

Abstract
The last 2 years have presented previously unforeseen challenges in pulmonary medicine. Despite the significant impact of the SARS‐CoV‐2 pandemic on patients, clinicians and communities, advances in the care and understanding of interstitial lung disease (ILD) continued unabated. Recent studies have led to improved guidelines, better understanding of the role for antifibrotics in fibrosing ILDs, prognostic indicators and novel biomarkers. In this concise contemporary review, we summarize many of the important studies published in 2021, highlighting their relevance and impact to the management and knowledge of ILD.
Keywords: fibroblast, fibrosis, interstitial lung disease, lung, respiratory
INTRODUCTION
Interstitial lung disease (ILD) refers to a heterogeneous group of over 200 disorders with varied clinical presentation and prognoses. Major advances have occurred in the last decade in this area, and despite the ongoing COVID‐19 pandemic and its related challenges, there were major advances in the research of ILD in 2021, including pathogenic mechanisms of disease, novel potential therapeutic targets and identification of novel biomarkers.
MECHANISMS OF DISEASE
Key points.
The genetics of ILD is complex, with modern GWAS identifying numerous candidate genes involved in pathogenesis and possible therapeutic targets.
Immune cells play a key role in fibrogenesis, and antifibrotics have been shown to reduce B‐cell‐induced fibroblast migration and activation.
Several novel potential therapeutic pathways have been described in IPF and ILD, including targeting fibrocytes, exosomes, matrix metalloproteinases, IL‐23 and fatty acids.
Genetics and epigenetics
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease, with a median survival of 3–5 years, and it is the archetypal form of fibrotic ILD. 1 , 2 Recent genome‐wide association studies (GWAS) have consistently demonstrated that the gain‐of‐function MUC5B promoter variant is the dominant genetic risk factor for the development of IPF. 3 , 4 In IPF patients, excessive MUC5B protein can be found in areas of lung fibrosis and in other cells and has been demonstrated to be measurable in bleomycin mouse models of fibrosis. 5 What remains unclear is the disconnection between relatively high prevalence of MUC5B mutations in the general population, yet a rarity of IPF. Dobrinskikh et al. examined separate diversity outbred strains of mice administered bleomycin. They identified a variety of phenotypes including strains with extensive fibrosis and high Muc5b expression, limited fibrosis and low expression, and strains that had no relationship between Muc5b and fibrosis. 6 Further cluster analysis indicated that other factors likely are at work in concert with or independent of Muc5b to promote fibrosis.
A rarer genetic association with fibrotic ILD is that of telomerase‐related gene (TRG) mutations and short telomere syndromes, and van der Vis et al. sought to examine the risk of pulmonary fibrosis in family members of TRG mutation carriers who did not inherit the mutation. 7 Following whole‐exome sequencing of 99 probands, they identified 20 probands with TRG variants and screened each proband's family and identified five family members with fibrosis and no TRG mutation. They further showed that non‐TRG variant carrier patients displayed short telomere length in the lung or in the blood, likely inherited from a TRG mutation carrier ancestor, suggesting that just because the TRG mutation was not inherited the risk may still be present.
To further understand the complex molecular landscape of IPF, Konigsberg et al. applied an integrative systems biology multiomic approach, utilizing DNA methylomic, coding and non‐coding transcriptomic and proteomic data from IPF lungs compared to control. 8 This analysis confirmed previously validated pathways known to be dysregulated in IPF and identified novel molecular features. Interestingly, this multiomic approach suggested that 18 regions of differential methylation and 10 long non‐coding RNAs may be important in the regulation of matrix metalloproteinase 7 (MMP7), 9 , 10 providing potential novel therapeutic avenues for this pathway.
Furthermore, McErlean et al. investigated epigenetic changes in alveolar macrophages in patients with IPF. 11 They identified a distinct DNA methylation pattern in alveolar macrophages compared to circulating monocytes and revealed differentially methylated positions (n = 11) and differentially methylated regions (n = 49) between IPF and controls. Interestingly, the differentially methylated positions encompassed genes involved in glucose and lipid metabolism and the changes were associated with IPF disease severity, and importantly the epigenetic heterogeneity was not associated with accelerated ageing. Moreover, a study by Cui et al. demonstrated the role of lactate in driving epigenetic alteration of macrophage function, 12 and that increased lactate of myofibroblast origins in fibrotic mouse lungs was taken up by macrophages and resulted in histone lactylation, resulting in the upregulation of profibrotic genes.
Fibroblast and epithelial cell biology
It has been previously shown that B cells when exposed to certain microbial antigens are activated and release profibrotic metalloproteases, cytokine and chemokines. 10 , 13 , 14 , 15 Ali et al. examined B cells from patients with IPF, 16 demonstrating that B cells secrete profibrotic and pro‐inflammatory proteins in response to microbial antigens (CpG or β‐glucan). Microbial antigens increased the secretion of IL‐6, IL‐8 and MMP7 in B cells; subsequently, the supernatant from the antigen‐exposed B cells was used to stimulate fibroblasts. They demonstrated that fibroblast migration and activation was significantly increased after exposure to these activated B cells compared to control. They further demonstrated that both nintedanib and pirfenidone altered the secretory profile of B cells and nintedanib, but not pirfenidone, reduced B‐cell‐induced fibroblast migration and activation, indicating separate mechanisms of action and differential effects (Figure 1).
FIGURE 1.
