

Abstract
An estimated 220 million people worldwide are chronically exposed to inorganic arsenic (iAs) primarily as a result of drinking iAs-contaminated water. Chronic iAs exposure is associated with a plethora of human diseases including skin lesions and multi-organ cancers. iAs is a known clastogen, inducing DNA double strand breaks (DSBs) in both exposed human populations and in vitro. However, iAs does not directly interact with DNA, suggesting that other mechanisms, such as inhibition of DNA repair and DNA Damage Response (DDR) signaling, may be responsible for iAs-induced clastogenesis. Recent RNA-sequencing data from human keratinocytes (HaCaT cells) indicate that mRNAs for phosphatases important for resolution of DDR signaling are induced as a result of chronic iAs exposure prior to epithelial to mesenchymal transition. Here, we report that phosphorylation of ataxia telengectasia mutated (ATM) protein at a critical site (pSer1981) important for DDR signaling, and downstream CHEK2 activation, are significantly reduced in two human keratinocyte lines as a result of chronic iAs exposure. Moreover, RAD50 expression is reduced in both of these lines, suggesting that suppression of the MRE11-RAD50-NBS1 (MRN) complex may be responsible for reduced ATM activation. Lastly, we demonstrate that DNA double strand break accumulation and DNA damage is significantly higher in human keratinocytes with low dose iAs exposure. Thus, inhibition of the MRN complex in iAs-exposed cells may be responsible for reduced ATM activation and reduced DSB repair by homologous recombination (HR). As a result, cells may favor error-prone DSB repair pathways to fix damaged DNA, predisposing them to chromosomal instability (CIN) and eventual carcinogenesis often seen resulting from chronic iAs exposure.
Keywords: Arsenic, keratinocytes, DNA Damage Response, ATM, Phosphatases, MRN
INTRODUCTION
Approximately 220 million people worldwide across 70 countries are chronically exposed to inorganic arsenic (iAs) primarily through ingestion of contaminated drinking water that exceeds the current Environmental Protection Agency and World Health Organization standards (10 μg/L) (Podgorski and Berg, 2020). Chronic iAs exposure can also occur as a result of diet, occupational, or iatrogenic circumstances (Mondal et al., 2010). Extensive literature links chronic iAs exposure to a myriad of health problems including skin lesions, cardiovascular disease, diabetes, and many types of cancer (Martinez et al., 2011). Furthermore, iAs is a known clastogen; strong experimental evidence from epidemiological studies in chronically exposed human populations as well as in vitro studies demonstrates that iAs exposure is associated with heightened occurrence of DNA double-strand breaks (DSBs) (Gonsebatt et al., 1994; Gebel, 2001; Mahata et al., 2004; Ghosh et al., 2007; Chakraborty and De, 2009; Roy et al., 2018). Misrepaired DSBs lead to chromosomal instability (CIN). In 1999, the National Research Council determined CIN as the most likely mode of action for iAs-induced carcinogenesis (National Research Council Subcommittee on Arsenic in Drinking, 1999). Importantly, iAs does not directly interact with DNA to induce point mutations, but can act as a co-mutagen (Li and Rossman, 1989; Hei et al., 1998). The molecular mechanisms by which chronic iAs exposure induces clastogenesis have yet to be defined.
DSBs can accumulate from induction of DNA damage and/or failure to repair basal endogenous DNA damage (Hoeijmakers, 2001). Several molecular mechanisms which contribute to iAs-induced clastogenesis have been intensely studied including but not limited to generation of reactive oxygen species (ROS), inhibition of several DNA repair pathways (i.e. nucleotide excision repair, base excision repair (BER), nucleotide excision repair (NER), interstrand crosslink repair (ICL)), chromatin remodeling, and transcriptional regulation of DNA repair genes (Tam et al., 2020). However, the majority of studies have been performed utilizing high dose-acute exposure regimens (μM-mM; hours to days). These experimental designs do not accurately recapitulate chronic iAs exposure in human populations. Mean blood and serum concentrations in iAs-exposed human populations are reported as typically around 100 nM (Wang et al., 1993; Pi et al., 2000; Wu et al., 2001).
DSBs are the most dangerous type of DNA lesion because they can lead to chromosomal rearrangements, a precursor of carcinogenesis (Aparicio et al., 2014; Vitor et al., 2020). Failure to repair DSBs leads to structural CIN (i.e. chromosomal rearrangements, micronuclei formation, and deletions or insertions). Loss or rearrangement of genetic information can further compromise specific components of DSB repair. Under such circumstances, other available DNA repair pathways, often error-prone, are employed to effect repair, leading to increased mutagenesis and eventually carcinogenesis (Hanahan and Weinberg, 2011; Kieffer and Lowndes, 2022). Importantly, genomic instability and mutations arising out of error-prone repair systems are hallmarks of carcinogenesis (Hanahan and Weinberg, 2011).
Structural CIN, or structural rearrangements of chromosomes, often occurs because of faulty DSB repair processes (Hanahan and Weinberg, 2011; Bakhoum and Cantley, 2018). To repair damaged DNA, cells have evolved a comprehensive cellular response known as the DNA damage response (DDR), a signal transduction pathway which serves to detect DNA damage, mediate DNA repair, and arrest the cell cycle to prevent mitotic entry of cells containing damaged DNA (Ciccia and Elledge, 2010). Proper activation of the DDR ultimately prevents passage of faulty genetic information to daughter cells and prevents accumulation of DNA damage that can put cells at higher risk for malignant transformation. The DDR is coordinated by three apical phosphoinositide 3-kinase (PI3K)-related kinases including DNA-dependent protein kinase catalytic subunit (DNA-PKcs), ATM Serine/Threonine kinase (ATM), ATR Serine/Threonine kinase (ATR), and members of the poly(ADP-ribose) polymerase (PARP) family (PARP1 and PARP2) (Blackford and Jackson, 2017). The apical kinase utilized to signal DNA damage is dependent on both the type of DNA break and stage of the cell cycle when damage occurs. While both ATM and DNA-PKcs respond to DSBs, ATR and PARP1/2 respond to a wider spectrum of DNA damage, including both DSBs and single-stranded breaks (SSBs) (Ciccia and Elledge, 2010).
ATM has multiple functions in cancer development including activation of cell cycle checkpoints, DSB Repair, metabolic regulation, cell migration, and chromatin remodeling (Jin and Oh, 2019). In absence of DSBs, ATM exists as a catalytically inactive dimer. Upon induction of DSBs, dimeric ATM is rapidly autophosphorylated on Ser1981 and dissociates into active monomers (Dupre et al., 2006) (Figure 1). Phosphorylated ATM (Ser-1981) in turn activates many downstream targets, including CHEK2, TP53, H2AX, and BRCA1 which are important for maintaining genomic integrity (Dupre et al., 2006). The MRN complex (a trimeric complex containing MRE11 Homolog, Double Strand Break Repair Nuclease (MRE11), RAD50 Double Strand Break Repair Protein (RAD50) and nibrin (NBN, formerly NBS1)) senses DSBs and is required upstream for ATM activation (Uziel et al., 2003). Following DDR activation and completion of repair, activation is resolved through dephosphorylation by DDR phosphatases, allowing cells to exit from cell cycle arrest (Shimada and Nakanishi, 2013; Campos and Clemente-Blanco, 2020).
Figure 1.
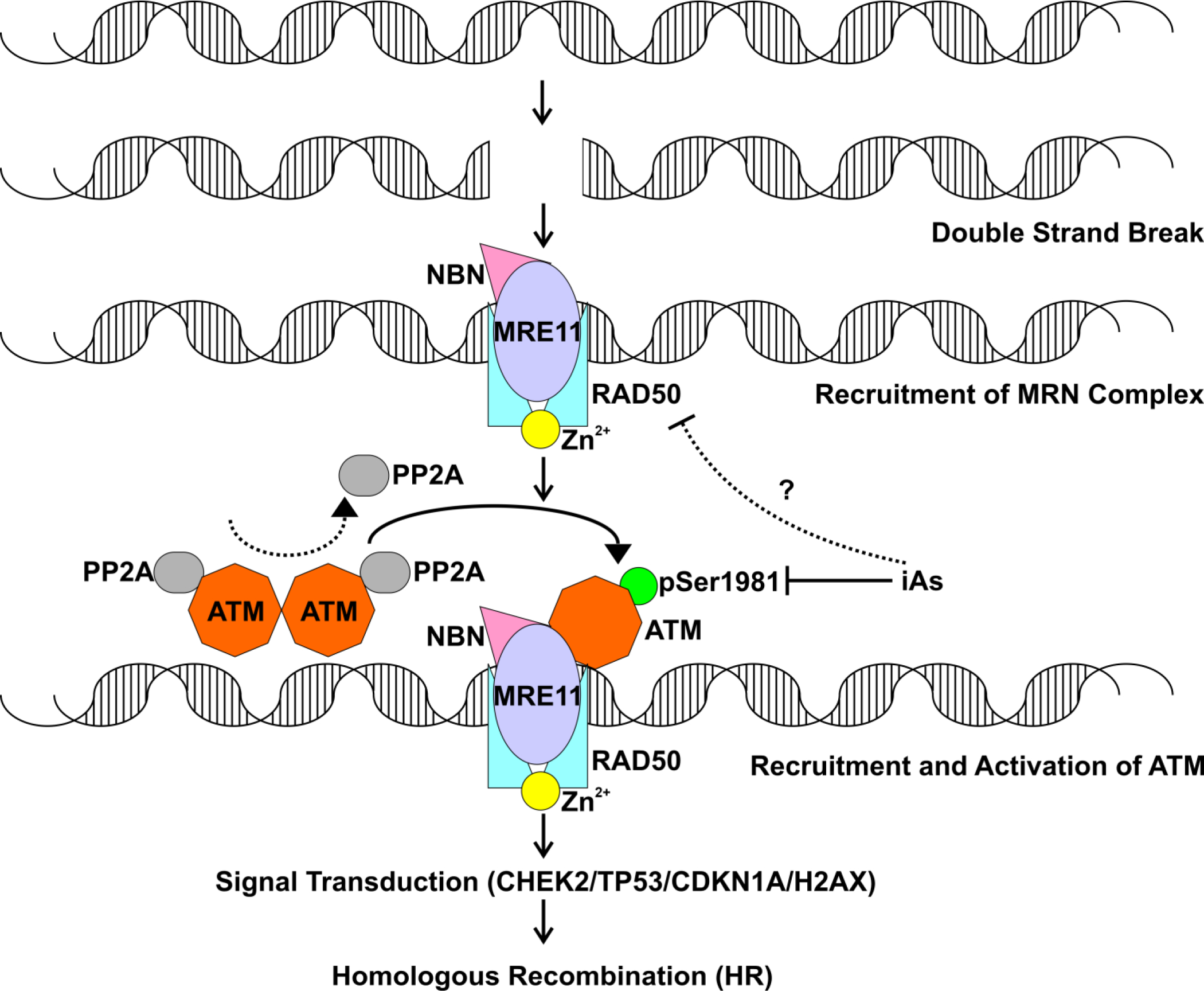
ATM activation by DNA double-strand breaks. Following DNA double strand breaks (DSBs) induced either by endogenous or exogenous agents, the MRN (MRE11-RAD50-NBN) complex is recruited to DSBs. In the absence of DNA damage, protein phosphatase A (PP2A) binds to ATM and prevents in activation. As a result, ATM is present as an inactive dimer. In the event of DSBs, ATM is recruited by the MRN to sites of DNA damage and undergoes rapid autophosphorylation of serine 1981 which dissociates ATM dimers and initiates ATM kinase activity. Downstream substrates of ATM include CHEK2 and P53, which induces expression of CDKN1A. ATM can also phosphorylate histone H2AX, which in turn can recruit more DNA repair factors to sites of DNA damage. The MRN complex promotes high fidelity DNA repair by homologous recombination (HR Repair).
