

Abstract
The lipase‐catalyzed polycondensation of azelaic acid and glycerol is investigated according to a Design‐of‐Experiment approach that helps to elucidate the effect of experimental variables on monomer conversion, M n and regioselectivity of acylation of glycerol. Chemometric analysis shows that after 24 h the reaction proceeds regardless of the presence of the enzyme. Accordingly, the biocatalyst was removed after a first step of synthesis and the chain elongation continued at 80 °C. That allowed the removal of the biocatalyst and the preservation of its activity: pre‐requites for efficient applicability at industrial scale. The experimental study, combined with docking‐based computational analysis, provides rational guidelines for the optimization of the regioselective acylation of glycerol. The process is scaled up to 73.5 g of monomer. The novelty of the present study is the rigorous control of the reaction conditions and of the integrity of the immobilized biocatalyst, which serve to avoiding any interference of free enzyme or fines released in the reaction mixture. The quantitative analysis of the effect of experimental conditions and the overcoming of some major technical bottlenecks for the scalability of enzymatic polycondensation opens new scenarios for industrial exploitation.
Keywords: biocatalysis, carboxylic acids, lipases, oligoesters, polycondensation
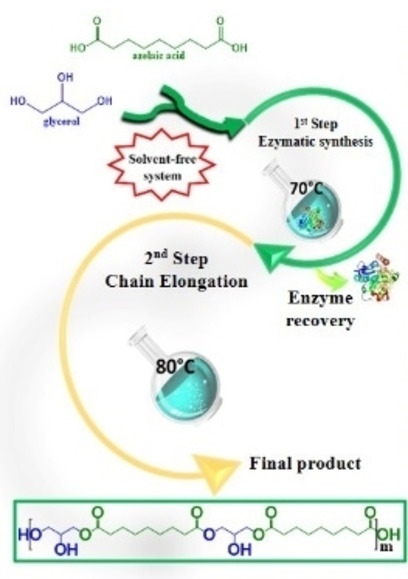
Two steps to scalability: A solvent‐free system is demonstrated for the two‐step synthesis of azelaic acid‐based oligoesters. The enzyme is removed after the first reaction step and chain elongation continues at 80 °C. This method permits the removal of the biocatalyst and the preservation of its activity, both of which are prerequisites for efficient applicability at industrial scale.
Introduction
The catalytic efficiency and selectivity of hydrolases enable the synthesis of novel polyesters under environmentally benign conditions. Enzyme selectivity minimizes branching and can be used at temperatures below 80 °C, thus enabling the synthesis of functionalized polyesters not achievable through the classical chemical routes when monomers prone to cross‐reactivity are used. [1] In most cases the enzymatic polycondensation leads to polyesters with low or moderate molecular weight, used both in coating and adhesive applications and in the cosmetic sector, which are also biodegradable. When higher molecular weights are desired further chemical processing is necessary for their elongation.[ 2 , 3 ] Different polyesters were also synthesized enzymatically by using renewable monomers for applications such as elastomers, sealants, and adhesives, which resulted hydrolysable in different media or in contact with tissues. [4]
Nevertheless, insufficient progresses have been reported towards the preparative and industrial applications of the enzymatic polycondensation. Notably, the polycondensation of polyols and diacids catalyzed by lipase B from Candida antarctica (CaLB) has been studied already in the 1990s and then transferred at industrial scale by Baxenden Chemicals (UK) for the production, later dismissed, of highly regular structures with low polydispersity. [5] Among the major bottlenecks for the sustainable and efficient scalability of enzymatic polycondensation the following ones were identified: i) the high viscosity of solvent‐less reaction mixtures, which prevents the efficient mass transfer and limits the chain extension; ii) the lack of stability of the immobilized enzymes when exposed to the mechanical stress caused by the viscosity and the mixing procedures that can cause both the detachment of the protein from the carrier and the abrasion of the carrier itself; iii) as a consequence of the previous problem, the impossibility to recover and reuse the immobilized biocatalyst.
Most scientific literature reports the commercially available formulation of CaLB (Novozyme 435), a lipase physically adsorbed on a macro‐porous organic resin that showed high efficiency in solvent‐free polycondensation applications at temperatures as high as 80 °C. However, this formulation demonstrated several important drawbacks, including the leaching of the enzyme and the fragility of the support under stirring, that prevent its industrial application for this specific scope. [6] In light of this, further strategies have been developed in recent years to use different hydrolases in polycondensation processes after improving their immobilization, either by physical or covalent methods.[ 7 , 8 , 9 , 10 ]
Several approaches have been reported also for the increase of the molecular weights of enzymatically synthesized polyesters, as the use of high‐boiling solvents. However, the scale‐up of procedures employing organic solvents is complicated due to the formation of cyclic products, [11] indicating that the use of solventless conditions is preferable. Temperatures above 80 °C decrease the viscosity by some extent but are detrimental for the biocatalyst stability.
In a previous study of the polycondensation of adipic acid (AA) and 1,4‐butanediol (BDO) at 50 °C, atmospheric pressure and in the presence of covalently immobilized CaLB we observed that the reaction proceeded even after the removal of the biocatalyst but by increasing the temperature to 80 °C and working under reduced pressure (70 mbar). [6] The enzyme (CaLB) was immobilized covalently on a solid carrier to assure the effective removal of the enzyme from the reaction mixture and prevent the contamination of the product with the protein. Moreover, the mixing was obtained by applying a thin‐film methodology that preserves the integrity of the carrier. Such conditions enabled a modest progress of the polycondensation after 72 h. Most probably, the polyesterification in the absence of the biocatalyst occurs as a result of the dissociation of the protons of the carboxyl groups leading to the self‐catalyzed esterification.[ 12 , 13 , 14 ] Reduced pressure was necessary as the uncatalyzed polycondensations of alcohols with carboxylic acids have an equilibrium constant below 10 (generally K C<10). [15] A more relevant elongation of the polyester chain was later observed when the same reaction was performed in a turbo‐reactor at pilot scale under thin‐film conditions and atmospheric pressure. [16] The first biocatalytic step at 60 °C yielded oligomers with a number average molecular weight (M n) of approximately 1800 Da, whereas the second catalyst‐free step at 90 °C led to an improvement of M n up to 2900 Da.
A more recent study [15] reported the thermal treatment of polyesters in vacuum after the removal of the biocatalyst, by filtration. The oligoesters poly(1,4‐butylene adipate) (PBA), poly(1,4‐butylene isophthalate) (PBI), poly(1,4‐butylene 2,5‐furandicarboxylate) (PBF) and 2,4‐pyridinedicarboxylate) (PBP) were synthesized starting from the diesters of the corresponding diacids and using the adsorbed formulation of CaLB (Novozyme 435). The weight average molecular weight (MW) of the enzymatically synthesized products was in the range of 600–2200 Da and upon treatment at temperatures ranging from 140 to 180 °C increased 8.5, 2.6, 3.3, and 2.7 folds, respectively. However, the use of the adsorbed enzyme as biocatalyst does not exclude the presence of residual active lipase in the reaction mixture even after the filtration of the immobilized biocatalyst, as previously demonstrated. [6]
In the light of the fragmented knowledge available and the limited understanding of the effect of the experimental variables on the enzymatic polycondensation of diacids and polyols here we report a systematic study of the process by using a design of experiment (DoE) approach integrated with statistical analysis and molecular modeling describing the enzyme‐substrate interactions.