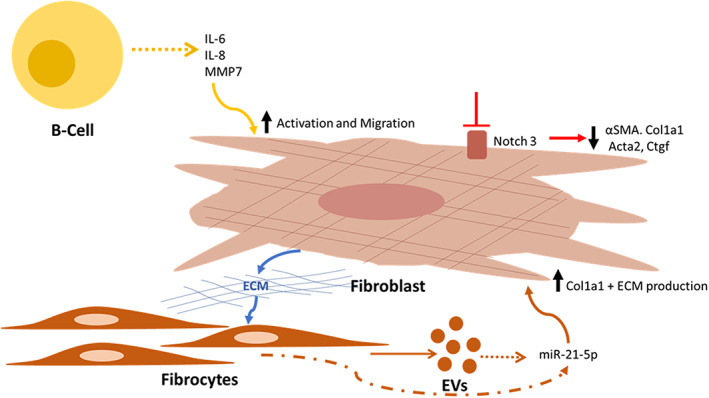
Recently reported pathogenic mechanisms in interstitial lung disease. B cells secrete profibrotic and pro‐inflammatory proteins in response to microbial antigens, and increased IL‐6, IL‐8 and matrix metalloproteinase 7 (MMP7) lead to activated fibroblasts and increased migration. 16 The Notch pathway is involved in myofibroblast differentiation, and reduced Notch3 signalling results in less α‐smooth muscle actin (αSMA), collagen type 1 (Col1a1) and other profibrotic gene expression from fibroblasts. 17 Abnormal extracellular matrix (ECM) in fibrotic lung tissue increases the expression of cellular and extracellular vesicle (EV) expressed miR‐21‐5p from fibrocytes, which exert pro‐fibrotic effects and upregulation of Col1a1 in fibroblasts leading to further ECM 19
Fibroblast differentiation to myofibroblasts and their activation is an essential element in the pathogenesis of fibrosis and the Notch pathway has been reported to be involved in myofibroblast differentiation. To determine the relevance of Notch3 (activity of which is present in collagen‐expressing cells of the lung), Vera et al. used a Notch3‐knockout mouse model. 17 Following bleomycin injury, Notch3‐knockout animals had significantly fewer α‐smooth muscle actin (αSMA)‐expressing myofibroblasts and less induction of profibrotic genes compared to controls. Further in vitro experiments demonstrated that Notch3 participates in fibroblast survival and differentiation. Hence, the Notch pathway appears to be involved in fibrogenesis in the lung and offers a potential novel target (Figure 1). While fibroblast activation is persistent in fibrotic disease, in other conditions such as wound healing, it is a transient process, and this is also seen in resolution of fibrosis in bleomycin injury models; hence, a clearer understanding of the deactivation of fibroblasts may give insight into novel therapeutic targets. Tan et al. characterized gene signature of fibroblasts isolated from control mice with those isolated at days 14 and 30 after bleomycin exposure, representing the peak of extracellular matrix (ECM) deposition and an early stage of fibrosis resolution. 18 RNA sequencing analysis of freshly sorted fibroblasts identified novel candidate antifibrotic genes and pathways. Aldh2 and Nr3c1 were two such genes that were decreased at day 14 and reversed at day 30, and enhanced expression of these genes in cultured fibroblasts resulted in reduced profibrotic gene expression, fibronectin deposition and collagen gel compaction, indicative of important roles in fibroblast deactivation. 18
Potential novel therapeutic pathways
Other novel pathways of fibrosis reported include the interplay between fibrocytes and fibroblasts. Sato et al. demonstrated that ECM in fibrotic lung tissue increases the expression of cellular and extracellular vesicles that expressed miR‐21‐5p from fibrocytes, which exert pro‐fibrotic effects and upregulation of collagen type 1 (Col1a1) in fibroblasts. 19 Melfin was demonstrated to be of importance to cell senescence and transforming growth factor beta (TGFβ) fibrogenesis, 20 and it was shown that alveolar type 2 (AT2) cells isolated from IPF demonstrate transcriptomic features of cellular senescence, and that senescence rather than loss of AT2 cells promotes progressive fibrosis, which occurs in a p53‐dependant manner. Targeting senescence‐related pathways may be a promising therapeutic potential in early fibrosis. 21
MMPs, IL‐23 and fatty acids were all recently examined as potential therapeutic targets in IPF. Espindola et al. reported the utility of targeting MMP9 with an anti‐MMP9 antibody, demonstrating increased MMP9 expression in airway cells from IPF lungs and blocking MMP9 with an anti‐MMP9 antibody showed mixed response in patient‐derived cells. This response was associated with interferon expression; thus, targeting MMP9 signalling may have benefit in a subset of IPF patients who have sufficient expression of type 1 interferons. 22 Zhang et al. demonstrated that IL‐23 upregulated αSMA and collagen‐I/III protein, reduced caveolin‐1 and induced invasion of AT1 cells, indicative of promoting epithelial‐to‐mesenchymal transition (EMT). They further revealed that IL‐23‐induced EMT was dependent on mammalian target of rapamycin (mTOR) signalling and could be reversed by rapamycin inhibition of mTOR. Transcriptional analysis of rheumatoid arthritis‐associated ILD lung tissue and fibroblasts identified increased IL‐23 expression, supporting a role for IL‐23 in driving fibrosis through mTOR‐driven EMT in epithelial cells. 23 Additionally, Senoo et al. showed that IL‐23 levels were higher in the bronchoalveolar lavage (BAL) fluid of IPF patients with acute exacerbation and treating mice with anti‐IL‐23 antibody attenuated airway inflammation and fibrosis, further supporting the potential for IL‐23 blockade as novel therapy. 24 It has been previously reported that long‐chain fatty acids are increased in IPF lungs 25 ; however, Kim et al. analysed IPF lungs with an unbiased lipidomic screen and identified that stearic acid levels were decreased and further demonstrated that stearic acid significantly reduced the TGFβ‐induced expression of αSMA and collagen type 1 in fibroblasts. 26 Moreover, stearic acid reduced fibrosis in a bleomycin mouse model, indicating that stearic acid may have novel antifibrotic properties 26 (Figure 2).
FIGURE 2.
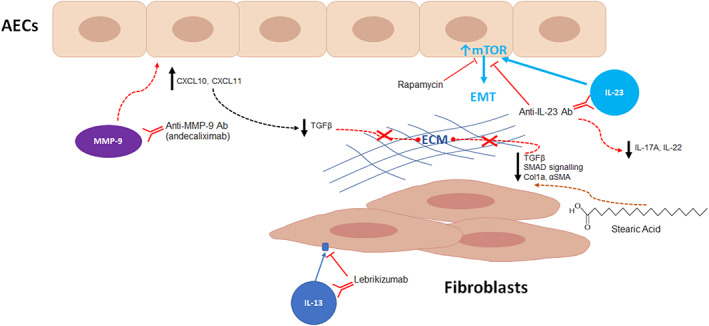
Novel potential therapeutic targets in interstitial lung disease. Matrix metalloproteinase 9 (MMP9) blockade with the monoclonal antibody, andecaliximab, led to response in lungs with a type I interferon‐rich environment with increased concentrations of CXCL10 (CXC motif chemokine ligand 10) and by reducing transforming growth factor beta (TGFβ) signalling and decreasing fibrotic extracellular matrix (ECM) deposition. Epithelial–mesenchymal transition (EMT) creates an environment facilitating fibrosis following alveolar epithelial cell injury. 22 IL‐23‐induced EMT is dependent on mammalian target of rapamycin (mTOR) signalling and can be reversed by rapamycin, 23 or potentially targeted blockade by anti‐IL‐23 antibody which can attenuate airway inflammation in acute exacerbations. 24 Stearic acid reduced the TGFβ‐induced expression of α‐smooth muscle actin (αSMA) and collagen type 1 (Col1a1) in fibroblasts, indicating potential novel antifibrotic properties. 26 Recent studies of IL‐13 blockade using the monoclonal antibody, lebrikizumab, was demonstrated to be well tolerated and safe, but lebrikizumab alone or in combination with pirfenidone did not reduce the rate of forced vital capacity 87
DIAGNOSIS AND PROGNOSTICATION
Key points.