Our understanding of molecular events that contribute to iAs-induced skin carcinogenesis following chronic iAs exposure have been largely studied using immortalized human keratinocytes (HaCaT) (Boukamp et al., 1988). Previous work from our lab and others has demonstrated that treatment of HaCaT cells with toxicologically relevant iAs concentrations (100 nM) results in epithelial to mesenchymal transition (EMT) after 28-wk continued exposure (Pi et al., 2008; Banerjee et al., 2021). EMTs occur in neoplastic cells that have previously undergone genetic and epigenetic changes which allow for favorable clonal outgrowth and the development of localized tumors (Kalluri and Weinberg, 2009). Recently, our group has provided a comprehensive picture of important molecular events at three stages of iAs-induced carcinogenesis in HaCaT cells chronically exposed to 100 nM iAs (pre-transition at 7 weeks, transition initiation at 19 weeks, and fully transitioned at 28 weeks) (Banerjee et al., 2021). We demonstrated that chronic iAs exposure (7 weeks) induced expression of two key phosphatase mRNAs, Protein Phosphatase 2 Regulatory Subunit Beta Gamma (PPP2R2C), a regulatory subunit for PP2A (Fan et al., 2013; Leong et al., 2020), and Protein Phosphatase 5 (PP5) (Banerjee et al., 2021). Both these phosphatases are known to play intergral roles in determining the phosphorylation mediated activation of the DDR pathway (Lee and Chowdhury, 2011; Shimada and Nakanishi, 2013).
Given the combined findings that chronic iAs exposure is known to induce CIN and higher phosphatase mRNA expression for select phosphatase genes, we evaluated the effects of chronic iAs exposure on expression and activation of DDR apical kinases, ATM and ATR in two different keratinocyte cell lines; HaCaT and Ker-CT at early pre-EMT stages of exposure (7 or 8 weeks, respectively)(Ramirez et al., 2003). We demonstrate that chronic iAs exposure suppressed activation of ATM, but not of ATR in both keratinocyte cell lines. In conjuction, chronic iAs exposure led to higher accumulation of DSBs in both cell lines. Our study highlights a new molecular mechanism that likely contributes to iAs-induced clastogenesis and CIN, and thus to carcinogenesis.
MATERIALS AND METHODS
Chemicals
Sodium arsenite (NaAsO2; CAS 7784-0698) was obtained from Thermofisher Scientific Inc. (Waltham, MA, USA). Single-thaw aliquots of sodium arsenite were prepared in UltraPure™ DNAse/RNase-Free Distilled Water (Thermo Fisher Scientific Inc.) and were thawed immediately before use. Trypsin-EDTA (0.05% trypsin-0.02% EDTA) used for passaging were obtained from Thermo Fisher Scientific Inc. Neocarzinostatin (NCS) was acquired from Sigma (N9162, St. Louis, MO).
Cell Culture
HaCaT cells were the kind gift of Dr. Tai Hao Quan, University of Michigan. Ker-CT cells were obtained from ATCC (CRL-4048, Manassas, VA). HaCaT and Ker-CT long term cultures were maintained as independent quadruplicates exposed to 0 or 100 nM NaAsO2 at 37°C in a humidified 5% CO2 atmosphere. iAs exposed cells were continuously maintained in 100 nM iAs-containing media throughout the passaging process and culturing. HaCaT cells were cultured for 7 weeks in MEM alpha modification media (Thermo Fisher Scientific Inc.) supplemented with 10% fetal bovine serum from Hyclone (Logan, UT, USA), 100 units/mL penicillin/100 μg/mL streptomycin and 2 mM glutamine. At each passage, 106 cells were plated per 100 mm dish. Ker-CT cells were cultured in KGM™ Gold Keratinocyte Growth Medium from Lonza (CC-3103, Basel, Switzerland) supplemented with the KGM™ SingleQuots™ Supplement Pack (CC-4152) and cultured continuously 8 weeks. At each passage, 4 × 105 cells were plated per 100 mm dish or 1.2 × 106 cells per 150 mm dish. Protein lysates or cells used for Neutral COMET assays (see below) were collected at 7 weeks for HaCaT cells, or 8 weeks for Ker-CT cells. For NCS experiments, HaCaT cells were plated at 1 × 106 per 100 mm dish and Ker-CT cells were plated at 4 × 105 cells per 100 mm dish. Subsequently, 24 h post-seeding the cells were treated with 0 or 20 nM NCS (in independent triplicates for HaCaT cells and independent quadruplicates for Ker-CT cells) and incubated at 37°C for 30 minutes. Following the 30 min incubation, cells were washed three times with Hanks Balanced Salts Solution (HBSS) prior to protein lysate collection.
Immunoblotting
Immunoblotting was performed to examine the expression and activation of proteins (marked by phosphorylation) involved in DDR. Sample preparation, estimation of protein content, immunoblotting, and image acquisition was performed as described previously with a few modifications (Banerjee et al., 2020). Details regarding the antibodies used and their dilutions are presented in Supplemental Table 1. Proteins were resolved by electrophoresis in SDS polyacrylamide gels of appropriate percentage (5% or 10%) or in 4 – 20% Mini-PROTEAN TGX™ Precast Protein Gels, 15-well from BioRad (4561096, Hercules, CA). Signals for phosphorylated and total forms of the same protein were always developed on the same membrane. Phosphorylated proteins were first visualized, followed by stripping of the membrane employing RestoreTM PLUS western blot stripping buffer (Thermo Fisher Scientific Inc.) at 37°C for 1h. To ensure removal of signal, stripped PVDF membranes were incubated with secondary antibody and inspected for persistence of signal. Successfully stripped membranes were blocked for 30 min at room temperature in 5% milk, incubated with primary and secondary antibodies for the corresponding total protein and developed. Raw data for densitometric analysis was generated from the images using Image J software (Schneider et al., 2012). For each protein of interest, total protein was normalized against a suitable housekeeping protein, while activation was calculated as a ratio of the phosphorylated/total signal for each sample.
Neutral COMET assays
HaCaT or Ker-CT cells were plated at 2.5 × 105 cells per 60 mm dish and grown for 48 h prior to performing neutral COMET assays using the CometAssay Single Cell Gel Electophoresis Assay (R&D Systems, Minneapolis, MN) according to manufacturer’s protocol. Briefly, cells were washed once either with PBSE (1 × Phosphate Buffered Saline with 5 mM Na2EDTA, HaCaT) or HBSS (Hank’s Balanced Salt Solution, Ker-CT), trypsinized, collected by centrifugation at 200 × g for 5 min at 4°C and subsequently resuspended in ice cold PBS. Cells were counted by Trypan blue assay and diluted to a concentration of 1 × 105 cells/mL and combined with 37°C low melting agarose at a ratio of 1:10 (v/v) and immediately pipetted onto prewarmed 37°C CometSlide slides. Slides were placed on a flat surface in the dark at 4°C for 30 minutes. The slides were then serially treated with lysis solution and neutral electrophoresis buffer for 60 minutes and 30 minutes each, respectively, at 4°C. Next, single cell electrophoresis was performed for 45 minutes at 4°C employing a potential difference of 1 V/cm. Slides were then treated with DNA precipitation solution for 30 minutes at room temperature, followed by dehydration by immersion in 70% ethanol for 30 minutes at room temperature and drying at 37 °C for 15 minutes. Dried slides were placed into a dessicator overnight to bring cells into a single plane to facilitate observation. Slides were stained using 0.003% SYBR Gold staining solution for 30 minutes at room temperature in the dark. Excess SYBR solution was removed and the slides were dried completely at 37 °C prior to scoring. Images were captured using an Olympus IX50 inverted fluorescence microscope using the FITC 488 filter (Olympus America Inc., Center Valley, PA) fitted with a QImaging Retiga EXi Fast 1394 12-bit cooled monochromatic camera (QImaging Corp., Burnaby, BC, Canada). Comet images were analyzed using ImageJ software using the OpenComet v1.3 plugin (Gyori et al., 2014). Only single cells were quantified as opposed to congregated cells. HaCaT and Ker-CT cells treated with a known DNA damaging agent hydrogen peroxide (100 μM; 20 min at 4°C) were used as positive control. Olive tail moment was used as a measure to assess DNA damage (Kumaravel et al., 2009).
Statistical Analyses
For iAs exposure experiments, each molecule was analyzed by immunoblotting, the mean of the unexposed samples was set to 100%, and data are expressed as % mean unexposed. For NCS treatment experiments, the mean of the untreated samples was set to 100% and data are expressed as % mean untreated. Densitometric analysis and bar graphs were generated using GraphPad Prism 9.0.1 (GraphPad Software, San Diego, CA, USA). The data are presented as mean ± SD for all analyses. Densitometric data were analyzed using unpaired two-tailed t-test; p-value ≤ 0.05 was considered significant. For neutral COMET assays, the Olive tail moment data were first tested for normal distribution using D’Agostino & Pearson test; p-value ≤ 0.05 was considered significant. Data that did not pass normality tests were statistically analyzed using the unpaired two-tailed Mann Whitney test; p-value ≤ 0.05 was considered significant.
RESULTS
Chronic iAs exposure results in reduced ATM activation in human keratinocytes
The effect of chronic iAs exposure on ATM activation and signal transduction are presented in Figure 2 (HaCaT cell line) and Figure 3 (Ker-CT cell line). Basal ATM activation, measured by the ratio of phosphorylated ATM (ATM-pSer1981) to total ATM (t-ATM) (Bakkenist and Kastan, 2003), was significantly reduced following chronic iAs exposure in both HaCaT and Ker-CT cells (Figure 2A, 2B and Figure 3A, 3B). Basal phosphorylation of Checkpoint Kinase 2 (CHEK2, formerly CHK2) indicates ATM activity (Figure 1). CHEK2 activation, measured by the ratio of phosphorylated CHEK2 (CHEK2-pThr68) to total CHEK2 (t-CHEK2), was also significantly decreased in iAs exposed HaCaT and Ker-CT cells consistent with decreased ATM activity (Figure 2A, 2C and Figure 3A, 3C). However, iAs exposure did not change basal ATR activation, as measured by the ratio of phosphorylated ATR (ATR-pSer428) to total ATR (t-ATR) (Liu et al., 2011), in either cell line (Figure 2A, 2D and Figure 3A, 3C). Interestingly, t-ATM protein remained unchanged in HaCaT cells, but was significantly reduced in Ker-CT cells; total protein levels for CHEK2 and ATR remained unchanged by iAs exposure in both cell lines (Supplemental Figure 1).
