

Abstract
Background
Bleeding is a clinically significant issue with all current anticoagulants. Safer antithrombotic strategies are required.
Objectives
To investigate the safety, pharmacodynamics, and pharmacokinetics of BAY 1831865, a humanized, factor XI (FXI)‐directed monoclonal antibody, after single intravenous (i.v.) or subcutaneous (s.c.) doses in healthy volunteers.
Patients/Methods
In a first‐in‐human, phase I study, 70 volunteers were randomly assigned (4:1) to receive single‐dose BAY 1831865 (3.5, 7, 17, 35, 75, or 150 mg i.v. or 150 mg s.c.) or placebo. Adverse events, pharmacodynamics, and pharmacokinetics were evaluated.
Results
In this study, no hemorrhage, or hypersensitivity or infusion‐/injection‐related reactions were reported. Drug‐related adverse events occurred in 3 (5.4%) of 56 volunteers; all were mild and self‐limited. Dose‐dependent prolongation of activated partial thromboplastin time (aPTT) and inhibition of FXI clotting activity was observed with BAY 1831865 i.v. (geometric mean maximum ratio‐to‐baseline: aPTT, range, 1.09–3.11 vs. 1.05 with placebo; FXI, range, 0.70–0.04 vs. 0.91 with placebo). Onset of effect was rapid after i.v. administration, with duration of effect (up to 55 days) determined by dose. BAY 1831865 s.c. had similar pharmacodynamic effects but a slower onset of action. Terminal half‐life increased continuously with increasing i.v. dose (range, 28–208 h), leading to strong and continuous increases in systemic exposure to BAY 1831865. Absolute bioavailability of BAY 1831865 s.c. was 47.2% (95% confidence interval, 30.2–73.7).
Conclusions
BAY 1831865 i.v. or s.c. was well tolerated, with no evidence of bleeding in healthy volunteers. BAY 1831865 exhibited pronounced, sustained dose‐dependent prolongation of aPTT and duration of FXI inhibition.
Keywords: antibodies, monoclonal, humanized; drug‐related side effects and adverse reactions; factor XI; pharmacokinetics; pharmacology
Essentials.
Targeting factor XI (FXI) is a novel anticoagulation strategy that is not expected to impact hemostasis.
FXI‐directed monoclonal antibody BAY 1831865 was investigated in a phase I study.
Single doses of BAY 1831865 were well tolerated in volunteers, with no evidence of bleeding.
BAY 1831865 prolonged contact clotting time with a dose‐dependent duration of effect.
1. INTRODUCTION
Anticoagulation is the cornerstone of treatment for arterial and venous thrombosis, the goal of which is to attenuate thrombosis without impairing hemostasis. 1 While the introduction of direct oral inhibitors of activated factor X and/or thrombin has improved both the safety and convenience of anticoagulation therapy, 2 these agents do not differentiate between thrombin generation contributing to thrombosis and that required for hemostasis. 3 Thus, bleeding remains a concern, particularly in at‐risk and vulnerable patients. 1 , 2
Following efforts to develop safer anticoagulants, factor XI (FXI) has been identified as a potential target. FXI is the zymogen of protease‐activated factor XI (FXIa) and an integral component of the intrinsic (contact) coagulation cascade. 4 Epidemiological studies of individuals with low FXI levels and animal models suggest that FXI plays an important role in thrombin generation while leaving the hemostatic response largely unaffected. 4 Several strategies to inhibit the activity or activation of FXI are being investigated, including antisense oligonucleotide knockdown of FXI, 5 monoclonal antibodies, 6 , 7 small molecules, 8 , 9 , 10 , 11 and aptamers, 12 although the mechanisms differ. Antisense oligonucleotides require up to 4 weeks to reduce FXI levels to within the therapeutic range and show efficacy. 1 Monoclonal antibodies, 6 , 7 aptamers 12 and small‐molecule inhibitors show a rapid onset of action, and the latter can be given orally. 8 , 10 , 11 The mechanism of action may be orthosteric (active site) or allosteric targeting mainly FXIa. 6 , 7 , 13 , 14 , 15 , 16 , 17 Allosteric inhibitors bind away from the active site and reduce FXIa catalytic activity through conformational changes. 7 , 13 , 15 , 16 It is currently unknown whether introducing zymogen inhibition together with modulation of the active site provides additional clinical benefits. 6 Proof‐of‐principle studies have shown that pharmacological inhibition of FXI clotting activity effectively prevents venous thromboembolism in patients undergoing elective knee arthroplasty compared to standard thromboprophylaxis, without an apparent increase in the risk of bleeding. 5 , 18 , 19 , 20
The coagulation cascade consists of several amplification loops and active site–specific inhibitors that may limit the effectiveness of current therapeutics. FXI‐directed monoclonal antibodies currently under investigation either bind directly to the FXI catalytic domain 18 or to a region adjacent to the active site. 7 Thus, an antibody combining FXI zymogen inhibition with active site–focused inhibition may have distinct properties. BAY 1831865 is a humanized, sequence‐optimized, monoclonal antibody that binds specifically to the apple domain 3 of primate FXI (data on file, Bayer AG), which is a binding site for several ligands, including the FXIa substrate factor IX (FIX). FXIa proteolytically cleaves FIX in an interaction requiring apple domain 3. Additionally, BAY 1831865 inhibits the factor XIIa (FXIIa)‐mediated activation of FXI by steric hindrance. Therefore, BAY 1831865 exhibits a dual mode of action, hindering the binding of FIX and generation of FIXa, and inhibiting FXI activation to FXIa, but not FXIa activity. BAY 1831865 prolonged contact clotting time in a concentration‐dependent manner in vitro and ex vivo and demonstrated antithrombotic efficacy in an animal model of arterial‐ and venous‐type thrombosis without increasing bleeding time (data on file, Bayer AG).
This first‐in‐human, phase I, dose‐escalation study aimed to evaluate the safety, pharmacodynamic properties, and pharmacokinetic properties of single intravenous (i.v.) or subcutaneous (s.c.) doses of BAY 1831865 in healthy volunteers. The rationale for developing an s.c. formulation is to provide a safe therapy that can be self‐administered by the patient or given in a home‐based environment for patients who require long‐term anticoagulation.
2. METHODS
2.1. Study population
Eligible participants were healthy White men aged 18–55 years with a body weight of 70–110 kg and a body mass index of 18–30 kg/m2. Key exclusion criteria were known disorders with an increased risk of bleeding, a history of bleeding, or known thrombophilia disorders; clinically relevant findings on electrocardiogram (ECG); current smoker (more than five cigarettes a day); and regular use of therapeutic medicines or recreational drugs.
The study met all local legal and regulatory requirements and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Each participant provided written informed consent prior to study entry.