The final objective is to provide solid rational basis for the efficient scalability of the enzymatic process by means of the optimal control of temperature, pressure and biocatalyst amount. Moreover, we intend to provide clear evidence of the feasibility of the two‐step polycondensation process that would finally enable the recovery of the enzyme at an early stage of the reaction when the mixture is still fluid, thus limiting the exposure of the biocatalyst to stressing environmental conditions. For this purpose, the experimental study was planned in such a way to avoid interference due to enzyme detachment from the support.
The model reaction selected for the study was the polyesterification of glycerol (1,2,3‐propanetriol, or GLO) and azelaic acid (AZA) in a solvent‐less system yielding the corresponding poly(glycerolazelate), a product of potential relevance for dermatological applications whose market is predicted to reach USD 160 million by 2023 due to its esters and polyesters use in plastics, biodegradable polymers, biolubricants, and materials for electronics. [17]
The pharmacological applications of azelaic acid were studied since 1980’s and it has been approved by both FDA (US Food and Drug Administration) and by EMA (European Medicines Agency) for external uses in the treatment of inflammatory acne vulgaris and in the treatment of skin pigmentation and melasma. [18] However, AZA suffers from low‐solubility, high melting point and large dosage requirement, which limit its wide application in cosmetics and pharmaceutical preparations. Moreover, high dosage pharmaceutical preparations lead to side effects associated to the acid character of the molecule. Therefore, the enzymatic synthesis of lauryl esters[ 19 , 20 ] have been described with the aim of improving its compatibility respect to other ingredients of cosmetic and dermatologic formulations. The enzymatic synthesis of poly(glycerolazelate) have been also reported in patent US005962624A [21] but in all cases the synthesis were catalyzed by means of the adsorbed immobilized lipase Novozyme 435 and the option of carrying out the process in two steps was not considered.
GLO was selected as polyol because of its wide availability at low cost [22] and especially because the enzyme selectivity enables the predominant acylation of the primary hydroxy groups leaving the secondary −OH unreacted. [23] The presence of a pendant hydroxy group confers specific solubility and viscosity properties to the polymeric chain and allows for the anchoring of different bioactive molecules. On this respect, the present study addresses also the factors affecting the regioselectivity of the enzymatic polycondensation, attempting to shed light on the substrate specificity of lipases.
Results and Discussion
One‐ and two‐step enzymatic syntheses of poly(glycerolazelate)
The enzymatic polycondensation of GLO and AZA was tested using two formulations of covalently immobilized lipase B from Candida antarctica (CaLB) and lipase from Thermomyces lanuginosus (TLL). Both lipases are widely employed in cosmetic and food industry as physically immobilized biocatalysts. [24] In the present study they were immobilized covalently on epoxy methacrylic resins to avoid the interference deriving from the detachment of the enzyme from the support.[ 6 , 23 ] The robustness of the covalent anchoring was verified after the immobilization procedure (see Experimental Section). A third industrially relevant lipase, from Ryzopus oryzae, was also considered but unfortunately the biocatalyst lost almost all the activity after the immobilization procedure.
Before starting the polycondensation, a preliminary experimental assay of the substrate specificity of CaLB and TLL was carried out to better define the amount of biocatalyst to be employed in the polycondensation and work under similar conditions. The hydrolysis of diesters containing saturated carbon chains of different lengths, both in even and odd numbers of carbon atoms was studied. They were diethylmalonate (C3); dimethyl adipate (C6); dimethyl pimelate (C7); dimethyl suberate (C8); diethyl azelate (C9); diethyl sebacate (C10). Because of the wide difference in terms of specific activity, substrate specificity was expressed for each lipase as the ratio between the specific activity measured with each ester and the highest value obtained for the enzyme (Figure 1). Among all the tested substrates the highest specificity of TLL was obtained for dimethyl suberate (C8), whereas for CalB the highest activity values were obtained for dimethyl adipate (C6).
Figure 1.
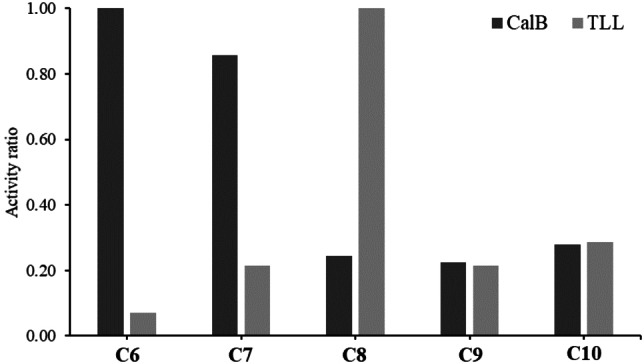
Comparison of substrate specificity of lipase B from Candida antarctica (CaLB) and lipase from Thermomyces lanuginosus.
The values are expressed as the ratio between each measured specific activity and the highest hydrolytic activity measured for each enzyme, namely diethyl adipate for CaLB and diethyl suberate for TLL. Both enzymes were unable to catalyze the hydrolysis of diethyl malonate probably due to the steric hindrance caused by the two ethyl groups at the center of the short carbon chain, which confer a globular shape to the molecule. Concerning the diethyl ester of azelaic acid, the activity ratio of both lipases was comparable. The covalently immobilized formulations of CaLB and TLL were employed in a first polycondensation of AZA and GLO performed at 70 °C, 70 mbar, for 24 h with 30 % of immobilized biocatalyst per gram of monomer (Scheme 1).
Scheme 1.

Polycondensation of glycerol with azelaic acid, catalyzed by covalently immobilized lipases.
The reactions were carried out under thin‐film conditions in a rotary evaporator to avoid the stress derived from mechanical mixing and promote efficient mass transfer and water removal upon formation.[ 6 , 23 ]
Surprisingly, no product was observed in the case of TLL, even when a different formulation was employed obtained by physical immobilization. That attempt was made to verify whether the covalent immobilization of TLL causes the obstruction of the active site as a consequence of the rigidification of the lid, namely the flexible dominion covering the active site entrance. In fact, unlike CaLB, TLL is a typical lipase that undergoes interfacial activation when approaching the hydrophobic substrates.
Therefore, the focus of the investigation was maintained exclusively on CalB. Table 1 gives M n and M w values for the enzymatically synthesized product after 24 h of reaction measured by means of microTOF and ESI‐MS analysis and calculated according to the equation reported in the Experimental section. After obtaining the enzymatically synthesized oligoester, the biocatalyst was removed by filtration and reused for a second step of polycondensation in the absence of the enzyme but increasing the temperature to 80 °C. The data indicate a 27 % of increase of M n and 24 % in the case of M w.
Table 1.
M n and M w obtained in the synthesis of poly(glycerolazelate) catalyzed by covalently immobilized CaLB after 24 h and after two steps of 24 h reaction without any enzyme at 80 °C.
|
Cat. |
T [°C] |
t [h] |
P [mbar] |
M n [a] [g mol−1] |
M n [b] [g mol−1] |
M w [a] [g mol−1] |
M w [b] [g mol−1] |
ĐM [c] |
|---|---|---|---|---|---|---|---|---|
|
CalB |
70 |
24 |
70 |
524 |
687 |
550 |
754 |
1.09 |
|
no |
80 |
24 (48) |
70 |
631 |
908 |
725 |
919 |
1.01 |
|
no |
80 |
48 (72) |
70 |
744 |
983 |
774 |
1072 |
1.09 |
[a] Molecular weights determined based on microTOF analysis. [b] Determined by ESI‐MS. [c] ĐM=dispersity, determined by ESI.
