

Abstract
Sepsis is a major cause of mortality and morbidity in trauma patients despite aggressive treatment. Traumatic injury may trigger infective or non-infective systemic inflammatory response syndrome (SIRS) and sepsis. Sepsis and SIRS are accompanied by an inability to regulate the inflammatory response but the cause of this perturbation is still unknown. The major pathophysiological characteristic of sepsis is the vascular collapse (i.e., loss of control of vascular tone); however, at the cellular level the final mediator of extreme vasodilatation has yet to be identified. After trauma, cellular injury releases endogenous damage-associated molecular patterns (DAMPs) that activate the innate immune system. Mitochondrial DAMPs express at least two molecular signatures, N-formyl peptides and mitochondrial DNA that act on formyl peptide receptors (FPRs) and Toll-like receptor 9, respectively. N-Formyl peptides are potent immunocyte activators and, once released in the circulation, they induce modulation of vascular tone by cellular mechanisms that are not completely understood. We have observed that N-formyl peptides from bacterial (FMLP) and mitochondrial (FMIT) sources induce FPR-mediated vasodilatation in resistance arteries. Accordingly, we propose that tissue and cellular trauma induces the release of N-formyl peptides from mitochondria triggering inflammation and vascular collapse via activation of FPR and contributing to the development of sepsis. The proposed hypothesis provides clinically significant information linking trauma, mitochondrial N-formyl peptides and inflammation to vascular collapse and sepsis. If our hypothesis is true, it may lead to new strategies in the management of sepsis that can help clinicians effectively manage non-infectious and infectious inflammatory responses.
Background and significance
The pathogenesis of sepsis
Trauma is a significant cause of death, with 5.8 million deaths per year globally [1]. Traumatic injury may trigger infectious or non-infectious systemic inflammatory response syndrome (SIRS) and sepsis. Sepsis is a major cause of mortality and morbidity in trauma patients despite aggressive treatment [1]. Sepsis and SIRS are accompanied by a failure to regulate the inflammatory response, but the cause of this malfunction is still unknown. An epidemiological study from North America found that the incidence of sepsis was 3 cases per 1000 hospitalized patients and approximately 750,000 cases per year [2]. The hallmark of sepsis is the loss of control of vascular tone; however, at the cellular level the final mediator of extreme vasodilatation has yet to be identified [3].
Previously, sepsis was thought to be a phenomenon related to the host’s response to infection [4]. However, many patients with sepsis do not have a documented infection. Clinically, the onset of sepsis is often insidious and symptoms may include fever, mental confusion, transient hypotension, diminished urine output and/or thrombocytopenia [5]. Moreover, septic patients manifest disseminated intravascular coagulation with consumption of platelets and prolongation of clotting times [6]. This altered hemostasis causes blood to clot, narrows blood vessels and reduces blood flow.
Initially, sepsis may be characterized by increases in pro-inflammatory mediators, but as sepsis persists, there is a shift toward an anti-inflammatory, immunosuppressive state [7]. Numerous trials have been conducted with agents that block the inflammatory cascade: corticosteroids, anti-endotoxin antibodies, tumor necrosis factor (TNF) antagonists, interleukin-1 receptor antagonists, and other agents [7]. The concomitant presence of pro-inflammatory mediators and failure of anti-inflammatory agents to control sepsis suggest that sepsis may result from uncontrolled inflammation. However, the molecular events linking trauma-induced inflammation to SIRS and sepsis remain poorly understood. Thus, patients will continue being treated with unnecessary and potentially deleterious antimicrobials while failing to identify appropriate molecular targets for therapy.
