

Abstract
Background:
The role of fatty acid ethyl esters (FAEEs) during human alcoholic pancreatitis is unknown. We compared FAEE levels to their non-esterified fatty acids (NEFA) precursors during alcohol intoxication and clinical alcoholic pancreatitis. The pathophysiology underlying FAEE increase and their role as diagnostic biomarkers for alcoholic pancreatitis was investigated.
Methods:
A prospective blinded study compared FAEE, NEFA and ethanol blood levels on hospitalization for alcoholic pancreatitis (n=31), alcohol intoxication (n=25) and in normal controls (n=43). Serum FAEEs were measured at admission for non-alcoholic pancreatitis (n=75). Mechanistic cell and animal studies were done.
Results:
Median FAEEs were similarly elevated during alcohol intoxication (205nM; CI 71.8-515nM, P<0.001) and alcoholic pancreatitis (103.1nM; CI 53-689nM, P<0.001), vs. controls (1.7nM; CI 0.02-4.3nM) or non-alcoholic pancreatitis (8nM; CI 1.1-11.5nM). Alcoholic pancreatitis increased serum NEFA (1024± 710 μM vs. 307±185 μM in controls, p<0.05). FAEEs comprised 0.1-2% of the parent NEFA concentrations. FAEES correlated strongly with NEFAs independent of ethanol levels in alcoholic pancreatitis but not during alcohol intoxication. On receiver operating characteristic (ROC) analysis for diagnosing alcoholic pancreatitis, serum FAEE’s area under the curve (AUC) was 0.87 (CI 0.78-0.95, P<0.001) In mice and cells, alcohol administration transiently increased all FAEEs. OAEE, was the only FAEE with a sustained increase up to 24 hours by intraperitoneal OA+ ethanol.
Conclusions:
The sustained, alcohol independent, large (20-50-fold) increase in circulating FAEEs during alcoholic pancreatitis results from their visceral release and mirrors the 2-4-fold increase in parent NEFA. The large AUC of FAEEs on ROC analysis supports their role as alcoholic pancreatitis biomarkers.
Keywords: alcohol, acute, marker, intoxication, fatty acid
Lay summary:
Fatty acid ethyl esters are biomarkers that can help diagnose alcoholic pancreatitis. They are released by breakdown of fat during pancreatitis, and parallel the parent fatty acids released during fat breakdown.
Graphical Abstract
INTRODUCTION:
Alcohol is a common cause for pancreatitis. However, the diagnosis of alcoholic pancreatitis can be challenging since it depends on a reliable history, with no objective way to support or refute the diagnosis. Alcohol blood levels peak within 1-2 hours of intake, followed by elimination within hours1, thus ethanol is commonly undetectable when a patient presents with alcoholic pancreatitis. Excess alcohol consumption can cause numerous diseases2, 3. However, alcohol may be undetected due to this rapid turnover, and therefore the diagnosis of alcohol related diseases may confounded by an inaccurate history4. Thus, diagnosis and management of diseases like alcoholic pancreatitis that are influenced by the patterns of alcohol consumed can be challanging5, 6.
Fatty acid ethyl esters (FAEE) are non-oxidative metabolites of non-esterified fatty acids (NEFA) and ethanol that can be present in the visceral fat of alcoholics even when they are not intoxicated7. While NEFA, which are released from fat necrosis8, 9 are elevated in human pancreatitis10, 11, the levels of FAEEs in human alcoholic pancreatitis are unknown and their role in human pancreatitis is debated based on animal studies12–14. High FAEEs were first reported in alcoholics dying in motor vehicle accidents7 whose pancreas, fat, liver, heart, brain, skeletal muscle, and testes7 had FAEEs in descending order of concentration. FAEE formation from esterification of NEFA by ethanol was first shown to be catalyzed by a lipase, which like pancreatic triglyceride lipase (the principal lipase mediating fat necrosis9) could also hydrolyze triglyceride (TG), and was colipase dependent15. FAEEs can also form via trans-esterification of TG by ethanol16 in the adipose of alcoholics without acute pancreatitis (AP)7. Recently carboxyl-ester lipase14, which is also present in the pancreas17 was proposed to esterify NEFA to FAEEs. FAEE synthase activity is also present in other tissues18, and fungal or bacterial lipases16. The most abundant FAEEs in the fat and pancreas of humans are ethyl esters of long chain fatty acids palmitic acid (PAEE), oleic acid (OAEE), and stearic acid (SAEE)7, which mimics the adipose tissue TG composition19, 20. A time course of blood FAEEs in human volunteers showed them to follow alcohol by 30 minutes1, perhaps due to rapid degradation or excretion21.
While FAEEs blood levels parallel those of ethanol in humans1, their levels during alcoholic pancreatitis are unknown, and the relationship of FAEE to the parent NEFA is also unexplored. A clinically relevant scenario is, that if fat necrosis 22–24 were to generate or release stored FAEEs7 during alcoholic AP; these could be compared to the NEFA 8–11, 25, 26 generated during AP as biomarkers for alcoholic pancreatitis. Similarly, if the FAEE levels in the circulation were in the range that cause cell injury 13, 27–29, these could be compared as potential therapeutic targets to their parent NEFA which are known to worsen AP 9, 30, 31. We therefore aimed to characterize the profile and behavior of FAEEs in human alcoholic AP versus alcohol intoxication and studied their utility as biomarkers to diagnose alcoholic pancreatitis. We also used animal and cellular models to understand the mechanisms underlying these patterns.
METHODS:
Patient recruitment: The studies were prospectively conducted between May 2017 and July 2020 at the Carl T. Hayden Veterans’ Administration (VA) Medical Center, Phoenix, AZ, and at the Mayo Clinic, Arizona. All protocols were reviewed and approved by the institutional review board of the VA (IRB; Project #1123) and that of the Mayo Clinic Foundation (16-002800, 15-08838). All consecutive VA patients were approached to be included in the study unless they met exclusion criteria. Normal controls were recruited by research study flyers. Patients presenting with acute alcoholic intoxication to the emergency room, or alcoholic pancreatitis within 24 hours admission were approached after chart review. Diagnosis of pancreatitis was made based on the presence of at least 2 out of 3 criteria as per the American Pancreatic Association/International Association of Pancreatology Guidelines32. Etiology of pancreatitis was determined based on physician documentation. Consent: Subjects were presented with the informed consent and after reading it, any questions or concerns were addressed before the subject signed the form. Exclusion Criteria: Acute pancreatitis, and acute alcoholic intoxication patients: History of chronic pancreatitis, pancreatic cancer or pancreatic surgery, Congestive heart failure with ejection fraction <35%, history of myocardial infarction, history of Stage 4 renal failure and/or on dialysis, pregnant women, patients enrolled in another research study, or if patient could not be enrolled within 24 hours of presentation. Controls: All the above, and body mass index >35, Chronic renal insufficiency, home oxygen use, diabetes with complications including retinopathy, neuropathy. Overall, 186 patients were recruited in this manner with 55 controls, 25 alcohol intoxicated, 31 alcoholic pancreatitis patients and 75 patients with non-alcoholic pancreatitis. Since the study was focused on alcohol; forty-three controls who were sex and body mass index matched were chosen. Acute pancreatitis patients’ residual samples collected as per approved IRB protocols (15-08838) at the time of admission via the emergency room at Mayo Clinic Hospital, Phoenix were analyzed for FAEEs and NEFA using methods described below. The admission alcohol blood levels, and lipase levels were retrieved.
Blood ethanol levels in patients: Ethanol levels at presentation were done at the discretion of the attending physician. Lack of documentation of alcohol intoxication and undetectable alcohol in lab values was taken as ethanol levels below detection (<10mg/dL). The clinical data were cataloged in a secure excel sheet. Staff at the Mayo Clinic doing the analysis were blinded to all patient and clinical data till after all lipidomic analyses (described below) were done.
Sample collection and transport: Samples from controls and alcoholic intoxicated patients were collected in an exam room or the emergency room respectively. Samples from alcoholic pancreatitis patients were collected within 24 hours of presentation, which could be the same time as presenting to the ER, or the next morning, in case a patient presented after regular work hours. The blood samples were collected in a serum container and stored on ice. De-identified, coded samples from the VA were placed on ice and packed in a Styrofoam container prior to being sent by courier service to the Research Team at the Mayo Clinic, Scottsdale, AZ. Samples were processed on ice within 18 hours of collection, aliquoted and stored at −80C until further use.
