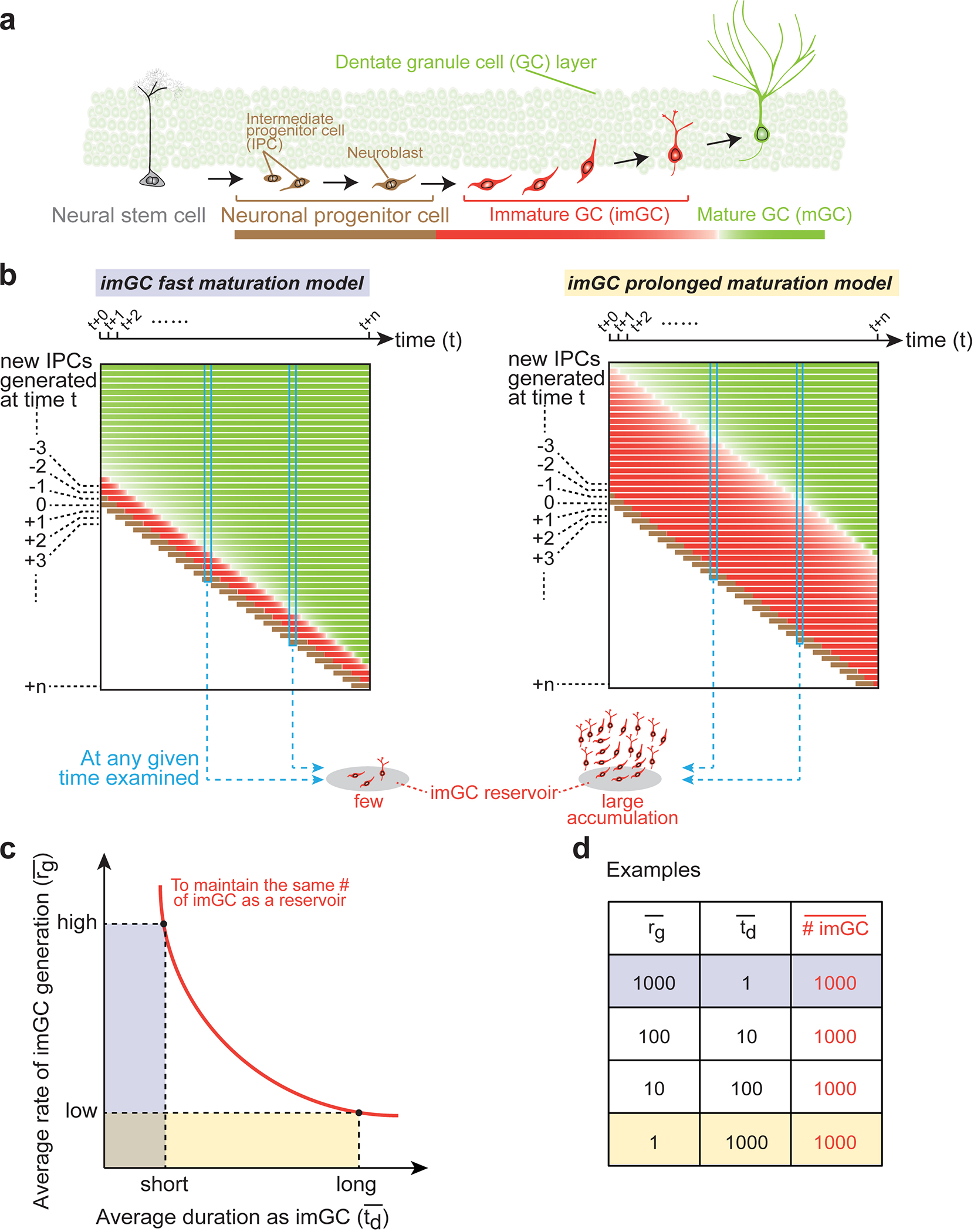
a, Process of adult hippocampal neurogenesis1,2. Proliferating intermediate progenitor cells (IPCs) and neuroblasts (brown) arising from activated neural stem cells (NSCs, grey) generate new post-mitotic immature dentate granule cells (imGCs, red), which develop over time into mature dentate granule cells (mGCs, lime-green). b, An “imGC protracted maturation” model explaining how low-rate, continuous IPC generation can lead to a large number of imGCs as a reservoir, as opposed to a “fast maturation” model. The size of the imGC reservoir in the adult hippocampus depends on a number of factors at the cellular level, such as the rate of stem cell activation and IPC generation, the number of progeny each IPC generates, the percentage of progeny that survives79, and the duration of imGCs remaining in the immature state, and these parameters may vary tremendously across species and ages80. Here we illustrate side-by-side two schematic models showing how changing one factor, the length of imGC maturation duration, alone while keeping all other parameters the same can lead to significant differences in the outcome on the number of imGCs at a given time. For IPCs in a newly generated cohort at a given time t, they go through stereotypical developmental stages to become imGCs and then mGCs (x-axis). At time t+1, a new IPC cohort is generated (y-axis). With all other parameters the same, if the imGCs mature fast, very few imGCs will be observed at any given time (left model). In contrast, if the average length of imGC maturation duration is substantially longer, imGCs in various maturation stages accumulate over time and are present as a large population in any “snapshot” (right model). Prolonged maturation duration of new neurons in the hippocampus has been demonstrated in non-human primates using nucleotide analog tracing analysis to be at least six months49 and over a year50. Furthermore, human induced pluripotent stem cell-derived transplanted neurons display significantly slower maturation compared to those of three non-human primates81. c, d, An indifference curve qualitatively depicting different combinations of two factors, the average rate of new neuron generation (r̅g) and the average duration of imGC maturation (t̅d), to achieve an equal size of imGC reservoirs (c). Hypothetical examples shown in d. A significantly longer t̅d in the adult human hippocampus spares the system from high demand of r̅g to maintain the same size of imGC reservoir, which is a potential model to explain the seemingly counterintuitive discrepancy between the few IPCs and a large number of imGCs in our results.