International clinical practice guidelines on hypersensitivity pneumonitis, sarcoidosis and Sjogren's‐related lung disease provide an invaluable adjunct for clinicians and highlight the need for further research in this area.
Both the proportion of adipose tissue and the vascular volume on CT imaging are associated with the development of interstitial lung abnormalities and may be modifiable risk factors.
Peripheral blood monocyte count predicts progression of IPF.
As the awareness of ILD subtypes increases and with therapies available, it is paramount that ILD is diagnosed early, correctly classified and there is a growing need to appropriately stratify and prognosticate. 27
Society guidelines and documents
The American Thoracic Society (ATS) published clinical practice guidelines on the diagnosis of hypersensitivity pneumonitis (HP) in 2020, 28 and the CHEST guidelines for ‘Diagnosis and Evaluation of Hypersensitivity Pneumonitis’ were published in 2021. 29 These guideline documents address several parallel diagnostic questions with the ultimate aim of providing guidance on the diagnosis of HP and both guidelines' diagnostic approaches share numerous similarities. Importantly, the CHEST guidelines address several diagnostic issues not covered by the ATS. The most noticeable difference is the probabilistic reasoning during the stepwise diagnostic process, leading to importance in establishing likelihood of antigen exposure and not dichotomizing as yes or no. The other notable difference is that the CHEST guidelines do not recommend BAL for lymphocyte analysis in all patients considered to have HP, especially in patients with a typical HRCT pattern and compelling exposure history. However, both documents reported that all evidence was of low quality and that the diagnosis of HP should be a patient‐centred approach with significant multidisciplinary input. 29
The European Respiratory Society published clinical practice guidelines on treatment of sarcoidosis, 30 with recommendations on treatment of pulmonary, cutaneous, cardiac and neurological sarcoidosis as well as constitutional symptoms, particularly fatigue. An update in guidelines for treatment of sarcoidosis has been long awaited, given that the prior iteration was published in 1999.31, 32 however, the authors of the current guideline felt that based on the available evidence the indications for treatment of pulmonary sarcoidosis remain unclear and are mostly on case‐to‐case basis; clearer outcome measures and better clinical trials are needed to adequately support future recommendations. Also published were the CHEST ‘Consensus Guidelines for Evaluation and Management of Pulmonary Disease in Sjogren's’. 33 While most of the evidence was considered low quality due to the lack of randomized, placebo‐controlled trials, the recommendations span topics of assessment and management of airway disease, ILD and lymphoproliferative disease in Sjogren's.
Screening and early diagnosis
Due to the progressive nature of most ILD subtypes, the need for earlier diagnosis and screening is increasingly apparent. Interstitial lung abnormalities (ILAs) refer to specific computed tomography (CT) findings that are potentially compatible with ILD. 34 The Multi‐Ethnic Study of Atherosclerosis (MESA) is an ongoing prospective cohort study that enrolled 6814 adults aged 45–84 years between 2000 and 2002 with numerous subsequent follow‐up examinations. Anderson et al. evaluated adipose tissue and the risk of ILD; while previous work examining the link between BMI and parenchymal lung disease had been inconsistent, the authors demonstrated that in the MESA cohort, for every doubling in pericardial adipose tissue volume there was 20% increase in the odds of ILAs and for visceral adipose tissue there was 30% increased odds and lower forced vital capacity (FVC) measures. 35 They concluded that increased pericardial and/or abdominal visceral adipose tissue were associated with early lung injury and thus, adipose tissue may represent a modifiable risk factor for ILAs and an early indicator of ILD risk. 35 Another large analysis of CT findings in the Framingham Heart Study found that lower pulmonary vascular volumes on CT were associated with greater odds of ILAs and restrictive spirometry, identifying another possible early predictor of significant ILD. 36
With the emergence of novel approaches, there remains the possibility that lung biopsy need can be further reduced in the diagnostic work up of ILDs. Richeldi et al. 37 reported that combining HRCT imaging findings with molecular transcription signatures from transbronchial biopsy could accurately identify a usual interstitial pneumonia (UIP) pattern and potentially avoid the need for surgical lung biopsy. The Envisia Genomic Classifier diagnostic test provides a binary result of either positive or negative for UIP and, when combined with HRCT patterns, had a sensitivity of 72.9% and specificity of 90.6%. Together with the growing use of cryobiopsy, this type of approach may further reduce the need for traditional surgical biopsies. 38 Moreover, prior preliminary studies demonstrated that endobronchial optical coherence tomography (EB‐OCT), a minimally invasive, real‐time, optical imaging modality that provides in vivo 3D imaging, had potential as a low‐risk method for evaluation of ILD without biopsy. 39 Nandy et al. examined EB‐OCT in 27 patients and reported that this was 100% sensitive and specific for diagnosing UIP and clinical IPF. 40 Due to the ability to image several areas of the lung during bronchoscopy and avoiding the need for general anaesthesia, this is an attractive future diagnostic option and warrants further study. Another emerging non‐invasive tool in pulmonary medicine is the analysis of exhaled breath volatile organic compounds, which are generated by cells during different metabolic and pathological states. 41 Moor et al. performed exhaled breath analysis on 370 individuals (322 ILD; 48 controls) using eNose technology. eNose sensors accurately differentiated ILD from controls and reliably distinguished IPF from other ILDs, with receiver operating characteristic analysis demonstrating areas under the curve of 0.91 and 0.87. Exhaled breath analysis may serve as a novel biomarker of ILD and may have future utility in diagnosis and screening. 42
Prognostication and novel biomarkers
Several approaches to predicting progression have been explored including traditional methods, novel modifications of these methods and experimental biomarkers. Robbie et al. demonstrated that a decline in CT measures of lung volume over time provides evidence of disease progression in IPF and can predict outcome better than FVC. 43 Noth et al. reported on the utility of home spirometry as a valid measure of FVC decline in IPF. 44 They demonstrated that the mean adherence to recording weekly home spirometry was 86% and there were strong correlations between home and clinic measurements indicating that home spirometry is a feasible and valid measure in IPF. However, estimates of rate of decline from home measurements correlated poorly with laboratory spirometry, hence its utility as a predictive tool remains unclear. 44 Furthermore, Oldham et al. demonstrated that lung FVC appears to vary considerably across ILD subtypes, even in those with a progressive fibrosing (PF) phenotype, hence caution needs to be applied when assessing FVC decline. 45
The inability to predict short‐term decline in FVC has led to the exploration of alternative biomarkers or progression. Huang et al. examined transcriptional changes in blood over 4 months in 74 IPF patients and compared stable to progressive patients. Single‐cell RNA sequencing analysis of peripheral blood monocytes identified natural killer cells as significantly correlated with progression. 46 Correspondingly, Kreuter et al. performed a retrospective pooled analysis in 2067 patients 47 from the phase III studies, ASCEND, 48 INSPIRE 49 and CAPACITY, 50 to examine whether peripheral monocyte count at baseline predicted progression. They reported that monocyte counts above 0.60 × 109 cells/L at baseline were associated with a significantly increased risk of IPF progression, hospital admission and death over 1 year. Interestingly, a study by Stewart et al. reported that circulating fibrocytes were not associated with IPF disease activity and elevated fibrocyte levels were not specific to disease status, hence they have limited prognostic value. 51 Other molecular and protein biomarkers have been recently examined as prognosticators of ILD, including collagen turnover products (Figure 3), KL‐6 and CYFRA‐21‐1 (Figure 4). 52 , 53 Clynick et al. demonstrated that aggregating known potential biomarkers into a progression index may provide additional utility in predicting disease course. 54 Combining osteopontin, MMP7, intercellular adhesion molecule‐1 and periostin in this progression index accurately predicted progression, stability and mortality in three independent IPF cohorts, 55 , 56 , 57 thus applying that a composite biomarker approach may be the future.