Reagents:
Fatty acids and their ethyl esters were purchased from Sigma-Aldrich (St. Louis, MO) and Cayman Chemical (Ann Arbor, MI) respectively. Ethanol and other chemicals were from Sigma-Aldrich. All reagents were of highest purity available (≥98%). Any solutions were prepared just before use.
FAEE analysis:
Methods for extraction and measurement of FAEEs were derived from Kulig et al33. For this 375 μL of patient plasma (neat or diluted in saline) was added to 3 mL of cold acetone containing 55 μM ethyl heptadecanoate as an internal standard. Samples were briefly vortexed and centrifuged at 1400 g for 10 minutes. Lipids were extracted in two subsequent additions of 3mL hexane, then were combined and dried under vacuum. Samples were reconstituted in 75 μL hexane containing 10ppm caffeine and 1μL was injected via Agilent 7693 Automatic Liquid Sampler into an inlet maintained at 260°C. Gas chromatography (GC) was performed on an Agilent 7890B instrument and HP-5ms (30m x 0.25 mm ID) (5%-phenyl)-methylpolysiloxane ultra inert column. Helium carrier gas was maintained at 1mL/min flow rate. Samples were introduced onto the column with the GC oven at 80°C, upon injection the temperature increased at 30°/min to 150°C and held there for 2 min, then slowly raised to 250°C at 4°/min, and finally increased to 300°C at 20°/min and held for 2 min. Samples were quantified using an Agilent 5977A mass spectrometer (MS) with the GC-MS interface maintained at 280°C. FAEEs were identified by retention time and quantified using Selected Ion Monitoring with the following m/z: Lauric acid ethyl ester- 88, 101, 183; Myristic acid ethyl ester- 88, 101, 183; Palmitoleic acid ethyl ester- 88, 101, 194; PAEE- 88, 101, 241; LAEE- 81, 95, 263; OAEE- 88, 101, 202, 265; SAEE- 88, 101, 269; Arachidonic acid ethyl ester - 91, 105, 203. The low limits of detection for PAEE and SAEE were 1 and 3 nM, respectively. The rest were all detectable at levels ≥ 10nM. Since free SA in high concentrations coelutes with and obstructs the base ion ratio for OAEE; samples were again dried and reconstituted in 200 μL hexane and free fatty acids were selectively esterified in a ‘soft’ derivatization reaction to form dimethylamides, previously described by Kangani et al34. This shifts the retention time of stearate, enabling detection of OAEE.
Assays:
NEFAs were measured using GC at the Hormone Assay and Analytical Services Core (Vanderbilt University Medical Center) as described previously35. Alcohol levels in mice was measured using the colorimetric method by Pointe Scientific (Canton, OH). The lower limit of detection was 10 mg/dL.
Evaluation and reporting of FAEES as biomarkers to diagnose alcoholic pancreatitis:
The Standards for Reporting of Diagnostic Accuracy Studies (STARD) guidelines36 were used for this purpose.
Statistical analysis and graphical representation:
Continuous variables are shown as bar-graphs (mice) or Box plots (human) with mean as “+” median (solid line), boxes (interquartile range), error bars (range) and points or circles (individual values). Categorical variables (sex) were compared across groups using a Fisher’s exact test. Linear regression with logarithm transformation (with base 10) on the dependent variable or Spearman correlation coefficient were used to assess the association between two continuous variables. Line graphs were used for correlation analysis of continuous variables with grey shaded area depicting 95% confidence intervals. The magnitude of the association estimated from the linear regression was denoted as ϐ (the slope of the fitted regression line) on the left upper part of the figures, and from the Spearman correlation; depicted on the right lower part of the figure as r. A positive value of ϐ or r indicated positive correlation between two variables. Significance was determined at a P<.05. Data for multiple groups were compared by the analysis of variance or Kruskal-Wallis test versus controls and values significantly different from controls were shown as (*) or with the p-value mentioned above the corresponding conditions. Graphing and statistical analysis was done using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com, and R4.0.3 (R Project for Statistical Computing).
The methods for animal and cell studies are described in the supplementary section.
RESULTS:
We first aimed to understand how circulating FAEEs relate to their parent NEFA and ethanol during alcohol intoxication and alcoholic pancreatitis in consecutive patients who presented to the emergency room, and in normal volunteer controls. Details are provided in the methods section. The following paragraphs describe these findings and the potential role of FAEEs as biomarkers to diagnose alcoholic pancreatitis. Mechanistic studies to understand how FAEEs are generated and released into the circulation are described in the end.
Patients with alcoholic pancreatitis have elevated FAEEs and NEFAs in the absence of significantly elevated blood alcohol levels:
As seen in figure 1A, all groups had similar body mass index (BMI). Sex distribution showed a higher proportion of males in alcohol intoxication than in alcoholic pancreatitis (96% vs. 71%, P=.03). This issue is addressed later in the third section of results. While controls were older than alcoholic AP patients, there was no difference in age between those with alcohol intoxication and alcoholic AP. Patients with alcoholic AP (Alc AP), had higher serum lipase (869±727 U/L) vs. controls (40±34U/L, P<.001), or the alcoholic intoxication group (Alc) whose levels were 37±25 U/L (Fig 1B). Conversely, while the alcohol intoxication group had elevated serum alcohol levels (205±95 mg/dL), only 6 of 31 patients with alcoholic AP had detectable alcohol levels (Fig 1C), which were not significantly elevated compared to controls. Serum NEFA (Fig 1D) in alcoholic pancreatitis were 1024± 710 μM, which were higher than during alcohol intoxication (535±332 μM, P=.007), which in-turn were higher than controls (307±185 μM, P<.05). Interestingly, FAEEs (Fig 1E) were similarly elevated in both the alcoholic intoxication (581±1234 nM, P<.001) and alcoholic AP groups (1787±3518nM, P<.001) vs. controls (3.0±3.8nM). Noting this large elevation of FAEEs in the alcoholic groups, we went on to study the relation of FAEEs to the levels of their precursor lipids and alcohol.