Supplementary Material
Acknowledgments
We thank all brain donors, patients, and their families for the tissue specimens used in this study and members of Ming and Song laboratories for discussion; B. Temsamrit, E. LaNoce, and A. Garcia for technical support; A. Angelucci and J. Schnoll for lab coordination; F.H. Gage, G. Kempermann, S. Jessberger, K.M. Christian for comments; O. Spicer at the NIH NeuroBioBank, J. Glausier at the University of Pittsburgh, A. LeFevre and J. Cottrell at the University of Maryland, and J. Hussain at the University of Pennsylvania for assistance with human tissue acquisition; K. McCanon at the Santa Cruz Biotechnology, Z. Yang at the Thermo Fisher Scientific, and H. Zhu and CDI Laboratories for providing antibody samples, and M. Llorens-Martin for suggestions on immunohistology. Some of the schematic illustrations were modified from images from https://togotv.dbcls.jp/ or https://biorender.com/. This work was supported by grants from the National Institutes of Health (K01MH125144 to A.M.B., R35NS097370 to G.-l.M., and R35NS116843 to H.S.), The Lieber Institute for Brain Development (to J.E.K., T.M.H. and D.R.W.), and Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to G.-l.M.).
Footnotes
Competing interests
The authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Code availability
All software used in this study are listed in the Reporting Summary along with their versions. The computational code used in this study is available at GitHub (https://github.com/zhoujoeyyi/humanImmatureNeurons) or upon request.
Additional information
Supplementary information is available for this paper.
Reprints and permissions information is available at http://www.nature.com/reprints.
Data availability
The snRNA-seq data are available at the GEO database (accession numbers: GSE185553, GSE185277, and GSE198323). Specimen information and sequencing statistics are described in Supplementary Tables 1, 2. Sources of the published scRNA-seq or snRNA-seq datasets used in this study are described in Supplementary Table 3.