2.2. Study design
This was a phase I, multicenter, randomized, single‐blind (participant), parallel‐group, placebo‐controlled, dose‐escalation investigation conducted at two centers in Germany (EudraCT: 2018–000787–28; Figure 1). The first participant first visit took place on November 19, 2018 and the last participant last visit on February 18, 2020. Eligible volunteers were randomized 4:1 to receive a single dose of BAY 1831865 or placebo. Participants who met the entry criteria were then randomized centrally by assignation of their unique number to one of the treatments according to a computer‐generated randomization list provided by Data Sciences and Analytics. Initially, BAY 1831865 was given as a 60‐min i.v. infusion in one of up to seven dose steps. The starting dose was 3.5 mg i.v. with subsequent dose escalation planned in up to six sequential steps (i.e., 7, 17, 35, 75, 150, and 300 mg i.v.). Each dose step proceeded only if the previous dose had shown an acceptable safety profile and acceptable tolerability. The highest dose that was judged to have an acceptable safety profile and be well tolerated with i.v. administration was then administered by s.c. injection in a separate cohort.
FIGURE 1.
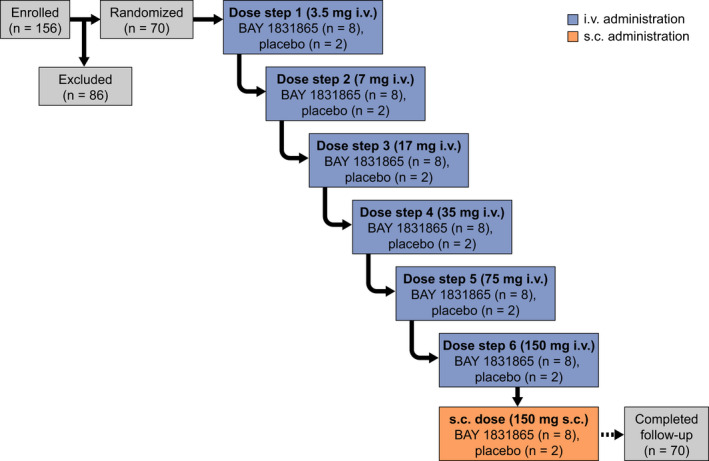
Study schema and participant flow. i.v., intravenous; s.c., subcutaneous
The study included an 8‐day in‐house treatment period from 24 h before to 6 days after study drug administration (days −1 to 7). Volunteers returned for three outpatient visits after i.v. doses (days 14, 21, and 28) and five outpatient visits after the s.c. dose (days 9, 11, 14, 21, and 28). There was a follow‐up visit on day 56 after both i.v. and s.c. doses. An additional visit was also added on day 84 for the BAY 1831865 150 mg dose owing to its long half‐life.
The primary endpoint was the safety and tolerability of BAY 1831865 after doses of 3.5 mg–300 mg administered as i.v. and s.c. doses, as determined by the number of participants with treatment‐emergent adverse events. The secondary endpoints were to investigate the pharmacodynamics (maximal to baseline ratio of activated partial thromboplastin time [aPTT]) and pharmacokinetics (area under the plasma concentration‐time curve from zero to infinity [AUC], AUC divided by dose [AUC/D], maximum observed drug concentration [Cmax], and Cmax divided by dose [Cmax/D]) of BAY 1831865 administered as i.v. and s.c. doses.
2.3. Safety
Adverse events were assessed from screening throughout the study period after BAY 1831865 administration. Adverse events were considered treatment‐emergent if they started or worsened from after study drug administration until day 105. The intensity of each adverse event was classified by the investigator according to intensity (mild, moderate, or severe), as well as seriousness. Protocol‐defined adverse events of special interest were hemorrhage, hypersensitivity and infusion‐ or injection‐related reactions, and antidrug antibodies. Development of antidrug antibodies was investigated in blood samples taken before dosing and on days 7, 14, 21, 28, and 56 after dosing. Plasma concentrations of antidrug antibodies were determined using an electrochemiluminescence ligand‐binding assay with polyethylene glycol precipitation of antidrug antibodies and sulfo‐tagged BAY 1831865 as the detection reagent. Confirmed positive samples showed a reduction in absorbance of more than 28.347% (specificity cut‐point based on a 99.9% confidence interval approach during validation) in the presence of excess BAY 1831865. Other safety examinations (physical examination, body temperature, blood pressure, heart rate, and 12‐lead ECG) were performed throughout the study, along with monitoring of clinical laboratory variables (hematology, serum chemistry, and urinalysis).
2.4. Pharmacodynamics
Citrated blood samples for the analysis of aPTT, FXI activity, FXIa activity, and prothrombin time were collected on day −1 (FXI activity and FXIa activity only); before dosing; at 1, 2, 4, 8, 12, 24, and 36 h after dosing; and on days 3–7, 9, 11, 14, 21, 28, and 56 after BAY 1831865 administration. Samples were centrifuged for plasma preparation within 10 min and stored at −70°C.
Activated partial thromboplastin time was measured using a validated method in which citrated plasma was recalcified in the presence of a standardized quantity of cephalin (a platelet substitute) and a FXII activator (kaolin). The analysis was performed using a STA‐C.K. Prest test kit on an STA Compact Max 3 analyzer (both Diagnostica Stago). The calibration range of the assay was 20–240 s and the analytical range determined was 26.6–107.0 s. Quality control samples with a mean of 30.0–54.0 s were determined with a precision of 2.0%–8.7% coefficient of variation (CV). FXI activity (clotting assay) was assessed using a modified aPTT‐based coagulation test with FXI‐deficient plasma and using reagents and hardware from Instrumentation Laboratory. In brief, plasma samples were diluted and mixed with human plasma that had been immunodepleted of FXI activity. Correction of the clotting time of the FXI‐deficient plasma to that of the mixture was proportional to the residual activity of FXI in the plasma sample, interpolated from a calibration curve. The calibration range of the assay was 0–150.0% and the analytical range determined was below lower limit of quantification (LLOQ) to 116.3%. Quality control samples with a mean of 5.1%–104.7% were determined with a precision of 14.7%–24.4% CV. FXIa activity was measured using a proprietary fluorogenic substrate assay. Prothrombin time was assessed using STA‐Neoplastine CI Plus reagent on an STA coagulation analyzer (both Diagnostica Stago).
Blood samples for thromboelastometry were taken before dosing, 2 h after dosing on day 1, and on days 7, 14, 21, 28, and 56 after BAY 1831865 administration. Assessments of clotting time, clot formation time, maximum clot firmness, and clot lysis at 60 min were performed using rotational thromboelastometry and INTEM reagents (ROTEM system; TEM International GmbH).