FIGURE 3.
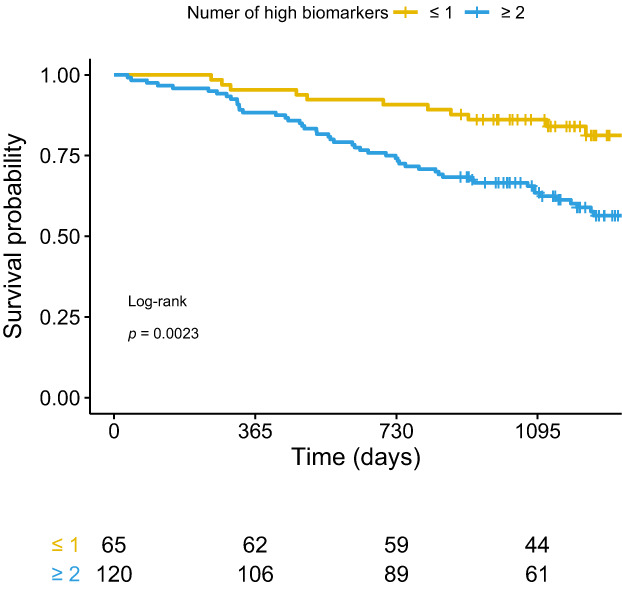
Collagen turnover products correlate with survival in pulmonary fibrosis. Neoepitope biomarkers of types III and VI collagen turnover (C3M, C6M, PRO‐C3 and PRO‐C6) were measured in 185 patients with newly diagnosed idiopathic pulmonary fibrosis. High baseline levels of C3M, C6M, PRO‐C3 and PRO‐C6 were associated with more advanced disease at the time of diagnosis. Figures show Kaplan–Meier curves of survival of participants in the Pulmonary Fibrosis Biomarker (PFBIO) cohort followed up for 3 years. Patients are divided according to the number of high neoepitope biomarkers (C3M, C6M, PRO‐C3 and PRO‐C6) at baseline. Reproduced from Hoyer et al., 52 with permission
FIGURE 4.
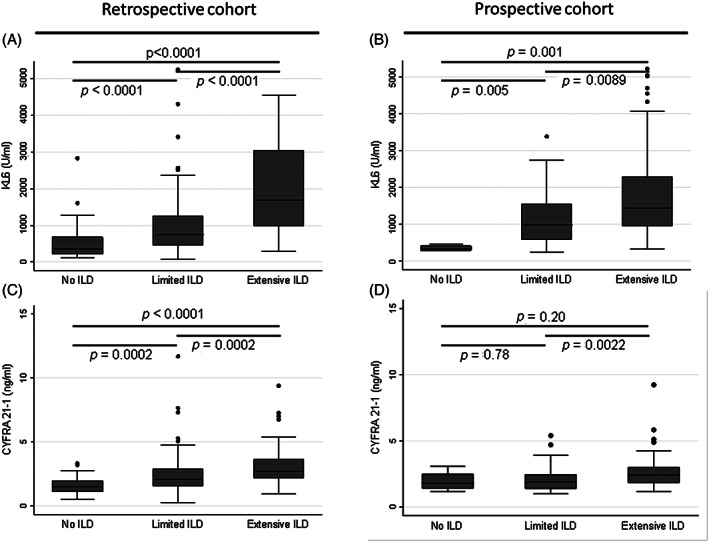
Serum KL‐6 and CYFRA 21‐1 are associated with the presence and extent of systemic sclerosis‐associated interstitial lung disease (SSc‐ILD). KL‐6 is a mucin‐like glycoprotein (MUC1) that is expressed by type II pneumocytes, and CYFRA 21‐1 is expressed by alveolar and bronchiolar epithelial cells. Serum KL‐6 and CYFRA 21‐1 levels were measured in a retrospective (n = 189) and a prospective (n = 118) cohort of SSc‐ILD patients. Serum KL‐6 and CYFRA 21‐1 correlated with the presence and severity of ILD, with higher levels in SSc‐ILD compared to SSc‐no ILD, and in extensive compared to limited ILD. Serum KL‐6 levels in no‐ILD, limited ILD and extensive ILD in the (A) retrospective and (B) prospective cohorts. Serum CYFRA 21‐1 levels in no‐ILD, limited ILD and extensive ILD in the (C) retrospective and (D) prospective cohorts. Central lines indicate median values with boxes showing the 25th and 75th percentiles, whiskers indicate upper and lower quartile + 1.5 × interquartile range. Reproduced from Stock et al., 53 with permission
Disease progression and mortality
The true burden of IPF has often been difficult to measure and accurately quantify 58 ; Jeganathan et al. addressed this in their study of mortality trends in the United States due to IPF. 59 They reported that from 2004 to 2017 the age‐adjusted mortality of males with IPF reduced by 4.1% from 75.5 deaths per million to 72.4 deaths and this was decreased in females by 13.4%. The reasons for reduction in mortality are unclear, whether it is related to smoking rates, earlier diagnosis, management, frailty levels or environmental/geographic factors remains to be seen. 60 , 61 Cutting et al. demonstrated that patient‐reported family history of fibrosis predicts reduced survival in both IPF and non‐IPF ILD, indicating that a positive family history should be considered in prognosticating risk. 62 Additionally, Lee et al. reported that the risk of several cancers in IPF is increased, with a significant risk of lung cancer (hazard ratio 5.89), thus demonstrating the need for clinicians to consider this risk when managing IPF. 63 An additional factor in mortality over the last 2 years has been COVID‐19 and while it was previously reported that ILD conferred increased risk of death, 64 Gallay et al. demonstrated that the case fatality rate in fibrotic ILD was 35% compared to 19% with other ILDs, hence the need for acute awareness of this risk in IPF and PF‐ILD patients. 65
THERAPEUTIC ADVANCES IN ILD
Key points.
