

Abstract
This work reports the first isolation and characterization of an alkaline phosphatase (AP) from a hyperthermophilic archaeon. An AP gene from Pyrococcus abyssi, a euryarchaeon isolated from a deep-sea hydrothermal vent, was cloned and the enzyme expressed in Escherichia coli. Analysis of the sequence showed conservation of the active site and structural elements of the E. coli AP. The recombinant AP was purified and characterized. Monomeric and homodimeric active forms were detected, with a monomer molecular mass of 54 kDa. Apparent optimum pH and temperature were estimated at 11.0 and 70°C, respectively. Thus far, P. abyssi AP has been demonstrated to be the most thermostable AP, with half-lives at 100 and 105°C of 18 and 5 h, respectively. Enzyme activity was found to be dependent on divalent cations: metal ion chelators inhibited activity, whereas the addition of exogenous Mg(II), Zn(II), and Co(II) increased activity. The enzyme was inhibited by inorganic phosphate, but not by molybdate and vanadate. Strong inhibitory effects were observed in the presence of thiol-reducing agents, although cysteine residues of the P. abyssi AP were not found to be incorporated within intra- or interchain disulfide bonds. In addition, P. abyssi AP was demonstrated to dephosphorylate linear DNA fragments with dephosphorylation efficiencies of 93.8 and 84.1% with regard to cohesive and blunt ends, respectively.
Alkaline phosphatases (APs) (orthophosphate monoester phosphohydrolases [alkaline optimum]; EC 3.1.3.1.) are classically described as homodimeric nonspecific metalloenzymes which catalyze phosphomonesterase reactions (45). The fact that they are widely found in nature, from bacteria to mammals indicates that APs are included in fundamental biochemical processes (38). Despite the fact that their physiological function is not clear, their induced production under inorganic phosphate starvation in many species (especially procaryotic organisms) indicates that they play a vital role in the phosphate metabolism. In mammals, they are linked with transport mechanisms (30).
Many APs have been characterized since the 1960s. The Escherichia coli AP has been widely studied in terms of biosynthesis (11, 26), structure, and catalytic properties (9). Numerous mammalian AP cDNAs have been cloned, and the corresponding enzymes have been characterized (5, 48, 49). Alignments of the deduced protein sequences have shown a strong conservation of the catalytic site, which involves a serine residue and three metal ions per monomer, two Zn(II) and one Mg(II) (27). However, mammalian APs differ from their E. coli counterpart in terms of Mg(II) secondary ligand (33), membrane anchoring (45), glycosylation (27), etc. APs represent a large research field as they are good models to study metal ion-dependent catalysis and are used in several application fields (molecular biology [3] and immunodetection [43]).
Although Archaea have been studied for a few decades, relatively few archaeal APs have been described. All of them are from halophilic species. APs have been isolated and characterized from three species of the genus Halobacterium (6, 15, 16). In 1990, Goldman et al. described an AP from the halophilic archaeon Haloarcula marismortui (17). So far, no APs from hyperthermophilic archaeons have been isolated and characterized. As studying thermostable enzymes appears interesting for the understanding of life at high temperatures, as well as for industrial processes, new APs exhibiting this property have been investigated. Thermostable APs from the following thermophilic bacteria have been described: Thermotoga neapolitana (12), Thermus caldophilus (36), Thermus thermophilus (37), and Bacillus stearothermophilus (32).
Pyrococcus abyssi is a heterotrophic hyperthermophilic euryarchaeon isolated from a deep-sea hydrothermal vent with an optimal growth temperature of 100°C (14). As its complete genome sequence is known and is publicly available (http://www.genoscope.fr), AP patterns were identified and allowed to isolate an archaeal AP gene. In this report, we describe the first characterization of a thermostable AP isolated from a hyperthermophilic archaeon.
MATERIALS AND METHODS
Organisms and growth conditions
P. abyssi (strain Orsay) was used in this study (14). The strain was grown in 2216S medium (4) at 95°C. E. coli HMS 174(DE3) harboring pLysS, purchased from Novagen, was used as the host strain for the recombinant plasmid of pARHS (10), and E. coli DH5α was used for the pBSK vector in dephosphorylation studies. E. coli strains were grown in 2×YT medium (Bacto Peptone, 16 g · liter−1; yeast extract, 10 g · liter−1; NaCl, 10 g · liter−1) on a rotary shaker at 37°C for various times. Ampicillin was added to 2× YT to give a final concentration of 100 μg · ml−1. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added to give a final concentration of 0.5 mM to induce gene expression (3).
Cloning of the AP gene.
Based on the P. abyssi genome sequence (http://www.genoscope.fr/Pab/, accession number 2366), the phoAP.abyssi gene was amplified by PCR on a DNA Thermal Cycler (Stratagene). Genomic DNA extraction was performed on a P. abyssi culture as previously described (3), and the DNA was used as a template. The two primers, including NdeI and BamHI restriction sites (in boldface type), were as follows: AP1 (5′-GTTTCCATTTGCTCATATGTCTCCAAGCGG-3′, sense) and AP2 (5′-CTCACCTCAAGGATCCTTAAGAAGAAGC-3′, antisense). By introducing a start codon at the beginning of the putative mature protein gene, AP1 was designed so that the putative signal peptide sequence of the P. abyssi AP would not be amplified. AP2 was targeted to the stop codon of the phoAP.abyssi gene, introducing the BamHI restriction site next to the TAA codon. In addition to the template, the 50-μl reaction mixture contained 100 pmol of each primer, 10 nmol of an equimolar deoxynucleoside triphosphate mix (Eurogentec S.A.), Taq DNA polymerase buffer containing 5 mM MgCl2 (Qbiogene), and 1.5 U of Taq DNA polymerase (Qbiogene). The mixture was subjected to eight cycles of amplification (30 s at 94°C, 30 s at 45°C, and 1 min 30 s at 72°C). A PCR product with the expected size was digested with NdeI and BamHI and cloned in a pARHS vector, producing the recombinant plasmid pPabAP, and transformed into E. coli HMS 174(DE3) pLysS using standard procedures (3). Using this strategy, the putative mature enzyme was produced in the cytoplasm of E. coli.
Expression and purification of the recombinant AP.