Blood samples for the analysis of thrombosis biomarkers (D‐dimer, thrombin‐antithrombin complex [TAT], and prothrombin fragment 1.2 [F1.2]) were taken before dosing and on days 2, 7, 14, 21, 28, and 56 after BAY 1831865 administration. D‐dimer levels were measured using a particle‐enhanced immunoturbidimetric method (STA‐Liatest D‐Di) on an STA coagulation analyzer (both Diagnostica Stago), and TAT and F1.2 levels were measured using commercially available enzyme‐linked immunosorbent assays (ELISAs; Enzygnost TAT micro ELISA and Enzygnost F1+2 monoclonal test kit; Siemens Healthcare Diagnostics Products GmbH).
Bleeding time was evaluated before dosing, and at 2 h and on day 7 after administration using the Surgicutt system (Accriva Diagnostics Inc.). A standard incision (5 mm length, 1 mm depth) was made on the forearm, and blood was absorbed from the incision using filter paper every 30 s until bleeding stopped. During the measurement, the blood pressure monitor was inflated to 40 mmHg.
2.5. Pharmacokinetics
Blood samples for pharmacokinetic analysis were taken before dosing; at 0.25, 0.5, 1, 2, 4, 8, 12, 24, and 36 h after dosing on day 1; and on days 3–7, 9, 11, 14, 21, 28, and 56 after BAY 1831865 administration. All samples were stored at or below −65°C and analyzed up to 113 days after sampling. Plasma concentrations of BAY 1831865 were determined using a ligand‐binding assay with electrochemiluminescence readout. BAY 1831865 was captured using a biotinylated anti‐idiotypic antibody to bind the BAY 1831865‐FXI complex and was detected using a sulfo‐TAG‐labeled antigen‐binding fragment (Fab) antibody. The calibration range of the procedure was from 1.0 mg/L (LLOQ) to 20.0 mg/L (upper limit of quantification). Quality control samples (blank plasma spiked with BAY 1831865 concentration range, 2.50–15.0 mg/L) were determined with an accuracy of 104%–110% and a precision of 6.21%–9.39%.
The main pharmacokinetic parameters were AUC, AUC/D, Cmax, and Cmax/D. Time to Cmax (tmax), half‐life associated with the terminal slope (t1/2), total body clearance of drug (CL), volume of distribution at steady state after intravascular administration, and apparent volume of distribution during terminal phase after extravascular administration (Vz/F) were also considered.
Pharmacokinetic parameters were calculated using the model‐independent, compartment‐free method with Phoenix 8.1 software in conjunction with the noncompartmental analysis tool plug‐in (release 1.0; Certara). Calculations were based on the actual sampling and dosing times, and only values above the LLOQ were used.
2.6. Statistics
No formal sample size determination was performed for this exploratory study. Data were presented by BAY 1831865 dose step and separated by route of administration (i.v. or s.c.).
Pharmacodynamic results were presented as the change from baseline and the ratio‐to‐baseline including all observed data. Maximal ratio‐to‐baseline of aPTT, the main pharmacodynamic parameter, was compared for each dose of BAY 1831865 versus placebo with an exact Wilcoxon rank‐sum test (one‐sided significance level α = 0.05) in sequential order (i.e., if all previous null hypotheses for the endpoint had been rejected, the next null hypothesis was tested) starting with the highest i.v. dose. The comparison between the s.c. dose and placebo was performed in the end in any case. Hodges‐Lehmann estimates of the location shift between treatment groups were calculated along with 90% confidence intervals (CIs).
Exploratory analyses of variance (ANOVA), including the factor treatment, were performed to detect deviations from dose‐proportionality of BAY 1831865 i.v. using log‐transformed values of AUC/D and Cmax/D. The absolute bioavailability of BAY 1831865 s.c. was also investigated using exploratory ANOVA, including the factor treatment, by comparing the log‐transformed values of AUC and Cmax for the same dose given via i.v. and s.c. administration.
Data were analyzed by descriptive statistical methods, and all planned statistical analyses were exploratory in nature. There was no imputation for missing data. Statistical evaluation was performed using SAS software, version 9.4 (SAS Institute Inc.).
3. RESULTS
3.1. Disposition and demographics
In total, 70 healthy male volunteers were randomized to study treatment (BAY 1831865 i.v., n = 48; placebo i.v., n = 12; BAY 1831865 s.c., n = 8; placebo s.c., n = 2; Figure 1). All 70 volunteers were treated as planned, completed the study, and were included in the safety analysis and pharmacodynamic analysis sets. Key baseline characteristics are shown in Table 1.
TABLE 1.
Demographics and baseline characteristics (safety analysis set, n = 70)
| BAY 1831865 | Placebo | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
3.5 mg i.v. n = 8 |
7 mg i.v. n = 8 |
17 mg i.v. n = 8 |
35 mg i.v. n = 8 |
75 mg i.v. n = 8 |
150 mg i.v. n = 8 |
150 mg s.c. n = 8 |
i.v. n = 12 |
s.c. n = 2 |
||
| Race, n (%) | ||||||||||
| White | 8 (100) | 8 (100) | 8 (100) | 8 (100) | 8 (100) | 8 (100) | 8 (100) | 12 (100) | 2 (100) | |
| Age, years | ||||||||||
| Mean (SD) | 37.0 (8.3) | 42.6 (6.5) | 39.6 (11.8) | 41.6 (8.6) | 34.0 (6.4) | 43.3 (10.5) | 28.0 (3.7) | 36.4 (10.3) | 48.0 (2.8) | |
| Range | 26–46 | 32–52 | 25–55 | 29–54 | 26–46 | 30–55 | 23–34 | 19–55 | 46–50 | |
| Weight, kg | ||||||||||
| Mean (SD) | 80.4 (9.0) | 87.4 (5.5) | 82.3 (6.6) | 84.9 (8.9) | 84.7 (7.5) | 84.4 (7.5) | 82.5 (9.5) | 83.0 (8.4) | 76.1 (5.4) | |
| Range | 70.6–95.5 | 78.5–98.0 | 72.2–90.7 | 71.3–101.2 | 74.4–96.2 | 71.9–93.7 | 73.9–98.7 | 70.8–95.8 | 72.2–79.9 | |
| Height, cm | ||||||||||
| Mean (SD) | 175.3 (8.2) | 180.9 (4.6) | 182.1 (6.4) | 182.3 (6.7) | 182.3 (7.1) | 183.0 (5.8) | 183.1 (5.5) | 182.5 (4.6) | 172.5 (3.5) | |
| Range | 160.0–184.0 | 176.0–188.0 | 171.0–191.0 | 171.0–191.0 | 175.0–198.0 | 171.0–191.0 | 176.0–193.0 | 178.0–195.0 | 170.0–175.0 | |
| BMI, kg/m2 | ||||||||||
| Mean (SD) | 26.2 (2.9) | 26.7 (1.6) | 24.9 (2.4) | 25.6 (2.7) | 25.5 (1.9) | 25.3 (2.5) | 24.7 (3.1) | 24.9 (2.5) | 25.6 (0.8) | |
| Range | 22.3–29.8 | 24.3–29.0 | 21.6–28.4 | 21.8–29.2 | 23.0–28.4 | 21.2–29.0 | 19.8–29.8 | 21.4–28.6 | 25.0–26.1 | |
Abbreviations: BMI, body mass index; i.v., intravenous; s.c., subcutaneous; SD, standard deviation.