Therefore, the data confirm that the enzyme is necessary for catalyzing the initial synthesis of short pre‐polymers, as also confirmed by the absence of product in the reactions using TLL as biocatalyst. Once the oligoesters are formed, the M n of the polyester can increase in the absence of the biocatalyst only driven by the removal of the by‐product, namely water.
A preliminary characterization of the oligoesters was carried out for better understanding its applicability for dermatological application. More precisely, we tested the possible hydrolysis of the oligomers at pH value close to that encountered in face skin affected by pathological conditions such as acne and rosacea. The results indicate that the oligomers undergo slow progressive hydrolysis (see the Supporting Information, Figure S1). Of course, the assay provides only preliminary indications and specific in vitro and in vivo release studies would be required to evaluate the applicability of the enzymatic polycondensation product. It should be emphasized that the hydrolysis of the oligomer could be favored within a formulation containing an aliquot of lipolytic enzymes. In this regard, lipases are already used in various cosmetic formulations, which would therefore allow the release in situ of the active principle starting from inactive precursors. [25] The product was also characterized in terms of free carboxylic acids exposed on the oligomers, which was 5.2 mmol per gram of product (see the Supporting Information).
Multivariate analysis of the effect of experimental variables on the enzymatic synthesis of poly(glycerolazelate)
Once demonstrated the feasibility of the two step polycondensation we proceeded with a systematic analysis of the factors affecting the enzymatic polycondensation of AZA and GLO to gather rational guidelines for the optimization of an industrial process. More specifically we monitored the reaction for 72 h taking samples every 24 h, to identify the optimal time for the removal of the biocatalyst.
The analysis of the parameters that influence the enzymatic polycondensation of AZA and GLO was performed using a Fractional Factorial Design and results were analyzed using MOODE software. The Fractional Factorial Design method consisted of 8 experiments, where the four experimental variables were studied at their highest (+1) and lowest (−1) level. Three variables, temperature (T), enzyme amount (E) and pressure (p) are continuous variables whereas the stepwise addition of the GLO (A) is a discontinuous variable. Five more experiments were added to the factorial design where the conditions correspond to the average value of the continuous variables and to each of the levels assumed by the discontinuous variable (GLO addition).
For the temperature, a range between 50 °C and 70 °C has been set by considering three aspects: (i) an increase of temperature decreases the viscosity of the medium and improves mass transfer and the accessibility of substrates to the biocatalyst; (ii) temperatures above 80 °C involve greater economic and energy expenditure; (iii) and lead to the gradual loss of enzyme activity. [26] The pressure was varied in a range 70–500 mbar, since operating at low values of pressure allows the removal of water produced during the reaction to prevent hydrolysis. However, the application of very low pressures translates into an increase of the process costs and complexity. The effect of the amount of biocatalyst immobilized was studied in the range of 10 and 30 % w/w of the monomer reagents (Figure 2), to identify the amount of biocatalyst sufficient to accelerate the polycondensation without affecting too much the overall process cost. The biocatalyst used in the study was formulated by immobilizing a low amount of protein on the carrier, since we have previously demonstrated that the homogeneous distribution of the enzyme in the viscous reaction mixture affects positively the course of the reaction, rather than the addition of a high amount of enzymatic units concentrated in a small volume. [23] Finally, the effect of the addition of GLO in two stages was studied to verify any possible detrimental effect of the polyols, as previous observed for 1,4‐butanediol. [23] Therefore, the glycerol was added at the starting of the reaction or in two aliquots, at the starting of the reaction and after 24 h.
Figure 2.

Response surface describing the effect of pressure and temperature on the conversion of the azelaic acid studied with three different amounts of biocatalyst.
The set of 13 experiments generated by the MOODE software are included in the Table 2 and were performed, as described in the Experimental Section. The dependent responses measured in each set of experimental conditions were the conversion of azelaic acid (determined by NMR spectroscopy) and the M n of the oligomers (determined by ESI‐MS).
Table 2.
Levels and values of the independent variables investigated in the experimental design. Conversion, expressed as the percentage of esterification of azelaic acid, obtained at different reaction times and evaluated also after the work up of the reaction mixture at 72 h.
|
Experiment No. |
T [°C] |
Enzyme amount [%w/w] |
P [mbar] |
Two‐step glycerol addition |
Conversion [%][a] |
|||
|---|---|---|---|---|---|---|---|---|
|
24 h |
48 h |
72 h |
72 h and workup |
|||||
|
|
50 |
10 |
70 |
No |
71.8 |
82.3 |
86.8 |
86.8 |
|
2 |
70 |
10 |
70 |
Yes |
80.3 |
86.8 |
89.0 |
89.0 |
|
3 |
50 |
30 |
70 |
Yes |
80.5 |
87.8 |
88.6 |
88.8 |
|
4 |
70 |
30 |
70 |
No |
89.3 |
92.0 |
93.0 |
92.8 |
|
5 |
50 |
10 |
500 |
Yes |
53.6 |
62.1 |
68.8 |
69.8 |
|
6 |
70 |
10 |
500 |
No |
62.8 |
68.6 |
73.8 |
74.0 |
|
7 |
50 |
30 |
500 |
No |
63.3 |
68.8 |
75.6 |
75.8 |
|
8 |
70 |
30 |
500 |
Yes |
66.6 |
70.3 |
78.0 |
78.8 |
|
9 |
60 |
20 |
285 |
No |
68.9 |
72.6 |
79.3 |
80.3 |
|
10 |
60 |
20 |
285 |
No |
68.1 |
79.3 |
90.0 |
86.3 |
|
11 |
60 |
20 |
285 |
Yes |
76.1 |
86.0 |
88.5 |
89.0 |
|
12 |
60 |
20 |
285 |
Yes |
75.6 |
85.8 |
86.5 |
89.8 |
|
13 |
60 |
20 |
285 |
Yes |
86.5 |
76.1 |
86.0 |
89.0 |
[a] The progress of each reaction was monitored by 1D and 2D NMR analyses.
Effect of independent variables on AZA conversion
The conversion was determined by 1H NMR spectroscopy, by monitoring increases in the integral values of the signals in the interval 2.40–2.30 ppm, which correspond to the esterified azelaic acid, and the concomitant decrease of the integral values in the range 2.30–2.2, assigned to the free azelaic acid. The conversion values determined based on the sample collected and measured after 24, 48 and 72 h are presented in Table 3. The 1H NMR spectrum of the reaction mixture leading to the highest conversion of AZA, namely EXP4, is shown in Figure 3 along with the signal assignments.
Table 3.
Effect of the independent variables and their interactions on the conversion. Statistically significant variables are in bold.
|
t [h] |
Temperature |
Enzyme amount |
Pressure |
Glycerol addition 2 steps |
Interactions 12+34 |
Interactions 13+24 |
Interactions 23+14 |
Standard deviation |
|---|---|---|---|---|---|---|---|---|
|
24 |
7.45 |
7.80 |
−18.90 |
−0.45 |
6.60 |
−4.96 |
−4,08 |
5.06 |
|
48 |
4.18 |
4.78 |
−19.78 |
−0.45 |
0.79 |
−0.21 |
−0.57 |
5.35 |
|
72 |
3.50 |
4.20 |
−15.30 |
−0.27 |
0.12 |
0.75 |
5.13 |
4.52 |
|
post workup |
3.35 |
4.15 |
−14.75 |
1.08 |
0.15 |
0.25 |
1.25 |
2.47 |
Figure 3.