Figure 2.
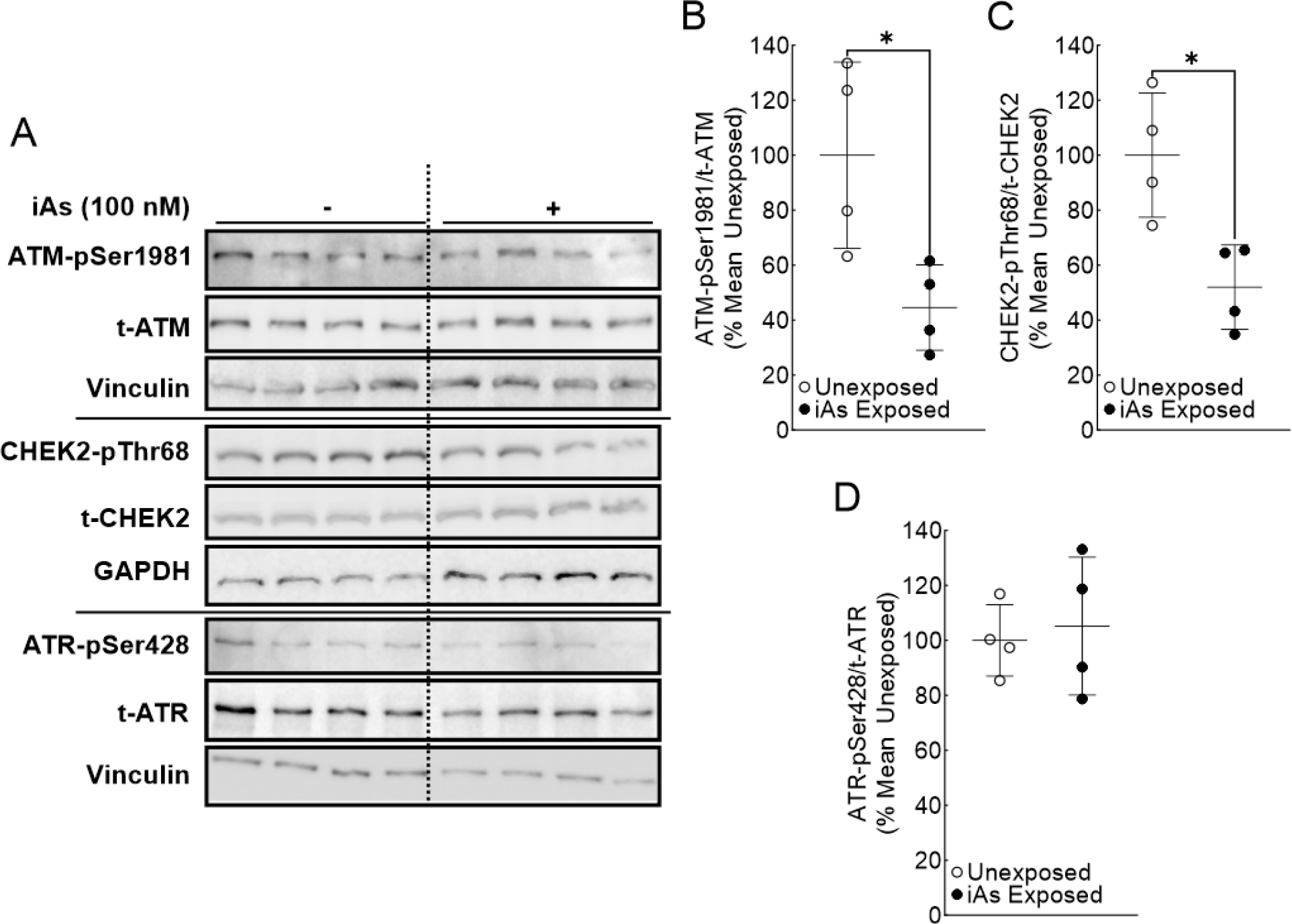
ATM activation is significantly reduced in iAs-exposed HaCaT cells. A. Immunoblot for ATM pathway signaling (ATM and CHEK2) and ATR activation in quadruplicate HaCaT cell cultures exposed to 0 or 100 nM sodium arsenite for 7 weeks. B. Densitometric analysis of ATM activation (ATM-pSer1981/t-ATM). C. Densitometric analysis of CHEK2 activation (CHEK-2pThr68/t-CHEK2). D. Densitometric analysis of ATR activation (ATR-pSer428/t-ATR). Statistical analysis completed by unpaired two-tailed t-test; *p ≤ 0.05
Figure 3.
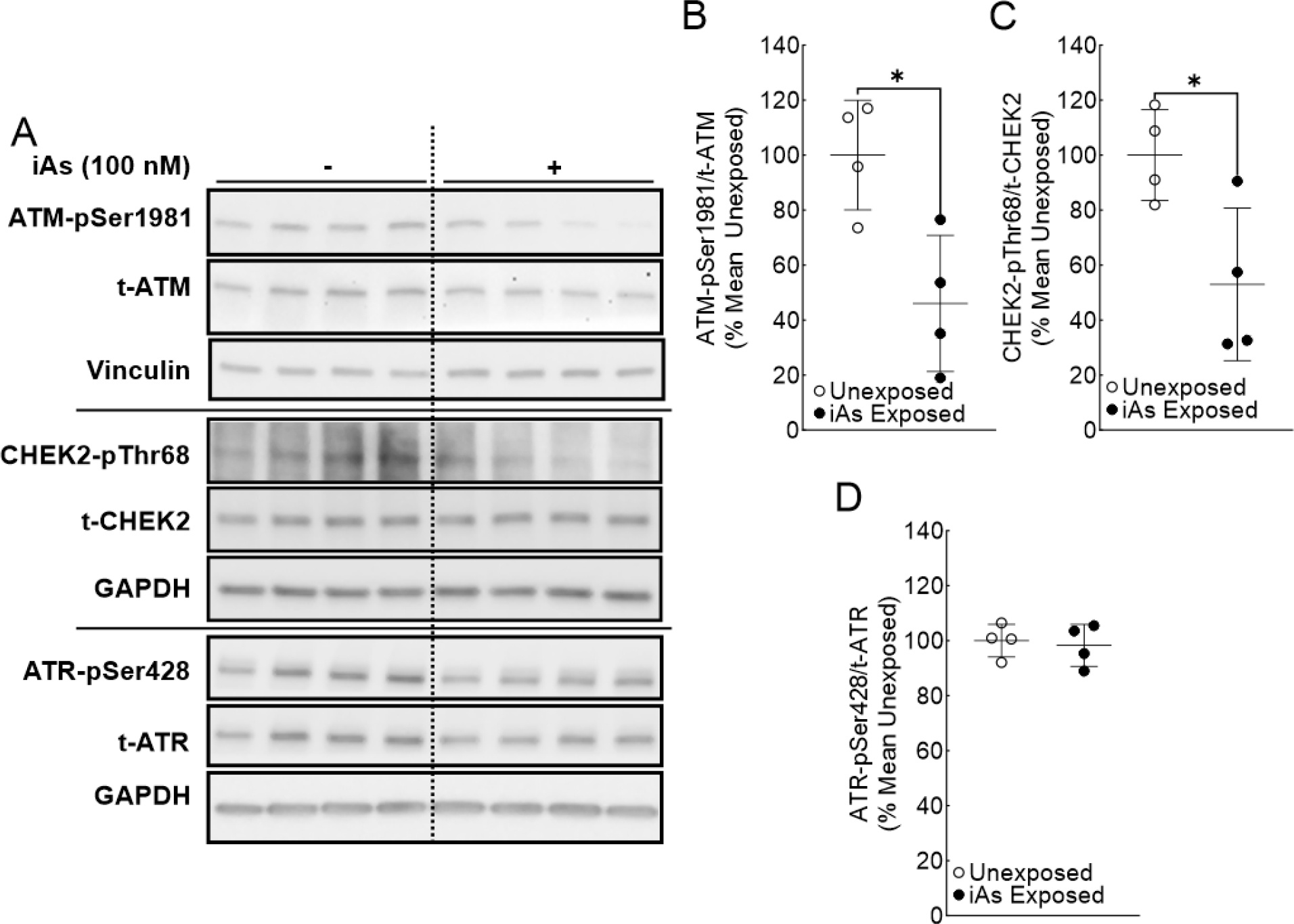
ATM activation is significantly reduced in iAs-exposed Ker-CT cells. A. Immunoblot for ATM pathway signaling (ATM and CHEK2) and ATR activation in quadruplicate Ker-CT cell cultures exposed to 0 or 100 nM sodium arsenite for 8 weeks. B. Densitometric analysis of ATM activation (ATM-pSer1981/t-ATM). C. Densitometric analysis of CHEK2 activation (CHEK-2pThr68/t-CHEK2). D. Densitometric analysis of ATR activation (ATR-pSer428/t-ATR). Statistical analysis was completed by unpaired two-tailed t-test; *p ≤ 0.05
Additionally, to test whether the ATM pathway could be induced in both of these cell lines, we treated either unexposed HaCaT or Ker-CT cells with 20 nM NCS. NCS is a radiomimetic drug which directly induces DSBs and induces ATM-pSer1981 autophosphorylation (Uziel et al., 2003; Kang et al., 2009; Vitor et al., 2020). NCS-treated HaCaT cells (Supplemental Figure 2A–C) and Ker-CT cells (Supplemental Figure 2D–E) showed a significant induction in the activation of ATM and downstream activation of CHEK2 compared to untreated cells. We did not observe any signal for CHEK2-pThr68 in untreated Ker-CT cells, thereby making these lanes unquantifiable (Supplemental Figure 2D). t-ATM and t-CHEK2 protein levels remained unchanged between untreated and NCS-treated cells (Supplemental Figure 3). These results demonstrate that the ATM pathway can be activated significantly above basal levels in the presence of damaging stimuli and that chronic iAs exposure actively suppresses ATM activation in both human keratinocyte cell lines tested. Taken together, chronic iAs exposure suppressed the basal levels of phosphorylated ATM and its downstream target CHEK2, a measure of ATM activity, in both HaCaT and Ker-CT cells.