An overnight culture of E. coli HMS 174(DE3) pLysS, harboring pPabAP, was diluted in a 1:20 (vol/vol) ratio and grown until the optical density at 600 nm reached 0.8 (Milton Roy Spectronic 401, from Bioblock Scientific). The culture was induced by the addition of IPTG, to give a concentration of 0.5 mM, and incubated for an additional period of 4 h. Cells were harvested by centrifugation, washed in 50 mM sodium phosphate buffer (pH 7.5) containing 50 mM NaCl, and then resuspended in 50 mM sodium citrate buffer (pH 5.5). After sonication (375 W; 40% amplitude; Vibracell sonifier), cell debris was removed by centrifugation (10,000 × g for 10 min). The resulting supernatant was heated for 20 min at 80°C, and precipitated proteins were removed by another centrifugation as described above. As E. coli HMS 174(DE3) is not a phoAE.coli deletion strain, controls were used to evaluate the extent of E. coli AP contamination. The supernatant was subsequently dialyzed against sodium phosphate buffer (50 mM, pH 7.5) and loaded on an anion exchange column (ResourceQ; Pharmacia) equilibrated with the same buffer. Bound proteins were eluted by a linear gradient of NaCl (0 to 1 M in sodium phosphate buffer pH 7.5). Active fractions were eluted with 250 mM NaCl. The latter were pooled, loaded on a second anion exchange column (MonoQ; Pharmacia), and eluted by a linear gradient of NaCl (0 to 0.4 M in sodium phosphate buffer, pH 7.5). All chromatographic steps were performed at 4°C.
Protein samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using the method of Laemmli (29). Protein samples for SDS-PAGE were prepared by heating them for 10 min at 100°C in a 1:5 ratio volume of sample buffer (60 mM Tris-HCl buffer, pH 6.8; 2% SDS [wt/vol], 14.4 mM 2-mercaptoethanol [MCE], 25% glycerol [vol/vol], and 1% bromophenol blue [wt/vol]). Proteins were observed by staining them with Coomassie brilliant blue. Molecular mass standard was obtained from Pharmacia.
AP assay and protein determination.
The standard assay for AP activity was carried out at 70°C, for 2.5 min, using 2.5 mM p-nitrophenylphosphate (pNPP) (Sigma Chemical Co.) as the substrate in 50 nM NaOH-glycine (pH 10.0) buffer, containing 0.5 mM CoCl2, except as otherwise indicated. The release of p-nitrophenol in the reaction mixture (1 ml) was continuously measured spectrophotometrically at 410 nm using a Uvikon XL spectrophotometer (from BIO-TEK Instruments), over the linear period. Enzyme and substrate blanks were also included. One unit of enzyme activity is defined as the amount of enzyme required to release 1 μmol of p-nitrophenol from pNPP in 1 min. Protein concentration was determined by the method of Bradford, using bovine serum albumin as the standard (7). For routine enzymatic characterization assays, 252 ng of purified AP was added to the 1-ml reaction mixture. Each value is the mean of at least three assays.
Quaternary structure analysis.
Native gel electrophoresis was performed using 4 to 12% gradient polyacrylamide minigels (Bio-Rad). Samples were added in a 4:5 ratio (vol/vol) to native electrophoresis buffer (60 mM Tris-HCl buffer, pH 6.8; 25% glycerol; 1% bromophenol blue). Zymograms were performed by incubation of minigels in 50 mM NaOH-glycine (pH 10.0) buffer, containing 0.5 mM CoCl2 and 2.5 mM 4-methylumbelliferylphosphate (MUFP) (Sigma Chemical Co.) as the substrate, for 20 min at 70°C. Active bands were observed under UV light, and proteins were visualized after staining with Coomassie brilliant blue. Molecular mass standard was obtained from Pharmacia.
Temperature and pH optima, thermal stability, and kinetic parameters.
The apparent optimum temperature was determined by running the standard assay at temperatures ranging from 20 to 90°C in 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) buffer (0.2 M; pH 11.0). The apparent optimum pH of the enzyme was determined by running the standard assay, using Tris-HCl buffer (0.2 M) and CAPS buffer (0.2 M) for the pH ranges 6.5 to 10.5 and 9.0 to 12.5, respectively. Thermostability was determined using a diluted enzyme (0.63 mg of purified enzyme per ml was diluted 1:10 [vol/vol] in 50 mM Tris-HCl buffer, pH 8.0) and incubated at 90, 100, 105, and 115°C. Samples were taken at time intervals, and the residual activity was determined by the standard assay. pH was adjusted at room temperature for all the buffers used. Michaelis-Hill constants were determined from data obtained by determining the initial rate of pNPP hydrolysis under the assay conditions described above with 1 mM CoCl2, using a range from 0.1 to 10 mM substrate.
Cation and inhibitor effect studies.
The effects of monovalent and divalent cations were examined for their influence on enzyme activity. Increasing concentrations of NaCl and KCl were added to 50 mM NaOH-glycine (pH 10.0) containing 2.5 mM pNPP, and enzymatic activity was measured by standard procedures. Several divalent cations were also tested under similar conditions. All of them were used in their chloride form. The effect of inhibitors was determined by carrying out the same procedure, minus addition of CoCl2.
DNA dephosphorylation procedure
pBSK was digested using EcoRI- or EcoRV-producing, respectively, cohesive and blunt-ended linearized vectors. Linearized vector (1.1 μg) was incubated in dephosphorylation buffer and 50 U of AP for 2 h. The recombinant AP from P. abyssi was used in 0.2 M Tris-HCl buffer (pH 10.0) at 70°C. Commercial calf intestinal AP (CIAP) (Roche) was used as a positive dephosphorylation control in its commercial dephosphorylation buffer at 37°C. The number of AP units was determined by carrying out the standard assay procedure under dephosphorylation conditions of buffer and temperature. Following this, 0.55 μg of AP-treated linear vector was ligated in the presence of 2 U of T4 DNA ligase along with its commercial buffer (Roche) to give a final volume of 20 μl, which was incubated overnight at 16°C. Finally, 50 μl of competent E. coli DH5α cells was transformed by 1 μl of the ligation mixture. Preparation of the competent cells and transformations were performed using the standard CaCl2 transformation protocol (3). Transformed cells were selected by plating on 2×YT agar medium containing ampicillin (100 μg · ml−1). After linearization and dephosphorylation, DNA fragments were purified using a Spin Column (Qiagen) and resuspended in ultrapure water. Negative dephosphorylation control was performed by avoiding the dephosphorylation step. The percent dephosphorylation efficiency [DE(%)] was calculated from the following equation:
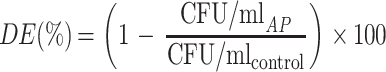 |
where CFU/ml is the number of CFU per milliliter of transformed cells, CFU/mlAP is CFU/ml after ligation performed on dephosphorylated vectors, and CFU/mlcontrol: is CFU/ml after ligation performed on nondephosphorylated vectors.
