

Abstract
Resolution of inflammation is an active process that is tightly regulated to achieve repair and tissue homeostasis. In the absence of resolution, persistent inflammation underlies the pathogenesis of chronic lung disease such as chronic obstructive pulmonary disease (COPD) with recurrent exacerbations. Over the course of inflammation, macrophage programming transitions from pro‐inflammatory to pro‐resolving, which is in part regulated by the nuclear receptor Peroxisome Proliferator‐Activated Receptor γ (PPARγ). Our previous work demonstrated an association between Fatty Acid Binding Protein 5 (FABP5) expression and PPARγ activity in peripheral blood mononuclear cells of healthy and COPD patients. However, a role for FABP5 in macrophage programming has not been examined. Here, using a combination of in vitro and in vivo approaches, we demonstrate that FABP5 is necessary for PPARγ activation. In turn, PPARγ acts directly to increase FABP5 expression in primary human alveolar macrophages. We further illustrate that lack of FABP5 expression promotes a pro‐inflammatory macrophage programming with increased secretion of pro‐inflammatory cytokines and increased chromatin accessibility for pro‐inflammatory transcription factors (e.g., NF‐κB and MAPK). And finally, real‐time cell metabolic analysis using the Seahorse technology shows an inhibition of oxidative phosphorylation in FABP5‐deficient macrophages. Taken together, our data indicate that FABP5 and PPARγ reciprocally regulate each other's expression and function, consistent with a novel positive feedback loop between the two factors that mediates macrophage pro‐resolving programming. Our studies highlight the importance of defining targets and regulatory mechanisms that control the resolution of inflammation and may serve to inform novel interventional strategies directed towards COPD.
Keywords: FABP5, macrophage polarization, PPARγ, resolution of inflammation
1. INTRODUCTION
Chronic inflammation underlies the pathogenesis of many chronic lung diseases. Thus, defining mechanisms by which pulmonary inflammation is regulated and resolved is highly significant to understanding disease processes. Macrophages are a hub for the resolution of inflammation, including engulfment of apoptotic cells and activation of anti‐inflammatory and repair processes. 1 , 2 Macrophages are known to generate cytokines (e.g., interleukin (IL)‐6, tumor necrosis factor (TNFα), etc.) that alert the body in response to infection or injury (pro‐inflammatory programming) but can also secrete anti‐inflammatory cytokines (e.g., IL‐10, IL‐33) that promote repair at the cellular and tissue level (pro‐resolving programming).
In chronic obstructive pulmonary disease (COPD), although cigarette smoking is the cardinal risk factor, repeated and sustained infections are clearly linked to non‐resolving inflammation and are responsible for COPD exacerbations. 3 Interestingly, COPD exacerbations are a risk factor for additional exacerbations, suggesting that un‐resolving chronic inflammation predisposes individuals for future episodes. 4 However, macrophage recruitment and function during COPD are a matter of debate. Several studies have demonstrated that COPD macrophages are not only increased in number, but also differ in function with an increased pro‐inflammatory over pro‐resolving programming. 5 , 6 Thus, understanding mechanisms of macrophage polarization could be important in the treatment of COPD.
Peroxisome Proliferator‐Activated Receptor γ (PPARγ) activation is required for the pro‐resolving programming of macrophages but has been shown to be impaired in COPD. 7 It is a ligand‐dependent transcription factor and a member of the nuclear receptor superfamily, 8 and has been shown to have pro‐resolving capabilities. 9 , 10 PPARγ agonists were shown to dampen macrophage activation in vitro, 11 , 12 and as such PPARγ has been targeted for therapeutic interventions. 13 However, PPARγ regulates a diverse spectrum of physiological processes and the use of synthetic PPARγ ligands has been associated with numerous off‐target effects. 14 , 15 Therefore, exploring alternative mechanisms of PPARγ activation could lead to novel, better‐targeted therapies available to patients with COPD and other diseases associated with chronic inflammation of the airways.
Fatty Acid Binding Proteins (FABPs) may promote anti‐inflammatory functions by transporting anti‐inflammatory mediators, among them ligands for PPARγ. 16 , 17 , 18 FABPs are a family of small, highly conserved, cytoplasmic proteins that bind long‐chain fatty acids and other hydrophobic ligands and are involved in fatty acid uptake, transport, and metabolism. 19 As such, they have the potential to contribute to either pro‐ or anti‐inflammatory properties of immune cells based on their binding partners. Fatty Acid Binding Protein 5 (FABP5), one of the FABPs expressed in macrophages, is suggested to sequester anti‐inflammatory mediators from their target, creating a pro‐inflammatory environment. 20 , 21 , 22 However, we have shown that FABP5‐deficient mice develop increased inflammation following influenza A infection that persists long after wild type mice recovery, 23 likely indicating a complex role for FABP5 in modulating inflammatory responses in vivo.
While exploring the potential for signaling between FABP5 and PPARγ, we have previously demonstrated that FABP5 expression is necessary to promote PPARγ activity and that FABP5 expression and PPARγ activity are positively correlated in human peripheral blood mononuclear cells (PBMCs). 24 However macrophages are the critical cell type in the resolution of inflammation. Here, we extend our previous work to identify and characterize a novel role for FABP5/PPARγ crosstalk in macrophage polarization using multiple in vitro and in vivo approaches. Our data demonstrate for the first time in macrophages that (1) FABP5 and PPARγ physically interact with one another, and that FABP5 expression increases PPARγ activity, (2) in turn, PPARγ increases FABP5 expression through direct transcriptional modulation, and (3) lack or reduction of FABP5 increases macrophage pro‐infammatory programming. Altogether, these results support FABP5 as a promising alternative therapeutic target to stimulate PPARγ‐induced macrophage pro‐resolution in chronic inflammatory lung diseases such as COPD.
2. MATERIALS AND METHODS
2.1. RNA in situ hybridization (RNAscope)
Advanced Cell Diagnostics (ACD) designed and generated probes and reagent kits for RNA in situ hybridization. The probes and reagents are based on ACD proprietary RNAscope technology that integrates probe design with signal amplification and detection to achieve single‐molecule detection. We followed the procedure instructions provided by the manufacturer.
2.2. Cell culture and patient tissue samples
BEAS‐2B cells (ATCC) were cultured in Dulbecco's Modified Eagle Medium (DMEM; Corning) containing l‐glutamine and 4.5 g/L glucose, supplemented with 10% fetal bovine serum (VWR) and 1% penicillin/streptomycin (Corning). The human monocytic THP‐1 cells (ATCC) were cultured in RPMI‐1640 supplemented with 10% heat‐inactivated fetal bovine serum (FBS), beta mercaptoethanol (0.05 mM), penicillin (100 U/ml), and streptomycin (0.1 mg/ml). Monocyte‐derived macrophages were obtained by differentiation with PMA (10 nM) for 72 h. Primary human alveolar macrophages were obtained from the Human Lung Tissue Consortium at National Jewish Health. Cells were plated on 150 mm plates to allow macrophages to adhere for 2 h in DMEM (Corning) containing l‐glutamine and 4.5 g/L glucose, supplemented with 10% fetal bovine serum (VWR), 1% penicillin/streptomycin (Corning) and 2.5 µg/ml Amphotericin B (Sigma).