The peripheral circulation in sepsis
The major pathophysiological characteristic of sepsis, and mainly septic shock (characterized by severe hypotension even with adequate fluid resuscitation), is the loss of control of vascular tone (i.e., vascular collapse). The circulatory responses to sepsis are characterized by an initial hyperdynamic phase, where a reduction in systemic vascular resistance due to peripheral vasodilation and unresponsiveness to vasoconstrictor drugs are observed [8]. Later, the myocardial dysfunction caused by pro-inflammatory mediators leads to a hypodynamic phase, where cardiac output significantly falls and both the systolic and the diastolic functions of the heart are greatly impaired [8]. The endothelium lining of the vascular walls is a major target of sepsis-induced events and damage of endothelial cells accounts for much of the pathology of septic shock [8,9]. During septic shock, the function of the endothelial barrier is impaired leading to fluid loss into the extravascular space and consequently, causing life-threatening edema in the lungs, kidney, and brain of septic patients [8,9]. The increase in endothelial permeability and dysfunction has been associated with hypoxia, oxidative stress and augmented TNF-α activity [9]. Also, exacerbated production of nitric oxide by the inducible form of nitric oxide synthase (iNOS) contributes to hypotension and vascular hyporeactivity in sepsis. Nevertheless, pharmacological interventions using NOS inhibitors have not been successful. This result suggests that nitric oxide may not be the major factor that leads to the exacerbated relaxation and hypotension in sepsis. Also, it was demonstrated that prostaglandin and thromboxane A2 concentrations correlate with the severity of organ failure in patients with severe sepsis [10], and indomethacin decreased the injury induced by sepsis in animals [11]. Moreover, it has been shown that sepsis induces apoptosis of the endothelium mediated by TNF-α [12].
Decreased vascular resistance and significant attenuation to vasoconstrictor agents in different vascular beds (aorta, pulmonary, mesenteric resistance arteries and others) occur in sepsis [13–15]. Nevertheless, the mechanism by which the sepsis modulates the vascular tone is not fully understood. Understanding these mechanisms may help clinicians treat patients with sepsis.
Mitochondria, N-formyl peptides receptors and vascular tone
Mitochondria not only have a key role in ATP synthesis but are crucial for performing various signaling functions, acting as transducers and effectors in multiple processes including cell death signaling, oxidant signaling, innate immunity, Ca2+ homeostasis, fatty acid synthesis and autophagy [16]. Mitochondria are a source of damage-associated molecular patterns (DAMPs), a characteristic that stems from their bacterial ancestry [16,17]. DAMPs are endogenous molecules that are released from cells following injury or death. These factors lead to activation of the innate immune system [16]. Mitochondrial DAMPs express at least two molecular signatures, formyl peptides and mitochondrial DNA (mitDNA) that act on formyl peptide receptors (FPRs) and Toll-like receptor 9, respectively [16,18,19]. FPR has been identified as a subfamily of G protein-coupled receptors. FPR-1, its variant FPRL-1 (FPRL-like-1) and FPR-2 are expressed at high levels on neutrophils and monocytes, and they function to mediate cell chemotaxis [20–22]. In neutrophils, activation of FPR-1 by binding of N-formyl–methionyl–leucyl–phenylalanine (fMLP), an FPR agonist derived from bacteria, stimulates heterotrimeric G proteins, which rapidly dissociate into α and βγ subunits, and release Ca2+ from intracellular stores [20–22]. Other intracellular effectors in the FPR signaling cascade include phospholipase A2 and mitogen-activated protein kinases (MAPKs) [22]. Hauser et al. [23] have shown that mitochondrial DAMPs from femoral reamings activate neutrophils via FPR and p44/42 MAPK.