Chronic iAs exposure significantly reduces PPP2R2C and PP5 expression
RNA-seq data from our previously published data indicated that PPP2R2C and PP5 mRNAs are significantly induced in HaCaT cells exposed to 100 nM arsenite for 7 weeks (Banerjee et al., 2021). Decreased ATM and CHEK2 phosphorylation could be due to increased phospatase activity. To investigate whether induction of phosphatase expression was involved in suppression of ATM activation in iAs exposed cells, we investigated expression of PPP2R2C and PP5 at the the protein level. Surprisingly, PPP2R2C protein expression was significantly suppressed in both HaCaT and Ker-CT cells (Figure 4A–B, 4D–E) upon chronic iAs exposure. PP5 protein expression was either unaffected or significantly reduced in HaCaT or Ker-CT cells, respectively, upon iAs exposure (Figure 4A, 4C, 4D, and 4F). These results are inconsistent with increased phospatase activity being responsible for decreased ATM and CHEK2 phosphorylation.
Figure 4.
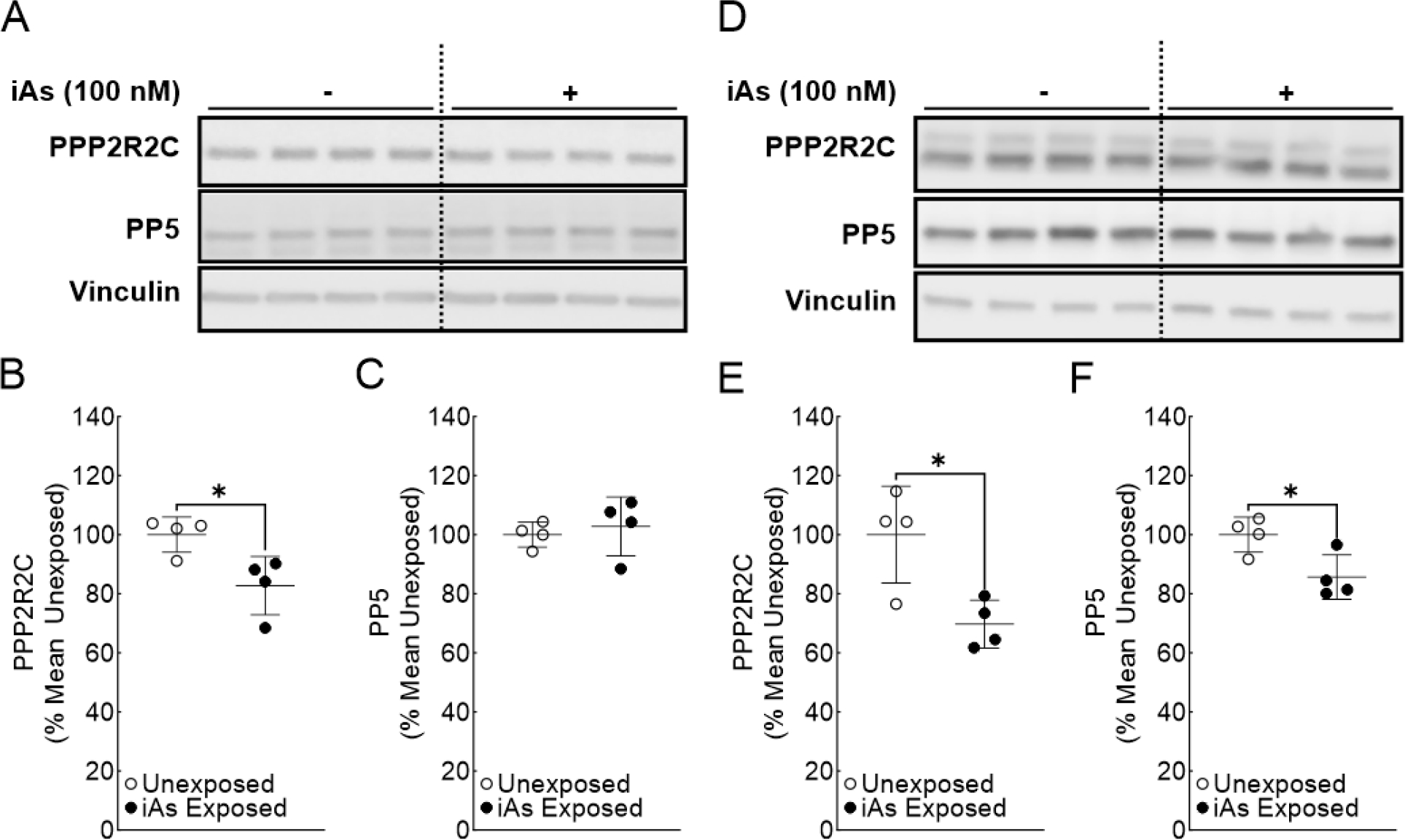
Phosphatase expression is significantly reduced in iAs-exposed human keratinocytes. A. Immunoblot for PPP2R2C and PP5 in quadruplicate HaCaT cell cultures exposed to 0 or 100 nM sodium arsenite for 7 weeks. B. Densitometric analysis of PPP2R2C expression (PPP2R2C/Vinculin) in HaCaT cells C. Densitometric analysis of PP5 expression (PP5/Vinculin) in HaCaT cells. D. Immunoblot for PPP2R2C and PP5 in quadruplicate Ker-CT cell cultures exposed to 0 or 100 nM sodium arsenite for 8 weeks. E. Densitometric analysis of PPP2R2C expression (PPP2R2C/Vinculin) in Ker-CT cells F. Densitometric analysis of PP5 expression (PP5/Vinculin) in Ker-CT cells. Statistical analysis was completed by unpaired two-tailed t-test; *p ≤ 0.05
RAD50 expression is reduced following chronic iAs exposure in human keratinocytes
MRN complex plays a pivotal role in regulating downstream ATM activation and recruitment of DSB repair machinery (Figure 1) (Uziel et al., 2003; Lee and Paull, 2007). Given that expression of ATM targeting phosphatases PPP2R2C and PP5 was suppressed (or unaffected) in our cell lines, we next investigated if iAs exposure affected the expression of proteins of the MRN complex (Figure 5). In both keratinocyte cell lines, RAD50 levels were suppressed in iAs exposed cells (Figure 5A–B and 5E–F). MRE11 levels were significantly suppressed in Ker-CT cells, but not in HaCaT cells upon iAs exposure (Figure 5A, C, E and G). Conversely, NBN levels were significantly induced in HaCaT cells chronically exposed to iAs, but remained unchanged in iAs exposed Ker-CT cells (Figure 5A, D, E, and H). Taken together, these results suggest that chronic iAs exposure dysregulates MRN complex protein stoichiometry in human keratinocytes.
Figure 5.
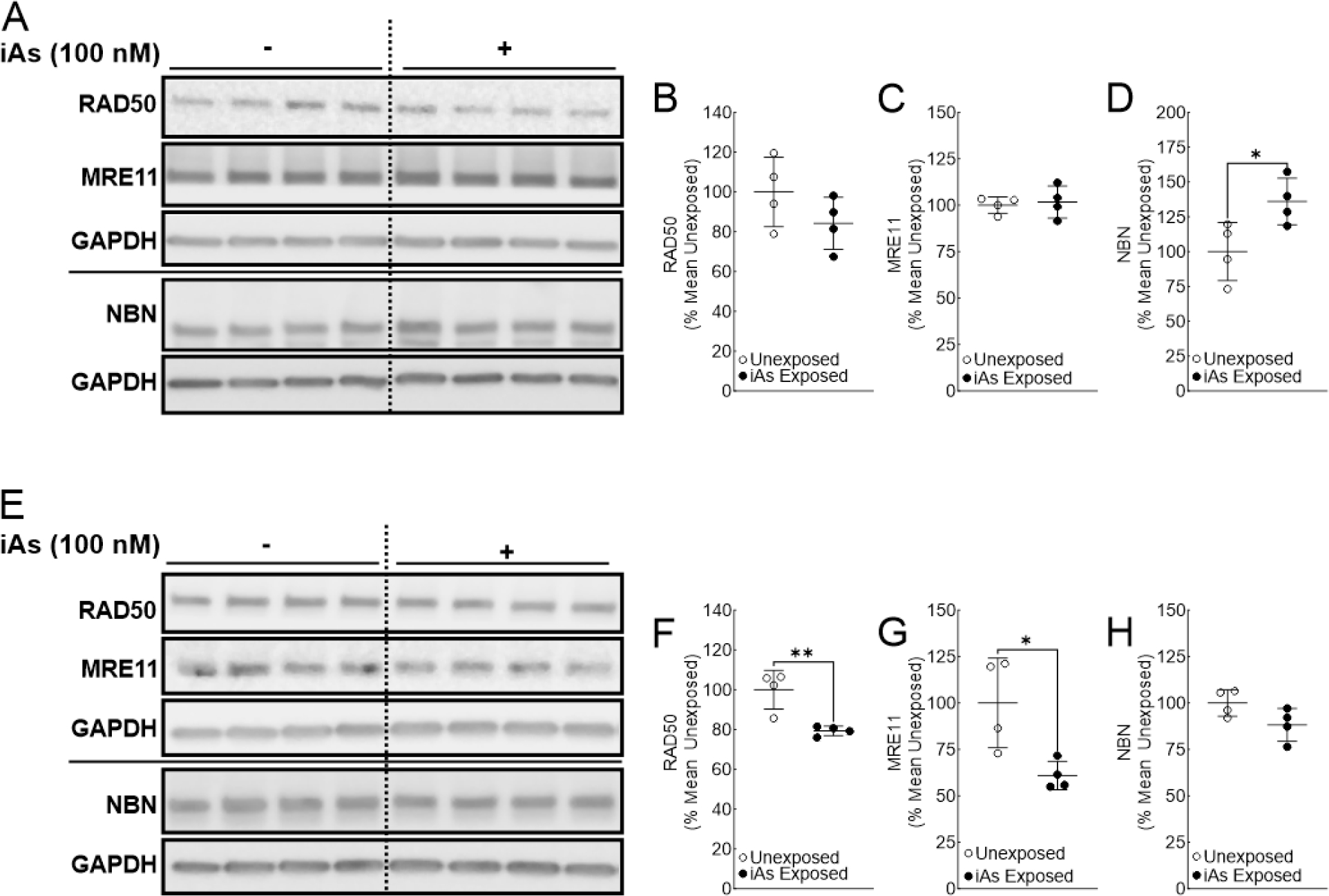
Rad50 expression is reduced in human keratinocytes chronically exposed to iAs. A. Immunoblot for RAD50, MRE11, and NBN in quadruplicate HaCaT cell cultures exposed to 0 or 100 nM sodium arsenite for 7 weeks. B. Densitometric analysis of RAD50 expression (RAD50/GAPDH) in HaCaT cells. C. Densitometric analysis of MRE11 expression (MRE11/GAPDH) in HaCaT cells. D. Densitometric analysis of NBN expression (NBN/GAPDH) in HaCaT cells. E. Immunoblot for RAD50, MRE11, and NBN in quadruplicate Ker-CT cell cultures exposed to 0 or 100 nM sodium arsenite for 8 weeks. F. Densitometric analysis of RAD50 expression (RAD50/GAPDH) in Ker-CT cells G. Densitometric analysis of MRE11 expression (MRE11/GAPDH) in Ker-CT cells. H. Densitometric analysis of NBN expression (NBN/GAPDH) in Ker-CT cells. Statistical analysis was completed by unpaired two-tailed t-test; *p ≤ 0.05, **p ≤ 0.01
Chronic iAs exposure leads to the accumulation of DSBs in human keratinocytes
Given that human keratinocytes chronically exposed to iAs have reduced ATM activation, we hypothesized that DSBs may accumulate in iAs-exposed keratinocytes. Neutral COMET assays were used to evaluate this hypothesis and the results indicated that chronic iAs exposure led to higher accumulation of DSBs in HaCaT cells (Figure 6A). In Ker-CT cells, there was no apparent difference in DNA damage between unexposed and iAs-exposed cells. However, we noticed that the DNA damage data for each group was in bimodal distribution (Figure 6B). Almost all the cells in each group either had olive tail moment value ≤6 arbitrary units or ≥17 arbitrary units. Thus, we stratified the data for Ker-CT cell lines into low DNA damage (≤6 arbitrary units) or high DNA damage (≥17 arbitrary units) groups. Statistical analysis (unpaired, two-tailed Mann-Whitney test) demonstrated that the iAs-exposed group had significantly higher damage in the high DNA damage (≥17 arbitrary units) group (Figure 6C) but there was no difference between unexposed and iAs-exposed groups in the low DNA damage category (stratified data not shown). These data suggest there may be subpopulations of Ker-CT cells responding differently to iAs-induced DSB accumulation. In conclusion, reduced ATM activation is associated with elevated accumulation of DSBs in human keratinocytes.