1H NMR spectrum and peak assignments of the product obtained after 72 h of reaction and workup of the reaction mixture of EXP4.
By comparing the difference between the value of the average of the factorial design runs and the average of the centers in relation to the global standard deviation, it can be stated that the response surface does not have curvatures. Therefore, the models to be used are linear, with the parameters reported in Table 3. Generally, the model will be of the type given by Equation 1:
| (1) |
where ‐y is the response, is the mean of the centers, k are the coefficients relating to the independent variables, P is pressure, %E is percentage (w/w) of immobilized enzyme on monomers basis, and T is temperature.
The highest conversion was obtained in EXP4, by employing the temperature and the enzyme at their highest level while the pressure is the lowest. Under such conditions, which correspond to the experiments reported in Table 1, three synthesis were performed at higher scale, up to 73.5 g of total monomers, always affording reproducible M n and conversions of azelaic acid ≥95 %. All reaction products were characterized by ESI‐MS and NMR spectroscopy (Table 4).
Table 4.
AZA conversion and M n values obtained at different scales working under conditions of EXP4.
|
No. |
Total mass of monomers [g] |
AZA conversion [%] |
M n [g mol−1] |
M w [g mol−1] |
ĐM |
|---|---|---|---|---|---|
|
1 |
18.29 |
95 |
938 |
1023 |
1.09 |
|
2 |
36.59 |
96 |
926 |
978 |
1.06 |
|
3 |
73.50 |
95 |
944 |
1036 |
1.10 |
The obtained data clearly indicate an excellent scalability of the process without the necessity of unreacted monomer separation. However, one major problem that prevents the scalability of the process is represented by the recovery and the reuse of the enzyme. Of course, ideally the biocatalyst should be recovered. when the viscosity is still low and in an early stage of the reaction so to prevent the detrimental action of temperature and other experimental factors.
It is interesting to note that at 24 h the model is affected negatively by the pressure and positively by the temperature and the amount of the biocatalyst. The corresponding model is given by Equation 2:
| (2) |
The behavior of the reaction is in line with previous studies reporting different polycondensations catalyzed by CaLB. One exception is represented by a study of the polycondensation of dimethyl adipate with 1,4‐butanediol, which reported a negative effect of the vacuum and the temperature on the efficiency of the enzymatic polycondensation catalyzed by cutinase 1 from Thermobifida cellulosilytica. [7] The result was explained by some molecular dynamics analysis that indicated how the active site of the cutinase is less accessible at high temperature and low pressure, whereas CaLB displays the maximum accessibility under the same conditions.
The most interesting result emerging from Table 3 is the negligible effect of the temperature and of the biocatalyst at higher reaction time. At 48 and 72 h the conversion increases, although moderately, in all reactions but there is no effect of the biocatalyst even when its amount is triplicated. Only the pressure appears to significantly affect the conversion (Figure 4). Therefore, the model at 48 h is given by Equation 3:
| (3) |
Figure 4.

Response surfaces describing the effect of independent variables on the conversion of azelaic acid after 48 h of reaction: a) Effect of pressure and enzyme at different temperatures levels; b) effect of pressure and temperature at different enzyme levels, indicting the negligible effect of the temperature.
Also, the mathematical models calculated for the data obtained after 72 h and after the workup of the reaction mixture (Figure S2) underline the negative effect of the pressure whereas the percentage of the enzyme and the temperature are no significant (Figure 5). Therefore, after 72 h of reaction the model is given by Equation 4:
| (4) |
Figure 5.

Response surfaces describing the effect of the independent variables on the conversion of azelaic acid after 72 h of reaction: a) effect of pressure and enzyme at different temperature levels; b) effect of pressure and temperature at different enzyme levels.
which is consistent with the model calculated on the products after workup of the reaction mixture at 72 h [Eq. 5]:
| (5) |
The consistency of the two models confirms that neither the sampling procedure nor the workup protocol affected the composition of the oligomers analyzed and, conversely, the experimental results.
The results clearly indicate that, regarding the conversion of AZA, the more relevant variable is the pressure, which is necessary for removing the water and prevent the hydrolysis throughout the 72 h of the process, whereas the enzyme plays a relevant role only in the first 24 h of the reaction. Unexpectedly, also the temperature, which in principle decreases the viscosity and facilitate the removal of the water, affect the polycondensation only during the first 24 h. The chemometric analysis suggests that after catalyzing the fast and complete conversion of AZA the enzyme can be removed since it does not significantly affect the progress of the polycondensation. Indeed, the M n obtained after 48 h in the experiment carried out in two steps, as reported in Table 1, is even higher than that observed in EXP 4 where the biocatalyst was maintained in the reaction mixture for the whole course of the reaction. At the best of our knowledge this is the first demonstration of the feasibility of the two‐step approach based on the combination of the experimental evidence and the statistical analysis of the effect of the experimental variables. We previously reported that the covalently immobilized CaLB can be efficiently removed by filtration when the oligoesters are formed and then reused at least for 8 cycles without the need of any rinsing step, thus avoiding the use of any organic solvent throughout the process. [6]
Effects of experimental variables on M n of poly(glycerolazelate)
The analysis of the distribution of medium molecular weights was performed based on the assignments of the masses from the ESI‐MS spectra collected for the samples at 24, 48 h and after the workup of the reaction mixture at the end of the reaction (Table 6). The spectra were collected in the negative mode, as described in the Experimental Section. Figure 6 reports the ESI mass spectra collected for the sample EXP4 after workup and they indicate the formation of 3 types of reaction products: [AB], [AB]A and B[AB] where A represents the azelaic acid and B the glycerol molecule. The range of the molecular weights reached after 72 h of reaction is between 756 and 1969 Da.
Table 6.
Effect of the experimental variables on the regioselectivity of the acylation of glycerol, expressed as the ratio of the percentage of the desired product (1,3‐acylated) to all the others after 24 and 72 h.
|
EXP No. |
T [°C] |
Enzyme amount [%] |
P [mbar] |
Ratio 1,3 to others 24 h |
Ratio 1,3 to others 72 h |
|---|---|---|---|---|---|
|
1 |
50 |
10 |
70 |
0.94 |
1.44 |
|
2 |
70 |
10 |
70 |
1.15 |
1.32 |
|
3 |
50 |
30 |
70 |
0.96 |
0.95 |
|
4 |
70 |
30 |
70 |
1.10 |
0.88 |
|
5 |
50 |
10 |
500 |
0.58 |
0.97 |
|
6 |
70 |
10 |
500 |
0.77 |
0.77 |
|
7 |
50 |
30 |
500 |
0.61 |
0.53 |
|
8 |
70 |
30 |
500 |
0.57 |
0.79 |
|
9 |
60 |
20 |
285 |
0.67 |
0.77 |
|
10 |
60 |
20 |
285 |
0.66 |
0.88 |
|
11 |
60 |
20 |
285 |
0.68 |
0.84 |
|
12 |
60 |
20 |
285 |
0.77 |
0.89 |
|
13 |
60 |
20 |
285 |
0.98 |
0.78 |
Figure 6.