Figure 1: Biometrics and serum parameters of patients included in the study:
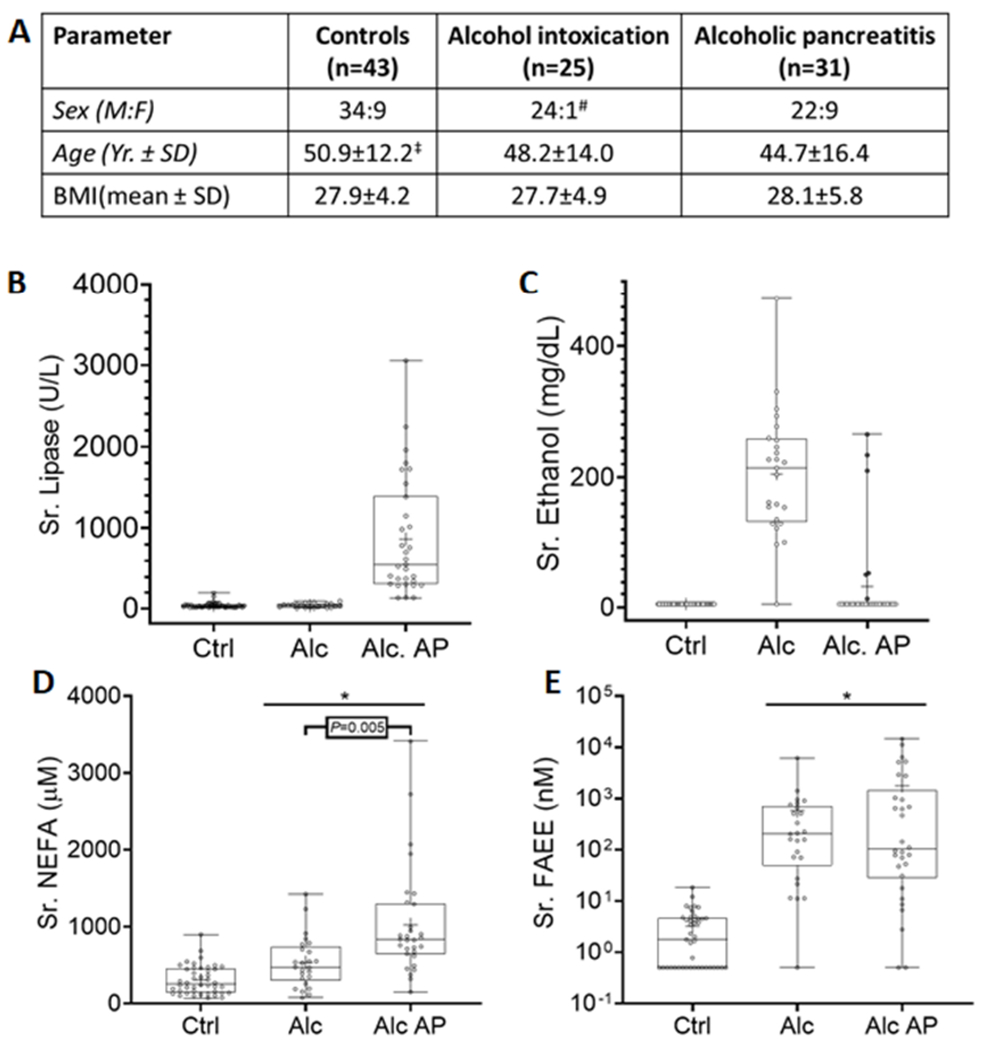
A: Table comparing biometric parameters of controls, alcohol intoxication and alcoholic pancreatitis patients. Each box shows data for the category listed at the top of the column with the mean ± standard deviation (SD) of the parameter compared in the row. # Significant difference in sex distribution was observed when comparing Alcohol intoxication vs. other groups (p=0.05), and Alcohol intoxication vs. AP (P=.03). ‡ Significant difference in age was observed when comparing with alcohol pancreatitis and control patients (P=.02). B-D: Box plots of serum parameters in controls (Ctrl), acute alcoholic intoxication patients (Alc) and or alcoholic pancreatitis (Alc AP) patients. Serum lipase (B), Ethanol (C), non-esterified fatty acids (NEFA; D) and fatty acid ethyl esters (FAEE; E) for each individual patient are shown as dots, the median as the horizontal line, mean as “+”, the boxes show the interquartile range and error bars show the range. The “*” indicates a P value <.05 vs. controls on ANOVA.
During alcohol intoxication FAEE concentrations correlate with blood alcohol, but not NEFA levels.
In alcoholics without pancreatitis total serum FAEEs, including the three principal FAEEs in serum, i.e. palmitic acid ethyl ester (PAEE), stearic acid ethyl ester (SAEE) and OAEE increased with alcohol concentrations (ϐ = 0.005-0.007, P<.001; r=0.53-0.58, P-values all significant; Fig 2A–D). Please note that this correlation was reported in the past1. We note the same despite the difference in timing of ethanol vs. FAEE samples. Interestingly, FAEEs did not correlate with NEFAs analyzed on the same sample (Fig 2E–H). Thus serum FAEE in alcoholics are related to alcohol blood levels as described previously1, but not NEFA levels.
Figure 2: Graphs showing correlation between total and individual fatty acid ethyl ester (FAEE) concentrations and serum parameters during alcohol intoxication.
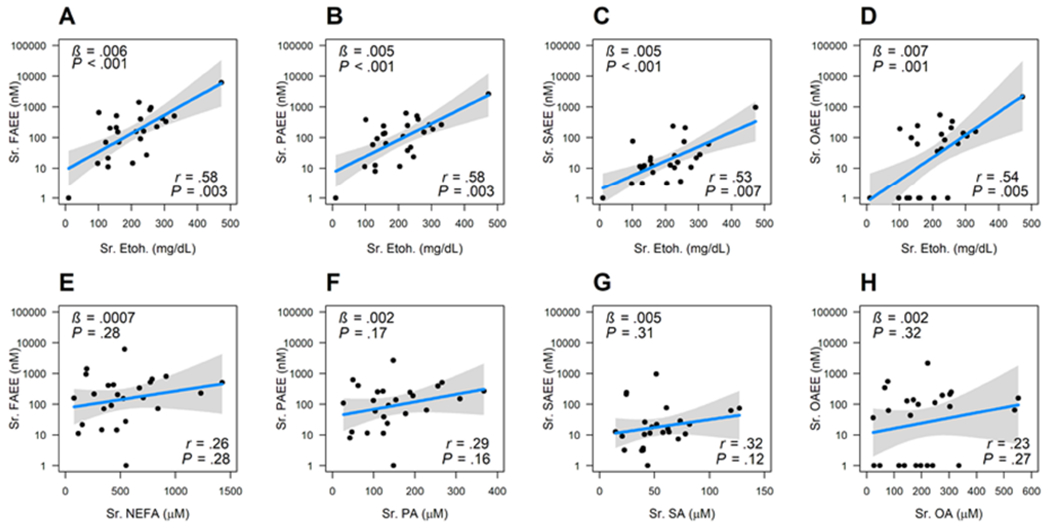
A-D show relation of blood ethanol (Etoh; on the x-axis), with total FAEE in A, palmitic acid ethyl ester (PAEE) in B, stearic acid ethyl ester (SAEE) in C, and oleic acid ethyl ester (OAEE) in D. E-H show the relationships with total non-esterified fatty acid (NEFA) in E, with Palmitic acid (PA) in F, with Stearic acid (SA) in G, with Oleic acid (OA) in H. The variable on the y-axis is treated as a dependent variable and the variable in the x-axis is treated as an independent variable. All the y-axes are plotted in logged scale with base 10. Black dots are the actual data; blue lines are the slope from the linear regression, and gray band shows the 95% confidence interval. At the bottom right are shown the correlation coefficients (r), along with the p values. On the upper left are the slope (β) of the regression line and p value are shown on the left upper corner.
FAEE concentrations in alcoholic pancreatitis parallel serum NEFA independent of blood alcohol concentrations.
We then studied FAEEs in patients with alcoholic AP. FAEEs were highest on the first day of abdominal pain (3174±4499 nM), vs ≥2 days (341±829 nM, P<.04). While FAEEs decreased with time (Fig 3A), these were detectable till 7 days into the attack. The 6 AP patients who had detectable alcohol blood levels (138±110 mg/dL; red dots) had higher FAEEs than the 25 in whom alcohol was below detection limits (black dots, Fig 3B). Blood alcohol levels in these 6 patients correlated with the FAEE concentrations (Fig 3C) as in alcohol intoxication (Fig 1C, 2A–D). Interestingly, while including these AP patients with elevated blood alcohol (red dots) diminished the association between serum FAEEs and corresponding NEFA concentrations (r=0.54, Fig 3D; r=0.47-0.53, Supplementary figure 1A–C); exclusion of the elevated blood alcohol group strengthened the associations between FAEEs (total and specific types) and their corresponding NEFA (r=0.50-0.78, Fig 3E–H). Lastly, since 24 of the 25 alcoholics were male, we went on to determine the relationship between FAEEs and NEFA in males (n=22) with alcoholic pancreatitis (supplementary figure 1D–E). In males this relationship between NEFA and FAEE was maintained (r=0.46-0.63, P≤.04) despite the smaller number (n=22) vs. the total 31 alcoholic AP cases (Supplementary figure 1A–C). Therefore, FAEE concentrations and type in alcoholic pancreatitis parallel those of the circulating NEFA independent of the blood alcohol concentrations or sex of a patient.
Figure 3: Graphs describing serum fatty acid ethyl ester concentrations during alcoholic pancreatitis.