Figure 6.
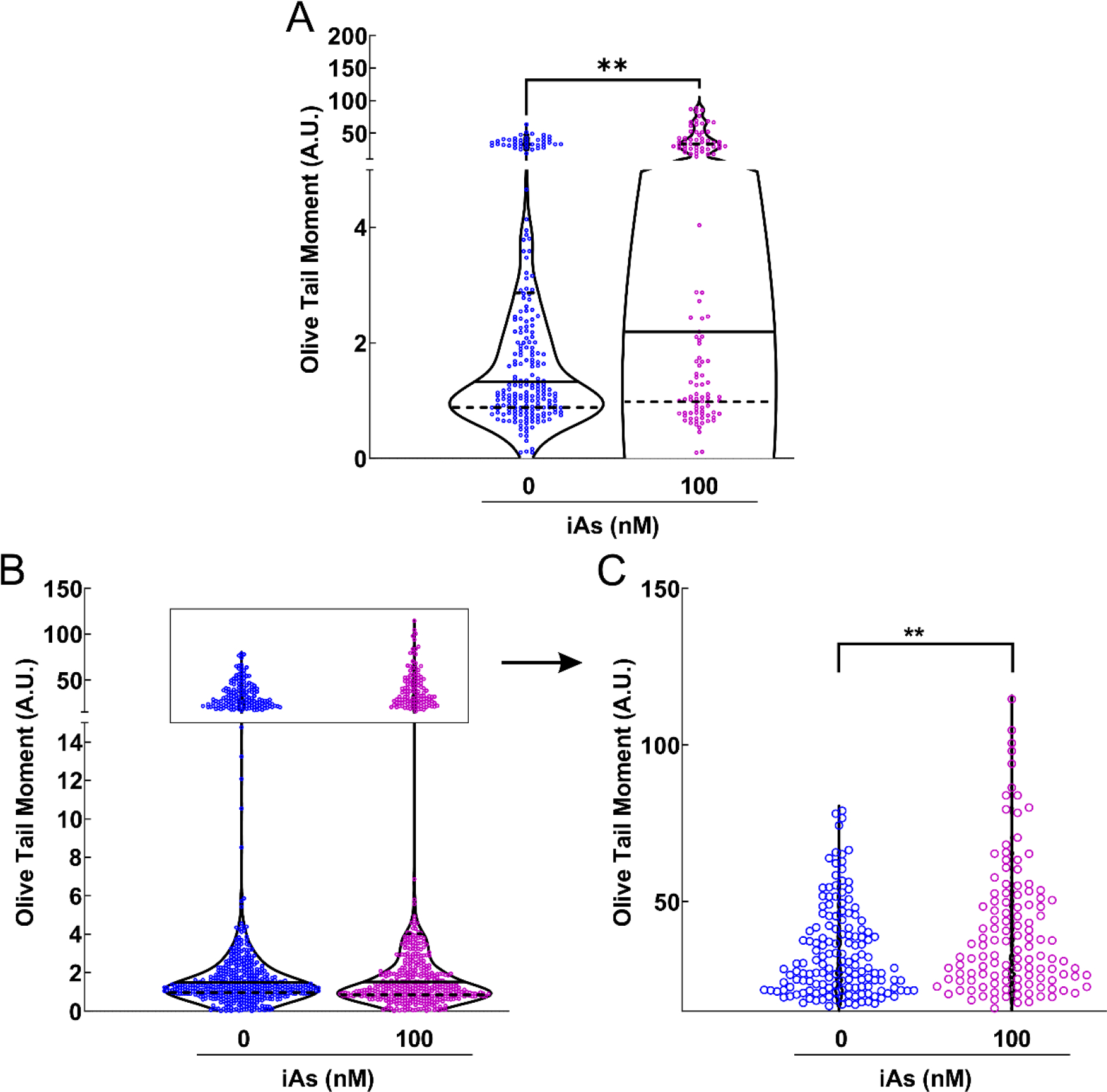
DNA double strand breaks accumulate in human keratinocytes chronically exposed to iAs. Neutral COMET assays were performed for human keratinocytes exposed to 0 or 100 nM sodium arsenite for 7 weeks. Significant DSB accumulation is measured by Olive Tail Moment (Amount of Tail DNA – Head DNA × Percent Tail DNA). A. HaCaT cells (unexposed n = 222, or iAs exposed n = 119). B. Ker-CT cells (unexposed n = 640, or iAs exposed n = 540). C. Expanded view of upper section of panel B. Statistical analysis was completed by Mann Whitney test **p ≤ 0.01
DISCUSSION
Genome integrity is consistently challenged by endogenous and exogenous agents that can induce DNA damage (Trenner and Sartori, 2019). DSBs are considered the most dangerous of DNA lesions because if left unrepaired, discontinuous chromosomes (i.e. micronuclei) can result in cell death due to misegregation of chromosomes during cell division (Kwon et al., 2020). Alternatively, suboptimal detection and repair of DSBs can induce CIN, facilitating carcinogenesis (Bakhoum and Cantley, 2018).
Cells normally repair DSBs by one of two competing pathways: classical non-homologous end joining (c-NHEJ) and homologous recombination (HR). Which pathway will be used for repair is determined by several factors including genetic background, DSB complexity (Schipler and Iliakis, 2013), chromatin state (Marini et al., 2019), and cell cycle phase (Trenner and Sartori, 2019). Unlike c-NHEJ which can operate throughout the cell cycle, HR relies on the presence of an undamaged sister chromatid to repair DSBs and is restricted to S/G2. Recognition of DSBs by the MRN complex in conjunction with CtIP (RBBP8, RB Binding Protein 8) is the first critical step for DSB repair by HR (Anand et al., 2016). The MRE11-RAD50 (MR) complex is evolutionarily conserved and is required for initial recognition and tethering to DNA lesions, nucleolytic processing, and later recruitment of ATM for DDR signaling (Rojowska et al., 2014). Morales and colleagues identified skewing of pathway choice towards NHEJ versus HR following arsenic trioxide exposure (Morales et al., 2016). Our studies add support to Morales’ findings because we find that chronic iAs exposure reduces ATM activation (pSer1981) and downstream ATM signaling (CHEK2 pThr68), which may impact downstream cell cycle checkpoint activation required for cells to repair DSBs prior to cell division (Figure 7). Arsenite is well-known to disrupt mitosis and suppression of p53 or CDKN1A (downstream of CHEK2) causes a shift towards centromere negative micronuclei in daughter cells indicating clastogenesis (Salazar et al., 2010; States, 2015). We also identified a significant reduction in RAD50 protein expression suggesting compromised MRN activity and MRN-dependent DSB repair (i.e. HR) in cells exposed chronically to iAs (Figure 7). Interestingly, a separate study conducted by the Panda group identified that RAD50 mRNA expression is significantly reduced in human urinary bladder carcinoma tissue containing high iAs levels (Basu et al., 2020). Furthermore, they also found that 50% of bladder carcinomas (n = 21) with high iAs levels contain deleted RAD50 loci. Taken together, results from Basu et al. and our study suggest that RAD50 expression is significantly reduced in cells chronically exposed to iAs.
Figure 7.