Antifibrotics are associated with reduced all‐cause mortality in fibrosing ILDs, and are safe and well tolerated in older populations, those with multiple comorbidities and in those with telomerase gene mutations.
Inhaled N‐acetylcysteine may lead to a poorer outcome in IPF, and proton pump inhibitors have no specific benefit in IPF.
Non‐pharmacological management remains a keystone in the care of ILD, and studies on cough 66 , 67 and peripheral muscle training continue to be conducted, 68 without any clear effect demonstrated yet. Exercise induces thoracoabdominal asynchrony and is associated with dyspnoea and desaturation in fibrotic lung disease 69 and daily physical activity may predict disease progression. 70 Marillier et al. demonstrated that exercise‐induced hypoxaemia was associated with fatigue and this could be reversed with ambulatory supplementary oxygen. 71 Separately, Dowman et al. showed that interval exercise can facilitate ILD patients to complete high‐intensity exercise without worsening dyspnoea, fatigue or desaturation. 72
Antifibrotics in fibrosing ILDs
The use of antifibrotics in IPF 48 , 73 and PF‐ILDs 74 has become standard of care and with clear efficacy in reducing the rate of lung function decline. Many questions remain regarding treatment initiation, safety 75 and effectiveness on mortality. Indeed, the initial report of the INBUIILD trial demonstrated that nintedanib slowed FVC decline over 1 year, and the recent report of the entire trial period supported the evidence that nintedanib reduced progression, exacerbations and death. 76 The SENSCIS trial examined the efficacy of nintedanib compared to placebo in a cohort of 576 systemic sclerosis‐associated ILD (SSc‐ILD) patients. Nintedanib was associated with a lower risk for FVC decline and a trend towards reduced death, 77 further confirming the utility of nintedanib in SSc‐ILD. Independently, Roofeh et al. demonstrated that tocilizumab (a humanized monoclonal antibody targeting IL‐6 receptor) administered to early SSc‐ILD stabilized FVC over 48 weeks regardless of radiological severity, reinforcing the need to manage SSc‐ILD in multidisciplinary team with rheumatologists and pulmonologists. 78
A meta‐analysis by Petnak et al. of 12,956 patients from 26 separate studies revealed that antifibrotics were associated with reduced all‐cause mortality across subgroups, underpinning their utility in PF‐ILD. 79 Whether antifibrotics should be commenced in advanced disease or in older patients were also addressed. Durheim et al. examined disease progression and mortality following initiation of antifibrotics in patients with advanced IPF (n = 66) compared to mild to moderate IPF (n = 436). 80 They identified that patients with advanced IPF did not demonstrate more rapid lung function decline but did have higher mortality, indicating that prospective studies in high‐risk groups are required. Glaspole et al. assessed the efficacy of nintedanib in patients with IPF who were elderly and had multiple comorbidities from pooled data of five separate clinic trials. 81 They showed that the efficacy of nintedanib on FVC decline was consistent regardless of comorbidities or age, but the rate of drug discontinuation was markedly higher in patients over 75 years and with more comorbidities. 81 Justet et al. addressed antifibrotic safety in patents with TRG mutations, demonstrating that both pirfenidone and nintedanib appeared safe in a cohort of 89 patients and reduced FVC decline. 82
Many studies have examined the role of N‐acetylcysteine in specific clusters of patients with IPF. 83 Podolanczuk et al. conducted a randomized open label study of 81 patients assigned to receive pirfenidone or pirfenidone plus N‐acetylcysteine, and showed that the rate of FVC decline over 48 weeks in the N‐acetylcysteine plus pirfenidone group was more than twice that in the pirfenidone group alone, indicating that N‐acetylcysteine may lead to worse outcomes in IPF. 84 Also of interest has been the concomitant treatment of pulmonary hypertension (PH) with sildenafil. Behr et al. reported data on 177 patients in a randomized study of sildenafil or placebo added to patients on pirfenidone with advanced IPF and at risk of PH. 85 Addition of sildenafil to pirfenidone did not provide a treatment benefit versus pirfenidone plus placebo, with no difference in the proportion of patients with disease progression or adverse events, indicating that currently there is no evidence to suggest sildenafil has treatment effect in IPF.
Alternative therapeutic strategies in ILD
It has been suggested that gastroesophageal reflux disease may contribute to IPF progression and thus proton pump inhibitors (PPIs) as a possible therapy. Tran et al. identified a cohort of IPF patients from the UK Clinical Practice Research Datalink and matched 1852 PPI users to 1852 non‐users. 86 PPI use was not associated with a lower mortality or hospital admission rate and concluded that PPIs appear to have no specific benefit in treating IPF. A separate therapeutic strategy is blocking IL‐13 signalling with lebrikizumab. Two cohorts were included, a treatment‐naïve group (n = 154) and a cohort receiving pirfenidone (n = 351). Lebrikizumab was demonstrated to be well tolerated and safe, but lebrikizumab alone or in combination with pirfenidone was not associated with reducing FVC decline. 87
FUTURE DIRECTIONS
Progress in ILD research continues at pace; much remains unclear and further studies are needed in all key areas reviewed here, including studies to identify ILD earlier, better predict progression, validate the clinical utility of novel biomarkers, further explore how to enable patients to monitor disease at home and continue to explore novel therapeutic pathways.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENT
Open access funding provided by IReL.