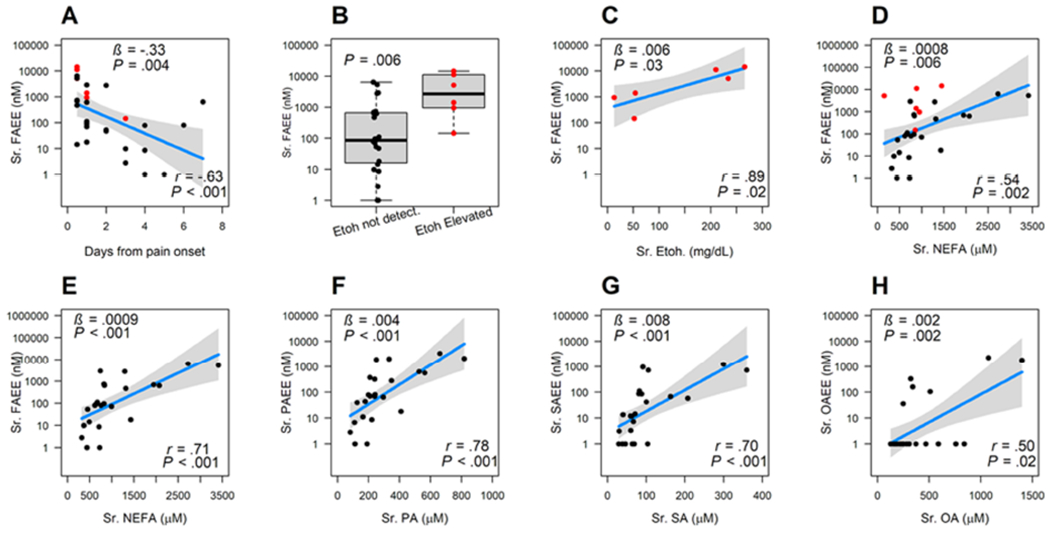
A: Change in FAEE levels over time from onset of pain. B: Box plots comparing FAEE concentrations in patients with elevated ethanol (Etoh.) levels (red dots, right side) to those in whom ethanol was not detectable (black dots, left side). P-value is computed from Wilcoxon rank sum test. C: Relationship between FAEEs and serum ethanol levels. D: Relationship between total FAEEs and NEFA in all patients with alcoholic pancreatitis including those with elevated alcohol (red dots). E-H: Correlations in patients who did not have elevated blood alcohol showing relationship between total non-esterified fatty acid (NEFA) to total FAEEs (E), individual FAEEs to NEFA (on the x-axis), which are F: Palmitic acid (PA) G: Stearic acid (SA) H: Oleic acid (OA). At the bottom right are shown the correlation coefficients, along with the p values. In A and C-H, the variable on the y-axis is treated as a dependent variable and the variable in the x-axis is treated as an independent variable. All the y-axes are plotted in logged scale with base 10. Dots are the actual data; blue lines are the slope from the linear regression, and gray band shows the 95% confidence interval. On the upper left are the slope (β) of the regression line and p value are shown on the left upper corner.
Circulating FAEEs are potential diagnostic biomarkers of alcoholic AP, despite being non-toxic and forming a minute proportion of circulating NEFA.
Since FAEEs and NEFA can be biologically active13, 37 in pancreatitis, we went on to compare their amounts in the circulation. Consistent with the median FAEE concentrations during alcoholic intoxication being 205 nM (95% confidence interval; CI: 72-515nM), and 103 nM (CI: 53-689 nM) during alcoholic pancreatitis vs. 1.7 nM (CI 0.02-4.3 nM) in controls. (Fig 1E); FAEEs averaged at ≈ 0.15% of the corresponding serum NEFA concentrations irrespective of the type of FAEE, or group of alcoholic patients (Figure 4A). In contrast, median serum NEFA concentrations were much higher, being 256 μM (CI: 211-367 μM) in controls, 475μM (CI: 349-673 μM) in alcoholics, and 831 μM (CI: 712-947 μM) during alcoholic pancreatitis. At these concentrations we noted the principal unsaturated NEFA, oleic acid (OA, blue lines) to dose dependently cause necrosis, seen as propidium iodide (PI) uptake and lactate dehydrogenase (LDH) leakage from pancreatic acini and the kidney cell line HEK293 (Supplementary figure 2A–C). This was however not seen with OAEE, PAEE or PA (a saturated NEFA), as shown previously8, 12, 38. Thus, despite the >50 fold increase over controls in both alcoholic AP and alcohol intoxication, FAEE concentrations are typically <1% of the parent NEFA, and in the biologically inactive range.
Figure 4: Serum FAEE concentrations as a proportion of NEFA, and the diagnostic accuracy of serum FAEEs for alcoholic pancreatitis.
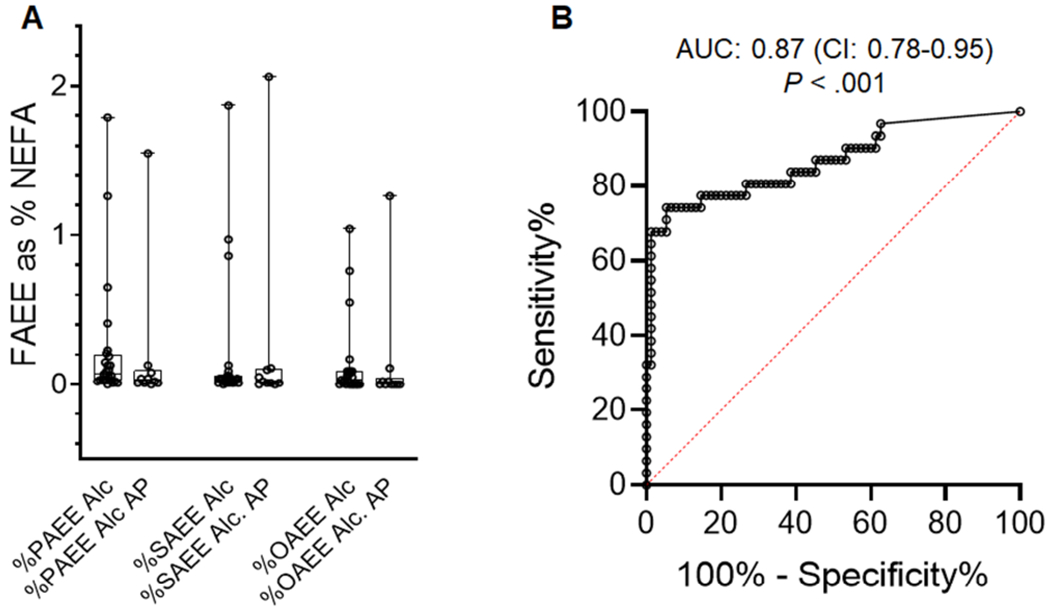
A) Concentrations of FAEEs as percentage of parent NEFA are shown as box plots. The boxes show the interquartile range and error bars show the range. Values for each individual patient are shown as circles. Ale; Alcohol intoxication, Ale AP; Alcoholic acute pancreatitis. The error bars depict SD, and * indicate a P <0.05 between OA and OAEE on Student’s t-test. B: Receiver operating characteristic (ROC) curve for serum FAEEs to diagnose alcoholic pancreatitis. ROC was performed for serum FAEE concentrations (nM) (n=106 acute pancreatitis patients, with 31 having a diagnosis of alcoholic pancreatitis). Area under the curve (AUC) for FAEE was 0.87, with a P<.001)
We then studied the utility of FAEEs as biomarkers to diagnose alcoholic pancreatitis. For this we measured their concentrations at hospital admission in consecutive patients diagnosed with non-alcoholic pancreatitis (n=75) during the same period. While the sex distribution (29 females, 46 males) and BMIs (30.5±6.3) of these patients were similar to those of alcoholic pancreatitis patients, those with non-alcoholic pancreatitis were significantly older (55.1±16.2 years; supplementary figure 3). Median levels FAEE levels of the non-alcoholic pancreatitis patients were 8nM (CI 1.1-11.5nM), which were significantly lower than those in alcoholic AP (103.1nM; CI 53-689nM, P<0.001). On receiver operating characteristic (ROC) curve analysis, the area under the curve (AUC) for FAEEs as a diagnostic biomarker for alcoholic pancreatitis was 0.87 (CI: 0.78-0.95) with a P<.001. FAEEs >30nM has a sensitivity of 77.4% (CI 60.1-88.6%) and specificity of 85.3% (CI 75.6-91.6%) for alcoholic AP, with a likelihood ratio of 5.3.
Visceral FAEE release causes an autonomous, sustained, and specific increase in serum FAEEs, unlike that due to alcohol alone.