RESULTS
P. abyssi AP gene presents classical and unique features
Among the 1,765 open reading frames deduced from the complete genome sequence of P. abyssi (available at http://www.genoscope.fr/Pab/), the phoAP.abyssi gene encoding an AP was identified as a 1,485-bp fragment located between positions +1001913 and +1003397 on the P. abyssi chromosome. Its putative genomic environment is composed of five genes encoding various phosphate transporting proteins, suggesting the existence of a pho regulon, as previously observed in other species (22, 44). The deduced product of phoAP.abyssi is a protein of 495 amino acids, with a theoretical molecular mass and isoelectric point of 54.5 kDa and 4.90, respectively. Hydropathy analysis of the N-terminal end of the protein revealed a domain with a high hydrophobicity (data not shown), suggesting that it is a signal peptide. In accordance with the “−3, −1 rule,” the most probable cleavage site for the signal peptidase is between A25 and S26 (35, 46). Significant peptide sequence similarities were found with other APs already described, ranging from less than 30% for eucaryotic counterparts to approximately 35% for procaryotic APs, with the exception of Pyrococcus furiosus AP, which is 80% similar. In addition, alignment of peptide sequences from various APs with the mature product of the phoAP. abyssi gene (Fig. 1) shows conservation of well-defined regions which are conserved throughout the entire family. These regions contain the amino acid residue S102 (mature E. coli AP numbering), which has been shown to be incorporated within the phosphoseryl intermediate, in addition to the residues involved in the coordination of the two Zn(II) and the Mg(II) ions. However, the secondary ligands of the Mg(II), i.e., D153 and K328 (mature E. coli AP numbering), are replaced by H108 and H228 in the P. abyssi AP. These amino acid substitutions have been shown to be specific to mammalian APs (33). By comparison of AP sequences from P. abyssi and E. coli, it was concluded that the 10 strands of central β-sheet are conserved (data not shown). Thus, the topology of the E. coli AP central β-sheet is most likely conserved in the archaeal enzyme. Also, a putative Ca(II)-binding domain (Ef-hand) was deduced from the C-terminal sequence analysis and found to be specific to P. abyssi and P. furiosus AP sequences.
FIG. 1.

Conservation of the active site. Shown is the peptide sequence alignment of P. abyssi AP with other members of the AP family. Abbreviations are as follows (sequences were obtained from the accession numbers indicated in parentheses): Eco, E. coli (pir, PAECA); Pab, P. abyssi (pir, E75081); Pfu, P. furiosus (Pf, 962748); Tma, T. maritima (pir, 72410); Sgr, Streptococcus griseus (pir, S17780); Bsu, Bacillus subtilis AP IV (pir, B69676); Bli, Bacillus licheniformis (genpept, AAG10093); Sce, Saccharomyces cerevisiae (pir, S69648); Ant, Antarctic bacterium (emb, CAB82508.1); Hb, Halobacterium sp. (genpept, AAG20936); Tca, T. caldophilus (genpept, AAF13361); Rno, Rattus norvegicus (pir, A28114); Mmu, Mus musculus (pir, A40172); Bta, Bos taurus (pir, A29600); Fca, Felis catus (pir, S66467); Hsa, Homo sapiens placental (pir, PAHUA); Bmo, Bombyx mori (pir, S19607). Strictly conserved residues and residues conserved more than 60% are highlighted in black and gray, respectively. Symbols above the sequences indicate the conserved residues which are involved in the active site, according to the following legend: ↓, metal ion coordination (Zn1, Zn2, Mg primary ligand); ⇓, metal ion coordination (Mg secondary ligand); ●, phosphoseryl intermediate formation; ○, phosphate coordination. Numbering of the sequences is given for precursors. Alignments were performed using the GAP program (Genetics Computer Group Wisconsin Package) in pairwise comparison.
Expression and purification of P. abyssi AP.
A 1,410-bp fragment was cloned in pARHS vector, encoding a 470-amino-acid protein which corresponds to the putative mature protein. The recombinant AP was expressed in E. coli HMS 174(DE3) pLysS cells harboring the pPabAP plasmid. The E. coli strain harboring the plasmid pPabAP was grown until mid-log phase and induced by IPTG. SDS-PAGE analysis of cell-free heat-treated extract of induced cells revealed an additional band of 52 kDa, corresponding to the calculated molecular mass of the phoAP. abyssi product without its putative signal peptide (data not shown). The 52-kDa band was absent in extracts of E. coli HMS 174(DE3) pLysS (data not shown). A low expression rate was observed (approximately 5% of the soluble cellular protein fraction). Optimization of the expression was attempted. Neither IPTG concentration (0.1 to 2.0 mM) nor induction time (4 h to overnight) variations showed significant changes in the final recombinant enzyme concentration. Negligible AP activity was observed in the insoluble cellular fraction. Moreover, cytotoxicity of the recombinant enzyme was observed as cell lysis after induction of expression. Therefore, an induction time of 4 h was preferred to overnight induction. Nevertheless, recombinant AP could be easily purified to homogeneity by a three-step purification procedure consisting of heat incubation (80°C, 20 min, pH 5.5), which results in denaturation of the majority of E. coli proteins, followed by two anion exchange chromatography steps (data not shown). In particular, it appears that the heat incubation at pH 5.5 completely denatured E. coli AP as shown by zymogram and standard assay using MUFP and pNPP as substrates, respectively. Purification was estimated at approximately 450-fold, and the enzyme was determined to be at least 95% pure by SDS-PAGE (Fig. 2). Finally, purified AP solution (0.63 mg · ml−1) was stored in phosphate buffer (pH 7.5) containing 50% glycerol at −20°C.
FIG. 2.

Purification of the recombinant P. abyssi AP. Results of SDS–12% PAGE, showing the different steps of AP purification, are presented. Recombinant AP was purified from the soluble cellular fraction of E. coli HMS 174(DE3) pLysS, harboring pPabAP plasmid, induced cells. Lanes M, molecular mass standard; lane 1, soluble cellular crude extract; lane 2, crude extract after thermal treatment; lane 3, ResourceQ pooled active fractions; lane 4, purified fractions after MonoQ chromatography.