3.2. Safety
Overall, 30 (53.6%) of 56 volunteers who received single doses of BAY 1831865 i.v. or s.c. reported at least one treatment‐emergent adverse event (TEAE) compared to 6 (42.9%) of 14 volunteers who received placebo i.v. or s.c. (Table 2). All TEAEs were of mild to moderate intensity and had resolved by study end. There were no serious adverse events or deaths reported with any dose of BAY 1831865. The most frequently observed TEAEs with BAY 1831865 i.v. or s.c. were nasopharyngitis (n = 11, 19.6% vs. n = 3, 21.4% with placebo), headache (n = 4, 7.1% vs. n = 1, 7.1% with placebo), back pain (n = 4, 7.1% vs. n = 1, 7.1% with placebo), and oropharyngeal pain (n = 3, 5.4% vs. no cases with placebo; Table S1 in supporting information). Increases in C‐reactive protein and liver function tests were each reported in two volunteers (3.4%; Table S1). All other TEAEs (n = 22) with BAY 1831865 i.v. or s.c. were each reported in one volunteer only.
TABLE 2.
Safety summary (safety analysis set, n = 70)
| BAY 1831865 | Placebo | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
3.5 mg i.v. n = 8 |
7 mg i.v. n = 8 |
17 mg i.v. n = 8 |
35 mg i.v. n = 8 |
75 mg i.v. n = 8 |
150 mg i.v. n = 8 |
150 mg s.c. n = 8 |
i.v. n = 12 |
s.c. n = 2 |
|
| Any TEAE, n (%) | 5 (62.5) | 7 (87.5) | 3 (37.5) | 4 (50.0) | 4 (50.0) | 4 (50.0) | 3 (37.5) | 4 (33.3) | 2 (100.0) |
| Mild | 4 (50.0) | 7 (87.5) | 3 (37.5) | 3 (37.5) | 4 (50.0) | 3 (37.5) | 3 (37.5) | 4 (33.3) | 2 (100.0) |
| Moderate | 1 (12.5) | 0 | 0 | 1 (12.5) | 0 | 1 (12.5) | 0 | 0 | 0 |
| Treatment‐related TEAE, n (%) | 0 | 1 (12.5) | 0 | 1 (12.5) | 0 | 0 | 1 (12.5) | 0 | 0 |
| Mild | 0 | 1 (12.5) | 0 | 1 (12.5) | 0 | 0 | 1 (12.5) | 0 | 0 |
| TEAE related to procedures required by protocol | 1 (12.5) | 1 (12.5) | 0 | 1 (12.5) | 0 | 0 | 0 | 0 | 0 |
| TEAE leading to discontinuation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Serious adverse event, n (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAE of special interest, a n (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Abbreviations: i.v., intravenous; s.c., subcutaneous; TEAE, treatment‐emergent adverse event.
Hemorrhage, hypersensitivity and infusion‐ or injection‐related reactions, and antidrug antibodies.
TEAEs considered to be related to BAY 1831865 by the investigator were reported in 3 (5.4%) of 56 volunteers. One volunteer received BAY 1831865 7 mg i.v. and reported elevated white blood cells, elevated C‐reactive protein, elevated liver function tests, and muscle pain in both upper legs. Each of the events were of mild intensity and resolved without any treatment. Another volunteer reported mild urinary urgency 30 min after administration of BAY 1831865 35 mg i.v., which resolved without treatment. The third volunteer reported mild redness at the injection site after s.c. administration of BAY 1831865 150 mg. The event lasted for 45 min and resolved without treatment.
At least one occurrence of antidrug antibody formation was confirmed in nine volunteers (BAY 1831865 35 mg i.v., n = 1; 75 mg i.v., n = 1; 150 mg i.v., n = 4; placebo i.v., n = 3). As positive antibody titers did not coincide with altered plasma concentrations of BAY 1831865 or any unexpected pharmacodynamic or safety findings, they were not classified as adverse events. No other TEAEs of special interest (hemorrhage, and hypersensitivity and infusion‐ or injection‐related reactions) were observed. Vital signs and ECG parameters remained within physiological ranges during the study. With the exception of aPTT (see Pharmacodynamics section in this article), no general trends for BAY 1831865 on any specific laboratory parameters, including hemoglobin, were observed.
3.3. Pharmacodynamics
3.3.1. aPTT
Dose‐dependent increases in aPTT and duration of aPTT prolongation were observed with BAY 1831865 i.v. (Figure 2). Geometric mean values for the maximum ratio‐to‐baseline for aPTT, the main pharmacodynamic variable, ranged from 1.09 (3.5 mg i.v.) to 3.11 (150 mg i.v.) compared to 1.05 for placebo; increases were significant for all i.v. doses of BAY 1831865 vs. placebo (Wilcoxon rank‐sum test P < .05). The interindividual variability of this effect was low (coefficient of variation [CV] range, 3.17%–10.01%). Onset of effect with BAY 1831865 i.v. was rapid, with pronounced increases in aPTT being evident within 1 h of initiating BAY 1831865 infusion for doses of 7 mg or greater (Figure 2). Offset of effect and duration of aPTT prolongation was dose‐dependent, with geometric mean aPTT ratio‐to‐baseline values returning to baseline levels between 2 days (7 mg) and 55 days (75 mg and 150 mg) after BAY 1831865 infusion (Figure 2).
FIGURE 2.
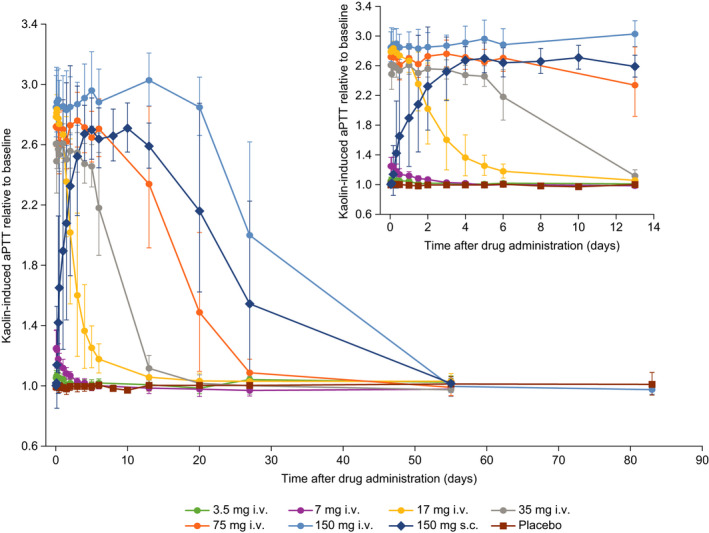
Effects of increasing doses of BAY 1831865 on activated partial thromboplastin time (aPTT) in healthy male volunteers. i.v., intravenous; s.c., subcutaneous
In contrast to i.v. administration, aPTT increased slowly with BAY 1831865 150 mg s.c. until about 5 days after dosing (Figure 2). The maximum geometric mean ratio‐to‐baseline with BAY 1831865 150 mg s.c. was 2.78 (CV, 5.54%) which was significant versus placebo (Wilcoxon rank‐sum test P < .05). There were pronounced interindividual differences in the time when the plateau of drug effect was reached (range, from about 8 h to 4 days). Geometric mean aPTT ratio‐to‐baseline remained elevated at 27 days but had returned to baseline at 55 days after s.c. administration (Figure 2).