We then compared the relative contributions of acute alcohol intoxication to serum FAEE levels, vs. release of FAEE from visceral stores, as may happen in acute pancreatitis. For this we studied the kinetics of serum FAEE elevation in mice. Administration of OAEE intraperitoneally (dashed blue line, Fig 5A–C) did not affect PAEE, or SAEE levels (Fig 5A, B), but caused a gradual and sustained increase in serum OAEE (Fig 5C.), which comprised >95% of all FAEEs (Fig 5D, E). OAEE levels peaked at 8 hours and remained elevated up to 24 hours, equivalent to the highest quartile of FAEEs noted in humans (Fig 1D). However, alcohol induced FAEE increase had a different pattern. Mice given a single dose of alcohol (2.2ml/kg), which is in the range of blood levels found in alcoholics (Fig 1C, 2A; up to 4.5 gm/Kg), increased blood alcohol to 104±30 mg/dL at 2 hours, similar to previous studies14. This caused a peak of all 3 principal ethyl esters at 2 hours (black line Fig 5A–C) which were similar in proportions (Fig 5F), and in the highest quartile of FAEEs noted in alcoholics (Fig 1E). While these approached baseline 8 hours later, PAEE and SAEE remained significantly elevated over OAEE (Fig 5G), consistent with the slower elimination of saturated FAEEs described previously39. Notably, LAEE was significantly lower at both 2 and 8 hours. Co-administration of OA at 0.3% body weight, which is a dose relevant to severe pancreatitis9, significantly elevated OAEE above others (Fig 5H, I). This shows that the FAEE elevated in serum during pancreatitis represents of the parent NEFA in the abdominal fat involved in the disease.
Figure 5: FAEE levels in the sera of mice given alcohol, oleic acid, or its ethyl ester.
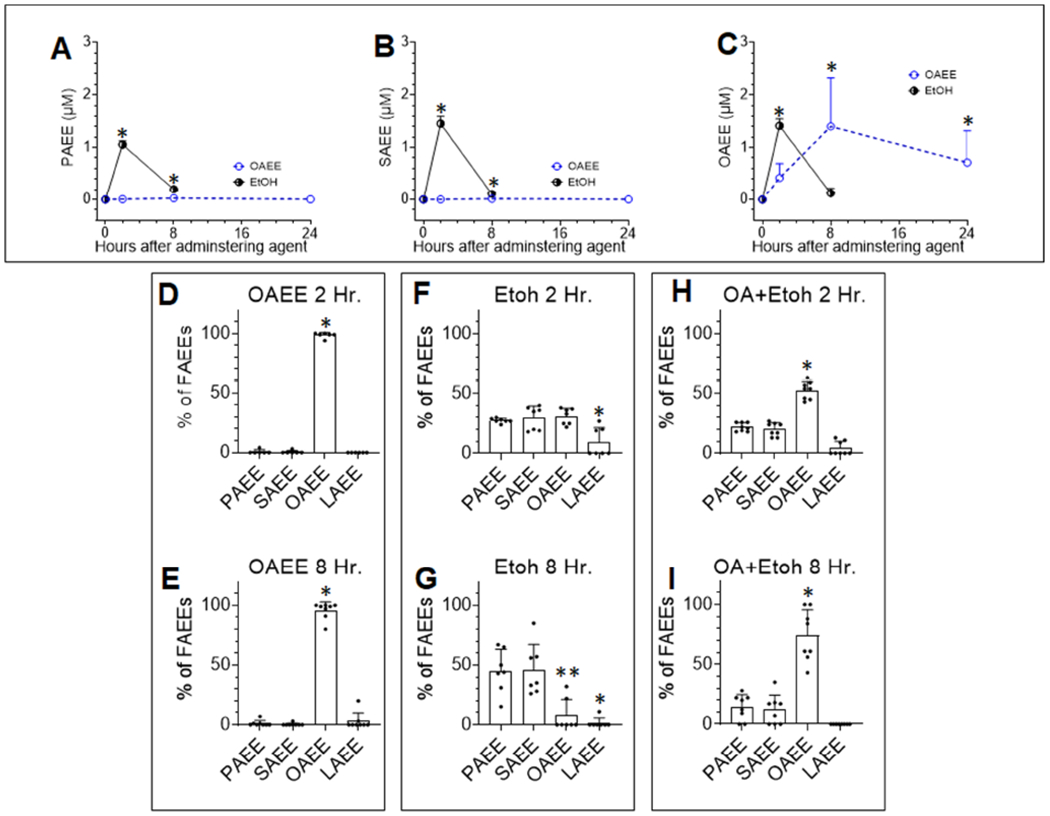
A-C: Time course of FAEE elevation in mice given 2.2gm/Kg ethanol (Etoh; black line) or 4.0 gm/Kg oleic acid ethyl ester (OAEE; dashed blue line). Shown are A: Serum PAEE, B: Serum SAEE, C: Serum OAEE. The “*” indicates a P value <.05 vs. values in control mice (time zero) using a Mann-Whitney or Student’s t-test depending on normality of distribution. Error bars are standard error of mean. Bar graphs showing proportions of the 4 principal ethyl esters in the sera of mice given OAEE (D, E), ethanol (F, G) or ethanol with oleic acid 0.3% body weight(H, I) at 2 and 8 hours. The value for each mouse is shown as an individual point. The error bars are standard deviation, * indicates a p<0.05 on ANOVA vs. other ethyl esters. In case of E, the ** indicates significantly lower values for OAEE than PAEE or SAEE.
In vitro, ethanol concentrations (≥250 mg/dL) in the upper range of alcohol intoxication (Fig 1B) injured pancreatic acini (Fig 6A) and HEK293 cells (Fig 6G) and increased FAEE formation in the absence of exogenous NEFA (Fig 6B, C, H) in the medium of both types of cells. Thus, ethanol induced membrane damage is a likely source of the lipid precursors of FAEEs (Figure 7, left panel). Alcohol with a clinically relevant NEFA 8, 9, 40, i.e. oleic acid (OA) worsened injury (Fig 6D), and increased OAEE (Fig 6E, 6I) in both cell types, but not PAEE or SAEE compared to alcohol or OA alone (Fig 6F). This increase in OAEE, by OA+ ethanol is consistent with FAEEs in alcoholic AP paralleling the parent NEFA (Fig 3E–H) and the In vivo findings in mice (Fig 5, I).
Figure 6: cell injury and FAEE generation from cells exposed to ethanol and/or the parent NEFA:
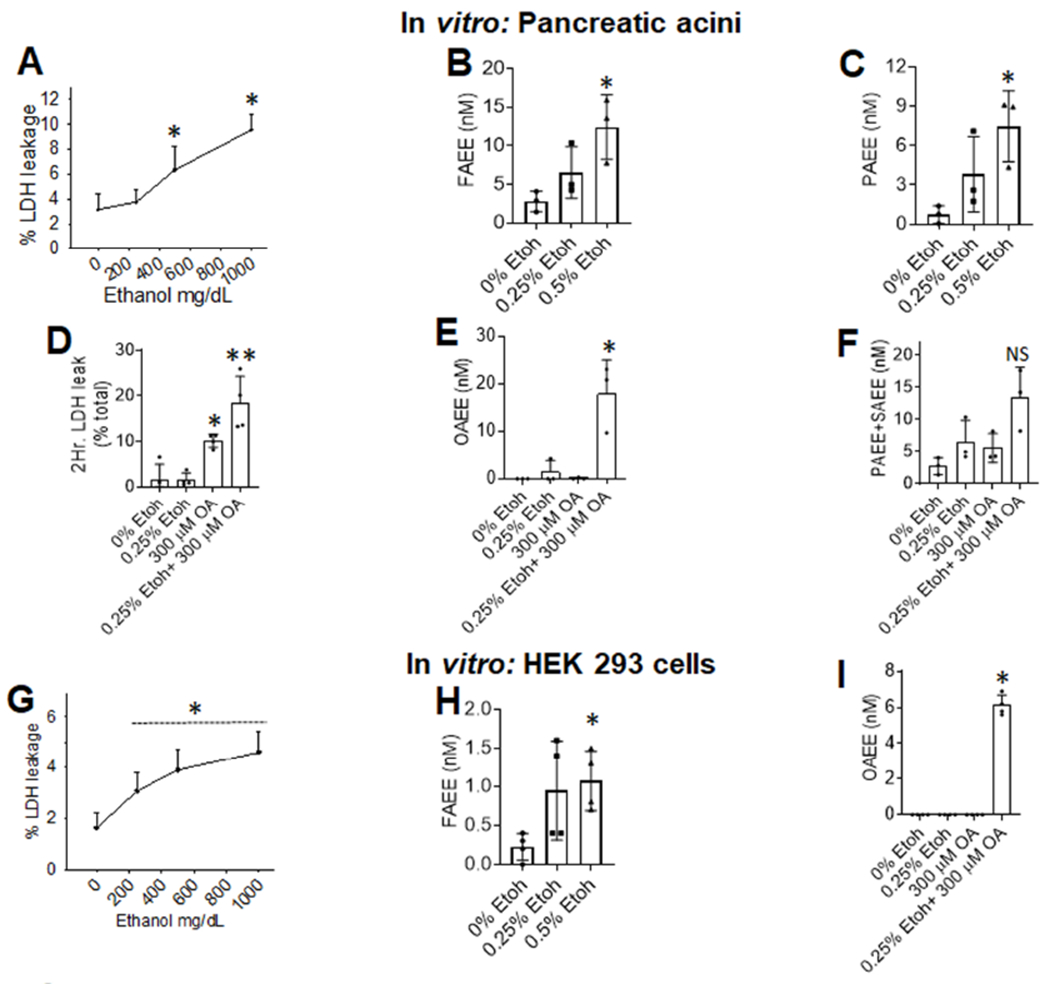
A: Dose response of ethanol induced LDH leakage from acini with line depicting the mean. Bar graphs of FAEE (B) or PAEE (C) concentrations in the medium. Bar graphs comparing the effect of 0.25% ethanol on 300μM OA induced LDH leakage (D) Levels of OAEE (E) or other FAEEs (F) in the medium. G: Dose response of ethanol induced LDH leakage from HEK 293 cells. Bar graphs of total FAEE (H) concentrations in the medium at different ethanol concentrations. Bar graphs comparing the effect of 0.25% ethanol on 300μM OA induced changes in OAEE concentrations (I) in the medium. Each experiment was done 3-4 times, with each point representing an average of duplicates collected at 2 or 4 hours, unless specified on the graph. Each. The error bars depict SD, and * indicate a P <0.05 vs. control (0% ethanol) on ANOVA. NS means not significant.