ESI‐MS spectrum of the EXP4 reaction products obtained after 72 h and workup of the reaction mixture obtained from the enzymatic polycondensation of azelaic acid (A, molecular weight 188.22 g mol−1) and glycerol (B, molecular weight 92.09 g mol−1), catalyzed by covalently immobilized CaLB. Target 1200 m/z, range between 100 and 2200 m/z, negative ions.
The data thus obtained were analyzed with the MODDE program, using the multiple linear regression (MRL) algorithm [27] to estimate the effect of individual variables at different times. From the comparison of the difference between the value of the average of the factorial design runs and the average of the centers in relation to the global standard deviation it can be observed that the response surface has no curvature.
Results reported in Tables S1 and S2 indicate that the single variables have no significant effect whereas there is a slightly significant effect of the variables interaction. However, when the effect of pressure and the enzyme amount on M n at 70 °C were analyzed more in detail (Figure 7) it become evident that in the first 24 h these two variables affect the M n in an opposite way as compared to what observed at 48 h and 72 h. This result is in agreement with the observation that after 24 h the polycondensation proceeds even without the catalytic action of the enzyme. Therefore, at reaction time >24 h, the increase in M n is mainly the result of the chemical polycondensation and the enzyme plays a negligible role.
Figure 7.

Effects of pressure and enzyme amount on M n of poly(glycerolazelate) analyzed at after 24 (a), 48 (b), and 72 h (c) of reaction always in the presence of the biocatalyst at 70 °C.
The highest M n was obtained in EXP2, which was carried out at high temperature and low pressure but with the lowest amount of the enzyme (Table 5), whereas the highest conversion of the azelaic acid was observed when the enzyme was used at the highest level (Table 2). Therefore, the concomitant optimization of the two responses requires the identification of a compromise, which could be identified by applying a desirability function approach. [28]
Table 5.
Distribution of medium molecular weights expressed as g mol−1 obtained at 24 h, 48 h and after 72 h and workup of the reaction mixture.
|
Experiment |
M n [g mol−1] |
||
|---|---|---|---|
|
24 h |
48 h |
72 h and workup |
|
|
1 |
682 |
978 |
940 |
|
2 |
837 |
967 |
1051 |
|
3 |
787 |
858 |
872 |
|
4 |
795 |
886 |
926 |
|
5 |
640 |
792 |
756 |
|
6 |
683 |
861 |
860 |
|
7 |
681 |
824 |
781 |
|
8 |
757 |
878 |
848 |
|
9 |
771 |
825 |
885 |
|
10 |
714 |
936 |
949 |
|
11 |
787 |
934 |
997 |
|
12 |
791 |
915 |
903 |
|
13 |
918 |
968 |
915 |
Analysis of the regioselectivity of the acylation
The study focused the attention also on the effect of the reaction conditions on the regioselectivity of the acylation of GLO, since hydrophilicity is a desired property of the final product that increases when the secondary alcohol of GLO is not acylated. That result is not achievable using conventional chemical polycondensations that lead to extensive acylation and branching.
The analysis of the product obtained after 24 and after the workup of the reaction mixture at 72 h was carried out by 2D DQCOSY and TOCSY NMR spectra (Figures S3–S7) that allowed for the identification of the specific signals of the glycerol acylated at a different extent and in different positions. The percentage isomer distribution (Figure 8) was calculated based on 1H NMR spectra and the results clearly indicate that in each experiment the highest content of the products is represented by the 1,3 diacylated form followed by the monoacylated form.
Figure 8.
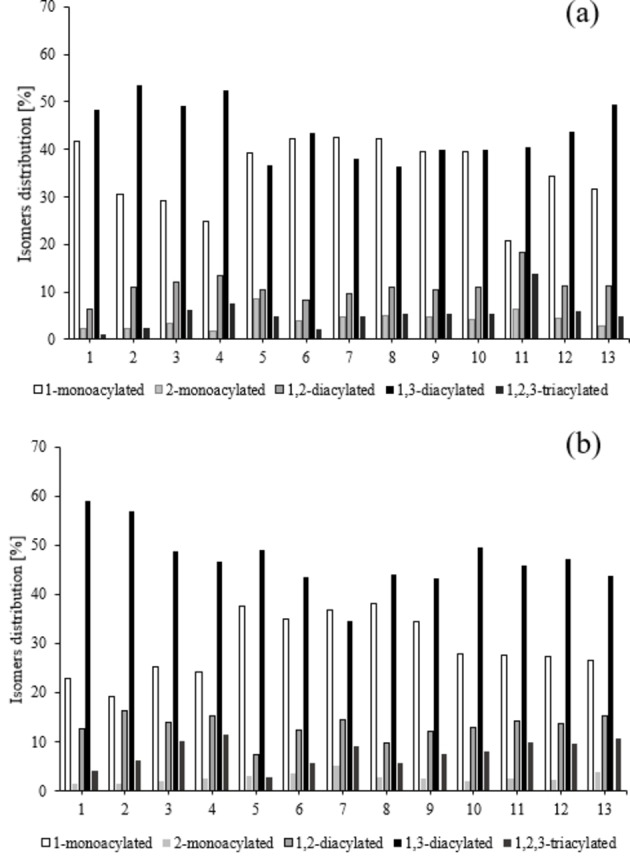
The percentage product distribution after 24 h (a) and 72 h (b) in the samples considered in the experimental design.
Analysis of the effects of the variables on the acylation of GLO indicates that at higher pressure the formation of the monoacylated glycerol is favored, probably because of the competing action of water that promotes the hydrolytic reactions (Table S3). Moreover, to obtain the selective acylation of the primary −OH groups it is preferable the use of a lower amount of biocatalyst.
To better understand the global effect of the variables on the regioselective acylation of glycerol, Table 6 gives the percentage of the desired product (1,3‐diacylated) respect to all the others. Data clearly indicate that at 72 h the highest percentages of the desired product are obtained by working at low pressure and with a low amount of biocatalyst. It must be underlined that, in the light of the observation that after 24 h the polycondensation occurs even in the absence of the enzyme, the results obtained at 72 h reflect not only the regioselectivity of the enzymatic synthesis but also the course of the chemical process occurring at higher reaction time. However, the observation that a lower amount of enzyme affects positively the product composition both at 24 and at 72 h indicates that the planning of the biocatalytic step can be optimized to influence the final outcome of the process.
Computational analysis of the selectivity of CalB and TLL by means of docking
To rationalize the reactivity of lipases against primary and secondary alcohols, [23] explain the unexpected lack of activity of TLL in the polycondensation reactions, and get more insights about CaLB and TLL substrate specificity and regioselectivity and the 1‐and 2‐monoacyl dimers, 1,2‐ and 1,3‐diacyl trimers and 1,2,3‐triacylated products (Figure 9) were docked to the catalytic site of CaLB and TLL to generate 500 bound conformation by means of rapid Lamarckian genetic algorithm search method and free energy estimation based on an empirical free‐energy force field, as implemented in Autodock.
Figure 9.

Chemical structures of the substrates considered for the docking studies. Schematic representation of the distances measured for the C5 atom of the azelaic acid and the angle considered between the C atom and the O of the catalytic Ser residue of the lipases. (a) Near attack conformation.