3.3.2. FXI and FXIa activities
Dose‐dependent inhibition of FXI activity (Figure 3) and FXIa activity (Figure S1 in supporting information) was observed with increasing i.v. doses of BAY 1831865. The onset of effect of BAY 1831865 was rapid, with pronounced decreases in the activity of both FXI and FXIa being evident shortly after administration of any i.v. dose. The maximum geometric mean ratio‐to‐baseline of FXI activity ranged from 0.70 (3.5 mg) to 0.04 (17 mg) compared to 0.91 for placebo; the interindividual variability of this effect was low to moderate (CV range, 4.76%–43.17%). The maximum geometric mean ratio‐to‐baseline of FXIa activity ranged from 0.55 (3.5 mg) to 0.01 (17 mg) compared to 0.86 for placebo; the interindividual variability of this effect was low to high (CV range, 10.79%–148.03%). FXI activity returned to baseline between 13 days (3.5 mg) and 55 days (35 mg, 75 mg, and 150 mg) after BAY 1831865 i.v. administration. Further dose escalation of BAY 1831865 above 150 mg was not considered because the extrapolated mean FXI activity was expected to drop to below 20% from baseline for 45 days or more with doses greater than 150 mg (protocol‐defined stopping rule for dose escalation). For FXIa activity, values returned to baseline after 13 days with the 3.5 mg i.v. dose and were almost, but not fully, restored to baseline during the observation period for other i.v. doses; the geometric mean ratios to baseline 55 days after administration of BAY 1831865 35, 75, and 150 mg i.v. were 0.98, 0.97, and 0.82, respectively.
FIGURE 3.
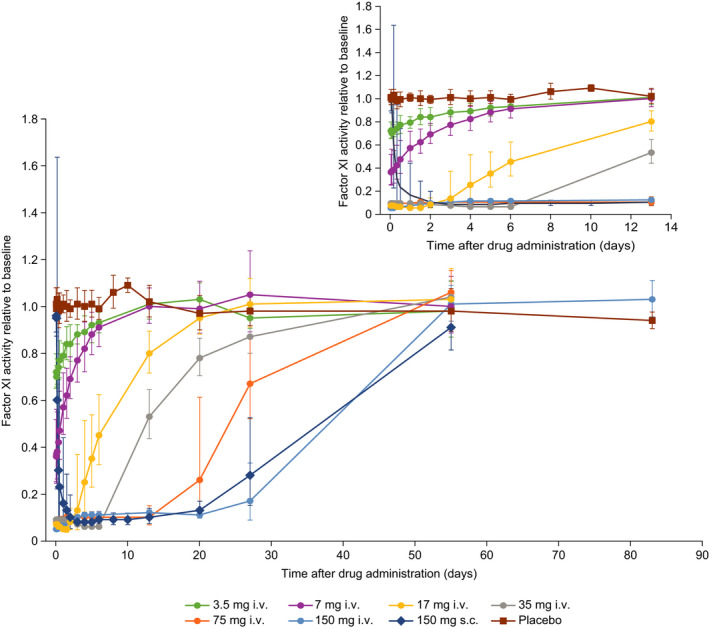
Effects of increasing doses of BAY 1831865 on factor XI activity in healthy male volunteers. i.v., intravenous; s.c., subcutaneous
After s.c. administration of BAY 1831865 150 mg, FXI activity and FXIa activity declined slowly, and the minimum activity of both variables was reached about 3 days after dosing. The maximum geometric mean ratios to baseline for FXI activity and FXIa activity were 0.06 (CV, 16.86%) and 0.02 (CV, 23.91%), respectively. Baseline FXI activity values were attained at 55 days, but FXIa activity remained below baseline at this time (last sampling time point).
3.3.3. Rotational thromboelastometry
Following i.v. administration of BAY 1831865, clotting time and clot formation time were increased in a dose‐dependent manner, whereas maximum clot firmness and clot lysis were not influenced by BAY 1831865. For clotting time, the maximum geometric mean ratio‐to‐baseline ranged from 1.2 (3.5 mg i.v.) to 3.4 (150 mg i.v.) compared to 1.2 for placebo (Figure S2 in supporting information). For clot formation time, the maximum geometric mean ratio‐to‐baseline ranged from 1.1 (3.5 mg i.v.) to 1.9 (75 mg and 150 mg i.v.) compared to 1.1 for placebo. The onset of effect after s.c. administration of BAY 1831865 150 mg on both clotting time and clot formation time was slower, with respective maximum geometric mean ratios to baseline of 2.9 and 1.7.
3.3.4. Other pharmacodynamic variables
Plasma levels of D‐dimer, TAT, and F1.2 were not clearly influenced by BAY 1831865 i.v. or s.c. (Table S2 in supporting information). In addition, BAY 1831865 i.v. or s.c. did not induce any changes in prothrombin time (Table S2). An analysis of absolute values and ratios to baseline for bleeding time revealed no changes with BAY 1831865 i.v. or s.c. compared to placebo (data not shown).
3.4. Pharmacokinetics
The pharmacokinetic analysis set included 48 volunteers who received BAY 1831865 i.v. and eight volunteers who received BAY 1831865 s.c. A summary of the pharmacokinetics of BAY 1831865 after i.v. or s.c. administration is provided in Table 3. In the lowest dose group (3.5 mg i.v.), plasma concentrations were only measurable in three volunteers, and geometric mean values could not be calculated for all variables.
TABLE 3.