References
- 1.Gage FH Adult neurogenesis in mammals. Science 364, 827–828, doi: 10.1126/science.aav6885 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Ming GL & Song H Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702, doi: 10.1016/j.neuron.2011.05.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terreros-Roncal J et al. Impact of neurodegenerative diseases on human adult hippocampal neurogenesis. Science 0, doi:: 10.1126/science.abl5163 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christian KM, Song H & Ming GL Functions and dysfunctions of adult hippocampal neurogenesis. Annual review of neuroscience 37, 243–262, doi: 10.1146/annurev-neuro-071013-014134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreno-Jimenez EP et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nature medicine 25, 554–560, doi: 10.1038/s41591-019-0375-9 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Sorrells SF et al. Positive Controls in Adults and Children Support That Very Few, If Any, New Neurons Are Born in the Adult Human Hippocampus. J Neurosci 41, 2554–2565, doi: 10.1523/JNEUROSCI.0676-20.2020 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreno-Jimenez EP, Terreros-Roncal J, Flor-Garcia M, Rabano A & Llorens-Martin M Evidences for Adult Hippocampal Neurogenesis in Humans. J Neurosci 41, 2541–2553, doi: 10.1523/JNEUROSCI.0675-20.2020 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kempermann G et al. Human Adult Neurogenesis: Evidence and Remaining Questions. Cell stem cell 23, 25–30, doi: 10.1016/j.stem.2018.04.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hochgerner H, Zeisel A, Lonnerberg P & Linnarsson S Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nature neuroscience 21, 290–299, doi: 10.1038/s41593-017-0056-2 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Eriksson PS et al. Neurogenesis in the adult human hippocampus. Nature medicine 4, 1313–1317, doi: 10.1038/3305 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Spalding KL et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227, doi: 10.1016/j.cell.2013.05.002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst A et al. Neurogenesis in the striatum of the adult human brain. Cell 156, 1072–1083, doi: 10.1016/j.cell.2014.01.044 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Schmidt-Hieber C, Jonas P & Bischofberger J Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 429, 184–187, doi: 10.1038/nature02553 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Ge S, Yang CH, Hsu KS, Ming GL & Song H A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron 54, 559–566, doi: 10.1016/j.neuron.2007.05.002 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marin-Burgin A, Mongiat LA, Pardi MB & Schinder AF Unique processing during a period of high excitation/inhibition balance in adult-born neurons. Science 335, 1238–1242, doi: 10.1126/science.1214956 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knoth R et al. Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PloS one 5, e8809, doi: 10.1371/journal.pone.0008809 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boldrini M et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell stem cell 22, 589–599 e585, doi: 10.1016/j.stem.2018.03.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobin MK et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell stem cell 24, 974–982 e973, doi: 10.1016/j.stem.2019.05.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ammothumkandy A et al. Altered adult neurogenesis and gliogenesis in patients with mesial temporal lobe epilepsy. Nature neuroscience 25, 493–503, doi: 10.1038/s41593-022-01044-2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorrells SF et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555, 377–381, doi: 10.1038/nature25975 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cipriani S et al. Hippocampal Radial Glial Subtypes and Their Neurogenic Potential in Human Fetuses and Healthy and Alzheimer’s Disease Adults. Cereb Cortex 28, 2458–2478, doi: 10.1093/cercor/bhy096 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Flor-Garcia M et al. Unraveling human adult hippocampal neurogenesis. Nat Protoc 15, 668–693, doi: 10.1038/s41596-019-0267-y (2020). [DOI] [PubMed] [Google Scholar]
- 23.Kempermann G, Jessberger S, Steiner B & Kronenberg G Milestones of neuronal development in the adult hippocampus. Trends Neurosci 27, 447–452 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 14, 955–958, doi: 10.1038/nmeth.4407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayhan F et al. Resolving cellular and molecular diversity along the hippocampal anterior-to-posterior axis in humans. Neuron 109, 2091–2105 e6, doi: 10.1016/j.neuron.2021.05.003 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davila-Velderrain J et al. Single-cell anatomical analysis of human hippocampus and entorhinal cortex uncovers early-stage molecular pathology in Alzheimer’s disease. 2021.2007.2001.450715, doi: 10.1101/2021.07.01.450715 %J bioRxiv (2021). [DOI] [Google Scholar]