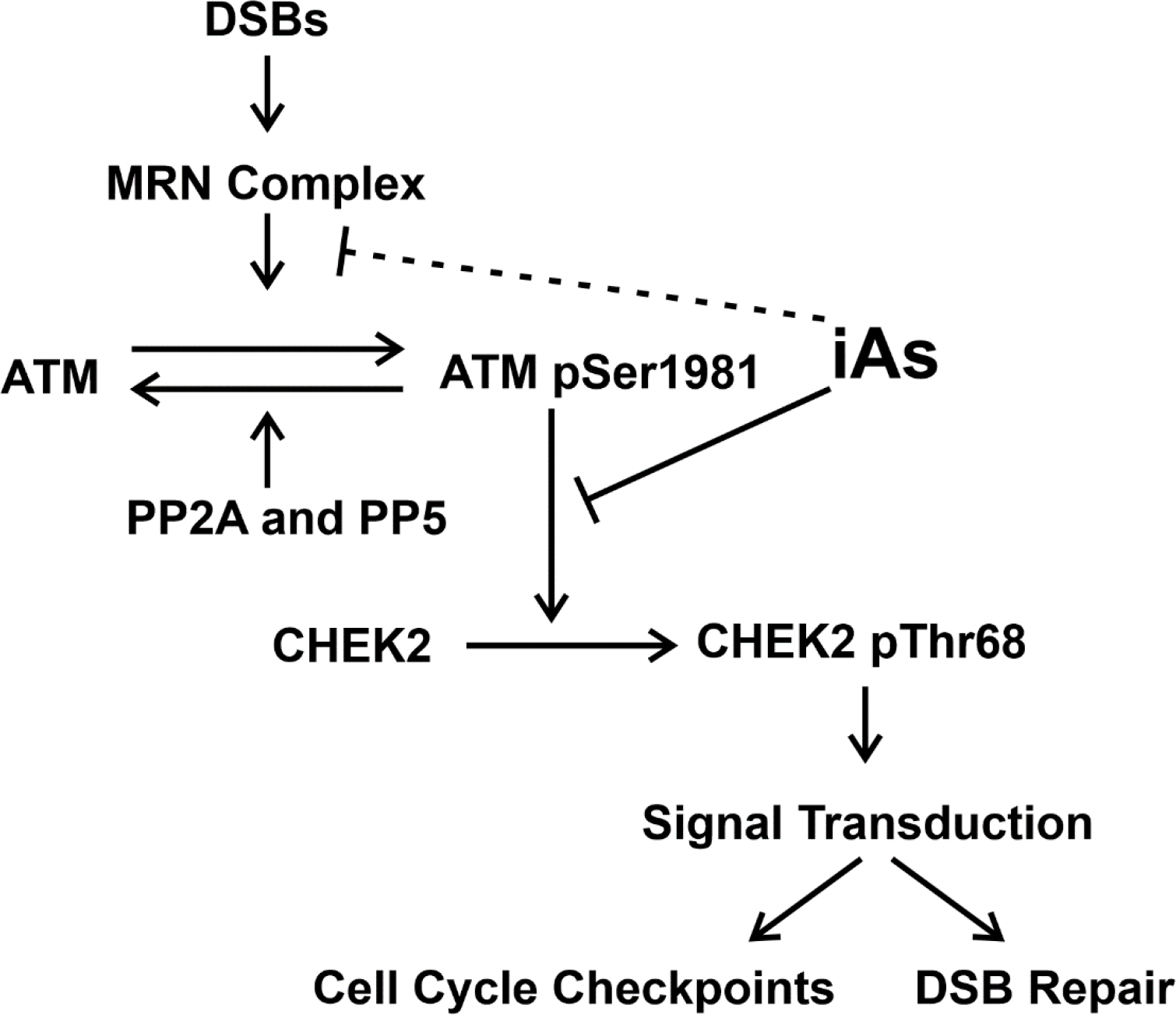
Chronic iAs exposure inhibits ATM activation (ATM-pSer1981) and downstream ATM signaling (measured by CHEK2-pThr68). Reduction of CHEK2 activation (CHEK-pThr68) may lead to reduced downstream signaling which mediates cell cycle checkpoints required for DSB repair prior to cell division. Chronic iAs exposure may reduce ATM activation by inhibition of MRN activity (dotted inhibitory line) which is required for initial ATM activation. Protein Phosphatase 2A (PP2A) and 5 (PP5) are required for ATM dephosphorylation.
Chronic iAs exposure is associated with an increased risk for non-melanoma cancers including basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) (Yager et al., 2016). Skin lesions are characteristic of exposure to iAs in drinking water (Rossman et al., 2004) and iAs-induced skin cancers usually occur on the sun-protected areas (Yu et al., 2006). Additionally, iAs can act as a cocarcinogen exacerbating development of non-melanoma skin cancer (SCC) in mice exposed to both iAs and ultraviolet radiation (UV) (Rossman et al., 2001). Positive associations between iAs exposed human populations, UV, and non-melanoma skin cancer are reported (Rossman et al., 2004). In skin, UV lesions are normally repaired by the nucleotide excision repair pathway (UV-induced pyrimidine dimers) (Ray et al., 2016) and the presence of UV-induced DNA damage activates ATR signaling (Musich et al., 2017). Here we demonstrate inhibition of ATM signaling whereas ATR activation is unaffected by iAs chronic exposure. These results are consistent with the etiology of iAs-induced skin lesions being independent of UV-photodimer induced mutations: since iAs does not suppress ATR protein expression or it’s activation, ATR is available for DDR signaling in UV-exposed skin.
As noted previously, most in vitro arsenic toxicology studies have used high dose and acute exposure conditions (Yih et al., 1997; Radha and Natarajan, 1998; Mahata et al., 2004; Azizian-Farsani et al., 2014; Liu et al., 2016), which do not faithfully replicate chronic iAs exposure. This point is important as iAs dose response for a variety of outcomes is not linear but J-shaped (Snow et al., 2005; Bodwell et al., 2006; Ahn et al., 2020). As such, inferences drawn from a high dose-short duration iAs exposure study may not translate to chronically exposed populations. In addition, few epidemiological studies have investigated the mechanism of iAs-induced DNA damage in chronically exposed populations (Basu et al., 2005; Jimenez-Villarreal et al., 2017). Together, these observations highlight the emergent need for investigations into the molecular mechanisms by which toxicologically relevant chronic iAs exposure (~100 nM) can lead to heightened accumulation of DNA damage, induction of CIN, and eventual carcinogenesis. Furthermore, because cell models are characterized by multiple structural and behavioral features dependent upon their genetic makeup, the use of keratinocyte cell models are important for specifically identifying molecular mechanisms governing iAs-induced carcinogenesis in skin.
The study design of the present work addresses the abovementioned issues. Historically, HaCaT cells either acutely or chronically exposed to iAs have been used to identify molecular mechanisms that may contribute to iAs-induced carcinogenesis (Yu et al., 2006; Sun et al., 2009; Banerjee et al., 2021). Here we present a new model for iAs-chronic exposure utilizing Ker-CT cells. Importantly, Ker-CT cells exhibit several features of normal keratinocytes because they are derived from human foreskin keratinocytes and immortalized by expression of human telomerase and mouse Cdk4 (Ramirez et al., 2003; Vaughan et al., 2004). As such, Ker-CT cells are well suited for studies involving CIN because they maintain near diploid chromosome number easily allowing chromosomal aberration identification (in comparison to HaCaT cells which are severely aneuploid), yet are able to proliferate indefinitely unlike primary keratinocytes and retain many properties of primary keratinocytes (Ramirez et al., 2003). Furthermore, by including this new model, we add robustness to our study by identifying a common phenomenon of reduced ATM activation in two human keratinocyte cell lines. We demonstrated that chronic iAs exposure leads to higher accumulation of DSBs in HaCaT cells, while elevated DSB accumulation was apparent in a subset of iAs exposed Ker-CT cells (Figure 6). This difference between the two cell lines could reflect: (i) genomic instability in HaCaT cells making them more susceptible to DSB accumulation with shorter exposure time, (ii) clonal variability in Ker-CT cells where some cells are preferentially susceptible to iAs-induced DSB accumulation, (iii) unique responses of HaCaT and Ker-CT to ATM suppression-mediated DSB accumulation, leading to differences in residual DNA damage when measured as a snapshot. In the future, it will be imperative to investigate which DNA repair pathways are being invoked by each cell line to cope with suppressed HR mediated DNA repair upon chronic iAs exposure.
DDR phosphatases are highly specialized complexes that exhibit substrate specificity based on their ability to form stable complexes with regulatory subunits. Protein phosphatase 2A (PP2A) and protein phosphatase 5 (PP5) are important for resolving both ATM and ATR pathway activation (Figure 7, ATR not shown) (Goodarzi et al., 2004; Campos and Clemente-Blanco, 2020). Interestingly, we found that PPP2RC and PP5 protein levels were reduced in spite of increased expression of PPP2R2C and PP5 mRNAs in HaCaT cells after chronic iAs exposure (7 weeks). Importantly, these results cannot rule out the possibility of heightened PP2A and PP5 phosphatase activity, and suggest post-transcriptional (i.e. miRNAs) or post-translational (i.e. protein degradation) regulation of protein phosphatases as a result of chronic iAs exposure. A multitude of miRNAs are dysregulated as a result of iAs exposure in arsenic-induced skin lesions (Al-Eryani et al., 2018) and HaCaT cells (Banerjee et al., 2021). Interestingly, we have found that four miRNAs induced after 7 weeks of chronic iAs exposure in HaCaT cells (hsa-miR-589, hsa-miR-4786, hsa-miR-5001, hsa-miR-3661), are predicted to target PPP2R2C using STarMirDB (Rennie et al., 2016). Thus, the reduced protein expression in spite of increased mRNA expression may be due to suppression of translation by the miRNAs.
PP2A is a heterotrimeric complex that is composed of a catalytic subunit, scaffold subunit, and regulatory subunit (Amin et al., 2021). PP2A regulatory subunits determine substrate specificity and there are four different types of B subunits that display several isoforms. PPP2R2C, or the B/B55γ subunit, is important for inhibiting Src tyrosine kinase protooncogenic activity (Eichhorn et al., 2007; Belli et al., 2020). Previous work demonstrated that iAs induces c-Src activation (Simeonova and Luster, 2002). Here we present that PPP2R2C is significantly reduced following iAs exposure in both keratinocyte lines tested. Therefore, reduced PPP2R2C activity due to reduced PPP2R2C expression may represent another mechanism for c-Src activation specifically in keratinocytes. Taken together, identifying dysregulation of protein phosphatase regulatory and scaffold subunits following iAs exposure may help in identifying apical mechanisms for arsenic-induced carcinogenesis.
The present work identifies a new molecular mechanism that can explain iAs-induced CIN employing two different human keratinocyte models representing the primary iAs target organ, skin. Our data provide strong evidence that suppression of ATM activation is an early iAs-induced cellular change in the process of iAs-induced clastogenesis. Reduced iAs-mediated ATM activation is further associated with increased accumulation of DSBs, an essential step in clastogenesis(Lobrich and Jeggo, 2007; Li et al., 2008). This work opens up avenues for future research to investigate how iAs exposure modulates cellular choice between NHEJ versus HR pathways in response to accumulating DSBs. Additionally, it would also be important to investigate if the effects of chronic iAs exposure on posttranslational modifications of members of the MRN complex and their downstream targets (Lee and Paull, 2007) contribute to clastogenesis. Lastly, larger population-based studies are needed to identify whether DDR activation is impaired in iAs-induced skin lesions.
