

Abstract
Objective
To analyze the efficacy and safety of nintedanib in patients with fibrosing autoimmune disease–related interstitial lung diseases (ILDs) with a progressive phenotype.
Methods
The INBUILD trial enrolled patients with a fibrosing ILD other than idiopathic pulmonary fibrosis, with diffuse fibrosing lung disease of >10% extent on high‐resolution computed tomography, forced vital capacity percent predicted (FVC%) ≥45%, and diffusing capacity of the lungs for carbon monoxide percent predicted ≥30% to <80%. Patients fulfilled protocol‐defined criteria for progression of ILD within the 24 months before screening, despite management deemed appropriate in clinical practice. Subjects were randomized to receive nintedanib or placebo. We assessed the rate of decline in FVC (ml/year) and adverse events (AEs) over 52 weeks in the subgroup with autoimmune disease–related ILDs.
Results
Among 170 patients with autoimmune disease–related ILDs, the rate of decline in FVC over 52 weeks was −75.9 ml/year with nintedanib versus −178.6 ml/year with placebo (difference 102.7 ml/year [95% confidence interval 23.2, 182.2]; nominal P = 0.012). No heterogeneity was detected in the effect of nintedanib versus placebo across subgroups based on ILD diagnosis (P = 0.91). The most frequent AE was diarrhea, reported in 63.4% and 27.3% of subjects in the nintedanib and placebo groups, respectively. AEs led to permanent discontinuation of trial drug in 17.1% and 10.2% of subjects in the nintedanib and placebo groups, respectively.
Conclusion
In the INBUILD trial, nintedanib slowed the rate of decline in FVC in patients with progressive fibrosing autoimmune disease–related ILDs, with AEs that were manageable for most patients.
Short abstract
INTRODUCTION
Interstitial lung disease (ILD) is a common manifestation of systemic autoimmune diseases including rheumatoid arthritis (RA) (1), systemic sclerosis (SSc) (2), and mixed connective tissue disease (MCTD) (3). Some patients with autoimmune disease–related ILD develop a progressive fibrosing phenotype characterized by increasing lung fibrosis on high‐resolution computed tomography (HRCT), decline in lung function, worsening symptoms and quality of life, and early mortality, despite immunomodulatory therapy (2, 3, 4, 5, 6, 7, 8, 9). Decline in forced vital capacity percent predicted (FVC%) is a predictor of mortality in patients with autoimmune disease–associated ILDs (2, 10, 11).
Immunosuppressants and disease‐modifying antirheumatic drugs (DMARDs) are the standard of care for systemic autoimmune diseases, but their efficacy in slowing the progression of ILD remains unclear. Tocilizumab, an antagonist of the interleukin‐6 receptor, and nintedanib, an intracellular inhibitor of tyrosine kinases, have been approved by the US Food and Drug Administration for slowing lung function decline in patients with SSc‐associated ILD (SSc‐ILD). Data from a subgroup of patients with SSc‐ILD from a randomized placebo‐controlled trial in patients with diffuse cutaneous SSc and elevated markers of inflammation suggested that tocilizumab slowed decline in FVC% (12). Nintedanib inhibits processes fundamental to the progression of lung fibrosis (13, 14, 15, 16, 17, 18). In clinical trials, nintedanib had a consistent effect on reducing the rate of decline in FVC in patients with idiopathic pulmonary fibrosis (IPF) (19), SSc‐ILD (20), and progressive fibrosing ILDs other than IPF (21), with an adverse event (AE) profile characterized mainly by gastrointestinal events. No significant effect of nintedanib on health‐related quality of life was observed in these studies. No heterogeneity was detected in the relative effect of nintedanib versus placebo on reducing the rate of FVC decline across diagnostic subgroups (19, 20, 21, 22). Here, we present further analyses of the efficacy and safety of nintedanib in patients with progressive autoimmune disease–related ILDs in the INBUILD trial.
PATIENTS AND METHODS
INBUILD trial design
The INBUILD trial (ClinicalTrials.gov identifier: NCT02999178) was a randomized, double‐blind, placebo‐controlled trial conducted in 15 countries (21). The trial was conducted in accordance with the protocol and principles of the Declaration of Helsinki and the Harmonized Tripartite Guideline for Good Clinical Practice from the International Conference on Harmonisation and was approved by local authorities. Written informed consent was obtained from all subjects before study entry.
The design of the INBUILD trial has been described, and the trial protocol and statistical analysis plan are publicly available (21). Briefly, eligible subjects had an ILD other than IPF that was diagnosed by an investigator according to their usual practice, with reticular abnormality with traction bronchiectasis (with or without honeycombing) of >10% extent on HRCT, FVC% ≥45%, and diffusing capacity of the lungs for carbon monoxide percent predicted (DLco%) ≥30% to <80%. Subjects met ≥1 of the following criteria for ILD progression within the 24 months before screening, despite management deemed appropriate in clinical practice: relative decline in FVC% ≥10%; relative decline in FVC% ≥5% to <10% and worsened respiratory symptoms; relative decline in FVC% ≥5% to <10% and increased extent of fibrosis on HRCT; worsened respiratory symptoms and increased extent of fibrosis on HRCT.