Although FPR and FPRL1 were initially detected in phagocytic leukocytes, other cell types also express these receptors [21,22]. Thus, it was demonstrated that FPR is expressed in endothelium, epithelia, microglia, spleen, lung, liver and skeletal muscle [21,22]. Also, we have observed that formyl peptide receptors 1 and 2 are present in mesenteric resistance arteries and heart from Wistar rats (Fig. 1). Nevertheless, its biological function in the cardiovascular system is not fully understood. Previous studies have shown that FPR modulates vascular tone even in the absence of leukocytes [24–26]; nonetheless, these results are controversial. Accordingly, Bode et al. [24] showed that fMLP activates cells of unknown identity in the adventitia or media of human coronary arteries to produce vasoconstrictor cyclooxygenase products. In addition, Keitoku et al. [25] showed that fMLP produced transient tension (contraction with subsequent relaxation) in human coronary arteries, mainly via generation of thromboxane A2 and prostacyclin. This effect of fMLP did not appear to be mediated by the activation of densely accumulated intimal and/or adventitial macrophages, but by the activation of unidentified medial tissue cells, which might have functional fMLP receptor homologues [25]. On the other hand, Laplante et al. [26] have demonstrated that fMLP has a relaxant effect on rabbit vascular strips (portal veins and pulmonary arteries). These studies suggest that the activation of FPR changes vascular function in a vessel-dependent manner. Preliminary data in our laboratory (Fig. 2) demonstrate that fMLP has a powerful relaxant effect in isolated mesenteric resistance arteries from Wistar rats, and cyclosporin H (FRP antagonist: CsH, 1 μM) inhibits the fMLP response (Fig. 2). Collectively, these data suggest that FPR has an important dilatory role in resistance arteries. However, it is noteworthy that fMLP (peptide from bacteria) was used as an agonist for FPR in all studies cited above. It is known that formylated peptides from mitochondria, corresponding to the N-terminus of mitochondria NADPH dehydrogenase subunits 4 (fMLKLIV) and 6 (fMMYALF), are equally potent to activate the FPR in human promyelocytic leukemia cells [27]. In this context, it seems reasonable to assume that formyl peptides from mitochondria, released from damaged cell, may activate FPR leading to changes in vascular function.
Fig. 1.
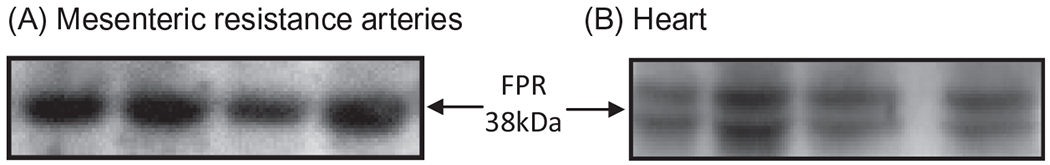
Representative image of the formyl peptide receptor (FPR) total protein expression in mesenteric resistance arteries (A) and heart (B) from non-treated Wistar rats. To obtain this data, Western blot technique was performed.
Fig. 2.
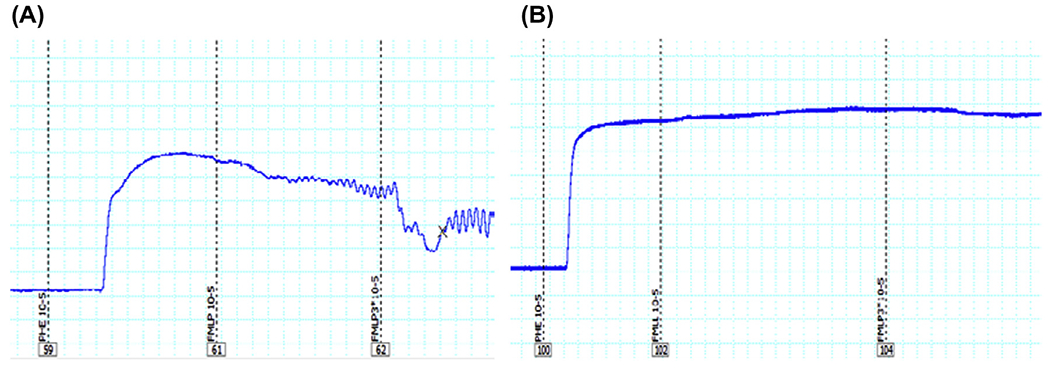
Representative tracing of formyl peptide from bacteria (FMLP)-induced relaxation in isolated resistance arteries (third order mesenteric arteries) from Wistar rats. The segments were initially contracted with phenylephrine (PE, 10 μM). Subsequently, FMLP (N-formyl–methionyl–leucyl–phenylalanine, 10 or 30 μM) was added in the absence or presence of cyclosporin H (FRP antagonist: CsH, 1 μM). As shown in (A), both concentrations of FMLP (10 or 30 μM) induced relaxation, and the pre-incubation with CsH inhibited this response (B). Wire myograph was used to analyze vascular function.
Hypothesis
We propose that trauma/injury releases N-formyl peptides from mitochondria (FMIT) that trigger inflammation and vascular dysfunction via FPR activation, contributing to the loss of control of vascular tone and to the development of sepsis (Fig. 3).