Pharmacokinetics of BAY 1831865 in plasma (pharmacokinetic analysis set, n = 56)
| Parameter | BAY 1831865 | ||||||
|---|---|---|---|---|---|---|---|
|
3.5 mg i.v. a n = 3 |
7 mg i.v. n = 8 |
17 mg i.v. n = 8 |
35 mg i.v. n = 8 |
75 mg i.v. n = 8 |
150 mg i.v. n = 8 |
150 mg s.c. n = 8 |
|
| AUC, mg.h/L | – | 85.9 (58.1) | 390 (32.4) | 1530 (28.1) | 3720 (32.6) | 10 900 (16.7) | 5160 (61.1) |
| AUC/D, h/L | – | 12.3 (58.1) | 23.0 (32.4) | 43.6 (28.1) | 49.6 (32.6) | 72.9 (16.7) | 34.4 (61.1) |
| Cmax , mg/L | 1.32 (24.9) | 2.15 (11.8) | 5.73 (26.2) | 13.7 (12.8) | 21.7 (13.7) | 48.3 (9.47) | 10.9 (67.4) |
| Cmax/D, /L | 0.378 (24.9) | 0.307 (11.8) | 0.337 (26.2) | 0.392 (12.8) | 0.289 (13.7) | 0.322 (9.47) | 0.0729 (67.4) |
| tmax , b h | 1.02 (1.00–2.00) | 1.00 (0.98–2.00) | 1.47 (1.00–3.98) | 2.00 (0.98–4.00) | 1.93 (1.00–1.95) | 1.94 (1.00–3.95) | 96.0 (48–239) |
| t1/2 , h | – | 28.4 (52.8) | 61.6 (31.2) | 107 (50.8) | 164 (68.0) | 208 (19.8) | 217 (36.5) |
| CL or CL/F, c L/h | – | 0.0815 (58.1) | 0.0435 (32.4) | 0.0229 (28.1) | 0.0202 (32.6) | 0.0137 (16.7) | 0.0291 (61.1) |
| VSS or Vz/F, c L | – | 3.36 (12.3) | 3.63 (25.5) | 3.35 (24.3) | 4.67 (25.0) | 4.17 (11.1) | 9.10 (68.6) |
Data presented as geometric mean (% coefficient of variance) unless otherwise stated.
Abbreviations: AUC, area under the plasma concentration‐time curve from zero to infinity; AUC/D, AUC divided by dose; CL, total body clearance of drug; CL/F, total body clearance of drug calculated after extravascular administration (e.g., apparent oral clearance); Cmax, maximum observed drug concentration; Cmax/D, Cmax, divided by dose; i.v., intravenous; s.c., subcutaneous; t1/2, half‐life associated with the terminal slope; tmax, time to Cmax; Vss, volume of distribution at steady state after intravascular administration; Vz/F, apparent volume of distribution during terminal phase after extravascular administration.
Plasma concentrations were only measurable in three volunteers, and not all parameters could be calculated.
Median (range).
CL and Vss (i.v. administration); CL/F and Vz/F (s.c. administration).
Following i.v. administration, there was a dose‐dependent increase in plasma concentration of BAY 1831865 (Figure 4). Geometric mean Cmax increased from 1.32 mg/L (3.5 mg) to 48.3 mg/L (150 mg). Dose‐normalized Cmax values varied slightly among the six i.v. dose steps, although no deviation from dose‐proportionality was indicated by the explorative ANOVA. Median tmax was similar for all i.v. doses, ranging from 1.0 h to 2.0 h after the start of infusion. However, there was an overproportional increase in exposure to BAY 1831865 with increasing i.v. dose. This was reflected by continuous and strong increases in AUC/D, with geometric mean values ranging from 12.3 h/L (7 mg) to 72.9 h/L (150 mg); the explorative ANOVA indicated a deviation from dose‐proportionality (P < .0001). Geometric mean values for CL decreased from 0.0815 L/h (7 mg) to 0.0137 L/h (150 mg), and t1/2 increased continuously from 28.4 h (7 mg) to 208 h (150 mg) across the i.v. dose range.
FIGURE 4.
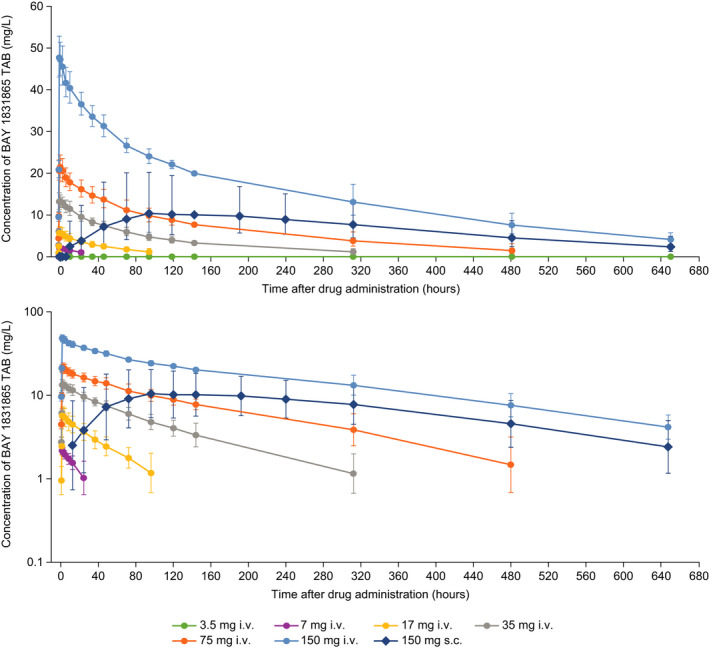
Plasma concentrations of BAY 1831865‐TAB presented with linear (upper panel) and semi‐logarithmic (lower panel) scales in healthy male volunteers. i.v., intravenous; s.c., subcutaneous
After s.c. administration of BAY 1831865 150 mg, AUC was about half that observed after i.v. administration of BAY 1831865 150 mg (geometric mean, 5160 mg.h/L vs. 10 900 mg.h/L), and Cmax was reduced by almost 80% (geometric mean, 10.9 mg/L vs. 48.3 mg/L). Median tmax after s.c. administration was 96.0 h. The t1/2 after s.c. administration was similar to that observed after i.v. administration (geometric mean, 217 h vs. 208 h). Geometric mean values for CL calculated after extravascular administration and Vz/F were 0.0291 L/h and 9.10 L, respectively. The absolute bioavailability of BAY 1831865 150 mg s.c. vs. 150 mg i.v. was 47.2% (95% CI, 30.2%–73.7%).
4. DISCUSSION
This first‐in‐human, phase I, dose‐escalation study demonstrated that single i.v. and s.c. doses of the FXI‐directed monoclonal antibody BAY 1831865 were well tolerated in healthy men and did not raise any unexpected safety signals. Drug‐related TEAEs were reported in three volunteers across all i.v. and s.c. doses. All the reported events were mild in intensity and resolved without treatment during the study period. Notably, there were no reports of bleeding or hemorrhage with BAY 1831865 during the study, and no changes in bleeding time, assessed using the Surgicutt test, were observed compared to placebo. There were also no hypersensitivity or infusion‐related reactions reported, suggesting that the risk of these events with BAY 1831865 appears to be low. While antidrug antibodies were documented in six volunteers who received BAY 1831865, their presence did not coincide with any changes in safety profile or pharmacodynamic properties and was not considered clinically relevant.