Figure 7: Schematic summarizing the different pathophysiology of FAEE generation during alcohol intoxication (left side) compared to alcoholic AP (right side).
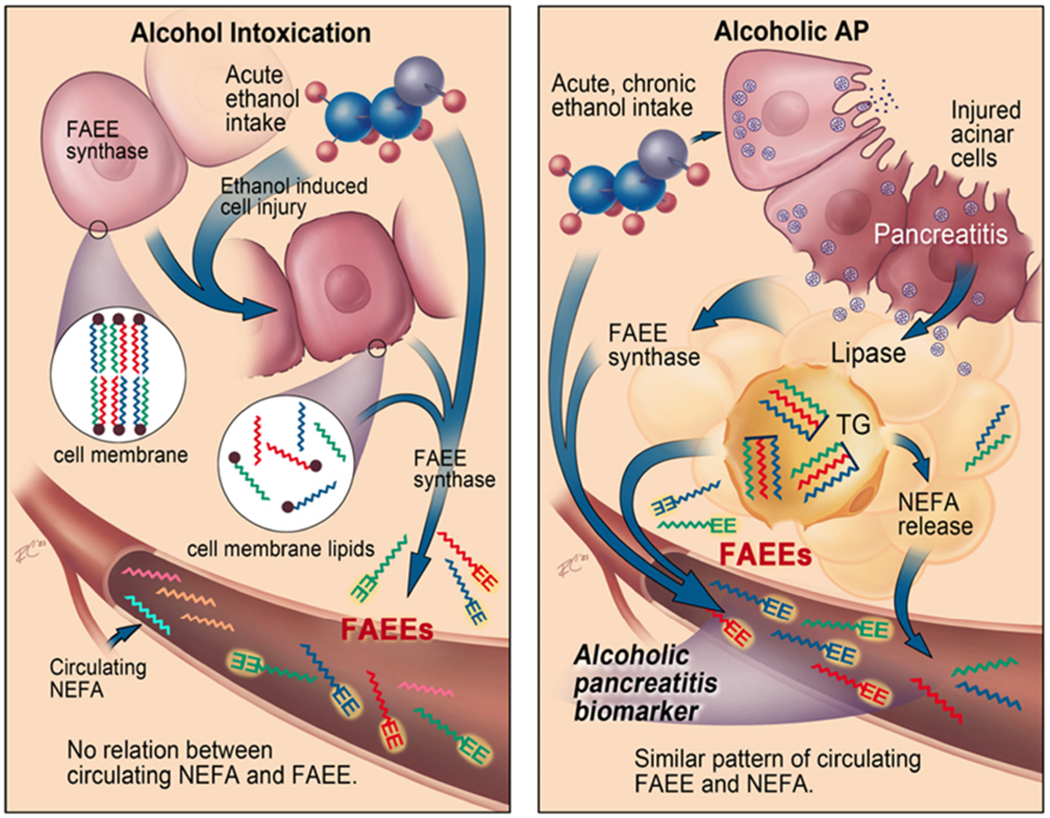
During alcohol intoxication, ethanol mediated injury to cells releases membrane lipids, that are (trans)esterified to FAEEs (zigzag lines with EE at the end) in the presence of ethanol by the FAEE synthase released. This FAEE increase has no relation to the circulating NEFA (only zigzag lines). During alcoholic AP, the FAEEs within adipose tissue, which were generated from the pancreatic lipase mediated esterification of NEFA (as shown in figure 5G–I) or trans-esterification of adipose triglyceride (TG) are released during fat necrosis. This FAEE increase in the circulation parallels that of the increase in parent NEFA released from fat necrosis.
DISCUSSION:
Here we note that during alcoholic AP: 1) The concentration and type of serum FAEEs match the parent NEFA (Fig 3E–H) and those in the visceral fat8, 9, 40 (Fig 5H, I). 2) Visceral FAEEs have a sustained release in mice (Fig 5C), and patients with ongoing alcoholic AP (Fig 3A). 3) During alcoholic AP the FAEEs increase is independent of blood alcohol levels (Fig 3E–H) or sex of the patient (supplementary figure 1). These features are unlike alcohol intoxication in which the serum FAEE increase is generic (Fig 5F, G), transient (Fig 5A–C), dependent on blood alcohol levels (Fig 2A–D), independent of circulating NEFA (Fig 2D–G), and mediated by injurious levels of alcohol reacting with lipid precursors (Fig 6A–C, 6H). Lastly, we note that FAEEs are potential biomarkers to diagnose alcoholic pancreatitis. These findings along with the fat necrosis present during AP 41, 42; collectively support FAEE elevation to have a unique pathophysiology and utility as a diagnostic biomarker for alcoholic pancreatitis. This is summarized in figure 7.
The clinical relevance of the FAEE elevation as a biomarker of alcoholic pancreatitis is supported by their large AUC on ROC analysis (Fig 4B), and correlation with circulating NEFAs being independent of alcohol blood levels (Fig 3E–H). We note the slope in Figure 3D (i.e. in the presence of alcohol) is 0.0008, and in Fig 3E (i.e. in the absence of alcohol) is 0.0009. This change in slope reflects the change of the FAEE in logged scale per unit (μM) increase in NEFA. A clinically meaningful interpretation evaluating FAEEs increase with NEFA shows that an increase of 1000 μM NEFA increases FAEE by 0.8 on the log scale in the presence of alcohol (i.e. 6.30-fold increase in fig 3D), and 0.9 on the log scale in the absence of alcohol (i.e. 7.94-fold increase in Fig 3E). Additionally, the Spearman correlation for improves from 0.54 to 0.71 in the absence of alcohol (Fig 3D, E). Thus, both the β coefficient, and the Spearman correlation show a consistent and stronger association between FAEEs and NEFA in the absence of alcohol.
Previous studies showed that the visceral fat of alcoholics without detectable blood ethanol had higher FAEE concentrations7 than the pancreas, liver or other tissues. It was also proposed that these FAEEs form intracellularly by the action of carboxylesterase on fatty acyl-CoA in the presence of ethanol43 over the duration alcohol is consumed. Visceral fat necrosis is highly prevalent in AP 22, 42 due to early adipose involvement42, which is commonly reported as “peripancreatic stranding” on imaging studies44. Thus, as shown in the right panel of figure 7, the parallel increase in NEFA and FAEEs (Fig 3E–H) likely result from their generation, storage and release during pancreatitis associated fat necrosis (Fig 5H). The intraperitoneal OAEE mediated sustained increase in serum OAEE (Fig 5C–E), but not in PAEE or SAEE supports that the FAEEs measured in the circulation are the same as those released from visceral fat. Therefore, the similarity of FAEE and NEFAs (Fig 3E–H) during alcoholic AP (Fig 1D), is consistent with both being released from a common source; i.e. fat necrosis during alcoholic pancreatitis8, 9, 25, 40.