P. abyssi AP exists in mono- and homodimeric forms.
Electrophoresis of the purified recombinant AP using a 4 to 20% gradient of polyacrylamide gel under native conditions revealed the presence of two active forms (Fig. 3). Their respective molecular masses were estimated at 54 and 106 kDa. Since the 54-kDa value is consistent with the calculated molecular mass of the cloned gene product, the recombinant P. abyssi AP appears to exist in two forms, monomeric and homodimeric, both of which are active (Fig. 3). As one cysteine residue is present in the deduced amino acid sequence, the hypothesis of a dimeric form maintained with an interchain disulfide bond was clarified by treating the P. abyssi AP with SDS before running the electrophoresis experiment. Treatment of the AP with SDS yielded only the monomeric form (Fig. 3), and it was therefore concluded that the homodimeric form is maintained by noncovalent interactions, such as ion pairs and/or hydrophobic interactions, rather than an interchain disulfide bond.
FIG. 3.

Quaternary structure analysis. Results of 4 to 20% gradient PAGE, showing active forms and denaturation studies of the purified recombinant AP, are presented. Lanes M, molecular mass standard; lane 1, purified AP after Coomassie blue staining; lane 2, purified AP after incubation of the gel under enzymatic reaction conditions, using MUFP as substrate; lane 3, purified AP after 10% SDS treatment (10 min, 100°C) and Coomassie blue staining.
P. abyssi AP presents high pH and temperature activation and high thermostability.
Apparent optimum pH of the AP was determined to be 11.0 in 0.2 M CAPS buffer. The enzyme exhibited at least 60% of its optimal activity over a rather narrow pH range, from 10.0 to 11.5 (Fig. 4A). Significant activity was observed over a pH range from 9.0 to 12.0. Furthermore, thermal activation studies enabled the determination of an apparent optimum temperature of 70°C in 0.2 M CAPS buffer (pH 11.0) (Fig. 4B). 25% of the maximum activity was found at 37°C. Moreover, the enzyme exhibited at least 80% of its optimal activity over a range of temperature from 60 to 80°C. Thermal stability was determined from enzyme incubation over a range of temperatures (90 to 115°C) (Fig. 4C). Half-lives of the P. abyssi AP at 100 and 105°C were estimated at 18 and 5 h, respectively. Eighty percent of the activity was recovered after incubation at 90°C for 36 h. Denaturation of the enzyme was observed to occur in less than 30 min at 115°C. Activation energy of the thermal denaturation was estimated at 262.1 kJ · mol−1, according to the Arrhenius relationship.
FIG. 4.

(A) Optimum pH. Enzymatic activity was assayed at 70°C in the presence of 2.5 mM pNPP as a substrate, using 0.2 M Tris-HCl buffer, pH 7.0 to 10.5(▵), and 0.2 M CAPS buffer, pH 9.0 to 12.5 (○). (B) Optimum temperature. Enzymatic activity was assayed at 22 to 85°C in the presence of 2.5 mM pNPP as a substrate, using 0.2 M CAPS buffer, pH 11.0. (C) Thermal stability. Residual activity versus incubation time at 90°C (▵), 100°C (□), 105°C (⋄), and 115°C (×) is shown. The insert graph shows lnk versus 10−3 · 1/T, where k is the rate constant from the Arrhenius relationship and T is the temperature of incubation (K).
P. abyssi AP is strongly dependent on divalent cations.
As APs are classically described as metalloenzymes (38), the effect of divalent cations was examined (Table 1). A strong inhibition in the presence of 1 mM EDTA in the reaction mixture was observed, whereas the enzyme exhibited a twofold activation in the presence of 1 mM Mg(II), Mn(II), Zn(II), or Co(II). Such results are a signature of strong dependency on divalent cations and confirm the metalloenzymatic nature of the P. abyssi AP. Increasing concentrations of Mg(II) in the reaction mixture provided a linear activation of the enzyme in the 0 to 2 mM range, followed by a plateau in the 2 to 10 mM range (data not shown). This is consistent with the presence of a Mg(II)-binding site which has become saturated with the addition of exogenous Mg(II). Despite the elucidation of a Ca(II)-binding site in the deduced peptide sequence, this cation was found to have no significant effect on enzymatic activity. Cu(II) and Ni(II) showed inhibitory effects. The influence of ionic strength was tested. Increasing concentrations of NaCl and KCl had an inhibitory effect on AP activity. At 2 M of NaCl or KCl, 60 and 90% of the enzyme activity, respectively, was recovered (data not shown).
TABLE 1.
Effect of divalent cations
| Effector (1 mM) | Relative activity (%)a |
|---|---|
| None | 100.0 A |
| Mg(II) | 223.1 A |
| Mn(II) | 206.1 B |
| Zn(II) | 204.0 B |
| Co(II) | 186.6 |
| Ca(II) | 82.8 B |
| Cu(II) | 61.9 |
| Ni(II) | 44.5 A |
| EDTA | 10.9 A |
Standard deviations (SD) were <5% except for values with an A (5% ≤ SD < 10%) and those with a B (10% ≤ SD < 15%).
P. abyssi AP catalytic properties: effect of substrate concentration and inhibitors.
Michaelian kinetic parameters were estimated using the Lineweaver-Burk representation (Fig. 5). The curve is made up of two linear parts which allowed the calculation of two (Vmax, Km) doublets. Thus, from 0.1 to 0.7 mM, obtained values were as follows: Vmax1 = 4.07 μmol · min−1 and Km1 = 166.33 μM; from 0.8 to 10 mM, obtained values were as follows: Vmax2 = 8.31 μmol · min−1 and Km2 = 1204.98 μM. Moreover, inhibitors were tested (Table 2): classical phosphatase inhibitors (inorganic phosphate, molybdate, and vanadate), denaturing agents (SDS, urea), metal ion chelators (EDTA, EGTA), and thiol-reducing agents (dithiothreitol [DTT], MCE). Inorganic phosphate inhibited AP activity with only 22.2% of the control activity recovered in the presence of 10 mM Na2HPO4. This is in accordance with the general inhibitory effect of inorganic phosphate on APs. Molybdate and vanadate were shown to have slight inhibitory effects, with 86.3 and 73.4% of the control activity retained, respectively, at an added concentration of 10 mM, respectively. SDS exhibited a strong inhibitory effect, whereas 2 and 4 M urea did not appear to have any effect. Metal ion chelators greatly inhibited enzymatic activity. Thiol-reducing agents (DTT and MCE) showed strong inhibitory effects. Upon addition of 2 mM DTT or MCE to the reaction mixture, residual AP activity was 1.6 and 58.5%, respectively.