In this study, BAY 1831865 was found to influence the intrinsic pathway, as evidenced by increases in aPTT, without affecting prothrombin time, a screening test for the extrinsic pathway. Administration of BAY 1831865 i.v. was associated with dose‐dependent increases in aPTT and pronounced dose‐dependent inhibition of FXI and FXIa activities. Near‐total inhibition of FXI and FXIa activities was achieved with BAY 1831865 doses of 17 mg or greater. Onset of effect following administration of BAY 1831865 i.v. on aPTT, FXI, and FXIa activity was rapid and evident within the first hour of completing drug infusion. Offset of effect was slower and dictated by BAY 1831865 dose, with aPTT and FXI activity returning to baseline levels within 55 days for all doses. Restoration of FXIa activity was almost, but not fully, achieved during the study period. From these observations, further dose escalation of BAY 1831865 above 150 mg was not considered because the mean FXI activity was predicted to drop to below 20% from baseline for 45 days or more. The pharmacodynamic profile of BAY 1831865 after s.c. administration was similar but with a delayed onset of action. A maximal effect on aPTT was evident approximately 5 days after s.c. administration, with return to baseline observed after 55 days. BAY 1831865 also increased clotting time and clot formation time in a dose‐dependent manner as expected, but it did not appear to influence biomarkers of coagulation (D‐dimer, TAT, and F1.2) or other clotting parameters (prothrombin time, maximum clot firmness, and clot lysis).
The clinical pharmacokinetic profile of BAY 1831865 was characterized by a long t1/2 and low volume of distribution, properties typically observed with parenterally administered monoclonal antibodies. 21 Notably, the t1/2 of BAY 1831865 increased continuously (from 28 h to 208 h) and CL decreased (from 0.08 L/h to 0.01 L/h) over the 7–150 mg dose range. This, in turn, led to a strong and continuous increase in systemic exposure to BAY 1831865 after i.v. administration over the same dose range. A possible explanation for this dose‐dependent (nonlinear) pharmacokinetic behavior is the involvement of a saturable metabolic process, that is, target‐mediated elimination that becomes saturated because of finite quantities of target antigen. 21 In support of this explanation, typical plasma concentrations of FXI dimers are approximately 5 mg/L (30 nM). 22 Under the assumption of one‐to‐one binding of BAY 1831865 to FXI, FXI is expected to be fully saturated by BAY 1831865 at plasma concentrations of 5 mg/L which occur at a dose of 17 mg i.v. (mean Cmax, 5.73 mg/L). These pharmacokinetic properties support an intermittent dosing schedule (possibly monthly), as increased exposure would be anticipated after repeat dosing of BAY 1831865. After s.c. administration, absorption of BAY 1831865 into the systemic circulation was slow (tmax, 96 h) and overall exposure was lower than after i.v. administration of the same dose (150 mg). The absolute bioavailability of s.c. BAY 1831865 was relatively low (47.2%), which is probably attributable to proteolytic degradation in the interstitial fluid or lymphatic system. 21
The rapid onset of action and long t1/2 of BAY 1831865 are consistent with other FXI/FXIa‐directed monoclonal antibodies 6 , 7 but are distinct from antisense oligonucleotides, which require up to 4 weeks to reduce FXI levels to within the therapeutic range, 1 and small‐molecule inhibitors, which have a relatively short t1/2. 10 The clinical implications of the near‐total inhibition of FXI activity provided by BAY 1831865 have yet to be investigated, but it is of interest that abelacimab, an FXI‐directed antibody that achieves a similar level of inhibition of FXI activity, has antithrombotic efficacy but without an increased risk of bleeding. 18 Similarly, inhibition of FXI via an antisense oligonucleotide or inhibition of FXIa with the monoclonal antibody osocimab before surgery in a total knee arthroplasty setting did not result in an excess in bleeding. 5 , 19 Thus, no restrictions were made to the daily routines of the participating volunteers during the out‐of‐hospital phase of this first‐in‐human study with no bleeding adverse events reported, providing additional reassurance of low bleeding risk. Moreover, a commercially available formulation of recombinant factor VIIa (NovoSeven) has been shown to reverse the effects of BAY 1831865 on aPTT in vivo, 23 although it is debated whether there is clinical need for such a reversal agent with long‐acting antithrombotics for emergency situations. 1 It is anticipated that the sustained anticoagulation provided by BAY 1831865 could be relevant for conditions when fluctuating drug levels due to missed doses or low trough levels are not tolerable (e.g., for patients with mechanical heart valves). The optimal dose and schedule for BAY 1831865 have yet to be investigated in clinical studies.
Several limitations inherent to phase I studies apply to this study. This study was designed to investigate single doses of BAY 1831865 in a population of healthy volunteers. The study population was small, excluded women, and consisted exclusively of White men. Therefore, data collected may not apply to other populations. We recognize that women should be included as early as possible in clinical research targeting diseases that are prevalent in both sexes. However, no clinical data from any FXI‐directed monoclonal antibodies were available when this study was conducted. Furthermore, while no clear association between plasma FXI levels and excessive menstrual bleeding has been found in patients with FXI deficiency, 24 FXI deficiency has been associated with an increased risk of heavy menstrual bleeding. 25 Thus, we considered it an unacceptable risk to include healthy women in this initial clinical trial of BAY 1831865 until some safety, pharmacokinetic, and pharmacodynamic data had been generated in men. Further studies are therefore required to characterize the pharmacology, safety, and immunogenicity of BAY 1831865 after multiple doses, and to understand the effects of this agent in broader populations. Furthermore, although participants were blinded to treatment, investigators were not, and treatment evaluations were nonblinded. An evaluation of FXI concentrations was performed in this study, but the data were inconclusive owing to technical problems and have not been presented for this reason.
In conclusion, in this first‐in‐human study in 70 healthy White men, BAY 1831865 was well tolerated when given as single i.v. or s.c. doses of up to 150 mg, with no evidence of bleeding or increased bleeding time. BAY 1831865 exhibited rapid and sustained dose‐dependent prolongation of aPTT and inhibition of FXI clotting activity, when the duration of effect was determined by dose. Further clinical studies are warranted to investigate the anticoagulation efficacy of BAY 1831865 and determine the optimal dose and schedule for clinical use.
CONFLICTS OF INTEREST
B. Nowotny, D. Thomas, S. Schwers, S. Wiegmann, W. Prange, and A. Yassen are all employees of Bayer AG. Additionally, W. Prange holds shares in Bayer AG and S. Schwers holds a patent for Bayer AG. S Boxnick is an employee of CRS Clinical Research Services Wuppertal GmbH.