McCarthy C, Keane MP. Contemporary Concise Review 2021: Interstitial lung disease. Respirology. 2022;27(7):539–548. 10.1111/resp.14278
Handling Editor: Philip Bardin
REFERENCES
- 1. Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18‐64 years old. Eur Respir J. 2016;48:179–86. [DOI] [PubMed] [Google Scholar]
- 2. Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty‐first century. Ann Am Thorac Soc. 2014;11:1176–85. [DOI] [PubMed] [Google Scholar]
- 3. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allen RJ, Guillen‐Guio B, Oldham JM, Ma S‐F, Dressen A, Paynton ML, et al. Genome‐wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2020;201:564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun. 2018;9:5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dobrinskikh E, Estrella AM, Hennessy CE, Hara N, Schwarz MI, Kurche JS, et al. Genes, other than Muc5b, play a role in bleomycin‐induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2021;321:L440–L50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Vis JJ, van der Smagt JJ, van Batenburg AA, Goldschmeding R, van Es HW, Grutters JC, et al. Pulmonary fibrosis in non‐mutation carriers of families with short telomere syndrome gene mutations. Respirology. 2021;26:1160–70. [DOI] [PubMed] [Google Scholar]
- 8. Konigsberg IR, Borie R, Walts AD, Cardwell J, Rojas M, Metzger F, et al. Molecular signatures of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2021;65:430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaminski N. Microarray analysis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2003;29:S32–6. [PubMed] [Google Scholar]
- 10. Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McErlean P, Bell CG, Hewitt RJ, Busharat Z, Ogger PP, Ghai P, et al. DNA methylome alterations are associated with airway macrophage differentiation and phenotype during lung fibrosis. Am J Respir Crit Care Med. 2021;204:954–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cui H, Xie N, Banerjee S, Ge J, Jiang D, Dey T, et al. Lung myofibroblasts promote macrophage profibrotic activity through lactate‐induced histone lactylation. Am J Respir Cell Mol Biol. 2021;64:115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ali MF, Dasari H, Van Keulen VP, Cornec D, Vasmatzis G, Peikert T, et al. Microbial antigens stimulate metalloprotease‐7 secretion in human B‐lymphocytes using mTOR‐dependent and independent pathways. Sci Rep. 2017;7:3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gill SE, Nadler ST, Li Q, Frevert CW, Park PW, Chen P, et al. Shedding of syndecan‐1/CXCL1 complexes by matrix metalloproteinase 7 functions as an epithelial checkpoint of neutrophil activation. Am J Respir Cell Mol Biol. 2016;55:243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ali MF, Dasari H, Van Keulen VP, Carmona EM. Canonical stimulation of the NLRP3 inflammasome by fungal antigens links innate and adaptive B‐lymphocyte responses by modulating IL‐1β and IgM production. Front Immunol. 2017;8:1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ali MF, Egan AM, Shaughnessy GF, Anderson DK, Kottom TJ, Dasari H, et al. Antifibrotics modify B‐cell‐induced fibroblast migration and activation in patients with idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2021;64:722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vera L, Garcia‐Olloqui P, Petri E, Vinado AC, Valera PS, Blasco‐Iturri Z, et al. Notch3 deficiency attenuates pulmonary fibrosis and impedes lung‐function decline. Am J Respir Cell Mol Biol. 2021;64:465–76. [DOI] [PubMed] [Google Scholar]
- 18. Tan Q, Link PA, Meridew JA, Pham TX, Caporarello N, Ligresti G, et al. Spontaneous lung fibrosis resolution reveals novel antifibrotic regulators. Am J Respir Cell Mol Biol. 2021;64:453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sato S, Chong SG, Upagupta C, Yanagihara T, Saito T, Shimbori C, et al. Fibrotic extracellular matrix induces release of extracellular vesicles with pro‐fibrotic miRNA from fibrocytes. Thorax. 2021;76:895–906. [DOI] [PubMed] [Google Scholar]
- 20. Nakahara Y, Hashimoto N, Sakamoto K, Enomoto A, Adams TS, Yokoi T, et al. Fibroblasts positive for meflin have anti‐fibrotic properties in pulmonary fibrosis. Eur Respir J. 2021;58:2003397. [DOI] [PubMed] [Google Scholar]
- 21. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med. 2021;203:707–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Espindola MS, Habiel DM, Coelho AL, Stripp B, Parks WC, Oldham J, et al. Differential responses to targeting matrix metalloproteinase 9 in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2021;203:458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang C, Wang S, Lau J, Roden AC, Matteson EL, Sun J, et al. IL‐23 amplifies the epithelial‐mesenchymal transition of mechanically conditioned alveolar epithelial cells in rheumatoid arthritis‐associated interstitial lung disease through mTOR/S6 signaling. Am J Physiol Lung Cell Mol Physiol. 2021;321:L1006–L22. [DOI] [PubMed] [Google Scholar]
- 24. Senoo S, Taniguchi A, Itano J, Oda N, Morichika D, Fujii U, et al. Essential role of IL‐23 in the development of acute exacerbation of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2021;321:L925–L40. [DOI] [PubMed] [Google Scholar]
- 25. Chu SG, Villalba JA, Liang X, Xiong K, Tsoyi K, Ith B, et al. Palmitic acid‐rich high‐fat diet exacerbates experimental pulmonary fibrosis by modulating endoplasmic reticulum stress. Am J Respir Cell Mol Biol. 2019;61:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim HS, Yoo HJ, Lee KM, Song HE, Kim SJ, Lee JO, et al. Stearic acid attenuates profibrotic signalling in idiopathic pulmonary fibrosis. Respirology. 2021;26:255–63. [DOI] [PubMed] [Google Scholar]
- 27. McLean AEB, Webster SE, Fry M, Lau EM, Corte P, Torzillo PJ, et al. Priorities and expectations of patients attending a multidisciplinary interstitial lung disease clinic. Respirology. 2021;26:80–6. [DOI] [PubMed] [Google Scholar]
- 28. Raghu G, Remy‐Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2020;202:e36–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fernandez Perez ER, Travis WD, Lynch DA, Brown KK, Johannson KA, Selman M, et al. Diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest. 2021;160:e97–e156. [DOI] [PubMed] [Google Scholar]
- 30. Baughman RP, Valeyre D, Korsten P, Mathioudakis AG, Wuyts WA, Wells A, et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. 2021;58:2004079. [DOI] [PubMed] [Google Scholar]
- 31. Statement on sarcoidosis. Joint statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999;160:736–55. [DOI] [PubMed] [Google Scholar]
- 32. Hunninghake GW, Costabel U, Ando M, Baughman R, Cordier JF, du Bois R, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16:149–73. [PubMed] [Google Scholar]