The >50 fold elevation in circulating FAEE’s in alcoholic AP (Fig 1E), unlike the 2-4 fold increase in NEFA10, 11 over controls (Fig 1D) allows for a large signal-to-noise ratio in favor of using FAEEs as biomarkers of alcoholic pancreatitis. We note this as a large AUC of 0.87 (CI: 0.78-0.95) with a P <0.001 in the 106 patients with pancreatitis, of which 31 had alcoholic pancreatitis.
The lack of correlation between serum FAEEs and NEFAs during alcohol intoxication (Fig 2E–H) suggests an alternate source of the precursor lipids for FAEEs. The ethanol induced increase of all FAEEs in vivo (Fig 5A–C, F–G), and by injurious ethanol concentrations in both acini and HEK293 cells (Fig 6A–C. 6G–H) is consistent with alcohol induced cell membrane damage providing the lipid precursors, and enzymes (FAEE synthase)1, 16 for generating FAEEs in the presence of alcohol15 (Fig 7, left panel). Since FAEEs are found in non-pancreatic tissues18, and are produced in HEK 293 cells, though in lesser amounts than in acini (Fig 6B,D,H,I), the identity of FAEE synthase remains unclear.
Interestingly, OAEE elevations in alcoholic AP are not influenced by alcohol blood levels (Fig 3H, Supp Fig 1C). This agrees well with the sustained OAEE elevation noted in mice sera (dashed blue line Fig 5C) irrespective of alcohol administration. This could be potentially explained by unsaturation making a NEFA like OA (C18:l) or its ethyl ester more aqueous stable than saturated NEFA like PA (C16:0)38 and therefore less influenced by alcohol blood levels.
We note FAEEs are present in much lower amounts (0-1%) compared to circulating NEFA (Fig 4), with typical FAEE concentrations in our patients being in the 0.01-1μM range (Fig 1E) compared to the NEFA concentrations being in the 300-1000 μM range. FAEEs are therefore unlikely to mediate disease since, 1) FAEE concentrations are relatively smaller (nanomole) vs. NEFA (Fig 1D–E), 2) the concentration of FAEEs are equivalent in alcoholics with or without pancreatitis (Fig 1E), and 3) FAEEs result in lower cell injury compared to the parent NEFA12 (Supplementary figure 2A–C).
Our study has some limitations. Patients with alcoholic pancreatitis were younger than those with non-alcoholic pancreatitis (Supplementary figure 3). However, since several previous studies have shown this pattern45–47, our population is consistent with the normal profile noted in pancreatitis. This study did not have sufficient patient enrollment for a validation cohort, but future studies are planned to validate these findings in a larger population. However, by identifying FAEEs as biomarkers of alcoholic pancreatitis, we have set a unique target to be tested in future studies. Our reference standard to compare the diagnostic accuracy of FAEEs is clinical history. As mentioned in the introduction, this can be unreliable. A more thorough gold standard may require multiple controls such as excluding other diseases related to alcohol intake (e.g. alcoholic hepatitis), excluding use of behavioral therapy (e.g. alcohol rehabilitation) or pharmacologic measures to prevent alcohol intake, and undetectable alcohol blood levels during other hospitalizations, before a case is labelled “non-alcoholic AP”. While these can be pursued in the future, the current study describes a unique pathophysiology, and the potential utility of FAEEs as diagnostic biomarkers for alcoholic pancreatitis. The timing of the blood draws in some of our patients may have lagged the first clinical sample used to make a diagnosis of AP or measure alcohol blood levels. Soderberg et al showed that during acute alcohol intoxication, FAEEs were undetectable by the end of the first day1. All our samples were collected within 24 hours of presentation. Interestingly, we note a significant (Fig 2A–D) association between blood alcohol and FAEEs during alcohol intoxication despite the variable delay, but not FAEEs and NEFA which were measured on the same sample.
In summary, we identify FAEEs as potential biomarkers for diagnosing alcoholic AP and identify a unique pathophysiology for their elevation in contrast to alcoholic pancreatitis. FAEE elevation during intoxication parallels alcohol blood levels, however during alcoholic AP, serum FAEEs parallel the precursor NEFA type and serum NEFA concentrations independent of alcohol levels. While FAEE levels are low compared to their parent NEFA which worsen AP; the large signal to noise ratio, and ROC analysis make FAEEs an attractive diagnostic biomarker to be tested in future studies of alcoholic acute pancreatitis and perhaps other alcohol related acute diseases.
Supplementary Material
What you need to know:
| Background and context | Alcohol related diseases are difficult to diagnose. Fatty acid ethyl esters (FAEEs) are known to be elevated during alcohol intoxication, but their levels and roles in diseases like alcoholic pancreatitis is unknown. |
| New Findings | The FAEE increase during alcoholic pancreatitis parallel their source, i.e. fatty acids released from fat breakdown. However, during alcohol intoxication, FAEEs come from alcohol mediated damage to cell membranes. |
| Limitations | We only sampled the blood fatty acids, and not the fat affected in patients with pancreatitis. There was no validation cohort to verify accuracy of FAEEs as biomarkers of alcoholic pancreatitis. |
| Impact | FAEEs may be useful as biomarkers of alcoholic pancreatitis. Their role in diagnosing other acute alcoholic diseases, such as alcoholic hepatitis can be explored. |
Acknowledgements:
We greatly appreciate the help of the Bryan Remuto at the Carl T. Hayden Veterans’ Administration Medical Center, and the Mayo Clinic hospital processing lab staff including Dasey Diaz, David Grabek, Diana Lopez, Jana Stieben, Rocio Flores, Sheila Sandolo, and of the Mayo Clinic Scottsdale Biospecimens Accessioning and Processing Core, for the efficient procurement of the patient samples used in this study.
Grant support:
Supported by Grant number W81XWH-16-1-0667 (SV), W81XWH-16-1-0668 from the Department of Army (DOA) (VPS), award number R01DK092460, R01DK119646 (VPS) from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The contents of the manuscript are solely the responsibility of the authors and do not necessarily represent the official view of DOA, NIDDK.
Abbreviations used in this paper:
- AP
acute pancreatitis
- CI
confidence interval
- FAEE
fatty acid ethyl esters
- GC
gas chromatography
- LDH
lactate dehydrogenase
- NEFA
non-esterified fatty acids
- OAEE
oleic caid ethyl ester
- PAEE
palmitic acid ethyl ester
- SAEE
stearic acid ethyl ester
- TG
triglyceride
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: VPS is a consultant for Arctx Medical Inc. and received royalties from Lamassu Pharma.