FIG. 5.

Kinetic parameters: 1/Vi versus 1/S. Enzymatic activity was assayed under standard condition with 1 mM CoCl2 using a range of pNPP concentrations from 0.1 to 10 mM. Abbreviations: Vi, initial velocity; S, pNPP concentration.
TABLE 2.
Spectrum of inhibitors
| Inhibitor | Concn (mM) | Relative activity (%)a |
|---|---|---|
| None | 100.0∗ | |
| SDS | 30 (1%) | 16.4∗ |
| Urea | 2,000 | 107.8 |
| 4,000 | 106.5∗ | |
| Molybdate | 1 | 101.2∗ |
| 10 | 86.3 | |
| Vanadate | 1 | 88.2∗ |
| 10 | 73.4∗ | |
| Inorganic phosphate | 1 | 74.8 |
| 10 | 22.2 | |
| EDTA | 1 | 10.7 |
| 5 | 5.6∗ | |
| EGTA | 1 | 17.8 |
| 5 | 4.6 | |
| DTT | 0.2 | 79.7 |
| 1 | 2.2 | |
| 2 | 1.6 | |
| MCE | 2 | 58.5∗ |
| 5 | 32.1 | |
| 10 | 14.1 |
Standard deviations were <5% except for values with an asterisk (5% ≤ standard deviation <10%).
P. abyssi recombinant AP is effective for DNA dephosphorylation.
APs are classically used in molecular biology to remove 5′-terminal phosphate from DNA fragments. As such, P. abyssi AP was tested against cohesive (EcoRI-linearized pBSK) and blunt (EcoRV-linearized pBSK) ends. The efficiency of dephosphorylation was estimated by comparing transformation rates after ligation of dephosphorylated and nondephosphorylated plasmids. Complete ligation of non-AP-treated fragments was observed when the experiment was carried out using cohesive ends as observed on agarose gel (Fig. 6A, lane 5). However, P. abyssi AP- and CIAP-treated fragments were generally found to be nonligated (Fig. 6, lanes 6 and 7, respectively). Dephosphorylation efficiency [DE(%)] was estimated at 93.8% for P. abyssi AP and 99.7% for CIAP. Similar results were observed on experiments using blunt ends, however, with partial ligation of non-AP-treated fragments (Fig. 6B). DE(%) was estimated at 84.1% for P. abyssi AP and 98.7% for CIAP. Obtained DE(%) values indicate the ability of P. abyssi AP to dephosphorylate linear DNA fragments, thus preventing self-ligation.
FIG. 6.

DNA dephosphorylation. Shown are results of 0.8% agarose gel electrophoresis analysis of the DNA dephosphorylation procedure. pBSK vector was used as a substrate after being digested by EcoRI (A) or EcoRV (B). Lanes M, DNA ladder (Eurogentec S.A.); lanes 1, circular pBSK; lanes 2, linearized pBSK; lanes 3, P. abyssi AP-treated linear pBSK (2 h, 70°C); lanes 4, CIAP-treated linear pBSK (2 h, 37°C); lanes 5, non-AP-treated plasmid after ligation reaction; lanes 6, P. abyssi AP-treated plasmid after ligation reaction; lanes 7, CIAP-treated plasmid after ligation reaction.
DISCUSSION
The P. abyssi phoA gene encoding an AP was cloned and the recombinant enzyme expressed in E. coli. The present work reports the first characterization of an AP from a hyperthermophilic archaeon. AP activity has already been found in Pyrococcus horikoshii, but the enzyme has not been isolated and characterized (18). In addition, comparison of the P. abyssi and P. horikoshii genomes was performed, and the P. abyssi AP sequence found no homologue in the P. horikoshii genome. Analysis of the deduced peptide primary structure revealed highly conserved patterns with other APs sequences. Comparison with the E. coli enzyme enabled identification of the strong conservation of both the catalytic site (metal ion coordination, phosphoseryl intermediate, and phosphate coordination) and secondary structure. This is consistent with previous studies on other APs (23, 27). This enzyme was expressed in E. coli HMS 174(DE3) harboring pLysS and recombinant pPabAP plasmids. Low expression levels were obtained, and experiments showed cytotoxicity of the recombinant AP. In addition, analysis of P. abyssi AP nucleotide sequences showed the presence of rare arginine-encoding codons AGG and AGA, occurring at 2.8 and 1.5%, respectively, whereas in E. coli they do so at only 0.14 and 0.21% (25). Thus, the level of expression might be improved by coexpressing the recombinant AP and tRNAUCU (28, 47).