2.3. Flag and Myc immunoprecipitation
BEAS‐2B cells were transfected with mPPARγ‐flag [PCS4 3XFlag‐PPARgamma1 was a gift from Jaewhan Song (Addgene plasmid # 78769)] 25 and mFABP5‐c‐Myc (OriGene plasmid #MR200811) constructs using Lipofectamine 2000 (Invitrogen). Cells were washed with ice‐cold PBS and lysed in lysis buffer (50 mM HEPES‐KOH, pH 7.4; 1 mM EDTA; 150 mM NaCl; 10% glycerol; 0.5% Triton X‐100) supplemented with protease inhibitors (Thermo Scientific) for 30 min at 4°C followed by centrifugation at 15 000 rpm for 15 min at 4°C. Cleared protein lysate was quantified using BCA assay (Thermo Scientific). One hundred µg of proteins were immunoprecipitated overnight at 4°C using flag (Sigma) or Myc conjugated beads (Pierce). Beads were washed 3 times before elution with Laemmli buffer, and Western blot was performed as outlined below.
2.4. Western blot
Cells were washed with ice‐cold PBS and lysed in lysis buffer (50 mM HEPES‐KOH, pH 7.4; 1 mM EDTA; 150 mM NaCl; 10% glycerol; 0.5% Triton X‐100) supplemented with protease inhibitors (Thermo Scientific) for 30 min at 4°C followed by centrifugation at 15 000 rpm for 15 min at 4°C. Cleared protein lysate was quantified using BCA assay (Thermo Scientific). Protein extracts were denatured in Laemmli buffer (Bio‐rad) for 10 min at 70°C and loaded onto precast 4%–12% bis‐tris protein gels (Invitrogen). Proteins were transferred onto PVDF membranes (Bio‐rad) using Novex Minicell (Invitrogen) per manufacturer's instructions. Membranes were blocked using 5% BSA in TBST for 1 h and incubated overnight at 4°C with the appropriate antibody in 5% BSA in TBST. Membranes were washed 3 times and incubated with HRP‐conjugated secondary antibodies unless using HRP‐conjugated primary antibody. Membranes were visualized using Luminata Forte Western HRP substrate (Millipore) on a Biorad ChemiDoc imaging system. Densitometry was performed using grayscale measurements in ImageJ software (NIH) and normalized to the loading control when appropriate. A list of the antibodies used can be found in Table S1.
2.5. Mouse models
Fabp5−/− mice, on a C57BL/6J background, were kindly provided by Dr. Gokhan Hotamisligil at Harvard University (Boston, MA). All experimental animals used in this study were covered under protocols approved by the Institutional Animal Care and Use Committee of National Jewish Health.
To reconstitute FABP5 in vivo in alveolar macrophages, we used our previously described mouse FABP5 lentiviral vector. 24 Intranasal administration of either the lenti‐FABP5 or lenti‐control vectors was performed on 6‐week old Fabp5−/− mice. The amount of 1 × 108 TU of lenti‐FABP5 or lenti‐control vectors were administered per mouse as described previously. 24
Two weeks after intranasal treatment, Fabp5−/− mice were exposed to smoke from non‐filtered research cigarettes (2R4; University of Kentucky, Lexington, Kentucky, USA), 5 h a day for 6 months using a Teague‐10 smoke chamber. The mice were exposed to a mixture of mainstream (11%) and sidestream (89%) cigarette smoke with a carbon monoxide (CO) concentration of 190 to 300 ppm and a total suspended particle (TSP) of 85 to 120 mg/m3. 26 Between exposures, mice were housed in a holding room with circulating filtered air and given free access to water and standard rodent chow. Control mice were exposed to filtered air. On the last day of air or cigarette smoke exposure, animals under anesthesia were infected with Pseudomonas aeruginosa (106 CFU/mouse) or saline (control) by oropharyngeal aspiration and euthanized 16 h later by intraperitoneal injection of Fatal Plus (2 µl/g of body weight) and tracheotomized. The lungs were perfused with 10 ml phosphate buffered saline (PBS). Right lung lobes were used for PPARγ activity assay. Left lung lobes were inflated and fixed in 10% phosphate‐buffered formalin for immunofluorescence and confocal microscopy.
2.6. Immunofluorescence and confocal microscopy
Inflated and fixed mouse left lung lobes were paraffin‐embedded and cut into 5 μm sections. Human lungs were obtained from the Human Lung Tissue Consortium at National Jewish Health from transplant donors with lungs unsuitable for transplantation. Excised human lungs were dissected from segmental bronchi to distal airways. Each dissected lung tissue specimen was fixed in 10% neutral buffered formalin for 24–36 h, paraffin‐embedded and cut into 5 μm sections. Sections were deparaffinized, rehydrated, and antigen retrieval performed, which consisted of boiling slides in a microwave pressure cooker (Tender Cooker; NordicWare) for 10 min in 0.01 M citrate buffer (pH 6.0). After blocking with 10% normal goat serum (Sigma) in PBS for 1 h, tissue sections were incubated with primary antibodies. Secondary antibodies were applied for 1 h. Sections were mounted with Vectashield medium containing DAPI, and cells were analyzed using a Zeiss LSM 700 confocal microscope and Zen Black software package (Carl Zeiss MicroImaging) with 10 × 63 magnification. A list of antibodies used can be found in Table S1.
2.7. PPARγ activity assay
Frozen lung tissues were pulverized and homogenized in nuclear protein extraction buffer to extract nuclear proteins following manufacturer's instructions (Active Motif, Carlsbad, CA). Nuclear proteins (20 µg per sample) were used to perform PPARγ ELISA (Active Motif, Carlsbad, CA) to quantify PPARγ activation. The results are expressed as absorbance read at 450 nm with a reference wavelength at 650 nm.
2.8. KEGG pathway analysis
We accessed RNA‐seq data available in the NCBI Gene Expression Omnibus (GEO Accession #GSE57148) from a study examining lung tissues of healthy and COPD patients. 27 We separated the samples into two groups based on FABP5 mRNA levels (Figure S1) (FABP5 high, 10 highest; FABP5 low, 10 lowest), and then performed differential expression analysis between the two groups across the genome‐wide dataset. Genes with significantly greater expression (≥2‐fold, p < .05) in FABP5 high vs FABP5 low samples were then subjected to the KEGG pathway analysis using the DAVID Functional Annotation Tool (v6.8; david.ncifcrf.gov).
2.9. Quantitative PCR
RNA was isolated from cell pellets using the QIAGEN RNeasy Plus kit per manufacturer's instructions. 10 ng/µl RNA was used with the Taqman RNA‐to‐Ct 1‐Step kit (Applied Biosystems) to perform qRT‐PCR as previously described 23 for FABP5 (Hs02339439_g1, amplicon length 91) and GAPDH (Hs03929097_g1, amplicon length 58) as an internal control. The comparative threshold cycle method was used to calculate the relative mRNA expression level of FABP5.