REFERENCES:
- 1.Soderberg BL, Sicinska ET, Blodget E, et al. Preanalytical variables affecting the quantification of fatty acid ethyl esters in plasma and serum samples. Clin Chem 1999;45:2183–90. [PubMed] [Google Scholar]
- 2.Clemens DL, Wells MA, Schneider KJ, et al. Molecular mechanisms of alcohol associated pancreatitis. World J Gastrointest Pathophysiol 2014;5:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shield KD, Parry C, Rehm J. Chronic diseases and conditions related to alcohol use. Alcohol Res 2013;35:155–73. [PMC free article] [PubMed] [Google Scholar]
- 4.Haeny AM, Littlefield AK, Sher KJ. False negatives in the assessment of lifetime alcohol use disorders: a serious but unappreciated problem. J Stud Alcohol Drugs 2014;75:530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maruyama K, Otsuki M. Incidence of alcoholic pancreatitis in Japanese alcoholics: survey of male sobriety association members in Japan. Pancreas 2007;34:63–5. [DOI] [PubMed] [Google Scholar]
- 6.Yadav D, Whitcomb DC. The role of alcohol and smoking in pancreatitis. Nat Rev Gastroenterol Hepatol 2010;7:131–45. [DOI] [PubMed] [Google Scholar]
- 7.Laposata EA, Lange LG. Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 1986;231:497–9. [DOI] [PubMed] [Google Scholar]
- 8.Navina S, Acharya C, DeLany JP, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Science translational medicine 2011;3:107ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Oliveira C, Khatua B, Noel P, et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J Clin Invest 2020;130:1931–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domschke S, Malfertheiner P, Uhl W, et al. Free fatty acids in serum of patients with acute necrotizing or edematous pancreatitis. Int J Pancreatol 1993;13:105–10. [DOI] [PubMed] [Google Scholar]
- 11.Sztefko K, Panek J. Serum free fatty acid concentration in patients with acute pancreatitis. Pancreatology 2001;1:230–6. [DOI] [PubMed] [Google Scholar]
- 12.Patel K, Durgampudi C, Noel P, et al. Fatty Acid Ethyl Esters Are Less Toxic Than Their Parent Fatty Acids Generated during Acute Pancreatitis. Am J Pathol 2016;186:874–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Criddle DN, Murphy J, Fistetto G, et al. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 2006;130:781–93. [DOI] [PubMed] [Google Scholar]
- 14.Huang W, Booth DM, Cane MC, et al. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 2014;63:1313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riley DJ, Kyger EM, Spilburg CA, et al. Pancreatic cholesterol esterases. 2. Purification and characterization of human pancreatic fatty acid ethyl ester synthase. Biochemistry 1990;29:3848–52. [DOI] [PubMed] [Google Scholar]
- 16.Uthoff S, Broker D, Steinbuchel A. Current state and perspectives of producing biodiesel-like compounds by biotechnology. Microb Biotechnol 2009;2:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khatua B, Trivedi RN, Noel P, et al. Carboxyl Ester Lipase May Not Mediate Lipotoxic Injury during Severe Acute Pancreatitis. Am J Pathol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calabrese V, Scapagnini G, Catalano C, et al. Effects of acetyl-L-carnitine on the formation of fatty acid ethyl esters in brain and peripheral organs after short-term ethanol administration in rat. Neurochem Res 2001;26:167–74. [DOI] [PubMed] [Google Scholar]
- 19.Thomas LW. The chemical composition of adipose tissue of man and mice. Quarterly journal of experimental physiology and cognate medical sciences 1962;47:179–88. [DOI] [PubMed] [Google Scholar]
- 20.Scott RF, Lee KT, Kim DN, et al. Fatty Acids of Serum and Adipose Tissue in Six Groups Eating Natural Diets Containing 7 to 40 Per Cent Fat. Am J Clin Nutr 1964;14:280–90. [DOI] [PubMed] [Google Scholar]
- 21.Saghir M, Werner J, Laposata M. Rapid in vivo hydrolysis of fatty acid ethyl esters, toxic nonoxidative ethanol metabolites. Am J Physiol 1997;273:G184–90. [DOI] [PubMed] [Google Scholar]
- 22.Kloppel G vGR, Dreyer T. Pathomorphology of acute pancreatitis. Analysis of 367 autopsy cases and 3 surgical specimens. Amsterdam, New York, Oxford, 1984. [Google Scholar]
- 23.Kloppel G, Maillet B. Pseudocysts in chronic pancreatitis: a morphological analysis of 57 resection specimens and 9 autopsy pancreata. Pancreas 1991;6:266–74. [PubMed] [Google Scholar]
- 24.Kloppel G, Dreyer T, Willemer S, et al. Human acute pancreatitis: its pathogenesis in the light of immunocytochemical and ultrastructural findings in acinar cells. Virchows Arch A Pathol Anat Histopathol 1986;409:791–803. [DOI] [PubMed] [Google Scholar]
- 25.Patel K, Trivedi RN, Durgampudi C, et al. Lipolysis of visceral adipocyte triglyceride by pancreatic lipases converts mild acute pancreatitis to severe pancreatitis independent of necrosis and inflammation. The American journal of pathology 2015;185:808–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durgampudi C, Noel P, Patel K, et al. Acute Lipotoxicity Regulates Severity of Biliary Acute Pancreatitis without Affecting Its Initiation. The American journal of pathology 2014;184:1773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Criddle DN, Sutton R, Petersen OH. Role of Ca2+ in pancreatic cell death induced by alcohol metabolites. J Gastroenterol Hepatol 2006;21 Suppl 3:S14–7. [DOI] [PubMed] [Google Scholar]
- 28.Mukherjee R, Criddle DN, Gukovskaya A, et al. Mitochondrial injury in pancreatitis. Cell calcium 2008;44:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Werner J, Laposata M, Fernandez-del Castillo C, et al. Pancreatic injury in rats induced by fatty acid ethyl ester, a nonoxidative metabolite of alcohol. Gastroenterology 1997;113:286–94. [DOI] [PubMed] [Google Scholar]
- 30.El-Kurdi B, Khatua B, Rood C, et al. Mortality From Coronavirus Disease 2019 Increases With Unsaturated Fat and May Be Reduced by Early Calcium and Albumin Supplementation. Gastroenterology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khatua B, Yaron JR, El-Kurdi B, et al. Ringer’s Lactate Prevents Early Organ Failure by Providing Extracellular Calcium. J Clin Med 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology : official journal of the International Association of Pancreatology 2013;13:e1–15. [DOI] [PubMed] [Google Scholar]
- 33.Kulig CC, Beresford TP, Everson GT. Rapid, accurate, and sensitive fatty acid ethyl ester determination by gas chromatography-mass spectrometry. J Lab Clin Med 2006;147:133–8. [DOI] [PubMed] [Google Scholar]
- 34.Kangani CO, Kelley DE, Delany JP. New method for GC/FID and GC-C-IRMS analysis of plasma free fatty acid concentration and isotopic enrichment. J Chromatogr B Analyt Technol Biomed Life Sci 2008;873:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ko H, Royer ME. A gas-liquid chromatographic assay for plasma free fatty acids. J Chromatogr 1974;88:253–63. [DOI] [PubMed] [Google Scholar]
- 36.Cohen JF, Korevaar DA, Altman DG, et al. STARD 2015 guidelines for reporting diagnostic accuracy studies: explanation and elaboration. BMJ Open 2016;6:e012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Criddle DN, Raraty MG, Neoptolemos JP, et al. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proceedings of the National Academy of Sciences of the United States of America 2004;101:10738–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khatua B, El-Kurdi B, Patel K, et al. Adipose saturation reduces lipotoxic systemic inflammation and explains the obesity paradox. Sci Adv 2021;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Best CA, Sarkola T, Eriksson CJ, et al. Increased plasma fatty acid ethyl ester levels following inhibition of oxidative metabolism of ethanol by 4-methylpyrazole treatment in human subjects. Alcohol Clin Exp Res 2006;30:1126–31. [DOI] [PubMed] [Google Scholar]
- 40.Noel P, Patel K, Durgampudi C, et al. Peripancreatic fat necrosis worsens acute pancreatitis independent of pancreatic necrosis via unsaturated fatty acids increased in human pancreatic necrosis collections. Gut 2016;65:100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banks PA, Bollen TL, Dervenis C, et al. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013;62:102–11. [DOI] [PubMed] [Google Scholar]
- 42.Nordback I, Lauslahti K. Clinical pathology of acute necrotising pancreatitis. Journal of clinical pathology 1986;39:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bencharit S, Edwards CC, Morton CL, et al. Multisite promiscuity in the processing of endogenous substrates by human carboxylesterase 1. J Mol Biol 2006;363:201–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koo BC, Chinogureyi A, Shaw AS. Imaging acute pancreatitis. Br J Radiol 2010;83:104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Farrell A, Allwright S, Toomey D, et al. Hospital admission for acute pancreatitis in the Irish population, 1997 2004: could the increase be due to an increase in alcohol-related pancreatitis? J Public Health (Oxf) 2007;29:398–404. [DOI] [PubMed] [Google Scholar]
- 46.Artaza Varasa T, Talavera Fabuel A, Legaz Huidobro M, et al. [Epidemiologic aspects of 116 acute pancreatitis prospectively studied]. Rev Esp Enferm Dig 1994;85:27–30. [PubMed] [Google Scholar]
- 47.Svensson JO, Norback B, Bokey EL, et al. Changing pattern in aetiology of pancreatitis in an urban Swedish area. Br J Surg 1979;66:159–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.