Properties of the recombinant enzyme have been elucidated, and since the folding conditions and the posttranslational modifications are different between the native and the recombinant AP biosynthesis, properties reported here do not allow any conclusions about the native AP. An apparent optimum pH was found to be 11.0 in 0.2 M CAPS buffer. P. abyssi AP is among the most alkaline APs characterized. APs from thermophilic bacteria showed high pH optima, e.g., 9.9 for the T. neapolitana enzyme (12) and 10.0 and 11.5 for the T. thermophilus APs (37). Mammalian APs possess higher pH optima than the E. coli enzyme, and it has been proposed that substitutions of D153 and K328 residues into corresponding histidine for the mammalian enzymes are responsible for this increase in pH (20, 33). The high optimum pH observed may be explained by the similarity of P. abyssi AP to that found in mammalian APs. The apparent optimum temperature was found to be 70°C in the 0.2 M CAPS (pH 11.0) buffer. Because P. abyssi's optimal growth temperature is near 100°C (14), the determined thermoactivation optimum value appeared lower than expected. Nevertheless, other recombinant proteins from P. abyssi present optimum temperature activity far below 100°C. Thermal stability was determined, and half-lives at 100 and 105°C were estimated at 18 and 5 h, respectively. Approximately 80% of the activity is conserved after a 36-h incubation at 90°C. These results show that the P. abyssi AP is the most thermostable AP described so far, in comparison to the T. neapolitana AP, which exhibits a half-life of about 4 h at 90°C in the presence of CoCl2 (12). Catalytic properties of the P. abyssi AP were determined. Phosphotransferase activity is commonly observed by using Tris-HCl or ethanolamine buffers (9, 31), which increases enzyme activity by promoting the phosphotransferase reaction. A fourfold increase in activity in Tris-HCl relative to NaOH-glycine buffer was observed with the P. abyssi AP in the same conditions of temperature, pH, and substrate. Michaelis constants were found to be Vmax1 = 4.07 μmol · min−1 and Km1 = 166.33 μM and Vmax2 = 8.31 μmol · min−1 and Km2 = 1204.98 μM. P. abyssi AP presents Km values which are consistent with those found for several APs (33). Lineweaver-Burke representation allowed the observation of two behaviors. Vmax2 is approximately twofold the Vmax1 value, and Km2 is about sevenfold the Km1 value. Since zymogram analysis of P. abyssi AP highlighted two active forms, the kinetic observations could correspond to the presence of both the monomer and the homodimer in the enzymatic reaction mixture. However, this hypothesis has to be confirmed by isolating the two enzyme forms and studying them separately. A spectrum of inhibitors was tested on the P. abyssi AP, using molybdate and vanadate as phosphotyrosylphosphatase inhibitors (1). More precisely, vanadate is defined as an inhibitor of phosphoryltransfer reactions, especially if they involve a stable-covalent phosphoseryl-enzyme intermediate (6). Vanadate appeared to be a weak inhibitor of P. abyssi AP. This is not consistent with previous studies on other APs (1, 6, 21) and may be a result of the experimental conditions (pH 10.0, 70°C) or of an original catalytic mechanism. In contrast, thiol-reducing agents exhibited a strong inhibitory effect on AP activity. Although P. abyssi AP contains no intra- or interchain disulfide bonds, cysteine residues (one per monomer) seem critically implied in the enzyme activity. Strong inhibition in the presence of 1% SDS and lack of inhibition in the presence of 2 and 4 M urea are consistent with properties of other APs (40).
APs are metalloenzymes (38), and they are all inhibited by metal ion chelators such as EDTA. They are classically considered to be Zn(II)- and Mg(II)- dependent enzymes, especially E. coli and mammalian APs (27). However, activation following Mn(II), Co(II), or other metal ion addition has already been observed among other APs (6, 8, 17, 31, 32, 37, 41, 50). The P. abyssi AP was found to be activated by Zn(II), Mg(II), Mn(II), and Co(II). Increasing the concentrations of Mg(II) results in an increase in enzymatic activity followed by a plateau (data not shown). Activation following the addition of Mg(II) has been observed with mammalian APs and largely characterized using E. coli AP mutants. Studies of E. coli AP concerning the secondary ligands of Mg(II) (D153 and K328 [mature E. coli AP sequence numbering]) have led to the conclusion that replacement of these residues with histidine results in the transformation of an Mg(II)-binding site into a Zn(II)-binding site. The affinity between Mg(II) and its binding site became lower for the (D153H, K328H) mutant AP, and optimal activity was recovered by the addition of Mg(II) (20, 24, 33, 34). The residues D153 and K328 in E. coli AP correspond to the residues H108 and H228 in P. abyssi AP. Both are similar to corresponding residues found in mammalian APs. This appears to be consistent with P. abyssi AP behavior towards Mg(II). Furthermore, the purified enzyme is active when no cations are added and inhibited by metal ion chelators. This may suggest that strong metal-enzyme interactions exist and that the purified AP is partially metalated. Therefore, P. abyssi AP may possess several metal binding sites. Such a hypothesis is consistent with the analysis of the sequence and other APs (17, 27, 42). Finally, reactivation of the P. abyssi AP apoenzyme was attempted by incubation of the latter in the presence of various divalent cations, at various temperatures. No reactivation was observed under these experimental conditions. Reactivation of AP apoenzymes has already been observed with varied efficiency (2, 12). However, drastic treatment of the purified P. abyssi AP using metal ion chelators appeared irreversible, suggesting important catalytic and structural roles of metal ions. Under present experimental conditions, P. abyssi AP presented two active forms: monomer and homodimer. APs are classically described as being homodimeric, which is the case with E. coli and mammalian APs. However, many monomeric (13, 19, 40) and oligomeric (17) forms have been described. Further studies may provide clarification of the parameters governing the equilibrium between the two forms.
APs are commonly used in molecular biology for the construction of recombinant plasmids. APs remove 5′-terminal phosphate from DNA fragments and, as such, AP-treated linearized plasmids are unable to exhibit self-ligation and are more likely to integrate an exogenous gene insert. P. abyssi AP was tested under plasmid dephosphorylation conditions using a linear cohesive or blunt-ended pBSK vector. Dephosphorylation efficiency with regard to cohesive and blunt ends was estimated at 93.8 and 84.1%, respectively. Bovine AP was found to be more effective than the P. abyssi AP; however; dephosphorylation conditions of the latter were not optimized. Although many APs have been isolated and characterized, only commercial E. coli AP (bacterial AP) and CIAP are routinely used in molecular biology. Such properties are rarely reported. DNA dephosphorylation has been performed with the Antarctic bacterium strain TAB5 AP (39). As P. abyssi AP presented great thermostability and good efficiency in DNA dephosphorylation, further studies may be required to elucidate its potential use in such applications.
ACKNOWLEDGMENTS
This work was supported by the Biotechnology Program grant 98 C 0173 from the Ministère de l'Education Nationale, de la Recherche et de la Technologie (MENRT), Paris, France.