2.10. Chromatin immunoprecipitation and qPCR
Primary human alveolar macrophages were treated with Rosiglitazone (10 µM) or vehicle (DMSO) for 1 h. Cells were then fixed with 1% formaldehyde, lysed, and chromatin was sonicated to obtain chromatin fragments of 200 to 1000 base pairs as previously described. 28 Chromatin immunoprecipitations (ChIP) were performed using 3 µg of PPARγ antibody (Diagenode C15410367) or Rabbit Polyclonal IgG (BioLegend, clone Poly29108) and nutated for 2 h at 4°C with ChIP‐Grade Protein G Magnetic Beads. Following cross‐link reversal, DNA was purified using the ChIP DNA Clean and Concentrator kit from Zymo Research. Enrichment of target DNA sequences was analyzed by quantitative PCR (qPCR) using SYBR Green. ChIP‐qPCR primer sequences can be found in Table S2.
2.11. Lentivirus‐mediated FABP5 knockdown and bone marrow‐derived macrophages
THP‐1 cells were transduced with either GFP‐tagged pLL3.7‐shFABP5 or pLL3.7‐shFirefly luciferase as previously described. 29
BMDM were generated in vitro by flushing bone marrow from mouse tibias and femurs. Progenitor cell suspensions were cultured for 7 days in DMEM containing 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin and 20 ng/ml M‐CSF (Peprotech). Macrophage differentiation was confirmed by flow cytometry (>95% positive for F4/80 and CD11b).
2.12. Macrophage polarization
For THP‐1 cells, following shRNA transduction which itself promotes pro‐inflammatory programming, the cells were treated with IFNγ + LPS (20 ng/ml and 10 pg/ml, respectively) for 72 h. Cells were then scraped, counted using trypan blue exclusion, and stained using HLA‐DR APC (BioLegend 307610). To identify the pro‐inflammatory macrophage population, we gated on GFP+ live cells (positive for shRNA transduction), excluded doublets, and finally gated on HLA‐DR+ cells. Samples were acquired using a LSR Fortessa Flow Cytometer (BD), and data analyzed using the FlowJo 10.6 software (Tree Star, Inc.).
For BMDM, after 6 days of culture, cells were scraped, counted and plated at 100 000 cells/well in a 96‐well plate, and treated with IL‐4 (20 ng/ml) or IFNγ + LPS (20 ng/ml and 10 pg/ml, respectively) for 72 h. Supernatants were removed and frozen at −80°C until cytokine determination.
2.13. Cytokine determination
Human cytokines from THP‐1 cell supernatants were measured using ELISA MAX (BioLegend) for IL‐10, and human IL‐6 Elisa kit (abcam). Cytokine concentrations in BMDM cell supernatants were measured using the MSD Pro‐inflammatory panel 1 (mouse) kit (Meso Scale Discovery) per manufacturer's instructions.
2.14. Assay for transposase‐accessible chromatin by sequencing and computational analysis
Bone marrow‐derived macrophages (BMDM) were washed twice with 1X PBS and collected by scraping prior to counting. Approximately 50 000 cells were pelleted and processed in duplicate for Omni‐ATAC‐seq as described previously. 30 Uniquely indexed libraries were pooled and sequenced on an Illumina NovaSeq 6000 using 150 bp paired‐end reads at the Genomics Shared Resource at the University of Anschutz Medical Campus.
Assay for transposase‐accessible chromatin by sequencing (ATAC‐seq) reads were trimmed for adapters, length and quality using the bbduk tool from the BBMap Suite (v. 38.05) with arguments ‘ref = adapters.fa ktrim = r qtrim = 10 k = 23 mink = 11 hdist = 1 ftr = 36 maq = 10 minlen = 20’. Quality control was monitored both pre‐ and post‐trim for all samples using FastQC (v. 0.11.8). Trimmed reads were mapped to the mouse genome (mm10; downloaded from http://igenomes.illumina.com.s3‐website‐us‐east‐1.amazonaws.com/Mus_musculus/UCSC/mm10/Mus_musculus_UCSC_mm10.tar.gz on December 21, 2021, with corresponding hisat2 index files) using hisat2 (v. 2.1.0) in ‘‐‐very‐sensitive’ mode. Resulting SAM files were converted to sorted BAM files using samtools (v. 1.3.1) and then passed to MarkDuplicates (Picard v. 2.6.0) with setting ‘REMOVE_DUPLICATES = true’ to remove PCR and sequencing duplicates. Deduplicated BAM files were converted to bedGraph format and sorted using genomeCoverageBed and sortBed, respectively, from the BEDTools suite (v. 2.25.0). Read coverage was normalized to reads per million mapped using a custom python script and normalized bedGraphs were converted to TDF format using to TDF from igvtools (v. 2.3.75) for visualization in the Integrative Genomics Viewer (IGV). Peak calling was performed using MACS2 (v. 2.1.1) callpeak with ‘‐‐SPMR ‐f BAMPE ‐q 0.00001’ arguments on sorted deduplicated BAM files for each pair of replicates, after which ENCODE‐blacklisted regions (downloaded from https://github.com/Boyle‐Lab/Blacklist/blob/master/lists/mm10‐blacklist.v2.bed.gz on December 20, 2021) were removed using bedtools intersect ‘‐v’ to yield clean peak files. Clean peak files were subjected to Transcription Factor Enrichment Analysis (TFEA; v. 1.1.4; https://github.com/Dowell‐Lab/TFEA 31 ) to detect differential central enrichment of consensus binding motifs represented in the HOCOMOCOv11 core mouse database (downloaded from https://hocomoco11.autosome.ru/final_bundle/hocomoco11/core/MOUSE/mono/HOCOMOCOv11_core_MOUSE_mono_meme_format.meme on December 24, 2021; mapped with a p‐value cutoff of 1e‐5), as described in detail in Ref. [32]. Enrichment scores (E‐scores) for each motif were calculated and corrected for sequence content to reduce known biases associated with local GC enrichment and p‐values are determined using Z‐scores.
2.15. Metabolic phenotyping
Oxygen consumption rate (OCR) was measured using the Agilent Seahorse XFe96 Bioanalyzer. Macrophages (5 × 104 per well) were plated in quadruplets onto Seahorse 96‐well plates and pre‐incubated in Seahorse XF media (DMEM, 10 mM glucose, 1 mM sodium pyruvate 2 mM glutamine, pH 7.4) at 37°C for 1 h in a non‐CO2 incubator. OCR was measured under basal conditions and after sequential addition of Oligomycin, Carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP) and Rotenone/Antimycin A following manufacturer's instructions. Each measured value was reported on Wave software (Agilent Technologies) and normalized to the number of cells in each well. The cell count per well was determined by fluorescent cell counting using the BioTek Cytation 1/5 instrument.