- 33. Lee AS, Scofield RH, Hammitt KM, Gupta N, Thomas DE, Moua T, et al. Consensus guidelines for evaluation and management of pulmonary disease in Sjogren's. Chest. 2021;159:683–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hatabu H, Hunninghake GM, Richeldi L, Brown KK, Wells AU, Remy‐Jardin M, et al. Interstitial lung abnormalities detected incidentally on CT: a position paper from the Fleischner Society. Lancet Respir Med. 2020;8:726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anderson MR, Kim JS, Allison M, Giles JT, Hoffman EA, Ding J, et al. Adiposity and interstitial lung abnormalities in community‐dwelling adults: the MESA cohort study. Chest. 2021;160:582–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Synn AJ, Li W, Hunninghake GM, Washko GR, San Jose Estepar R, O'Connor GT, et al. Vascular pruning on CT and interstitial lung abnormalities in the Framingham Heart Study. Chest. 2021;159:663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richeldi L, Scholand MB, Lynch DA, Colby TV, Myers JL, Groshong SD, et al. Utility of a molecular classifier as a complement to high‐resolution computed tomography to identify usual interstitial pneumonia. Am J Respir Crit Care Med. 2021;203(2):211–20. doi: 10.1164/rccm.202003-0877OC. [DOI] [PubMed]
- 38. Cooper WA, Mahar A, Myers JL, Grainge C, Corte TJ, Williamson JP, et al. Cryobiopsy for identification of usual interstitial pneumonia and other interstitial lung disease features. Further lessons from COLDICE, a prospective multicenter clinical trial. Am J Respir Crit Care Med. 2020;203:1306–13. [DOI] [PubMed] [Google Scholar]
- 39. Hariri LP, Adams DC, Wain JC, Lanuti M, Muniappan A, Sharma A, et al. Endobronchial optical coherence tomography for low‐risk microscopic assessment and diagnosis of idiopathic pulmonary fibrosis in vivo. Am J Respir Crit Care Med. 2018;197:949–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nandy S, Raphaely RA, Muniappan A, Shih A, Roop BW, Sharma A, et al. Diagnostic accuracy of endobronchial optical coherence tomography for the microscopic diagnosis of usual interstitial pneumonia. Am J Respir Crit Care Med. 2021;204:1164–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ratiu IA, Ligor T, Bocos‐Bintintan V, Mayhew CA, Buszewski B. Volatile organic compounds in exhaled breath as fingerprints of lung cancer, asthma and COPD. J Clin Med. 2020;10:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moor CC, Oppenheimer JC, Nakshbandi G, Aerts J, Brinkman P, Maitland‐van der Zee AH, et al. Exhaled breath analysis by use of eNose technology: a novel diagnostic tool for interstitial lung disease. Eur Respir J. 2021;57:2002042. [DOI] [PubMed] [Google Scholar]
- 43. Robbie H, Wells AU, Fang C, Jacob J, Walsh SLF, Nair A, et al. Serial decline in lung volume parameters on computed tomography (CT) predicts outcome in idiopathic pulmonary fibrosis (IPF). Eur Radiol. 2021;32:2650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Noth I, Cottin V, Chaudhuri N, Corte TJ, Johannson KA, Wijsenbeek M, et al. Home spirometry in patients with idiopathic pulmonary fibrosis: data from the INMARK trial. Eur Respir J. 2021;58:2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oldham JM, Lee CT, Wu Z, Bowman WS, Vu Pugashetti J, Dao N, et al. Lung function trajectory in progressive fibrosing interstitial lung disease. Eur Respir J. 2021;2101396. 10.1183/13993003.01396-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang Y, Oldham JM, Ma SF, Unterman A, Liao SY, Barros AJ, et al. Blood transcriptomics predicts progression of pulmonary fibrosis and associated natural killer cells. Am J Respir Crit Care Med. 2021;204:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kreuter M, Lee JS, Tzouvelekis A, Oldham JM, Molyneaux PL, Weycker D, et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2021;204:74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. King TE Jr, Bradford WZ, Castro‐Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92. [DOI] [PubMed] [Google Scholar]
- 49. King TE Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, et al. Effect of interferon gamma‐1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo‐controlled trial. Lancet (London, England). 2009;374:222–8. [DOI] [PubMed] [Google Scholar]
- 50. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet (London, England). 2011;377:1760–9. [DOI] [PubMed] [Google Scholar]
- 51. Stewart ID, Nanji H, Figueredo G, Fahy WA, Maher TM, Ask AJ, et al. Circulating fibrocytes are not disease‐specific prognosticators in idiopathic pulmonary fibrosis. Eur Respir J. 2021;58:2100172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoyer N, Jessen H, Prior TS, Sand JMB, Leeming DJ, Karsdal MA, et al. High turnover of types III and VI collagen in progressive idiopathic pulmonary fibrosis. Respirology. 2021;26:582–9. [DOI] [PubMed] [Google Scholar]
- 53. Stock CJW, Hoyles RK, Daccord C, Kokosi M, Visca D, De Lauretis A, et al. Serum markers of pulmonary epithelial damage in systemic sclerosis‐associated interstitial lung disease and disease progression. Respirology. 2021;26:461–8. [DOI] [PubMed] [Google Scholar]
- 54. Clynick B, Corte TJ, Jo HE, Stewart I, Glaspole IN, Grainge C, et al. Biomarker signatures for progressive idiopathic pulmonary fibrosis. Eur Respir J. 2022;59:2101181. [DOI] [PubMed] [Google Scholar]
- 55. Jo HE, Glaspole I, Grainge C, Goh N, Hopkins PM, Moodley Y, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J. 2017;49:1601592. [DOI] [PubMed] [Google Scholar]
- 56. Navaratnam V, Fogarty AW, McKeever T, Thompson N, Jenkins G, Johnson SR, et al. Presence of a prothrombotic state in people with idiopathic pulmonary fibrosis: a population‐based case‐control study. Thorax. 2014;69:207–15. [DOI] [PubMed] [Google Scholar]
- 57. Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med. 2017;5:946–55. [DOI] [PubMed] [Google Scholar]
- 58. Suzuki Y, Aono Y, Kono M, Hasegawa H, Yokomura K, Naoi H, et al. Cause of mortality and sarcopenia in patients with idiopathic pulmonary fibrosis receiving antifibrotic therapy. Respirology. 2021;26:171–9. [DOI] [PubMed] [Google Scholar]
- 59. Jeganathan N, Smith RA, Sathananthan M. Mortality trends of idiopathic pulmonary fibrosis in the United States from 2004 through 2017. Chest. 2021;159:228–38. [DOI] [PubMed] [Google Scholar]
- 60. Farooqi MAM, O'Hoski S, Goodwin S, Makhdami N, Aziz A, Cox G, et al. Prevalence and prognostic impact of physical frailty in interstitial lung disease: a prospective cohort study. Respirology. 2021;26:683–9. [DOI] [PubMed] [Google Scholar]
- 61. Shull JG, Pay MT, Lara Compte C, Olid M, Bermudo G, Portillo K, et al. Mapping IPF helps identify geographic regions at higher risk for disease development and potential triggers. Respirology. 2021;26:352–9. [DOI] [PubMed] [Google Scholar]