- 27.Franjic D et al. Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron 110, 452–469 e414, doi: 10.1016/j.neuron.2021.10.036 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.La Manno G et al. Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 167, 566–580 e519, doi: 10.1016/j.cell.2016.09.027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Galen P et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 176, 1265–1281 e1224, doi: 10.1016/j.cell.2019.01.031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Telley L et al. Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 364, doi: 10.1126/science.aav2522 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Bielecki P et al. Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature 592, 128–132, doi: 10.1038/s41586-021-03188-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shekhar K et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323 e1330, doi: 10.1016/j.cell.2016.07.054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gavet O et al. The stathmin phosphoprotein family: intracellular localization and effects on the microtubule network. J Cell Sci 111 (Pt 22), 3333–3346 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Zhong S et al. Decoding the development of the human hippocampus. Nature 577, 531–536, doi: 10.1038/s41586-019-1917-5 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Zhong S et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528, doi: 10.1038/nature25980 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420, doi: 10.1038/nbt.4096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durante MA et al. Single-cell analysis of olfactory neurogenesis and differentiation in adult humans. Nature neuroscience 23, 323–326, doi: 10.1038/s41593-020-0587-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lake BB et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80, doi: 10.1038/nbt.4038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hodge RD et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68, doi: 10.1038/s41586-019-1506-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopez R, Regier J, Cole MB, Jordan MI & Yosef N Deep generative modeling for single-cell transcriptomics. Nature Methods 15, 1053–1058, doi: 10.1038/s41592-018-0229-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu X et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982, doi: 10.1038/nmeth.4402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15, 1484–1506, doi: 10.1038/s41596-020-0292-x (2020). [DOI] [PubMed] [Google Scholar]
- 43.La Rosa C, Parolisi R & Bonfanti L Brain Structural Plasticity: From Adult Neurogenesis to Immature Neurons. Front Neurosci 14, 75, doi: 10.3389/fnins.2020.00075 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacob F et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 180, 188–204 e122, doi: 10.1016/j.cell.2019.11.036 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar A et al. Transcriptomic analysis of the signature of neurogenesis in human hippocampus suggests restricted progenitor cell progression post-childhood. IBRO Rep 9, 224–232, doi: 10.1016/j.ibror.2020.08.003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cole JD et al. Adult-Born Hippocampal Neurons Undergo Extended Development and Are Morphologically Distinct from Neonatally-Born Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 40, 5740–5756, doi: 10.1523/JNEUROSCI.1665-19.2020 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trinchero MF, Herrero M, Monzon-Salinas MC & Schinder AF Experience-Dependent Structural Plasticity of Adult-Born Neurons in the Aging Hippocampus. Front Neurosci 13, 739, doi: 10.3389/fnins.2019.00739 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trinchero MF et al. High Plasticity of New Granule Cells in the Aging Hippocampus. Cell Rep 21, 1129–1139, doi: 10.1016/j.celrep.2017.09.064 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Kohler SJ, Williams NI, Stanton GB, Cameron JL & Greenough WT Maturation time of new granule cells in the dentate gyrus of adult macaque monkeys exceeds six months. Proc Natl Acad Sci U S A 108, 10326–10331, doi: 10.1073/pnas.1017099108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ngwenya LB, Heyworth NC, Shwe Y, Moore TL & Rosene DL Age-related changes in dentate gyrus cell numbers, neurogenesis, and associations with cognitive impairments in the rhesus monkey. Front Syst Neurosci 9, 102, doi: 10.3389/fnsys.2015.00102 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferreira PG et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nature communications 9, 490, doi: 10.1038/s41467-017-02772-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Terstege DJ, Addo-Osafo K, Teskey GC & Epp JR New neurons in old brains: A cautionary tale for the analysis of neurogenesis in post-mortem tissue. 2021.2011.2012.468443, doi: 10.1101/2021.11.12.468443 %J bioRxiv (2021). [DOI] [Google Scholar]