2.16. Statistical analysis
Data are expressed as mean ± SEM. Statistical tests were performed using Prism 8 software (GraphPad). One‐way analysis of variance was used for multiple comparisons, and Tukey's post‐hoc test was applied where appropriate. Student's t‐test was used when only two groups were compared. Differences were considered statistically significant when p < .05.
3. RESULTS
3.1. FABP5 colocalizes and interacts with PPARγ
To determine whether PPARγ and FABP5 are co‐expressed, we used the RNAscope assay to show that the mRNA of both PPARγ and FABP5 were detected in human lung macrophages (Figure 1A). Next, to determine whether PPARγ and FABP5 physically interact with one another, we transfected BEAS‐2B cells, a human airway epithelial cell line exhibiting low PPARγ and FABP5 expression under basal culture conditions, with expression plasmids for mouse Pparγ and mouse Fabp5 that were respectively tagged with Flag and Myc. Upon Fabp5‐Myc immunoprecipitation, Pparγ could be detected in the pull down (Figure 1B) and reciprocally, following Pparγ‐Flag immunoprecipitation, Fabp5 could be detected (Figure 1C), consistent with a physical interaction between FABP5 and PPARγ under these conditions. We further observed FABP5, PPARγ and CD68 colocalization in alveolar macrophages from human lung tissues using immunofluorescence (Figure 1D). Taken together, these data indicate that FABP5 and PPARγ physically interact and colocalize with one another in human lung cells.
FIGURE 1.
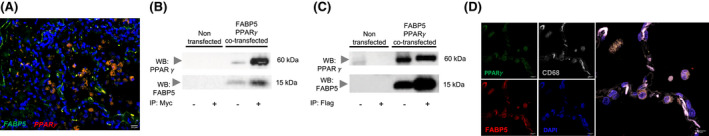
FABP5 and PPARγ are binding partners in cells from human lungs. (A) FABP5 and PPARγ mRNA expression were detected using RNAscope multiplex dual‐fluorescent assays in human lung tissue. Scale Bar 20 µm. Representative picture of 6 independent donor lungs. (B and C) BEAS‐2B cells were left non transfected or were co‐transfected with Fabp5‐Myc and Pparγ‐Flag. Cell lysates (−) or immunoprecipitated samples (+) were run on a SDS gel, transferred onto a nitrocellulose membrane and Western blotted for PPARγ (Top) or FABP5 (Bottom). (B) Myc immunoprecipitation. (C) Flag immunoprecipitation. (D) FABP5 and PPARγ colocalization in CD68‐positive cells in human lung tissue. Scale bar 10 µm. Representative picture of six independent donor lungs
3.2. FABP5 expression modulates PPARγ activity in vivo
We next asked whether FABP5 expression augments PPARγ activity in vivo. We took advantage of our previously described mouse model of FABP5 lentiviral re‐expression in FABP5‐deficient macrophages. 24 To mimic a COPD exacerbation, mice were exposed to 6‐months of cigarette smoke followed by P. aeruginosa infection and harvested 16 h later (Figure 2A). We first confirmed using immunofluorescence that PPARγ colocalized with FABP5 in mouse alveolar macrophages that received Lenti‐FABP5 (Figure 2B). We next compared PPARγ activity between mice that received Lenti‐control and mice that received Lenti‐FABP5. Interestingly, PPARγ activity was significantly enhanced in mouse lung tissue that received Lenti‐FABP5 (Figure 2C). In further demonstration of FABP5 modulation of PPARγ in COPD pathogenesis, we used previously deposited RNA‐seq data (GSE57148) (Figure S1) to show that the PPARγ pathway was the most significantly enriched pathway in human lungs with high levels of FABP5 expression, followed by lysosomal and phagosomal pathways along with lipid metabolism (Figure 2D). In aggregate, our data suggest that FABP5 can enhance PPARγ activity in vivo in lung macrophages.
FIGURE 2.
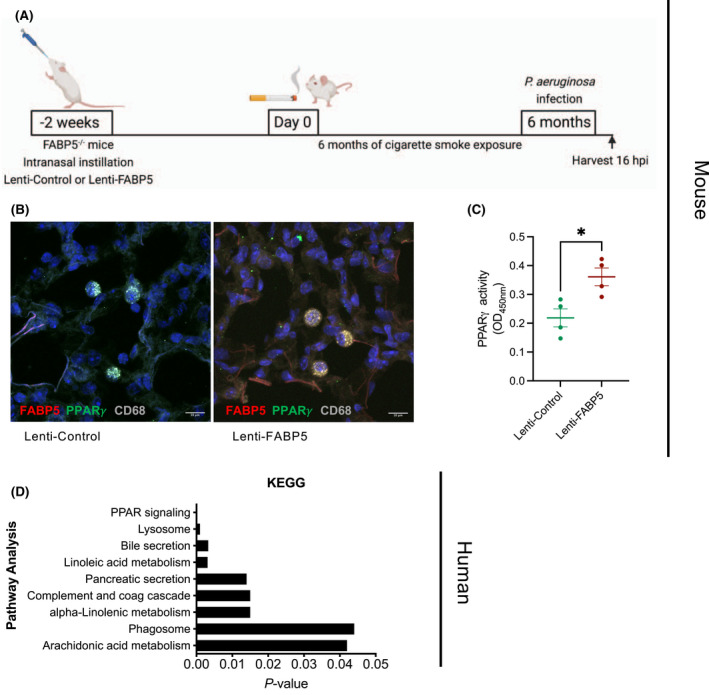
FABP5 promotes PPARγ activity. (A) Schematic of the reconstitution of FABP5 expression using a lentivirus in FABP5‐deficient mice by intranasal instillation. (B) FABP5 and PPARγ colocalization in CD68‐positive cells in lung tissues of FABP5‐deficient mice reconstituted with Lenti‐FABP5 and exposed to cigarette smoke and bacterial infection. Representative picture of two independent experiments with 4–5 mice in each group. (C) PPARγ activity measured by an ELISA‐based assay in whole lung of FABP5‐deficient mice reconstituted with Lenti‐Control (green) or Lenti‐FABP5 (red) and exposed to cigarette smoke and bacterial infection. *p < .05. n = 4 mice per group. (D) KEGG pathway analysis of genes significantly enriched by at least 2‐fold among FABP5high, compared to FABP5low expression among healthy human and COPD lung samples. Data were analyzed from a previous RNA‐seq study (GSE57148) 27
3.3. FABP5 is a direct transcriptional target of PPARγ
To examine whether FABP5 expression is reciprocally controlled by PPARγ, human monocyte‐derived macrophage THP‐1 cells were treated with the PPARγ agonist Rosiglitazone. Rosiglitazone increased both FABP5 mRNA (Figure 3A) and protein expression (Figure 3B). Furthermore, publicly available PPARγ ChIP‐seq peaks previously generated in mouse macrophages 33 were visualized in the UCSC Genome Browser at the Fabp5 locus (Figure S2). Two PPARγ ChIP‐seq peaks were identified within 10 kb down‐stream of the Fabp5 coding region (Figure S2A). Both peaks correspond to regions of moderate‐to‐high conservation with the human genome. Transcription factor binding motif analysis using the MatInspector software (Genomatix) uncovered matches to the PPARγ consensus binding sequence within both PPARγ ChIP‐seq peaks; however, only the binding site match within peak 2 exhibited high conservation between mice and humans (Figure S2B). Additional binding motif analysis indicated that the human ortholog of the conserved peak 2 region contains at least 3 matches to the PPARγ consensus binding motif (Figure S2C). We confirmed increased PPARγ occupancy at all three sites by performing ChIP‐qPCR in primary human alveolar macrophages (Figure 3C). Overall, these results demonstrate that PPARγ acts directly to increase FABP5 expression in primary human alveolar macrophages, and in context with the findings above, indicate that FABP5 and PPARγ exert reciprocal regulatory effects on one another.