- 62. Cutting CC, Bowman WS, Dao N, Pugashetti JV, Garcia CK, Oldham JM, et al. Family history of pulmonary fibrosis predicts worse survival in patients with interstitial lung disease. Chest. 2021;159:1913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee HY, Lee J, Lee CH, Han K, Choi SM. Risk of cancer incidence in patients with idiopathic pulmonary fibrosis: a nationwide cohort study. Respirology. 2021;26:180–7. [DOI] [PubMed] [Google Scholar]
- 64. Drake TM, Docherty AB, Harrison EM, Quint JK, Adamali H, Agnew S, et al. Outcome of hospitalization for COVID‐19 in patients with interstitial lung disease. An international multicenter study. Am J Respir Crit Care Med. 2020;202:1656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gallay L, Uzunhan Y, Borie R, Lazor R, Rigaud P, Marchand‐Adam S, et al. Risk factors for mortality after COVID‐19 in patients with preexisting interstitial lung disease. Am J Respir Crit Care Med. 2021;203:245–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Martinez FJ, Afzal AS, Smith JA, Ford AP, Li JJ, Li Y, et al. Treatment of persistent cough in subjects with idiopathic pulmonary fibrosis (IPF) with gefapixant, a P2X3 antagonist, in a randomized, placebo‐controlled clinical trial. Pulm Ther. 2021;7:471–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Guler SA, Clarenbach C, Brutsche M, Hostettler K, Brill AK, Schertel A, et al. Azithromycin for the treatment of chronic cough in idiopathic pulmonary fibrosis: a randomized controlled crossover trial. Ann Am Thorac Soc. 2021;18:2018–26. [DOI] [PubMed] [Google Scholar]
- 68. Nolan CM, Patel S, Barker RE, Walsh JA, Polgar O, Maddocks M, et al. Muscle stimulation in advanced idiopathic pulmonary fibrosis: a randomised placebo‐controlled feasibility study. BMJ Open. 2021;11:e048808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Santana PV, Cardenas LZ, Ferreira JG, de Carvalho CRR, de Albuquerque ALP, Caruso P. Thoracoabdominal asynchrony associates with exercise intolerance in fibrotic interstitial lung diseases. Respirology. 2021;26:673–82. [DOI] [PubMed] [Google Scholar]
- 70. Prasad JD, Paul E, Holland AE, Glaspole IN, Westall GP. Physical activity decline is disproportionate to decline in pulmonary physiology in IPF. Respirology. 2021;26:1152–9. [DOI] [PubMed] [Google Scholar]
- 71. Marillier M, Bernard AC, Verges S, Moran‐Mendoza O, O'Donnell DE, Neder JA. Oxygen supplementation during exercise improves leg muscle fatigue in chronic fibrotic interstitial lung disease. Thorax. 2021;76:672–80. [DOI] [PubMed] [Google Scholar]
- 72. Dowman LM, May AK, Cox NS, Morris NR, Nakazawa A, Parker L, et al. Attenuation of exertional desaturation and preference for interval exercise compared to continuous exercise in people with interstitial lung disease. Respirology. 2021;26:1076–9. [DOI] [PubMed] [Google Scholar]
- 73. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82. [DOI] [PubMed] [Google Scholar]
- 74. Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases‐subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double‐blind, placebo‐controlled, parallel‐group trial. Lancet Respir Med. 2020;8:453–60. [DOI] [PubMed] [Google Scholar]
- 75. Urushiyama H, Jo T, Hasegawa W, Ando T, Sakamoto Y, Uda K, et al. Preoperative use of pirfenidone and reduced risk of postoperative severe respiratory complications in patients with idiopathic pulmonary fibrosis: propensity score‐matched analysis using a nationwide database in Japan. Respirology. 2021;26:590–6. [DOI] [PubMed] [Google Scholar]
- 76. Flaherty KR, Wells AU, Cottin V, Devaraj A, Inoue Y, Richeldi L, et al. Nintedanib in progressive interstitial lung diseases: data from the whole INBUILD trial. Eur Respir J. 2021;59:2004538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maher TM, Mayes MD, Kreuter M, Volkmann ER, Aringer M, Castellvi I, et al. Effect of nintedanib on lung function in patients with systemic sclerosis‐associated interstitial lung disease: further analyses of a randomized, double‐blind, placebo‐controlled trial. Arthritis Rheumatol. 2021;73:671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Roofeh D, Lin CJF, Goldin J, Kim GH, Furst DE, Denton CP, et al. Tocilizumab prevents progression of early systemic sclerosis‐associated interstitial lung disease. Arthritis Rheumatol. 2021;73:1301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Petnak T, Lertjitbanjong P, Thongprayoon C, Moua T. Impact of antifibrotic therapy on mortality and acute exacerbation in idiopathic pulmonary fibrosis: a systematic review and meta‐analysis. Chest. 2021;160:1751–63. [DOI] [PubMed] [Google Scholar]
- 80. Durheim MT, Bendstrup E, Carlson L, Sutinen EM, Hyldgaard C, Kalafatis D, et al. Outcomes of patients with advanced idiopathic pulmonary fibrosis treated with nintedanib or pirfenidone in a real‐world multicentre cohort. Respirology. 2021;26:982–8. [DOI] [PubMed] [Google Scholar]
- 81. Glaspole I, Bonella F, Bargagli E, Glassberg MK, Caro F, Stansen W, et al. Efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis who are elderly or have comorbidities. Respir Res. 2021;22:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Justet A, Klay D, Porcher R, Cottin V, Ahmad K, Molina Molina M, et al. Safety and efficacy of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis and carrying a telomere‐related gene mutation. Eur Respir J. 2021;57:2003198. [DOI] [PubMed] [Google Scholar]
- 83. Rogliani P, Calzetta L, Cavalli F, Matera MG, Cazzola M. Pirfenidone, nintedanib and N‐acetylcysteine for the treatment of idiopathic pulmonary fibrosis: a systematic review and meta‐analysis. Pulm Pharmacol Ther. 2016;40:95–103. [DOI] [PubMed] [Google Scholar]
- 84. Podolanczuk AJ, Noth I, Raghu G. Idiopathic pulmonary fibrosis: prime time for a precision‐based approach to treatment with N‐acetylcysteine. Eur Respir J. 2021;57:2003551. [DOI] [PubMed] [Google Scholar]
- 85. Behr J, Nathan SD, Wuyts WA, Mogulkoc Bishop N, Bouros DE, Antoniou K, et al. Efficacy and safety of sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: a double‐blind, randomised, placebo‐controlled, phase 2b trial. Lancet Respir Med. 2021;9:85–95. [DOI] [PubMed] [Google Scholar]
- 86. Tran T, Assayag D, Ernst P, Suissa S. Effectiveness of proton pump inhibitors in idiopathic pulmonary fibrosis: a population‐based cohort study. Chest. 2021;159:673–82. [DOI] [PubMed] [Google Scholar]
- 87. Maher TM, Costabel U, Glassberg MK, Kondoh Y, Ogura T, Scholand MB, et al. Phase 2 trial to assess lebrikizumab in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2021;57:1902442. [DOI] [PMC free article] [PubMed] [Google Scholar]