REFERENCES
- 1.Ansai T, Awano S, Chen X, Fuchi T, Arimoto T, Akifusa S, Takehara T. Purification and characterization of alkaline phosphatase containing phosphotyrosyl phosphatase activity from the bacterium Prevotella intermedia. FEBS Lett. 1998;428:157–160. doi: 10.1016/s0014-5793(98)00514-6. [DOI] [PubMed] [Google Scholar]
- 2.Ásgeirsson B, Hartemink R, Chlebowski J F. Alkaline phosphatase from Atlantic cod (Gadus morhua). Kinetic and structural properties which indicate adaptation to low temperatures. Comp Biochem Physiol. 1995;110B:315–329. [Google Scholar]
- 3.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons Inc.; 1998. [Google Scholar]
- 4.Belkin S, Jannasch H W. A new extremely thermophilic sulfur-reducing heterotrophic marine bacterium. Arch Microbiol. 1985;141:181–186. [Google Scholar]
- 5.Berger J, Garattini E, Hua J-C, Udenfriend S. Cloning and sequencing of human intestinal alkaline phosphatase cDNA. Proc Natl Acad Sci USA. 1987;84:695–698. doi: 10.1073/pnas.84.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonet M L, Llorca F I, Cadenas E. Kinetic mechanism of Halobacterium halobium Mn2+-activated alkaline phosphatase. Biochem Mol Biol Int. 1994;34:1109–1120. [PubMed] [Google Scholar]
- 7.Bradford M M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 8.Chen Q-X, Zheng W-Z, Lin J-Y, Shi Y, Xie W-Z, Zhou H-M. Effect of metal ions on the activity of green crab (Scylla serrata) alkaline phosphatase. Int J Biochem Cell Biol. 2000;32:879–885. doi: 10.1016/s1357-2725(00)00026-1. [DOI] [PubMed] [Google Scholar]
- 9.Coleman J E. Structure and mechanism of alkaline phosphatase. Annu Rev Biophys Biomol Struct. 1992;21:441–483. doi: 10.1146/annurev.bb.21.060192.002301. [DOI] [PubMed] [Google Scholar]
- 10.De Moerlooze L, Struman I, Renard A, Martial J A. Stabilization of T7-promoter-based pARHS expression vectors using the parB locus. Gene. 1992;119:91–93. doi: 10.1016/0378-1119(92)90070-6. [DOI] [PubMed] [Google Scholar]
- 11.Derman A I, Beckwith J. Escherichia colialkaline phosphatase fails to acquire disulfide bonds when retained in the cytoplasm. J Bacteriol. 1991;173:7719–7722. doi: 10.1128/jb.173.23.7719-7722.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong G, Zeikus J G. Purification and characterization of alkaline phosphatase from Thermotoga neapolitana. Enzyme Microb Technol. 1997;21:335–340. doi: 10.1016/s0141-0229(97)00002-1. [DOI] [PubMed] [Google Scholar]
- 13.Eguchi M. Alkaline phosphatase isozymes in insects and comparison with mammalian enzyme. Comp Biochem Physiol. 1995;111B:151–162. doi: 10.1016/0305-0491(94)00248-s. [DOI] [PubMed] [Google Scholar]
- 14.Erauso G, Reysenbach A-L, Godfroy A, Meunier J-R, Crump B, Partensky F, Baross J A, Marteinsson V, Barbier G, Pace N C, Prieur D. Pyrococcus abyssisp. nov., a new hyperthermophilic archaeon isolated from a deep-sea hydrothermal vent. Arch Microbiol. 1993;160:338–349. [Google Scholar]
- 15.Fitt P S, Baddoo P. Separation and purification of the alkaline phosphatase and a phosphodiesterase from Halobacterium cutirubrum. Biochem J. 1979;181:347–353. doi: 10.1042/bj1810347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fitt P S, Peterkin P I. Isolation and properties of a small manganese-ion-stimulated bacterial alkaline phosphatase. Biochem J. 1976;157:161–167. doi: 10.1042/bj1570161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldman S, Hecht K, Eisenberg H, Mevarech M. Extracellular Ca2+-dependent inducible alkaline phosphatase from the extremely halophilic archaeabacterium Haloarcula marismortui. J Bacteriol. 1990;172:7065–7070. doi: 10.1128/jb.172.12.7065-7070.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.González J M, Masuchi Y, Robb F T, Ammerman J W, Maeder D L, Yanagibayashi M, Tamaoka J, Kato C. Pyrococcus horikoshiisp. nov., a hyperthermophilic archaeon isolated from a hydrothermal vent at the Okinawa Trough. Extremophiles. 1998;2:123–130. doi: 10.1007/s007920050051. [DOI] [PubMed] [Google Scholar]
- 19.Hauksson J B, Andrésson Ó, Ásgeirsson B. Heat-labile bacterial alkaline phosphatase from a marine Vibriosp. Enzyme Microb Technol. 2000;27:66–73. doi: 10.1016/s0141-0229(00)00152-6. [DOI] [PubMed] [Google Scholar]
- 20.Holtz K M, Kantrowitz E R. The mechanism of the alkaline phosphatase reaction: insights from NMR, crystallography and site-specific mutagenesis. FEBS Lett. 1999;462:7–11. doi: 10.1016/s0014-5793(99)01448-9. [DOI] [PubMed] [Google Scholar]
- 21.Holtz K M, Stec B, Kantrowitz E R. A model of the transition state in the alkaline phosphatase reaction. J Biol Chem. 1999;274:8351–8354. doi: 10.1074/jbc.274.13.8351. [DOI] [PubMed] [Google Scholar]
- 22.Hulett F M. The signal-transduction network for Pho regulation in Bacillus subtilis. Mol Microbiol. 1996;19:933–939. doi: 10.1046/j.1365-2958.1996.421953.x. [DOI] [PubMed] [Google Scholar]
- 23.Hulett F M, Kim E E, Bookstein C, Kapp N V, Edwards C W, Wyckoff H W. Bacillus subtilis alkaline phosphatases III and IV. Cloning, sequencing, and comparisons of deduced amino acid sequence with Escherichia colialkaline phosphatase three-dimensional structure. J Biol Chem. 1991;266:1077–1084. [PubMed] [Google Scholar]
- 24.Janeway C M L, Xu X, Murphy J E, Chaidaroglou A, Kantrowitz E R. Magnesium in the active site of Escherichia colialkaline phosphatase is important for both structural stabilization and catalysis. Biochemistry. 1993;32:1601–1609. doi: 10.1021/bi00057a026. [DOI] [PubMed] [Google Scholar]
- 25.Kane J F. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Opin Biotechnol. 1995;6:494–500. doi: 10.1016/0958-1669(95)80082-4. [DOI] [PubMed] [Google Scholar]