AUTHOR CONTRIBUTIONS
Concept and design: B. Nowotny, D. Thomas, S. Schwers, S. Wiegmann, W. Prange, A. Yassen, S. Boxnick. Analysis and/or interpretation of data: B. Nowotny, D. Thomas, S. Schwers, S. Wiegmann, W. Prange, A. Yassen, S. Boxnick. Critical writing or revising the intellectual content: B. Nowotny, D. Thomas, S. Schwers, S. Wiegmann, W. Prange, A. Yassen, S. Boxnick. Final approval of the version to be published: B. Nowotny, D. Thomas, S. Schwers, S. Wiegmann, W. Prange, A. Yassen, S. Boxnick.
Supporting information
App S1
ACKNOWLEDGMENTS
This study was funded by Bayer AG, Berlin, Germany. Editorial support was provided by Oxford PharmaGenesis, Oxford, UK with funding from Bayer AG.
Nowotny B, Thomas D, Schwers S, et al. First randomized evaluation of safety, pharmacodynamics, and pharmacokinetics of BAY 1831865, an antibody targeting coagulation factor XI and factor XIa, in healthy men. J Thromb Haemost. 2022;20:1684–1695. doi: 10.1111/jth.15744
Manuscript handled by: Zhi‐Cheng Jing
Final decision: Zhi‐Cheng Jing, 25 April 2022
Funding information
This study was funded by Bayer AG, Berlin, Germany.
REFERENCES
- 1. Weitz JI, Chan NC. Novel antithrombotic strategies for treatment of venous thromboembolism. Blood. 2020;135:351‐359. [DOI] [PubMed] [Google Scholar]
- 2. Bauer KA. How effective and safe is factor XI inhibition in preventing venous thrombosis? JAMA. 2020;323:121‐122. [DOI] [PubMed] [Google Scholar]
- 3. Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. 2015;13:1383‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gailani D, Gruber A. Factor XI as a therapeutic target. Arterioscler Thromb Vasc Biol. 2016;36:1316‐1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buller HR, Gailani D, Weitz JI. Factor XI antisense oligonucleotide for venous thrombosis. N Engl J Med. 2015;372:1672. [DOI] [PubMed] [Google Scholar]
- 6. Koch AW, Schiering N, Melkko S, et al. MAA868, a novel FXI antibody with a unique binding mode, shows durable effects on markers of anticoagulation in humans. Blood. 2019;133:1507‐1516. [DOI] [PubMed] [Google Scholar]
- 7. Thomas D, Thelen K, Kraff S, et al. BAY 1213790, a fully human IgG1 antibody targeting coagulation factor XIa: first evaluation of safety, pharmacodynamics, and pharmacokinetics. Res Pract Thromb Haemost. 2019;3:242‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perera V, Wang Z, Luettgen J, et al. First‐in‐human study to assess the safety, pharmacokinetics and pharmacodynamics of BMS‐986177/JNJ‐70033093, a direct, reversible, small molecule Factor XIa inhibitor in healthy volunteers. Eur Heart J. 2020;41:3362.32620942 [Google Scholar]
- 9. Pollack CV Jr, Kurz MA, Hayward NJ. EP‐7041, a factor XIa inhibitor as a potential antithrombotic strategy in extracorporeal membrane oxygenation: a brief report. Crit Care Explor. 2020;2:e0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perera V, Luettgen JM, Wang Z, et al. First‐in‐human study to assess the safety, pharmacokinetics and pharmacodynamics of BMS‐962212, a direct, reversible, small molecule factor XIa inhibitor in non‐Japanese and Japanese healthy subjects. Br J Clin Pharmacol. 2018;84:876‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thomas D, Kanefendt F, Schwers S, Unger S, Yassen A, Boxnick S. First evaluation of the safety, pharmacokinetics and pharmacodynamics of BAY 2433334, a small molecule targeting coagulation factor Xia. J Thromb Haemost. 2021;19(10):2407‐2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Woodruff RS, Ivanov I, Verhamme IM, Sun MF, Gailani D, Sullenger BA. Generation and characterization of aptamers targeting factor XIa. Thromb Res. 2017;156:134‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al‐Horani RA, Abdelfadiel EI, Afosah DK, et al. A synthetic heparin mimetic that allosterically inhibits factor XIa and reduces thrombosis in vivo without enhanced risk of bleeding. J Thromb Haemost. 2019;17:2110‐2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Al‐Horani RA, Afosah DK. Recent advances in the discovery and development of factor XI/XIa inhibitors. Med Res Rev. 2018;38:1974‐2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Al‐Horani RA, Desai UR. Designing allosteric inhibitors of factor XIa. Lessons from the interactions of sulfated pentagalloylglucopyranosides. J Med Chem. 2014;57:4805‐4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Argade MD, Mehta AY, Sarkar A, Desai UR. Allosteric inhibition of human factor XIa: discovery of monosulfated benzofurans as a class of promising inhibitors. J Med Chem. 2014;57:3559‐3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wong PC, Crain EJ, Bozarth JM, et al. Milvexian, an orally bioavailable, small‐molecule, reversible, direct inhibitor of factor XIa: in vitro studies and in vivo evaluation in experimental thrombosis in rabbits. J Thromb Haemost. 2022;20:399‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verhamme P, Yi BA, Segers A, et al. Abelacimab for prevention of venous thromboembolism. N Engl J Med. 2021;385:609‐617. [DOI] [PubMed] [Google Scholar]
- 19. Weitz JI, Bauersachs R, Becker B, et al. Effect of osocimab in preventing venous thromboembolism among patients undergoing knee arthroplasty: the FOXTROT randomized clinical trial. JAMA. 2020;323:130‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weitz JI, Strony J, Ageno W, et al. Milvexian for the prevention of venous thromboembolism. N Engl J Med. 2021;385:2161‐2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493‐507. [DOI] [PubMed] [Google Scholar]
- 22. Pathak M, Kaira BG, Slater A, Emsley J. Cell receptor and cofactor interactions of the contact activation system and factor XI. Front Med (Lausanne). 2018;5:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wallisch M, Buchmüller A, Johnson J, et al, editors. Activated (a) factor (F) VII for hemostasis enhancement in FXI‐ and FXIa inhibitor‐treated primates. Paper presented at: International Society on Thrombosis and Haemostasis Congress; 2017. [Google Scholar]
- 24. Wiewel‐Verschueren S, Mulder AB, Meijer K, Mulder R. Factor 11 single‐nucleotide variants in women with heavy menstrual bleeding. J Obstet Gynaecol. 2017;37:912‐918. [DOI] [PubMed] [Google Scholar]
- 25. Wiewel‐Verschueren S, Arendz IJ, M. Knol H, Meijer K. Gynaecological and obstetrical bleeding in women with factor XI deficiency ‐ a systematic review. Haemophilia. 2016;22:188‐195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
App S1