Out of the generated structure pool, productive docking poseswere selected by identifying those where the substrate assumes a near attack conformation (NAC) compatible with the productive attack of the catalytic serine (Ser105 of CalB or Ser 146 of TLL) to the carbon atom of the acyl groups of the ligands (either carboxylic acid leading to chain elongation, or ester causing the reverse of the reaction). In the NAC (Figure 9, structure a) the length of the single covalent bond between the carbon atom of the acyl groups and the oxygen is assumed to be between 1.43 and 2.15 Å, whereas the distance (d) between the serine oxygen and the acyl carbon was set at a maximum distance of 3.2 Å. Concerning the oxyanionic pocket, the stabilization of the oxyanion intermediate must occur at a maximum distance of 3 Å from the NH (H bond donor) of the residues forming the pocket. Finally, the orientation of the acyl carbons of the ligands towards the serine oxygen (ϑ) must be such as not to exceed a 90° angle. [23]
Figure 10 gives the percentage of productive poses identified by taking as a reference the two types of C atoms, carboxylic and ester groups, which lead either to the chain extension (carboxylic acid esterification) or to the reverse of the reaction (hydrolysis or alcoholysis of the formed ester bond) and their corresponding average free energies of each group (see Experimental Section for computational details) For all substrates there is a considerable percentage of productive poses for both enzymes. The docking free energies (red squares in Figure 10) correspond to the sum of intermolecular energies (including desolvation energies), internal energies, and torsional free energies. Large, negative, values are associated with favorable conformations. The high docking free energies of the 2‐monoacylated substrate make evident how the acylation and the hydrolysis at the 2 position are unfavored with respect to same reaction occurring on other sites.
Figure 10.
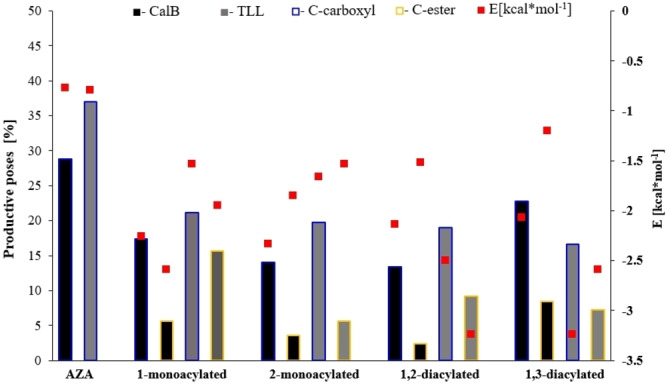
Comparison of the percentage of NAC productive poses obtained by analyzing the attack of the two different acyl groups (carboxylic acid=blue frame; ester group=yellow frame) using Autodock. The values are expressed as the percentage of the total number of generated poses (500) and refer to CaLB (black bars) and TLL (gray bars). Red squares correspond to the average of the docking free energies of the corresponding productive poses.
A detailed view of the poses that correspond to the lowest docking energies for the 1‐monoacylated is reported in Figure 11 for the CalB and TLL catalytic site. It is evident that in the catalytic site of TLL (on the right) assumes a conformation with a distance d incompatible with NAC (>3.2 Å) for chain elongation.
Figure 11.

1‐monoacylated substrate docked by Autodock in the active site of CaLB (left) and TLL (right). The catalytic Ser 105 of CalB and Ser 146 of TLL is in yellow, whereas the substrate is green.
To better evaluate the ability of CaLB to catalyze the chain elongation of the substrates, all poses corresponding to the lowest docking free energies for the attach of the free carboxylic group were analyzed in detail (Table 7) and were compared to the results obtained with a second computational method, implemented in Autodock Vina and based on Monte Carlo search and rapid gradient‐optimization conformational search coupled with a simple scoring function for free energy estimation. [29]
Table 7.
AutoDock and AutoDock Vina data relative to the docking free energies computed only for the poses corresponding to the lowest energy values (of the poses given in Figure 10). The distances and angles were calculated taking as a reference the C atom of the free ‐COOH group.
|
Lipase |
Ligand |
AutoDock |
AutoDock Vina |
||||
|---|---|---|---|---|---|---|---|
|
E*abs [kcal mol−1] |
d [Å] |
ϑ [°] |
E*abs [kcal mol−1] |
d [Å] |
ϑ [°] |
||
|
CalB |
Azelaic acid |
−3.70 |
3.1 |
70.0 |
−5.60 |
3.4 |
50.7 |
|
|
1‐monoacylated |
−4.24 |
3.2 |
65.7 |
−5.80 |
3.5 |
47.3 |
|
|
2‐monoacylated |
−3.96 |
3.2 |
65.8 |
−5.60 |
3.5 |
56.1 |
|
|
1,2‐diacylated |
−3.19 |
4.6 |
23.2 |
−4.40 |
2.8 |
67.4 |
|
|
1,3‐diacylated |
−4.61 |
4.6 |
35.8 |
−4.00 |
3.2 |
55.0 |
|
|
1,2,3‐triacylated |
n. d. |
n. d. |
n. d. |
−2.10 |
2.5 |
86.6 |
|
TLL |
Azelaic acid |
−1.59 |
3.2 |
75.3 |
−2.20 |
3.1 |
175 |
|
|
1‐monoacylated |
−3.10 |
3.1 |
139.9 |
−5.00 |
3.4 |
53.7 |
|
|
2‐monoacylated |
−3.76 |
6.0 |
126.4 |
−5.40 |
3.3 |
64.2 |
|
|
1,2‐diacylated |
−5.57 |
3.3 |
141.7 |
−5.30 |
3.6 |
42.7 |
|
|
1,3‐diacylated |
−5.51 |
3.2 |
114.3 |
−5.20 |
3.4 |
62.3 |
The results collected in Table 7 suggest that shorter ligands (monomers and dimers) can easily access the active site of CalB in productive conformations, as demonstrated by ϑ values compatible with NAC and d values within 0.2/0.3 Å as required. Concerning the larger ligands (trimers) the two docking protocols lead to opposite results, as shown in Figure 12 for the 1,3‐diacylated trimer. Indeed, AutoDock optimum poses presents angle compatibilities whereas the d values were larger than those expected for a NAC (by as much as 1.4 Å). On the contrary, for the same trimers, AutoDock Vina delivered both d and ϑ values compatible with NAC. Similar results were obtained for tetramers, in agreement with experimentally observed data, most probably because such larger substrates are characterized by higher flexibility and a larger number of torsions which can be more efficiently accounted for by a Monte Carlo‐based protocol.
Figure 12.

d values computed for the 1,3‐diacylated trimer docked in the active site of CaLB by using AutoDock (left) and Autodock Vina (right). The conformations of the substrates are in blue whereas the catalytic Ser 105 is in yellow.
In the case of TLL the docking free energies for AZA were much higher than CaLB energies, whereas the d values are comparable to those obtained for CalB. These results, holding for both docking protocols, support the experimental observations that TLL is unable to catalyze the esterification of GLO with AZA under the same conditions used for CaLB. Furthermore, in the case of the larger ligands, the values of d and ϑ determined at the lowest docking free energy poses are incompatible with NAC and therefore the esterification is not favored in the case of TLL.