Fig. 3.
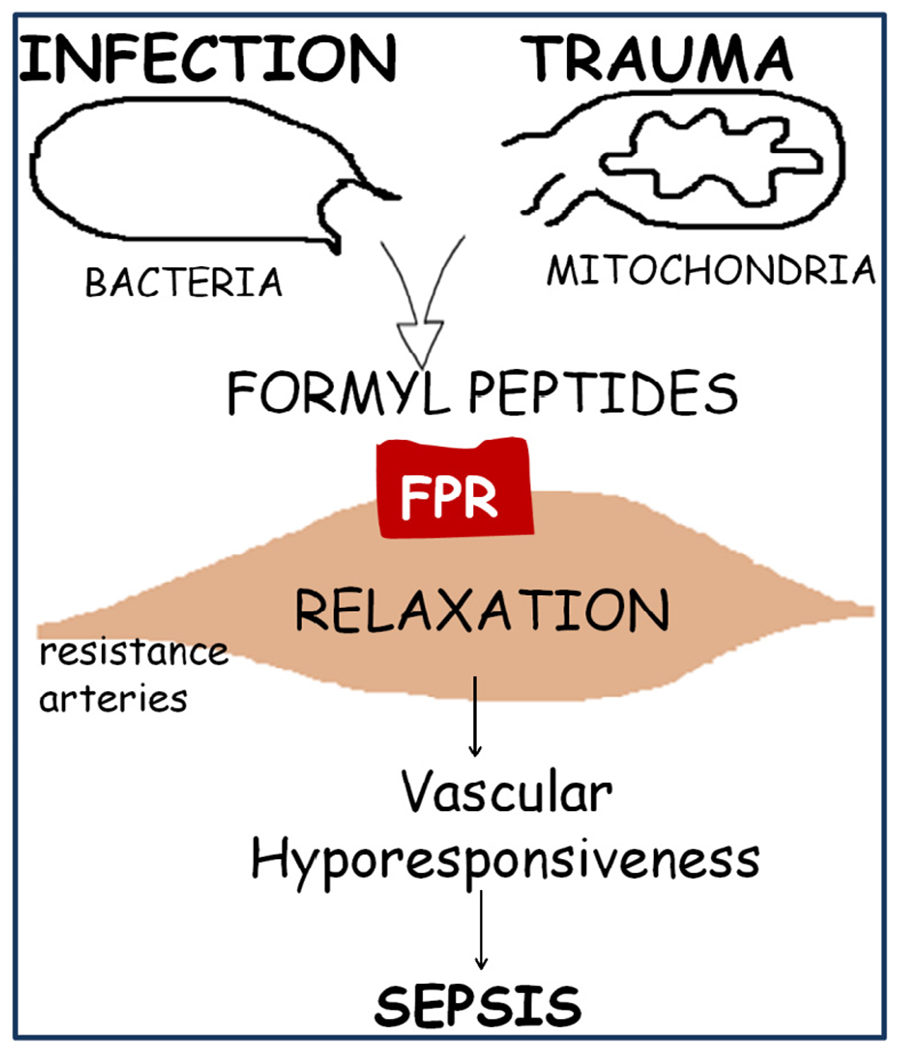
N-formyl peptides from mitochondria (FMIT) and from bacteria trigger inflammation and vascular dysfunction via formyl peptide receptor (FPR) activation, contributing to the loss of control of vascular tone and to the development of sepsis.
Evaluation of the hypothesis
To the best of our knowledge, we are the first to show that N-formyl peptides (FMIT) derived from mitochondria have a powerful relaxant effect in mesenteric resistance arteries from Wistar rats (Fig. 4). Based on this finding, it is reasonable to speculate that in conditions of enhanced FPR activation, (e.g., tissue damage/trauma) FPR-associated signaling leads to exacerbated relaxation and hypotension. This hypothesis has clinical relevance in the context of sepsis, where the FPR agonist released from tissue damage (cell injury/trauma) may induce exacerbated vasodilatation and uncontrolled systemic and local inflammation, leading to hypotension and septic shock (Fig. 3). This implies that FPR may be a target for the treatment of SIRS and sepsis.