Supplementary Material
Supplemental Figure 1. Total ATM expression is significantly reduced in iAs-exposed keratinocytes. A. Densitometric analysis of ATM expression (t-ATM/Vinculin) in HaCaT cells. B. Densitometric analysis of CHEK2 expression (t-CHEK2/GAPDH) in HaCaT cells. C. Densitometric analysis of ATR expression (t-ATR/Vinculin) in HaCaT cells. D. Densitometric analysis of ATM expression (t-ATM/Vinculin) in Ker-CT cells. E. Densitometric analysis of CHEK2 expression (t-CHEK2/GAPDH) in Ker-CT cells. F. Densitometric analysis of ATR expression (t-ATR/GAPDH) in Ker-CT cells. Statistical analysis was completed by unpaired two-tailed t-test; *p ≤ 0.05
Supplemental Figure 2. ATM pathway activation by acute DSB induction in human keratinocytes. A. Immunoblot for ATM pathway signaling (ATM and CHEK2) in HaCaT cells treated with 20 nM NCS. B. Densitometric analysis of ATM activation (ATM-pSer1981/t-ATM) in HaCaT cells. C. Densitometric analysis of CHEK2 activation (CHEK-2pThr68/t-CHEK2) in HaCaT cells. D. Immunoblot for ATM pathway signaling (ATM and CHEK2) in Ker-CT cells treated with 20 nM NCS. E. Densitometric analysis of ATM activation (ATM-pSer1981/t-ATM) in Ker-CT cells. Statistical analysis was completed by unpaired two-tailed t-test; **p ≤ 0.01, ***p ≤ 0.001
Supplemental Figure 3. Total ATM and CHEK2 protein levels are unchanged in cells with acute 20 nM NCS treatment. A. Densitometric analysis of ATM expression (t-ATM/Vinculin) in HaCaT cells. B. Densitometric analysis of CHEK2 expression (t-CHEK2/β-Actin) in HaCaT cells. C. Densitometric analysis of ATM expression (t-ATM/Vinculin) in Ker-CT cells. D. Densitometric analysis of CHEK2 expression (t-CHEK2/Vinculin) in Ker-CT cells. Statistical analysis was done by unpaired two-tailed t-test and no significant differences were found.
Supplemental Figure 4. DSBs significantly accumulate in human keratinocytes treated acutely with hydrogen peroxide. Neutral COMET assays were performed for unexposed cells either untreated or treated with 100 μM hydrogen peroxide (H2O2). A. HaCaT cells (unexposed n = 222, H2O2 n = 135) or B. Ker-CT cells (unexposed = 640, H2O2 n = 197) treated with H2O2 significantly accumulate DSBs compared to untreated controls. Statistical analysis was completed by Mann Whitney test ****p ≤ 0.0001
FUNDING
This work was supported by the National Institutes of Health R01ES027778, P30ES030283, T32ES011564, and R25CA134283. The views expressed are those of the authors and not the National Institutes of Health.
ABBREVIATIONS
- ATM
ATM Serine/Threonine kinase
- ATR
ATR Serine/Threonine kinase
- BCC
basal cell carcinoma
- BER
base excision repair
- CHEK2
Checkpoint Kinase 2
- CIN
chromosomal instability
- c-NHEJ
classical non-homologous end joining
- DDR
DNA damage response
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- DSBs
DNA double-strand breaks
- HR
homologous recombination
- ICL
interstrand crosslink repair
- iAs
inorganic arsenic
- MR
MRE11-RAD50
- MRE11
MRE11 Homolog, Double Strand Break Repair Nuclease
- MRN
MRE11-RAD50-NBN complex
- NaAsO2
sodium arsenite
- NBN
nibrin
- NCS
neocarzinostatin
- NER
nucleotide excision repair
- PARP
poly(ADP-ribose) polymerase
- PI3K
phosphoinositide 3-kinase
- ROS
reactive oxygen species
- PPP2R2C
Protein Phosphatase 2 Regulatory Subunit Beta Gamma
- PP5
Protein Phosphatase 5
- RAD50
RAD50 Double Strand Break Repair Protein
- SSBs
single-stranded breaks
- SCC
squamous cell carcinoma
- UV
ultraviolet radiation
Footnotes
CRediT authorship contribution statement
Alexandra N. Nail: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data Curation, Writing - Original Draft, Writing – Review and Editing, Visualization; Lakyn McCaffrey: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data Curation, Writing – Review and Editing; Mayukh Banerjee: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data Curation, Writing – Review and Editing; Ana P. Ferragut Cardoso: Methodology, Writing – Review and Editing; J. Christopher States: Conceptualization, Methodology, Resources, Data Curation, Writing – Review and Editing, Supervision, Project administration, Funding acquisition.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
J. Christopher States reports financial support was provided by National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Ahn J, Boroje IJ, Ferdosi H, Kramer ZJ, Lamm SH, 2020. Prostate Cancer Incidence in U.S. Counties and Low Levels of Arsenic in Drinking Water. Int J Environ Res Public Health 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Eryani L, Jenkins SF, States VA, Pan J, Malone JC, Rai SN, Galandiuk S, Giri AK, States JC, 2018. miRNA expression profiles of premalignant and malignant arsenic-induced skin lesions. PLoS One 13, e0202579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin P, Awal S, Vigneron S, Roque S, Mechali F, Labbe JC, Lorca T, Castro A, 2021. PP2A–B55: substrates and regulators in the control of cellular functions. Oncogene. [DOI] [PubMed] [Google Scholar]
- Anand R, Ranjha L, Cannavo E, Cejka P, 2016. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol Cell 64, 940–950. [DOI] [PubMed] [Google Scholar]
- Aparicio T, Baer R, Gautier J, 2014. DNA double-strand break repair pathway choice and cancer. DNA Repair (Amst) 19, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizian-Farsani F, Rafiei G, Saadat M, 2014. Impact of sodium arsenite on chromosomal aberrations with respect to polymorphisms of detoxification and DNA repair genes. Int J Toxicol 33, 518–522. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Cantley LC, 2018. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 174, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB, 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Banerjee M, Ferragut Cardoso A, Al-Eryani L, Pan J, Kalbfleisch TS, Srivastava S, Rai SN, States JC, 2021. Dynamic alteration in miRNA and mRNA expression profiles at different stages of chronic arsenic exposure-induced carcinogenesis in a human cell culture model of skin cancer. Arch Toxicol 95, 2351–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee M, Ferragut Cardoso AP, Lykoudi A, Wilkey DW, Pan J, Watson WH, Garbett NC, Rai SN, Merchant ML, States JC, 2020. Arsenite Exposure Displaces Zinc from ZRANB2 Leading to Altered Splicing. Chem Res Toxicol 33, 1403–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Som A, Ghoshal S, Mondal L, Chaubey RC, Bhilwade HN, Rahman MM, Giri AK, 2005. Assessment of DNA damage in peripheral blood lymphocytes of individuals susceptible to arsenic induced toxicity in West Bengal, India. Toxicol Lett 159, 100–112. [DOI] [PubMed] [Google Scholar]
- Basu M, Ghosh S, Roychowdhury A, Samadder S, Das P, Addya S, Roy A, Pal DK, Roychoudhury S, Ghosh A, Panda CK, 2020. Integrative genomics and pathway analysis identified prevalent FA-BRCA pathway alterations in arsenic-associated urinary bladder carcinoma: Chronic arsenic accumulation in cancer tissues hampers the FA-BRCA pathway. Genomics 112, 5055–5065. [DOI] [PubMed] [Google Scholar]
- Belli S, Esposito D, Servetto A, Pesapane A, Formisano L, Bianco R, 2020. c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve? Cancers (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP, 2017. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Bodwell JE, Gosse JA, Nomikos AP, Hamilton JW, 2006. Arsenic disruption of steroid receptor gene activation: Complex dose-response effects are shared by several steroid receptors. Chem Res Toxicol 19, 1619–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE, 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos A, Clemente-Blanco A, 2020. Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty T, De M, 2009. Clastogenic effects of inorganic arsenic salts on human chromosomes in vitro. Drug Chem Toxicol 32, 169–173. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ, 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Gautier J, 2006. Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat Struct Mol Biol 13, 451–457. [DOI] [PubMed] [Google Scholar]
- Eichhorn PJ, Creyghton MP, Wilhelmsen K, van Dam H, Bernards R, 2007. A RNA interference screen identifies the protein phosphatase 2A subunit PR55gamma as a stress-sensitive inhibitor of c-SRC. PLoS Genet 3, e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan YL, Chen L, Wang J, Yao Q, Wan JQ, 2013. Over expression of PPP2R2C inhibits human glioma cells growth through the suppression of mTOR pathway. FEBS Lett 587, 3892–3897. [DOI] [PubMed] [Google Scholar]
- Gebel TW, 2001. Genotoxicity of arsenical compounds. Int J Hyg Environ Health 203, 249–262. [DOI] [PubMed] [Google Scholar]
- Ghosh P, Banerjee M, De Chaudhuri S, Das JK, Sarma N, Basu A, Giri AK, 2007. Increased chromosome aberration frequencies in the Bowen’s patients compared to non-cancerous skin lesions individuals exposed to arsenic. Mutat Res 632, 104–110. [DOI] [PubMed] [Google Scholar]
- Gonsebatt ME, Vega L, Montero R, Garcia-Vargas G, Del Razo LM, Albores A, Cebrian ME, Ostrosky-Wegman P, 1994. Lymphocyte replicating ability in individuals exposed to arsenic via drinking water. Mutat Res 313, 293–299. [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK, 2004. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J 23, 4451–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV, 2014. OpenComet: an automated tool for comet assay image analysis. Redox Biol 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA, 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hei TK, Liu SX, Waldren C, 1998. Mutagenicity of arsenic in mammalian cells: role of reactive oxygen species. Proc Natl Acad Sci U S A 95, 8103–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH, 2001. Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374. [DOI] [PubMed] [Google Scholar]
- Jimenez-Villarreal J, Rivas-Armendariz DI, Pineda-Belmontes CP, Betancourt-Martinez ND, Macias-Corral MA, Guerra-Alanis AJ, Nino-Castaneda MS, Moran-Martinez J, 2017. Detection of damage on single- or double-stranded DNA in a population exposed to arsenic in drinking water. Genet Mol Res 16. [DOI] [PubMed] [Google Scholar]
- Jin MH, Oh DY, 2019. ATM in DNA repair in cancer. Pharmacol Ther 203, 107391. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA, 2009. The basics of epithelial-mesenchymal transition. J Clin Invest 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Lee JH, Hoan NN, Sohn HM, Chang IY, You HJ, 2009. Protein phosphatase 5 regulates the function of 53BP1 after neocarzinostatin-induced DNA damage. J Biol Chem 284, 9845–9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer SR, Lowndes NF, 2022. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front Genet 13, 793884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaravel TS, Vilhar B, Faux SP, Jha AN, 2009. Comet Assay measurements: a perspective. Cell Biol Toxicol 25, 53–64. [DOI] [PubMed] [Google Scholar]
- Kwon M, Leibowitz ML, Lee JH, 2020. Small but mighty: the causes and consequences of micronucleus rupture. Exp Mol Med 52, 1777–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Chowdhury D, 2011. What goes on must come off: phosphatases gate-crash the DNA damage response. Trends Biochem Sci 36, 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT, 2007. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 26, 7741–7748. [DOI] [PubMed] [Google Scholar]
- Leong W, Xu W, Wang B, Gao S, Zhai X, Wang C, Gilson E, Ye J, Lu Y, 2020. PP2A subunit PPP2R2C is downregulated in the brains of Alzheimer’s transgenic mice. Aging (Albany NY) 12, 6880–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Mitchell JR, Hasty P, 2008. DNA double-strand breaks: a potential causative factor for mammalian aging? Mech Ageing Dev 129, 416–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JH, Rossman TG, 1989. Mechanism of comutagenesis of sodium arsenite with n-methyl-n-nitrosourea. Biol Trace Elem Res 21, 373–381. [DOI] [PubMed] [Google Scholar]
- Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, Zou L, 2011. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell 43, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sun B, Wang X, Nie J, Chen Z, An Y, Tong J, 2016. Synergistic effect of radon and sodium arsenite on DNA damage in HBE cells. Environ Toxicol Pharmacol 41, 127–131. [DOI] [PubMed] [Google Scholar]
- Lobrich M, Jeggo PA, 2007. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 7, 861–869. [DOI] [PubMed] [Google Scholar]
- Mahata J, Ghosh P, Sarkar JN, Ray K, Natarajan AT, Giri AK, 2004. Effect of sodium arsenite on peripheral lymphocytes in vitro: individual susceptibility among a population exposed to arsenic through the drinking water. Mutagenesis 19, 223–229. [DOI] [PubMed] [Google Scholar]
- Marini F, Rawal CC, Liberi G, Pellicioli A, 2019. Regulation of DNA Double Strand Breaks Processing: Focus on Barriers. Front Mol Biosci 6, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez VD, Vucic EA, Becker-Santos DD, Gil L, Lam WL, 2011. Arsenic exposure and the induction of human cancers. J Toxicol 2011, 431287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal D, Banerjee M, Kundu M, Banerjee N, Bhattacharya U, Giri AK, Ganguli B, Sen Roy S, Polya DA, 2010. Comparison of drinking water, raw rice and cooking of rice as arsenic exposure routes in three contrasting areas of West Bengal, India. Environ Geochem Health 32, 463–477. [DOI] [PubMed] [Google Scholar]
- Morales ME, Derbes RS, Ade CM, Ortego JC, Stark J, Deininger PL, Roy-Engel AM, 2016. Heavy Metal Exposure Influences Double Strand Break DNA Repair Outcomes. PLoS One 11, e0151367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musich PR, Li Z, Zou Y, 2017. Xeroderma Pigmentosa Group A (XPA), Nucleotide Excision Repair and Regulation by ATR in Response to Ultraviolet Irradiation. Adv Exp Med Biol 996, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council Subcommittee on Arsenic in Drinking, W., 1999. Arsenic in Drinking Water. National Academies Press (US) [Google Scholar]
- Copyright 1999 by the National Academy of Sciences. All rights reserved., Washington (DC), pp.