Furthermore, the aptitude of CALB and TLL towards catalyzing the hydrolysis or alcoholysis of the formed oligoesters was also evaluated by measuring the ϑ and d referred to the C atom of the ester groups (Figure 9 and Table S4). Out of all the substrates, only 2 productive poses were obtained in the case of CalB for 1,2‐diacylated product (at C21) and for 1,3‐diacylated product (at C6), whereas all the other poses were considered nonproductive. The data suggest that in the case of CaLB, chain elongation is favored as compared to the reversal of the reaction. In the case of TLL, all poses proved unproductive (see the Supporting Information).
Conclusion
The present study represents the first systematic analysis of the experimental variables on the efficiency of an enzymatic polycondensation of a diacid and a polyol. The novelty of the approach is the rigorous control of the integrity of the enzyme to avoid release of the protein in the reaction mixture and the abrasion of the immobilization carrier as a consequence of the mechanical mixing of the viscous mixture. That allowed for the verification of the feasibility of a two‐step process employing the enzyme only for the initial formation of some short prepolymers. More importantly, the study highlighted that when the enzymatic polycondensation lasts more than 24 h, the nonenzymatic reaction prevails and the effects of the experimental variables on conversion, M n and regioselectivity change. The computational study provided solid rational guidelines for understanding and tuning the regioselectivity of the acylation of glycerol, which is crucial for the exploitation of this largely available bio‐based polyol. Overall, the investigation allowed for the optimization of the enzymatic synthesis of poly(glycerolazelate) – a product of interest for dermatological application – that was scaled up to 73.50 g, demonstrating the reproducibility of the method. In conclusion, this investigation helps to address some major problems that have hampered enzymatic polycondensation at industrial scale.
Experimental Section
Glycerol (99.5 %) provided by Carlo Erba e da A.C.E.F.. Diethyl malonate (99 %), dimethyl adipate (98 %), dimethyl pimelate (99 %), dimethyl suberate (99 %), diethyl azelate (90 %), diethyl sebacate (98 %) were purchased from Sigma‐Aldrich. CaLB was covalently immobilized on epoxy‐methacrylic resins Relizyme EP 113, kindly donated by Resindion (Mitsubishi Chemical Corporation), Binasco, Italy. The specific hydrolytic activity determined based on previously reported protocol was 350 U/g. Azelaic acid was kindly provided by Novamont S.p.A, Novara, Italy. All reagents were used without further purification
Covalent immobilization of lipases
Covalently immobilized CaLB was prepared using the epoxy acrylic resin Relizyme® EP113 (average pore diameter 20–50 nm, according to protocols of Resindion, Milano, Italy) according to protocols previously reported. [8] The loadings of the enzyme were of 10000 U per gram of resin. The hydrolytic activity was 526 U*g−1 dry respectively, determined via tributyrin lipase activity assay. More than 98 % of protein was immobilized and it was verified that there was no enzyme leaching from the support using a method previously reported. [30] The same protocol was used for the immobilization of TLL and ROL, by loading 50000 U per gram of dry resin.
Physical immobilization of lipase TLL
The physical immobilization of TLL was carried out on milled rice husk according to the method described previously. [30]
Activity assay for immobilized CalB lipase
The activity of covalently immobilized CalB was measured using the tributyrin as substrate as previously described. [30]
Enzymatic polycondensation of azelaic acid and glycerol using a thin‐film reaction system
The enzymatic polycondensations reactions were performed by using a thin‐film approach, as reported by Pellis et al. [6] Azelaic acid (5.0 g) and an equimolar amount of glycerol (2.4 g) were introduced into a 100 mL flask and different amount of lipases. The reactions were performed for 72 h at different temperatures and different values of the pressure and samples were withdrawn at 24 and 48 h. The reactions were performed using a Rotavapor R‐114 (BÜCHI) connected to a vacuum pump Vac® V‐500 (BÜCHI) and a pressure controllerV‐800 (BÜCHI). Temperature was controlled by means of a Waterbath B‐480 (BÜCHI). Samples were collected at pre‐set time intervals 24/48/72 h and analyzed by ESI‐MS spectrometry and NMR. At the end of reaction, methanol (40 mL) was added and the enzyme was removed by filtration and the product was recovered after methanol evaporation at 60 °C and stored at 4 °C until characterization without any further treatment. Control reactions, in the absence of enzyme were performed, and the formation of the product was not observed.
Experimental design and data analysis
The experimental design and results analysis were carried out using MOODE software [31] by a Fractional Factorial Design method consisting of 8 experiments, where the four experimental variables are studied at their highest (+1) and lowest (−1) level. Three variables, Temperature (T), enzyme amount (E) and pressure (p) are continuous variables whereas the stepwise addition of the glycerol (A) is a discontinuous variable. To this series of experiments, 5 experiments were added in which in which each continuous variable maintains its intermediate value, whereas the discontinuous varable changes. These experiments were performed in the random order defined by the MODDE software used to build the model. The fractional factorial design provides an adequate selection of the experimental conditions of the matrix of a full factorial design. In this way the experimental effort is reduced while the significance of the information is retained. However, the resolution of the analysis of the variable effects is decreased. In the present case the effect of each variable can be estimated separately, whereas the effect of their interactions is overlapped according to the following confounding pattern: TE+pA; Tp+EA; Ep+TA. To determine the significance of effects calculated for the variables using the MLR algorithm, it was necessary to determine the value of the standard deviation, both as an estimate within the various times and as a global data to which to compare the effects and interactions of the variables. This was calculated as the mean of the variances of the centers (at 24, 48, 72 h and post workup), from which the global standard deviation is derived, according to Equation 6:
| (6) |
where yi is the ith center of design, is the mean of the centers, and N‐1 represents the degrees of freedom of the system. The difference in design averages was also calculated by using Equation 7:
| (7) |
ESI‐MS analysis
The ESI‐MS analysis were performed as previously described by Corici et al. [23] About 1 mg of crude reaction mixture was dissolved in methanol (1 mL) and formic acid (0.1 % v/v) was added. The analyses were performed using an Esquire 4000 (Bruker) instrument in electrospray positive ionization mode by generating the ions in an acidic environment. The generated ions were positively charged with m/z ratio falls in the range of 200–1000. Weight average molecular weight (M w) and number average molecular weight (M n) were calculated according to Equations (8) and (9), respectively:
| (8) |
| (9) |
where Mi is the molecular weight of a chain and Ni is the number of chains of that molecular weight. The product dispersity (ĐM) was calculated by using Equation 10:
| (10) |
NMR spectroscopy
1H, 13C, and 2D NMR spectra were recorded by using a 400 MHz VarianR spectrometer using MeOD and CDCl3 as solvent.