Fig. 4.
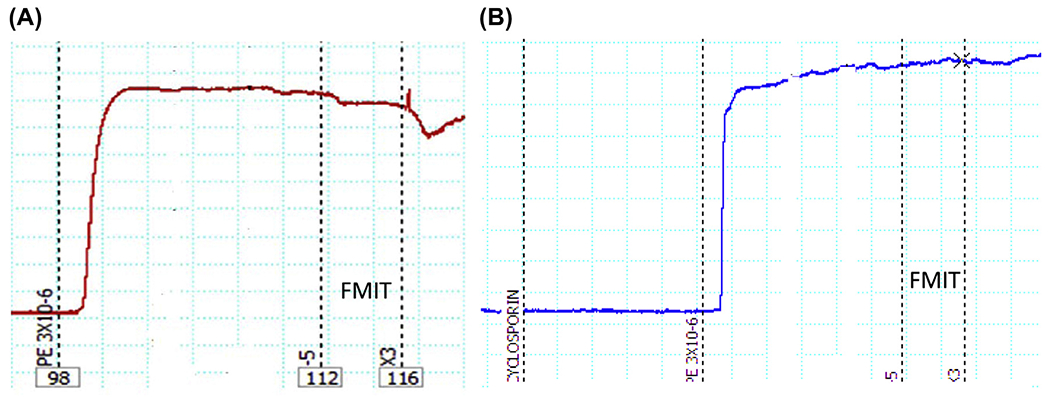
Representative tracing of formyl peptide mitochondrial (FMIT)-induced relaxation in resistance arteries (third order mesenteric arteries) from Wistar rats. The segments were initially contracted with phenylephrine (PE, 3 μM). Subsequently, FMIT (formyl–methionine–methionine–tyrosine–alanine–leucine–phenylalanine, 10 or 30 μM) was added in the absence or presence of cyclosporin H (FRP antagonist: CsH, 1 μM). As shown in (A), both concentrations of FMIT (10 or 30 μM) induced relaxation and the pre-incubation with CsH inhibited this response (B). Wire myograph was used to analyze vascular function.
Consequences of the hypothesis and discussion
Sepsis remains the chief cause of death in intensive care units in the United States and worldwide [1,3]. It is known that the major pathophysiological characteristic of sepsis is the loss of control of vascular tone. Therefore, understanding vascular function and the causes for this disruption would ultimately provide starting points for therapies designed to treat this devastating disease. Studies to test our hypothesis will generate new knowledge and concepts about the role of N-formyl peptides/FPR in the cardiovascular system. Also, such investigations may suggest a role of formyl peptide receptors as links between trauma, vascular collapse and sepsis, leading to novel strategies for the management of sepsis and its devastating consequences.
Future perspectives
Our group has investigated the role of released fragments from mitochondria (mitDAMPs, e.g., mitochondria DNA activating Toll like-9 [19] and formyl peptides activating FPR) in the genesis and maintenance of cardiovascular diseases. We have observed that one common aspect between these mitDAMPs is that both lead to a pro-inflammatory response; however, several questions still remain unanswered. Do the mitochondrial formyl peptides have an effect on blood pressure regulation? Do formyl peptides potentiate the actions of other vasodilators released during the septic response? Is it possible to use these mitochondrial formyl peptides as biomarkers for damaged cells? Several experiments and clinical trials need to be performed in order to answer these questions and promote this knowledge from bench to bedside.
Funding
This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) – Brazil and NIH.
Footnotes
Conflict of Interest
None.