- 53.Rosenberg AB et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182, doi: 10.1126/science.aam8999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qian X et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell stem cell 26, 766–781 e769, doi: 10.1016/j.stem.2020.02.002 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Su Y et al. Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat Neurosci 20, 476–483, doi: 10.1038/nn.4494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214, doi: 10.1016/j.cell.2015.05.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu P et al. Dissecting Cell-Type Composition and Activity-Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq. Mol Cell 68, 1006–1015 e1007, doi: 10.1016/j.molcel.2017.11.017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296, doi: 10.1186/s13059-019-1874-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pedregosa F et al. Scikit-learn: Machine Learning in Python. J Mach Learn Res 12, 2825–2830 (2011). [Google Scholar]
- 61.Marques S et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329, doi: 10.1126/science.aaf6463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Durinck S, Spellman PT, Birney E & Huber W Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191, doi: 10.1038/nprot.2009.97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin Y et al. Evaluating stably expressed genes in single cells. Gigascience 8, doi: 10.1093/gigascience/giz106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bindea G et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093, doi: 10.1093/bioinformatics/btp101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504, doi: 10.1101/gr.1239303 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287, doi: 10.1089/omi.2011.0118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu W, Clyne M, Khoury MJ & Gwinn M Phenopedia and Genopedia: disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics 26, 145–146, doi: 10.1093/bioinformatics/btp618 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Doncheva NT, Morris JH, Gorodkin J & Jensen LJ Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J Proteome Res 18, 623–632, doi: 10.1021/acs.jproteome.8b00702 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moran PA Notes on continuous stochastic phenomena. Biometrika 37, 17–23 (1950). [PubMed] [Google Scholar]
- 70.Khlghatyan J & Saghatelyan A Time-lapse imaging of neuroblast migration in acute slices of the adult mouse forebrain. J Vis Exp, e4061, doi: 10.3791/4061 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bardy C et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proceedings of the National Academy of Sciences of the United States of America 112, E2725–2734, doi: 10.1073/pnas.1504393112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berg DA et al. A Common Embryonic Origin of Stem Cells Drives Developmental and Adult Neurogenesis. Cell 177, 654–668 e615, doi: 10.1016/j.cell.2019.02.010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lange PS et al. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med 205, 1227–1242, doi: 10.1084/jem.20071460 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palmer TD et al. Cell culture. Progenitor cells from human brain after death. Nature 411, 42–43, doi: 10.1038/35075141 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Zhou Y et al. Autocrine Mfge8 Signaling Prevents Developmental Exhaustion of the Adult Neural Stem Cell Pool. Cell stem cell 23, 444–452 e444, doi: 10.1016/j.stem.2018.08.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun GJ et al. Tangential migration of neuronal precursors of glutamatergic neurons in the adult mammalian brain. Proceedings of the National Academy of Sciences of the United States of America 112, 9484–9489, doi: 10.1073/pnas.1508545112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun GJ et al. Latent tri-lineage potential of adult hippocampal neural stem cells revealed by Nf1 inactivation. Nature neuroscience 18, 1722–1724, doi: 10.1038/nn.4159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682, doi: 10.1038/nmeth.2019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Duque A, Arellano JI & Rakic P An assessment of the existence of adult neurogenesis in humans and value of its rodent models for neuropsychiatric diseases. Mol Psychiatry 27, 377–382, doi: 10.1038/s41380-021-01314-8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Snyder JS Recalibrating the Relevance of Adult Neurogenesis. Trends Neurosci 42, 164–178, doi: 10.1016/j.tins.2018.12.001 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Marchetto MC et al. Species-specific maturation profiles of human, chimpanzee and bonobo neural cells. Elife 8, doi: 10.7554/eLife.37527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The snRNA-seq data are available at the GEO database (accession numbers: GSE185553, GSE185277, and GSE198323). Specimen information and sequencing statistics are described in Supplementary Tables 1, 2. Sources of the published scRNA-seq or snRNA-seq datasets used in this study are described in Supplementary Table 3.