FIGURE 3.
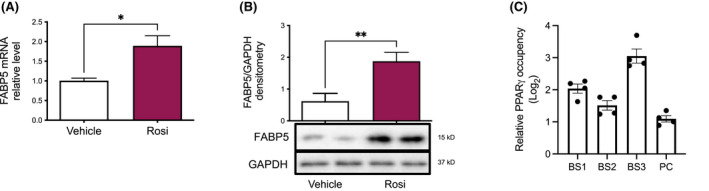
FABP5 transcription is a downstream target of PPARγ. (A) FABP5 mRNA measured by real time quantitative PCR in THP‐1 cells treated with PMA for 72 h and with DMSO (white) or Rosiglitazone (pink) for the last 24 h. *p < .05. (B) FABP5 protein expression quantified by Western blot and densitometry ratio to GAPDH in THP‐1 cells treated with PMA for 72 h and with DMSO (white) or Rosiglitazone (pink) for the last 48 h. **p < .01. (C) ChIP‐qPCR analysis of PPARγ occupancy within the three FABP5 gene binding sites (BS1‐3) and positive control (PC, Evl) in primary human alveolar macrophages. Data are representative of three independent experiments
3.4. FABP5 expression modulates macrophage polarization
To probe the biological significance of FABP5 and PPARγ interaction, we investigated macrophage polarization. Indeed, deletion of PPARγ in macrophages gravely impairs their ability to induce oxidative metabolism with reduced rates of fatty acid β‐oxidation. 34 Consequently, PPARγ null macrophages are unable to fully acquire the anti‐inflammatory phenotype in response to IL‐4 stimulation. 34 To determine whether FABP5‐deficiency would affect macrophage polarization, we first used THP‐1 cells that were transduced with FABP5‐shRNA and compared them to cells that were transduced with Luciferase‐shRNA as control. Upon LPS and IFNγ treatment, FABP5‐shRNA transduced cells demonstrated significantly increased expression of HLA‐DR and increased secretion of IL‐6 and TNFα, all markers of pro‐inflammatory polarization, compared to cells transduced with Luciferase‐shRNA (Figure 4A). Similarly, BMDM from FABP5‐deficient mice had heightened secretion of pro‐inflammatory cytokines, including IL‐6, KC and TNFα, in response to LPS and IFNγ treatment compared to BMDM from WT mice (Figure 4B). However, IL‐10 production, which is a marker of macrophage pro‐resolution polarization, was reduced in THP‐1 cells transduced with FABP5‐shRNA in comparison with cells transduced with Luciferase‐shRNA (Figure 4C). FABP5‐deficient BMDM also failed to induce IL‐10 and IL‐33 production in response to IL‐4 treatment as compared to WT BMDM (Figure 4D). Taken together, these data indicate that down‐regulation of FABP5 in macrophages inhibits IL‐10 and IL‐33 secretion while increasing pro‐inflammatory cytokines and HLA‐DR expression.
FIGURE 4.
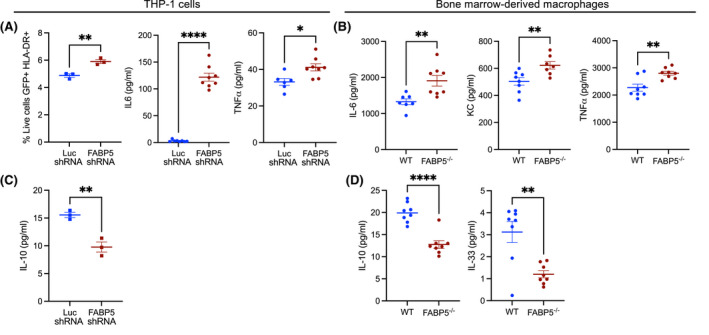
FABP5‐deficiency promotes pro‐inflammatory macrophage programming. (A) HLA‐DR surface expression determined by flow cytometry and inflammatory cytokines measured in the supernatant of THP‐1 cells transduced with Luciferase shRNA (blue) or FABP5 shRNA (red) and treated with IFNγ and LPS for 72 h. *p < .05, **p < .01. (B) Inflammatory cytokines measured in the supernatant of WT (blue) or FABP5−/− (red) BMDM treated with IFNγ and LPS for 72 h. **p < .01. (C) IL‐10 measured in the supernatant of THP‐1 cells transduced with Luciferase shRNA (blue) or FABP5 shRNA (red) and treated with IFNγ and LPS for 72 h. **p < .01. (D) IL‐10 and IL‐33 measured in the supernatant of WT (blue) or FABP5−/− (red) BMDM treated with IL‐4 for 72 h. ****p < .0001
3.5. FABP5‐deficiency increases pro‐inflammatory transcription factor signaling
To define transcriptional mediators of differential genomic responses to FABP5‐deficiency, we performed genome‐wide profiling of chromatin accessibility using the Assay for Transposase‐Accessible Chromatin using sequencing (ATAC‐seq) in unstimulated WT and FABP5‐deficient BMDM cultures. We then used a Transcription Factor Enrichment Analysis (TFEA) pipeline 35 to probe differential central enrichment for consensus transcription factor binding motifs in the ATAC‐seq peak sequences identified in FABP5‐deficient vs WT BMDM. This analysis identified significant differential enrichment for pro‐inflammatory transcription factors binding motifs, including Rela, NF‐κb, Fos, and Jun within open chromatin structures in FABP5‐deficient BMDM in comparison to WT BMDM (Table 1). These data strongly indicate that FABP5‐deficient macrophages exhibit an increased pro‐inflammatory phenotype with increased chromatin accessibility in regions enriched for pro‐inflammatory transcription factor binding motifs.
TABLE 1.
Significantly enriched pro‐inflammatory transcription factors in untreated FABP5‐deficient BMDM
| Gene | Corrected E‐score | Corrected p adj |
|---|---|---|
| TF65 | 0.074642542 | 1.00E‐08 |
| NFKB2 | 0.067278835 | 1.00E‐06 |
| NFKB1 | 0.067123189 | 1.00E‐05 |
| FOSB | 0.066874078 | 1.00E‐03 |
| JUNB | 0.066422989 | 1.00E‐03 |
| FOS | 0.054960969 | 1.00E‐04 |
| RELB | 0.050344295 | 1.00E‐02 |
| JUND | 0.046729207 | 1.00E‐02 |
Motif enrichment distributions of significantly enriched transcription factor binding motifs from TFEA of differentially regulated MACS2‐called ATAC‐Seq peaks between WT and FABP5‐deficient BMDM at baseline (untreated).