References
- [1].WHO. Global forum on trauma care. World Health Organization; 2009. [Google Scholar]
- [2].Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001;29:1303–10. [DOI] [PubMed] [Google Scholar]
- [3].Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med 2003;9:517–24. [DOI] [PubMed] [Google Scholar]
- [4].Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med 2001;29(Suppl.):S109–16. [DOI] [PubMed] [Google Scholar]
- [5].Cohen J The immunopsthogenesis os sepsis. Nature 2002;420:885–91. [DOI] [PubMed] [Google Scholar]
- [6].Remick DG. Pathophysiology of sepsis. Am J Pathol 2007;170(5):1435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003;348:138–50. [DOI] [PubMed] [Google Scholar]
- [8].Kotsovolis G, Kallaras K. The role of endothelium and endogenous vasoactive substances in sepsis. Hippokratia 2010;4(2):88–93. [PMC free article] [PubMed] [Google Scholar]
- [9].Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res 2003;60:49–57. [DOI] [PubMed] [Google Scholar]
- [10].Oettinger W, Berger D, Beger HG. The clinical significance of prostaglandins and thromboxane as mediators of septic shock. Klin Wochensch 1987;65:61–8. [DOI] [PubMed] [Google Scholar]
- [11].Short BL, Gardiner M, Walker RI, Jones SR, Fletcher JR. Indomethacin improves survival in gram-negative sepsis. Adv Shock Res 1981;6:27–36. [PubMed] [Google Scholar]
- [12].Haimovitz-Friedman A, Cordon-Cardo C, Bayoumy S, Garzotto M, McLoughlin M, Gallily R, et al. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med 1997;186(11):1831–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yaghi A, Paterson NA, McCormack DG. Vascular reactivity in sepsis: importance of controls and role of nitric oxide. Am J Respir Crit Care Med 1995;151:706–12. [DOI] [PubMed] [Google Scholar]
- [14].Chen SJ, Li SY, Shih CC, Liao MH, Wu CC. NO contributes to abnormal vascular calcium regulation and reactivity induced by peritonitis-associated septic shock in rats. Shock 2010;33(5):473–8. [DOI] [PubMed] [Google Scholar]
- [15].Aytekin B, Hunter LW, Sieck GC, Ouellette Y. Reduced Ca2+-sensitivity in mesenteric resistance arteries of mice with LPS-induced inflammation. FASEB J 2006;20:LB13. [Google Scholar]
- [16].Tait SWG, Green DR. Mitochondria and cell signaling. J. Cell Sci 2012;125:807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sagan L On the origin of mitosing cells. J Theor Biol 1967;14(3):255–74. [DOI] [PubMed] [Google Scholar]
- [18].Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 2011;32:157–64. [DOI] [PubMed] [Google Scholar]
- [19].Goulopoulou S, Matsumoto T, Bomfim GF, Webb RC. Toll-like receptor 9 activation: a novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin Sci 2012;123:429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boulay F, Tardif M, Brouchon L, Vignais P. The human N-formyl-peptide receptor. Characterization of two cDNA isolates and evidence for a new subfamily of G-protein-coupled receptors. Biochemistry 1990;29:11123–33. [DOI] [PubMed] [Google Scholar]
- [21].Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol 2002;23(11):541–8. [DOI] [PubMed] [Google Scholar]
- [22].Le Y, Oppenheim JJ, Wang JM. Pleiotropic roles of formyl-peptide receptors. Cytokine Growth Factor Rev 2001;12:91–105. [DOI] [PubMed] [Google Scholar]
- [23].Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial DAMPs from femoral reamings activate neutrophils via formyl peptide receptors and P44/42 MAP Kinase. J Orthop Trauma 2010;24(9):534–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bode SM, Kuhn M, Förstermann U. Chemotactic peptide FMLP contracts human coronary arteries via cyclooxygenase products. Am J Physiol 1990;258:H848–53. [DOI] [PubMed] [Google Scholar]
- [25].Keitoku M, Kohzuki M, Katoh H, Funakoshi M, Suzuki S, Takeuchi M, et al. FMLP actions and its binding sites in isolated human coronary arteries. J Mol Cell Cardiol 1997;29(3):881–94. [DOI] [PubMed] [Google Scholar]
- [26].Laplante C, Tremblay B, Marceau F. Relaxant effect of N-formyl–methionyl–leucyl–phenylalanine on rabbit vascular strips. J Pharmacol Exp Ther 1989;248(2):774–80. [PubMed] [Google Scholar]
- [27].Rabiet MJ, Huet E, Boulay F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur J Immunol 2005;35(8):2486–95. [DOI] [PubMed] [Google Scholar]