- 26.Karamyshev A L, Karamysheva Z N, Kajava A V, Ksenzenko V N, Nesmeyanova M A. Processing of Escherichia colialkaline phosphatase: Role of the primary structure of the signal peptide cleavage region. J Mol Biol. 1998;277:859–870. doi: 10.1006/jmbi.1997.1617. [DOI] [PubMed] [Google Scholar]
- 27.Kim E E, Wyckoff H W. Structure of alkaline phosphatases. Clin Chim Acta. 1989;186:175–188. doi: 10.1016/0009-8981(90)90035-q. [DOI] [PubMed] [Google Scholar]
- 28.Kim R, Sandler S J, Goldman S, Yokota H, Clark A J, Kim S H. Overexpression of archaeal proteins in Escherichia coli. Biotechnol Lett. 1998;20:207–210. [Google Scholar]
- 29.Laemmli U K. Cleavage of the structural proteins during the assembly of the head of the bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Mc Comb R B, Bowers G N, Posen S. Alkaline phosphatase. New York: Plenum Press; 1979. [Google Scholar]
- 31.Morales A C, Nozawa S R, Thedei G, Jr, Maccheroni W, Jr, Rossi A. Properties of a constitutive alkaline phosphatase from strain 74A of the mold Neurospora crassa. Braz J Med Biol Res. 2000;33:905–912. doi: 10.1590/s0100-879x2000000800006. [DOI] [PubMed] [Google Scholar]
- 32.Mori S, Okamoto M, Nishibori M, Ichimura M, Sakayama J, Endo H. Purification and characterization of alkaline phosphatase from Bacillus stearothermophilus. Biotechnol Appl Biochem. 1999;29:235–239. [PubMed] [Google Scholar]
- 33.Murphy J E, Tibbitts T T, Kantrowitz E R. Mutations at positions 153 and 328 in Escherichia colialkaline phosphatase provide insight towards the structure and function of mammalian and yeast alkaline phosphatases. J Mol Biol. 1995;253:604–617. doi: 10.1006/jmbi.1995.0576. [DOI] [PubMed] [Google Scholar]
- 34.Murphy J E, Xu X, Kantrowitz E R. Conversion of a magnesium binding site into a zinc binding site by a single amino acid substitution in Escherichia colialkaline phosphatase. J Biol Chem. 1993;268:21497–21500. doi: 10.2210/pdb1anh/pdb. [DOI] [PubMed] [Google Scholar]
- 35.Nielsen H, Engelbrecht J, Brunak S, Von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 36.Park T, Lee J-H, Kim H-K, Hoe H-S, Kwon S-T. Nucleotide sequence of the gene for alkaline phosphatase of Thermus caldophilusGK24 and characteristics of the deduced primary structure of the enzyme. FEMS Microbiol Lett. 1999;180:133–139. doi: 10.1111/j.1574-6968.1999.tb08787.x. [DOI] [PubMed] [Google Scholar]
- 37.Pantazaki A A, Karagiorgas A A, Liakopoulou-Kyriakides M, Kyriakidis D A. Hyperalkaline and thermostable phosphatase in Thermus thermophilus. Appl Biochem Biotechnol. 1998;75:249–259. doi: 10.1007/BF02787778. [DOI] [PubMed] [Google Scholar]
- 38.Posen S. Alkaline phosphatase. Ann Intern Med. 1967;67:183–203. doi: 10.7326/0003-4819-67-1-183. [DOI] [PubMed] [Google Scholar]
- 39.Rina M, Pozidis C, Mavromatis K, Tzanodaskalaki M, Kokkinidis M, Bouriotis V. Alkaline phosphatase from the Antarctic strain TAB5. Properties and psychrophilic adaptations. Eur J Biochem. 2000;267:1230–1238. doi: 10.1046/j.1432-1327.2000.01127.x. [DOI] [PubMed] [Google Scholar]
- 40.Sharipova M R, Balaban N P, Mardanova A M, Nekhotyaeva N V, Dementyev A A, Vershinina O A, Garusov A V, Leshchinskaya I B. Isolation and properties of extracellular alkaline phosphatase from Bacillus intermedius. Biochem (Moscow) 1998;63:1178–1182. [PubMed] [Google Scholar]
- 41.Sharipova M R, Balaban N P, Nekhotyaeva N V, Mardanova A M, Dementyev A A, Leshchinskaya I B. A novel Bacillus intermediusextracellular alkaline phosphatase: isolation, physico-chemical and catalytic characteristics. Biochem Mol Biol Int. 1996;38:753–761. [PubMed] [Google Scholar]
- 42.Spencer D B, Chen C P, Hulett F M. Effect of cobalt on synthesis and activation of Bacillus licheniformisalkaline phosphatase. J Bacteriol. 1981;145:926–933. doi: 10.1128/jb.145.2.926-933.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki C, Ueda H, Tsumoto K, Mahoney W C, Kumagai I, Nagamune T. Open sandwich ELISA with VH−/VL−alkaline phosphatase fusion proteins. J Immunol Methods. 1999;224:171–184. doi: 10.1016/s0022-1759(99)00020-4. [DOI] [PubMed] [Google Scholar]
- 44.Tommassen J, Lugtenberg B. PHO-Regulon of Escherichia coliK12: a minireview. Ann Microbiol (Inst Pasteur) 1982;133A:243–249. [PubMed] [Google Scholar]
- 45.Trowsdale J, Martin D, Bicknell D, Campbell I. Alkaline phosphatases. Biochem Soc Trans. 1990;18:178–180. doi: 10.1042/bst0180178. [DOI] [PubMed] [Google Scholar]
- 46.Von Heijne G. Patterns of amino acids near signal-sequence cleavage sites. Eur J Biochem. 1983;133:17–21. doi: 10.1111/j.1432-1033.1983.tb07424.x. [DOI] [PubMed] [Google Scholar]
- 47.Wakagi T, Oshima T, Imamura H, Matsuzawa H. Cloning of the gene for inorganic pyrophosphatase from a thermoacidophilic archaeon. Sulfolobus sp. strain 7, and overproduction of the enzyme by coexpression of tRNA for arginine rare codon. Biosci Biotechnol Biochem. 1998;62:2408–2414. doi: 10.1271/bbb.62.2408. [DOI] [PubMed] [Google Scholar]
- 48.Weiss M J, Henthorn P S, Lafferty M A, Slaughter C, Raducha M, Harris H. Isolation and characterization of a cDNA encoding a human liver/bone/kidney-type alkaline phosphatase. Proc Natl Acad Sci USA. 1986;83:7182–7186. doi: 10.1073/pnas.83.19.7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weissig H, Schildge A, Hoylaerts M F, Iqbal M, Millán J L. Cloning and expression of the bovine intestinal alkaline phosphatase gene: biochemical characterization of the recombinant enzyme. Biochem J. 1993;290:503–508. doi: 10.1042/bj2900503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamashita Y, Toyoshima K, Yamazaki M, Hanada N, Takehara T. Purification and characterization of alkaline phosphatase of Bacteroides gingivalis381. Infect Immun. 1990;58:2882–2887. doi: 10.1128/iai.58.9.2882-2887.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]