Abbreviations: ATAC‐Seq, assay for transposase‐accessible chromatin using sequencing; TFEA, transcription factor enrichment analysis.
3.6. Absence of FABP5 reduces macrophage oxidative metabolism
Macrophage activation and function are controlled by metabolic processes. 36 To determine whether FABP5 also controls macrophage metabolism, we used the Seahorse XF Cell Mito Stress Test. As illustrated in Figure 5A FABP5‐deficient BMDM display a sharp reduction of their mitochondrial oxidative capabilities in comparison with WT BMDM, shown by a large decline of the maximal respiration and spare respiratory capacity (Figure 5B). Since pro‐resolving macrophages are dependent on oxidative phosphorylation as a source of energy, the lack of FABP5 prevents the acquisition of macrophage pro‐resolving programming.
FIGURE 5.
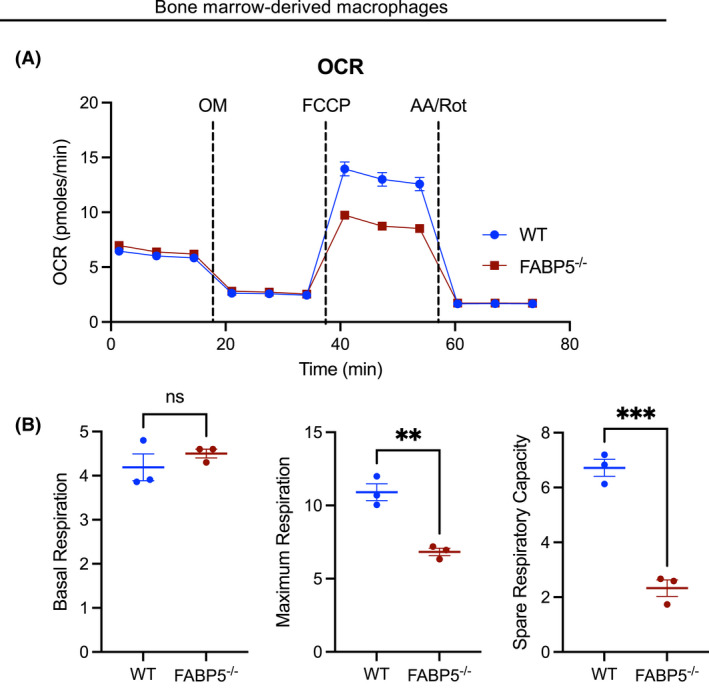
Metabolic differences promoted by FABP5‐deficiency in BMDM. (A) Oxygen Consumption Rate (OCR) measurements (in pmols/min) in WT (blue) and FABP5−/− (red) BMDM. AA, antimycin A; FCCP, Carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone; OM, oligomycin A; Rot, Rotenone. (B) Fundamental parameters of oxidative metabolism including basal respiration, maximum respiration, and spare respiratory capacity (SRC) in WT (blue) and FABP5−/− (red) BMDM. **p < .01, ***p < .001. Data are representative of three independent experiments
4. DISCUSSION
The present study not only confirms FABP5 and PPARγ interaction in lung macrophages, but our findings show that FABP5 plays a critical role in macrophage pro‐resolving programing where it induces the oxidative metabolism. Those results suggest that increasing the expression level of FABP5 could represent a metabolic switch that convert a pro‐inflammatory to a pro‐resolving macrophage (Figure 6). Our studies illustrate the importance of understanding the mechanisms controlling the resolution of inflammation in the lung and will likely lead to the identification of interventional strategies directed towards COPD.
FIGURE 6.
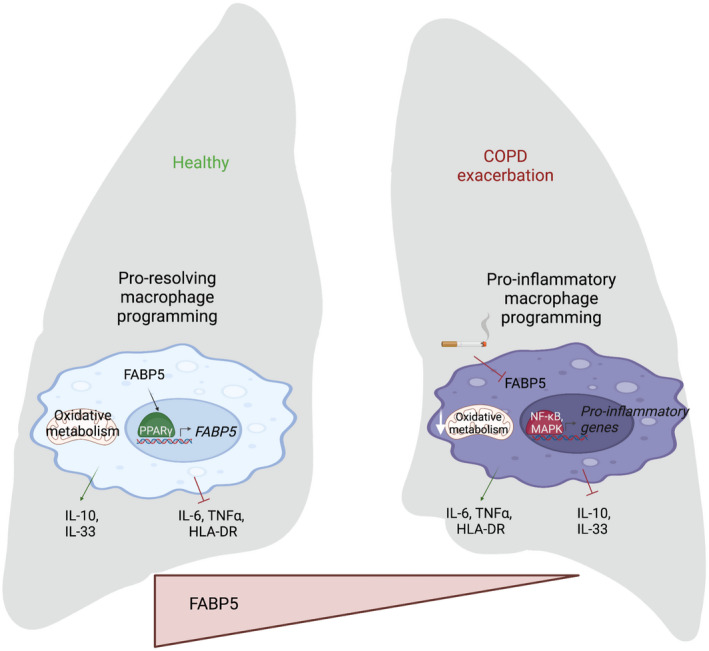
Model of FABP5‐PPARγ crosstalk and macrophage programming in healthy and COPD exacerbation lung. In a healthy lung, the presence of FABP5 increases PPARγ activity which in turn promotes FABP5 transcription and activates the pro‐resolving macrophage programming, including oxidative metabolism as a source of energy and secretion of IL‐10 and IL‐33, while pro‐inflammatory cytokine secretion and HLA‐DR expression are inhibited. In a COPD lung with exacerbation, the absence of FABP5 increases macrophage pro‐inflammatory programming by increasing the secretion of IL‐6 and TNF‐⍺ and chromatin accessibility for pro‐inflammatory transcription factor binding, while decreasing macrophage oxidative metabolism and secretion of pro‐resolving cytokines
Historically, in vitro macrophages have been divided into two distinct polarization states depending on exogenous stimulators: the classically activated phenotype, which is closely linked to pro‐inflammatory responses, and the alternatively activated phenotype that plays a key role in pro‐resolving responses. 37 Although the importance of PPARγ in macrophage reprogramming from a classical to an alternative phenotype is well described, 38 our data suggest that FABP5 and PPARγ regulate each other's expression in a novel positive feedback loop. Here, we demonstrate not only that FABP5 re‐expression in mouse lung tissue significantly increases PPARγ activity, but also that PPARγ agonism directly increases FABP5 expression in primary human alveolar macrophages. In addition, when FABP5 expression is reduced or absent, it leads to a dampening of macrophage pro‐resolving programming, similar to what was described in PPARγ‐deficient macrophages. 34 How this positive feedback loop between FABP5 and PPARγ is contained requires further investigation.