- Pi J, Diwan BA, Sun Y, Liu J, Qu W, He Y, Styblo M, Waalkes MP, 2008. Arsenic-induced malignant transformation of human keratinocytes: involvement of Nrf2. Free Radic Biol Med 45, 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Kumagai Y, Sun G, Yamauchi H, Yoshida T, Iso H, Endo A, Yu L, Yuki K, Miyauchi T, Shimojo N, 2000. Decreased serum concentrations of nitric oxide metabolites among Chinese in an endemic area of chronic arsenic poisoning in inner Mongolia. Free Radic Biol Med 28, 1137–1142. [DOI] [PubMed] [Google Scholar]
- Podgorski J, Berg M, 2020. Global threat of arsenic in groundwater. Science 368, 845–850. [DOI] [PubMed] [Google Scholar]
- Radha S, Natarajan AT, 1998. Sodium arsenite-induced chromosomal aberrations in the Xq arm of Chinese hamster cell lines. Mutagenesis 13, 229–234. [DOI] [PubMed] [Google Scholar]
- Ramirez RD, Herbert BS, Vaughan MB, Zou Y, Gandia K, Morales CP, Wright WE, Shay JW, 2003. Bypass of telomere-dependent replicative senescence (M1) upon overexpression of Cdk4 in normal human epithelial cells. Oncogene 22, 433–444. [DOI] [PubMed] [Google Scholar]
- Ray A, Blevins C, Wani G, Wani AA, 2016. ATR- and ATM-Mediated DNA Damage Response Is Dependent on Excision Repair Assembly during G1 but Not in S Phase of Cell Cycle. PLoS One 11, e0159344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie W, Kanoria S, Liu C, Mallick B, Long D, Wolenc A, Carmack CS, Lu J, Ding Y, 2016. STarMirDB: A database of microRNA binding sites. RNA Biol 13, 554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojowska A, Lammens K, Seifert FU, Direnberger C, Feldmann H, Hopfner KP, 2014. Structure of the Rad50 DNA double-strand break repair protein in complex with DNA. EMBO J 33, 2847–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman TG, Uddin AN, Burns FJ, 2004. Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol Appl Pharmacol 198, 394–404. [DOI] [PubMed] [Google Scholar]
- Rossman TG, Uddin AN, Burns FJ, Bosland MC, 2001. Arsenite is a cocarcinogen with solar ultraviolet radiation for mouse skin: an animal model for arsenic carcinogenesis. Toxicol Appl Pharmacol 176, 64–71. [DOI] [PubMed] [Google Scholar]
- Roy JS, Chatterjee D, Das N, Giri AK, 2018. Substantial Evidences Indicate That Inorganic Arsenic Is a Genotoxic Carcinogen: a Review. Toxicol Res 34, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar AM, Miller HL, McNeely SC, Sordo M, Ostrosky-Wegman P, States JC, 2010. Suppression of p53 and p21CIP1/WAF1 reduces arsenite-induced aneuploidy. Chem Res Toxicol 23, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipler A, Iliakis G, 2013. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res 41, 7589–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW, 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Nakanishi M, 2013. Response to DNA damage: why do we need to focus on protein phosphatases? Front Oncol 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonova PP, Luster MI, 2002. Arsenic carcinogenicity: relevance of c-Src activation. Mol Cell Biochem 234–235, 277–282. [PubMed] [Google Scholar]
- Snow ET, Sykora P, Durham TR, Klein CB, 2005. Arsenic, mode of action at biologically plausible low doses: what are the implications for low dose cancer risk? Toxicol Appl Pharmacol 207, 557–564. [DOI] [PubMed] [Google Scholar]
- States JC, 2015. Disruption of Mitotic Progression by Arsenic. Biol Trace Elem Res 166, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Pi J, Wang X, Tokar EJ, Liu J, Waalkes MP, 2009. Aberrant cytokeratin expression during arsenic-induced acquired malignant phenotype in human HaCaT keratinocytes consistent with epidermal carcinogenesis. Toxicology 262, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam LM, Price NE, Wang Y, 2020. Molecular Mechanisms of Arsenic-Induced Disruption of DNA Repair. Chem Res Toxicol 33, 709–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenner A, Sartori AA, 2019. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front Oncol 9, 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y, 2003. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22, 5612–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan MB, Ramirez RD, Brown SA, Yang JC, Wright WE, Shay JW, 2004. A reproducible laser-wounded skin equivalent model to study the effects of aging in vitro. Rejuvenation Res 7, 99–110. [DOI] [PubMed] [Google Scholar]
- Vitor AC, Huertas P, Legube G, de Almeida SF, 2020. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front Mol Biosci 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CT, Huang CW, Chou SS, Lin DT, Liau SR, Wang RT, 1993. Studies on the concentration of arsenic, selenium, copper, zinc and iron in the blood of blackfoot disease patients in different clinical stages. Eur J Clin Chem Clin Biochem 31, 759–763. [DOI] [PubMed] [Google Scholar]
- Wu MM, Chiou HY, Wang TW, Hsueh YM, Wang IH, Chen CJ, Lee TC, 2001. Association of blood arsenic levels with increased reactive oxidants and decreased antioxidant capacity in a human population of northeastern Taiwan. Environ Health Perspect 109, 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager JW, Erdei E, Myers O, Siegel M, Berwick M, 2016. Arsenic and ultraviolet radiation exposure: melanoma in a New Mexico non-Hispanic white population. Environ Geochem Health 38, 897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yih LH, Ho IC, Lee TC, 1997. Sodium arsenite disturbs mitosis and induces chromosome loss in human fibroblasts. Cancer Res 57, 5051–5059. [PubMed] [Google Scholar]
- Yu HS, Liao WT, Chai CY, 2006. Arsenic carcinogenesis in the skin. J Biomed Sci 13, 657–666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Total ATM expression is significantly reduced in iAs-exposed keratinocytes. A. Densitometric analysis of ATM expression (t-ATM/Vinculin) in HaCaT cells. B. Densitometric analysis of CHEK2 expression (t-CHEK2/GAPDH) in HaCaT cells. C. Densitometric analysis of ATR expression (t-ATR/Vinculin) in HaCaT cells. D. Densitometric analysis of ATM expression (t-ATM/Vinculin) in Ker-CT cells. E. Densitometric analysis of CHEK2 expression (t-CHEK2/GAPDH) in Ker-CT cells. F. Densitometric analysis of ATR expression (t-ATR/GAPDH) in Ker-CT cells. Statistical analysis was completed by unpaired two-tailed t-test; *p ≤ 0.05
Supplemental Figure 2. ATM pathway activation by acute DSB induction in human keratinocytes. A. Immunoblot for ATM pathway signaling (ATM and CHEK2) in HaCaT cells treated with 20 nM NCS. B. Densitometric analysis of ATM activation (ATM-pSer1981/t-ATM) in HaCaT cells. C. Densitometric analysis of CHEK2 activation (CHEK-2pThr68/t-CHEK2) in HaCaT cells. D. Immunoblot for ATM pathway signaling (ATM and CHEK2) in Ker-CT cells treated with 20 nM NCS. E. Densitometric analysis of ATM activation (ATM-pSer1981/t-ATM) in Ker-CT cells. Statistical analysis was completed by unpaired two-tailed t-test; **p ≤ 0.01, ***p ≤ 0.001
Supplemental Figure 3. Total ATM and CHEK2 protein levels are unchanged in cells with acute 20 nM NCS treatment. A. Densitometric analysis of ATM expression (t-ATM/Vinculin) in HaCaT cells. B. Densitometric analysis of CHEK2 expression (t-CHEK2/β-Actin) in HaCaT cells. C. Densitometric analysis of ATM expression (t-ATM/Vinculin) in Ker-CT cells. D. Densitometric analysis of CHEK2 expression (t-CHEK2/Vinculin) in Ker-CT cells. Statistical analysis was done by unpaired two-tailed t-test and no significant differences were found.
Supplemental Figure 4. DSBs significantly accumulate in human keratinocytes treated acutely with hydrogen peroxide. Neutral COMET assays were performed for unexposed cells either untreated or treated with 100 μM hydrogen peroxide (H2O2). A. HaCaT cells (unexposed n = 222, H2O2 n = 135) or B. Ker-CT cells (unexposed = 640, H2O2 n = 197) treated with H2O2 significantly accumulate DSBs compared to untreated controls. Statistical analysis was completed by Mann Whitney test ****p ≤ 0.0001