Computational analysis
The crystal structure of CaLB and TLL was retrieved from the Protein Data Bank (PDB); PDB ID: 1TCA (CALB) and 1DTE (TLL) The CaLB and TLL structure were pre‐processed by using Swiss‐PdbViewer. [32] Substrate molecules were generated with ChemDraw and minimized using MOPAC [33] with the AM1 semiempirical method prior docking. Both lipases were prepared by removing all the water molecules, heteroatoms, any co‐crystallized solvent and substrates and the polar hydrogens and partial charges were added to the structure using ADT 1.5.6. When AutoDock was used, for each experiment 500 dockings were calculated with docking box (60×60×60) Å, Grid Point Spacing 0.250 Å centered on t using a Lamarckian genetic algorithm. Auto‐Dock version 4.2 [34] was used for performing the docking calculations with Lamarckian genetic algorithm. GA population size was set at1500 and Maximum number of evaluations at 25000000. 500 docking generations, each one composed of 250 docking poses were generated. The same system was also docked with Autodock VINA by employing the same docking box with exhaustiveness 500 and energy range 50. Larger exhaustiveness ad energy ranges led to the same result. The best candidates were visualized and further analyzed using the software Pymol. [35]
Hydrolysis of poly(glycerol azelate)
The spontaneous hydrolysis test was performed by preparing an emulsion using milliQ water (23.4 mL), gum arabic suspension (5.1 mL; prepared as previously described for the tributyrin test [30] ) and oligoester sample (1.5 mL). 0.1 m pH 5 acetate buffer (2 mL) was added and the titration was performed for 8 h at 31 °C by using 0.1 m aqueous NaOH solution as titrant. A pH value of 5 was set as the end point.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors acknowledge Resindion (Mitsubishi Chemical Corporation) for the epoxy functionalized resin used for enzymes immobilization. This work has been partially financed by Regione Piemonte, project PRIME, POR FESR 2014/2020.
 This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement No 101029444, RenEcoPol. Open Access Funding provided by Universita degli Studi di Trieste within the CRUI‐CARE Agreement.
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement No 101029444, RenEcoPol. Open Access Funding provided by Universita degli Studi di Trieste within the CRUI‐CARE Agreement.
A. Todea, S. Fortuna, C. Ebert, F. Asaro, S. Tomada, M. Cespugli, F. Hollan, L. Gardossi, ChemSusChem 2022, 15, e202102657.
Data Availability Statement
Research data are not shared.
References
- 1. Guarneri A., Cutifani V., Cespugli M., Pellis A., Vassallo R., Asaro F., Ebert C., Gardossi L., Adv. Synth. Catal. 2019, 361, 2559–2573. [Google Scholar]
- 2. Kricheldorf H. R., Behnken G., Schwarz G., Polymer 2005, 46, 11219–11224. [Google Scholar]
- 3. Stavila E., Alberda van Ekenstein G. O. R., Loos K., Biomacromolecules 2013, 14, 1600–1606. [DOI] [PubMed] [Google Scholar]
- 4. Lang K., Sánchez-Leija R. J., Gross R. A., Linhardt R. J., Polymer 2020, 12, 2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Binns F., Harffey P., Roberts S. M., Taylor A., J. Chem. Soc.-Perkin Trans. 1999, 1, 2671–2676. [Google Scholar]
- 6. Pellis A., Corici L., Sinigoi L., D'Amelio N., Fattor D., Ferrario V., Ebert C., Gardossi L., Green Chem. 2015, 17, 1756–1766. [Google Scholar]
- 7. Pellis A., Ferrario V., Cespugli M., Corici L., Guarneri A., Zartl B., Herrero Acero E., Ebert C., Guebitz G. M., Gardossi L., Green Chem. 2017, 19, 490–502. [Google Scholar]
- 8. Cespugli M., Lotteria S., Navarini L., Lonzarich V., Del Terra L., Vita F., Zweyer M., Baldini G., Ferrario V., Ebert C., Gardossi L., Catalysts 2018, 8, 471–493. [Google Scholar]
- 9. Pellis A., Vastano M., Quartinello F., Herrero Acero E., Guebitz G. M., Biotechnol. J. 2017, 12, 1700322. [DOI] [PubMed] [Google Scholar]
- 10. Hevilla V., Sonseca A., Echeverría C., Muñoz-Bonilla A., Fernández-García M., Macromol. Biosci. 2021, 21, 2100156. [DOI] [PubMed] [Google Scholar]
- 11. Kricheldorf H. R., Rabenstein M., Maskos M., Schmidt M., Macromolecules 2001, 34, 713–722. [Google Scholar]
- 12. Salmi T., Paatero E., Nyholm P., Chem. Eng. Process. 2004, 43, 1487–1493. [Google Scholar]
- 13. Flory P. J., J. Am. Chem. Soc. 1939, 61, 3334–3340. [Google Scholar]
- 14. Tang A. U., Yao K. S., J. Polym. Sci. 1959, 35, 219–233. [Google Scholar]
- 15. Comerford J. W., Byrne F. P., Weinberger S., Farmer T. J., Guebitz G. M., Gardossi L., Pellis A., Materials 2020, 13, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.G. Cerea, L. Gardossi, L. Sinigoi, D. Fattor (Ambiente e Nutrizione SRL, Milan), WO2013110446 A1, 2013.
- 17. Todea A., Deganutti C., Spennato M., Asaro F., Zingone G., Milizia T., Gardossi L., Polymer 2021, 13, 4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gollnick H., J. Dermatolog. Treat. 1990, 1, 1–6. [Google Scholar]
- 19. Khairudin N., Basri M., Masoumi H. R. F., Sarah Samiun W., Samson S., RSC Adv. 2015, 5, 94909–94918. [Google Scholar]
- 20. Khairudin N., Basri M., Masoumi H. R. F., Samson S., Ashari S. E., Molecules 2018, 23, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.A. Vonderhagen, J. A. Gates, K. Hill, M. Lagarden, H. Tesmann (Henkel AG and Co KGaA, Düsseldorf), US005962624 A, 1999.
- 22. Metzger J. O., Bornscheuer U., Appl. Microbiol. Biotechnol. 2006, 71, 13–22. [DOI] [PubMed] [Google Scholar]
- 23. Corici L., Pellis A., Ferrario V., Ebert C., Cantone S., Gardossi L., Adv. Synth. Catal. 2015, 357, 1763–1774. [Google Scholar]
- 24. Cantone S., Spizzo P., Fattor D., Ferrario V., Ebert C., Gardossi L., Chim. Oggi 2012, 30, 22–26. [Google Scholar]
- 25. Ansorge-Schumacher M. B., Thum O., Chem. Soc. Rev. 2013, 42, 6475–6490. [DOI] [PubMed] [Google Scholar]
- 26. Skjøt M., De Maria L., Chatterjee R., Svendsen A., Patkar S. A., Østergaard P. R., Brask J., ChemBioChem 2009, 10, 520–527. [DOI] [PubMed] [Google Scholar]
- 27. Haaland P. D., in Experimental Design in Biotechnology, Marcel Dekker Inc, New York, 1992. [Google Scholar]
- 28. Costaa N. R., Lourençoa J., Pereira Z. L., Chemom. Intell. Lab. Syst. 2011, 107, 234–244. [Google Scholar]
- 29. Forli S., Huey R., Pique M., Nat. Protoc. 2016, 11, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spennato M., Todea A., Corici L., Asaro F., Cefarin N., Savonitto G., Deganutti C., Gardossi L., EFB Bioeconomy J. 2021, 1, 100008. [Google Scholar]
- 31. https://www.sartorius.com/en/products/process-analytical-technology/data-analytics-software/doe-software/modde, accessed 10.05.2019.
- 32. Guex N., Peitsch M. C., Electrophoresis 1997, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 33. Stewart J. J. P., in MOPAC2016, Colorado Springs, CO, USA: 2016. [Google Scholar]
- 34. Morris G. M., Ruth H., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J., J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delano W. L., in The PyMOL Molecular Graphics System, Schrödinger, LLC, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
Research data are not shared.