In accordance with the data presented here, we previously showed that in vivo FABP5‐deficient inflammatory macrophages produced increased amounts of iNOS in response to Listeria monocytogenes infection and FABP5‐deficient bone marrow derived‐macrophages (BMDM) produced higher amounts of NO2 − when stimulated with TNFα, IFNγ or LPS. 39 Additionally, we demonstrated that FABP5‐deficient macrophages are better equipped at killing L. monocytogenes than their WT counterpart. 39 In two additional studies, we showed that FABP5‐deficient mice have increased viral‐ and bacterial‐induced lung inflammation compared to WT mice. 23 , 24 The increased bactericidal activity of FABP5‐deficient macrophages suggests a pro‐inflammatory programming, while the persistence of lung inflammation suggests a deficit in pro‐resolving programming. The current study suggests that FABP5‐deficiency prevents macrophage polarization towards a pro‐resolving phenotype, favoring a pro‐inflammatory phenotype instead. Indeed, we demonstrate that polarized FABP5‐deficient macrophages using LPS and IFN‐γ secrete more inflammatory cytokines, while an IL‐4 polarization leads to decreased amounts of IL‐10 and IL‐33. Similar to IL‐10, IL‐33 has been shown to amplify the polarization of pro‐resolving macrophages. 40 Furthermore, using a completely agnostic approach that included ATAC‐seq followed by TFEA, we demonstrate that FABP5‐deficient macrophages, at baseline, exhibit differential chromatin accessibility for pro‐inflammatory transcription factor binding, including Rela, NF‐κB, Fos, and Jun. These data further support the notion that FABP5‐deficient macrophages display a pro‐inflammatory programing.
Interestingly, FABP5 was one of the gene signatures that was identified to be associated with a cluster of monocyte‐derived tumor associated macrophages (Mo‐TAM) that are actively phagocytosing cells, debris and lipid‐rich molecules. 41 TAM have been associated with a poor prognosis in cancer patients, likely due to their ability to suppress inflammation and therefore enhance tumor cell proliferation, promote metastasis, and stimulate angiogenesis. 42 This is another instance where FABP5 expression is shown to modulate anti‐inflammatory macrophages.
Macrophage activation and function are also controlled by metabolic reprogramming, which is now recognized as a new regulatory circuit that shapes immune responses. Indeed, pro‐resolving macrophages mainly rely on oxidative metabolism as a source of energy. 43 In agreement with this observation, we show here that FABP5‐deficient macrophages display greatly reduced mitochondrial activity with mitigated maximum respiration and limited spare respiratory capacity, which may inhibit their pro‐resolving programming. Given the fact that FABP5 is decreased in COPD airway epithelial cells, 44 but also in PBMCs of patients with COPD and further decreased in patients reporting one or more COPD exacerbation episode, 24 it is tempting to speculate that in COPD, FABP5 reduction may promote this chronic lung inflammation (Figure 6).
Macrophage polarization states in COPD are, however, less apparent than in vitro, even though macrophages have been described to play a key role in the development of the disease. 45 Macrophages and neutrophils are thought to be recruited by injured epithelial cells in response to cigarette smoke exposure and release of inflammatory cytokines and chemokines as well as damage‐associated molecular patterns. 46 Macrophages produce matrix metalloproteinases that contribute to tissue‐damage‐induced emphysema. 47 They also contribute to oxidative imbalance, which results in further tissue damage. 48 However, a clear macrophage polarization in COPD has never been proven, and cigarette smoke, rather than COPD was shown to be more influential on macrophage polarization, suggesting that the environment plays a role in macrophage polarization. 49 Additionally, impaired phagocytosis and efferocytosis have been described in COPD 50 , 51 and may, thus, additionally contribute to the lingering inflammation and non‐resolution due to persistence of infectious particles and apoptotic cells.
In aggregate, our data suggest that FABP5 may represent a valuable target for pharmacological intervention, in order to stimulate macrophage pro‐resolving programming. The question that remains is whether activation of FABP5, rather that PPARγ and its many side‐effects, can be a benefit to COPD patients suffering from persistent inflammation.
DISCLOSURES
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Anthony N. Gerber and Fabienne Gally designed the research studies; Manale El Kharbili, Katja Aviszus, Sarah K. Sasse, Xiaoyun Zhao and Fabienne Gally conducted the experiments; Manale El Kharbili, Katja Aviszus, Sarah K. Sasse, and Fabienne Gally acquired and analyzed the data; Karina A. Serban, Susan M. Majka and Anthony N. Gerber provided reagents; Fabienne Gally wrote the manuscript; all authors reviewed, edited and approved the final manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH R01HL141264 (MEK, KA, SKS, XZ, ANG, and FG). Some figures were produced using BioRender.
El Kharbili M, Aviszus K, Sasse SK, et al. Macrophage programming is regulated by a cooperative interaction between fatty acid binding protein 5 and peroxisome proliferator‐activated receptor γ. FASEB J. 2022;36:e22300. doi: 10.1096/fj.202200128R
DATA AVAILABILITY STATEMENT
The ATAC‐seq data that support the findings of this study have been deposited in NCBI's Gene Expression Omnibus, accession number GSE19374. All other data are available in the methods and/or Supporting Information of this article.
REFERENCES
- 1. Serhan CN. Resolution phase of inflammation: novel endogenous anti‐inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101‐137. [DOI] [PubMed] [Google Scholar]
- 2. Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673‐1682. [DOI] [PubMed] [Google Scholar]
- 3. Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355‐2365. [DOI] [PubMed] [Google Scholar]
- 5. Frankenberger M, Menzel M, Betz R, et al. Characterization of a population of small macrophages in induced sputum of patients with chronic obstructive pulmonary disease and healthy volunteers. Clin Exp Immunol. 2004;138:507‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kunz LI, Lapperre TS, Snoeck‐Stroband JB, et al.; Groningen Leiden Universities Corticosteroids in Obstructive Lung Disease Study, G . Smoking status and anti‐inflammatory macrophages in bronchoalveolar lavage and induced sputum in COPD. Respir Res. 2011;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lakshmi SP, Reddy AT, Zhang Y, et al. Down‐regulated peroxisome proliferator‐activated receptor gamma (PPARgamma) in lung epithelial cells promotes a PPARgamma agonist‐reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem. 2014;289:6383‐6393. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8. Auwerx J, Baulieu E, Beato M, et al. A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97:161‐163. [DOI] [PubMed] [Google Scholar]
- 9. Yoon YS, Kim SY, Kim MJ, Lim JH, Cho MS, Kang JL. PPARgamma activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol. 2015;8:1031‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gautier EL, Chow A, Spanbroek R, et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J Immunol. 2012;189:2614‐2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator‐activated receptor‐gamma is a negative regulator of macrophage activation. Nature. 1998;391:79‐82. [DOI] [PubMed] [Google Scholar]
- 12. Jiang C, Ting AT, Seed B. PPAR‐gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82‐86. [DOI] [PubMed] [Google Scholar]
- 13. Lakshmi SP, Reddy AT, Reddy RC. Emerging pharmaceutical therapies for COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:2141‐2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei W, Wang X, Yang M, et al. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone‐induced bone loss. Cell Metab. 2010;11:503‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferroni P, Della‐Morte D, Pileggi A, et al. Pleiotropic effects of PPARgamma agonist on hemostatic activation in type 2 diabetes mellitus. Curr Vasc Pharmacol. 2013;11:338‐351. [DOI] [PubMed] [Google Scholar]
- 16. Hertzel AV, Bernlohr DA. Regulation of adipocyte gene expression by polyunsaturated fatty acids. Mol Cell Biochem. 1998;188:33‐39. [PubMed] [Google Scholar]
- 17. Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator‐activated receptors alpha‐ and gamma‐mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc Natl Acad Sci USA. 2001;98:2323‐2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan NS, Shaw NS, Vinckenbosch N, et al. Selective cooperation between fatty acid binding proteins and peroxisome proliferator‐activated receptors in regulating transcription. Mol Cell Biol. 2002;22:5114‐5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zimmerman AW, Veerkamp JH. Fatty‐acid‐binding proteins do not protect against induced cytotoxicity in a kidney cell model. Biochem J. 2001;360:159‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Helledie T, Antonius M, Sorensen RV, et al. Lipid‐binding proteins modulate ligand‐dependent trans‐activation by peroxisome proliferator‐activated receptors and localize to the nucleus as well as the cytoplasm. J Lipid Res. 2000;41:1740‐1751. [PubMed] [Google Scholar]
- 21. Makowski L, Boord JB, Maeda K, et al. Lack of macrophage fatty‐acid‐binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reynolds JM, Liu Q, Brittingham KC, et al. Deficiency of fatty acid‐binding proteins in mice confers protection from development of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:313‐321. [DOI] [PubMed] [Google Scholar]
- 23. Gally F, Kosmider B, Weaver MR, Pate KM, Hartshorn KL, Oberley‐Deegan RE. FABP5 deficiency enhances susceptibility to H1N1 influenza A virus‐induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2013;305:L64‐L72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rao DM, Phan DT, Choo MJ, et al. Impact of fatty acid binding protein 5‐deficiency on COPD exacerbations and cigarette smoke‐induced inflammatory response to bacterial infection. Clin Transl Med. 2019;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim JH, Park KW, Lee EW, et al. Suppression of PPARgamma through MKRN1‐mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death Differ. 2014;21:594‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Teague SV, Pinkerton KE, Goldsmith M, et al. Sidestream cigarette smoke generation and exposure system for environmental tobacco smoke studies. Inhalation Toxicol. 1994;6:79‐93. [Google Scholar]
- 27. Kim WJ, Lim JH, Lee JS, Lee SD, Kim JH, Oh YM. Comprehensive analysis of transcriptome sequencing data in the lung tissues of COPD subjects. Int J Genomics. 2015;2015:206937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gally F, Sasse SK, Kurche JS, et al. The MUC5B‐associated variant rs35705950 resides within an enhancer subject to lineage‐ and disease‐dependent epigenetic remodeling. JCI Insight. 2021;6:e144294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gally F, Chu HW, Bowler RP. Cigarette smoke decreases airway epithelial FABP5 expression and promotes Pseudomonas aeruginosa infection. PLoS One. 2013;8:e51784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC‐seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rubin JD, Stanley JT, Sigauke RF, et al. Transcription factor enrichment analysis (TFEA) quantifies the activity of multiple transcription factors from a single experiment. Commun Biol. 2021;4:661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gupta A, Sasse SK, Gruca MA, Sanford L, Dowell RD, Gerber AN. Deconvolution of multiplexed transcriptional responses to wood smoke particles defines rapid aryl hydrocarbon receptor signaling dynamics. J Biol Chem. 2021;297:101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lefterova MI, Steger DJ, Zhuo D, et al. Cell‐specific determinants of peroxisome proliferator‐activated receptor gamma function in adipocytes and macrophages. Mol Cell Biol. 2010;30:2078‐2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Odegaard JI, Ricardo‐Gonzalez RR, Goforth MH, et al. Macrophage‐specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tripodi IJ, Allen MA, Dowell RD. Detecting differential transcription factor activity from ATAC‐Seq data. Molecules. 2018;23(5):1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Viola A, Munari F, Sanchez‐Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. 2019;10:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bashir S, Sharma Y, Elahi A, Khan F. Macrophage polarization: the link between inflammation and related diseases. Inflamm Res. 2016;65:1‐11. [DOI] [PubMed] [Google Scholar]
- 39. Rao DM, Phan DT, Choo MJ, Owen AL, Perraud AL, Gally F. Mice lacking fatty acid‐binding protein 5 are resistant to Listeria monocytogenes . J Innate Immun. 2019;11:469‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kurowska‐Stolarska M, Stolarski B, Kewin P, et al. IL‐33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469‐6477. [DOI] [PubMed] [Google Scholar]
- 41. Pombo Antunes AR, Scheyltjens I, Lodi F, et al. Single‐cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci. 2021;24:595‐610. [DOI] [PubMed] [Google Scholar]
- 42. Noy R, Pollard JW. Tumor‐associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu PS, Ho PC. Determining macrophage polarization upon metabolic perturbation. Methods Mol Biol. 2019;1862:173‐186. [DOI] [PubMed] [Google Scholar]
- 44. Green RM, Gally F, Keeney JG, et al. Impact of cigarette smoke exposure on innate immunity: a Caenorhabditis elegans model. PLoS One. 2009;4:e6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kapellos TS, Bassler K, Aschenbrenner AC, Fujii W, Schultze JL. Dysregulated functions of lung macrophage populations in COPD. J Immunol Res. 2018;2018:2349045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378:1015‐1026. [DOI] [PubMed] [Google Scholar]
- 47. Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379:1341‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Groot LES, van der Veen TA, Martinez FO, Hamann J, Lutter R, Melgert BN. Oxidative stress and macrophages: driving forces behind exacerbations of asthma and chronic obstructive pulmonary disease? Am J Physiol Lung Cell Mol Physiol. 2019;316:L369‐L384. [DOI] [PubMed] [Google Scholar]
- 49. Eapen MS, Hansbro PM, McAlinden K, et al. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci Rep. 2017;7:13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bewley MA, Belchamber KB, Chana KK, et al. Differential effects of p38, MAPK, PI3K or rho kinase inhibitors on bacterial phagocytosis and efferocytosis by macrophages in COPD. PLoS One. 2016;11:e0163139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Petrusca DN, Gu Y, Adamowicz JJ, et al. Sphingolipid‐mediated inhibition of apoptotic cell clearance by alveolar macrophages. J Biol Chem. 2010;285:40322‐40332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The ATAC‐seq data that support the findings of this study have been deposited in NCBI's Gene Expression Omnibus, accession number GSE19374. All other data are available in the methods and/or Supporting Information of this article.