Subjects receiving stable doses of approved medications to treat RA or connective tissue disease (CTD) could participate, but the protocol excluded those receiving azathioprine, cyclosporine, mycophenolate mofetil, tacrolimus, rituximab, cyclophosphamide, or oral glucocorticoids >20 mg/day. Investigators were asked not to consider patients with autoimmune disease that was managed using these therapies for participation in the trial. Patients who were receiving 1 of these therapies to treat their ILD, and whose ILD was progressing, could participate if the restricted therapy was discontinued. Washout periods for therapies used to treat ILD are described in the Supplementary Methods (available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). Initiation of these medications was allowed after 6 months of the trial in cases of deterioration of ILD or autoimmune disease.
Investigators documented an ILD diagnosis on the case report form based on the following options: idiopathic nonspecific interstitial pneumonia, unclassifiable idiopathic interstitial pneumonia, hypersensitivity pneumonitis, RA‐associated ILD (RA‐ILD), MCTD‐associated ILD (MCTD‐ILD), SSc‐ILD, exposure‐related ILD, sarcoidosis, and other fibrosing ILD. These diagnoses were not centrally reviewed. When the box for “other fibrosing ILD” was checked on the case report form, the investigator specified the diagnosis in a text box.
Subjects were randomized to receive nintedanib 150 mg twice a day or placebo, stratified by fibrotic pattern on HRCT (usual interstitial pneumonia [UIP]–like fibrotic pattern or other fibrotic pattern [21]) based on central review. The trial consisted of 2 parts: part A, which comprised 52 weeks of treatment, and part B, a variable period beyond week 52 during which subjects continued to receive blinded treatment until all subjects had completed the trial. Subjects who discontinued treatment were asked to attend all visits as planned, including an end‐of‐treatment visit and a follow‐up visit 4 weeks later. The second (final) database lock took place after all subjects had completed the follow‐up visit or had entered the open‐label extension study, INBUILD‐ON (ClinicalTrials.gov identifier: NCT03820726).
Outcomes
Here, we report analyses in the subgroup of subjects with autoimmune disease–related ILDs (RA‐ILD, SSc‐ILD, MCTD‐ILD, as well as subjects with an autoimmune disease noted in the “Other fibrosing ILDs” category of the case report form). We assessed the following: rate of decline in FVC (ml/year) over 52 weeks; absolute change from baseline in FVC (ml) at week 52; absolute change from baseline in FVC% at week 52; proportions of subjects with absolute declines or increases in FVC% >0% to ≤5%, >5% to ≤10%, >10% to ≤15%, and >15% at week 52; and change from baseline in the King's Brief ILD (K‐BILD) questionnaire (23) total score at week 52. The K‐BILD questionnaire consists of 15 items in 3 domains (breathlessness and activities, psychological factors, chest symptoms), and higher scores represent better health status (23). We report the time to absolute and relative declines from baseline in FVC% ≥5% and ≥10%, time to first acute exacerbation (21) of ILD or death, time to progression of ILD (absolute decline from baseline in FVC% ≥10%) or death, and time to death using data obtained over the course of the whole trial (i.e., up to the second database lock).
We conducted subgroup analyses of the rate of decline in FVC (ml/year) over 52 weeks in subgroups according to fibrotic pattern on HRCT (UIP‐like fibrotic pattern, other fibrotic patterns), according to ILD diagnosis (RA‐ILD, MCTD‐ILD, SSc‐ILD, other autoimmune disease–related ILDs), and according to use of DMARDs and/or glucocorticoids (any dose) at baseline.
AEs reported over the 52‐week period (irrespective of causality) and coded using preferred terms in the Medical Dictionary for Regulatory Activities are presented descriptively for all subjects with autoimmune disease–related ILDs and in subgroups according to use of DMARDs and/or glucocorticoids at baseline. DMARDs were defined based on World Health Organization standardized drug groupings.
Statistical analysis
Analyses were based on data from subjects who received ≥1 dose of the trial drug. The annual rate of decline in FVC was analyzed in all subjects with autoimmune disease–related ILDs using a random coefficient regression model (with random slopes and intercepts), including baseline FVC (ml), HRCT pattern (UIP‐like fibrotic pattern or other fibrotic patterns), and treatment, as well as treatment‐by‐time and baseline‐by‐time interactions. Subgroup analyses used the same model but with interaction terms for baseline‐by‐time, treatment‐by‐subgroup, and treatment‐by‐subgroup‐by‐time interaction. In subgroup analyses, the interaction P value was an indicator of the potential heterogeneity in the treatment effect of nintedanib versus placebo across the subgroups (i.e., a P value more than 0.05 indicated that there was no evidence that the treatment effect differed across the subgroups). Statistical analyses of other end points are described in the Supplementary Methods (available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). Hazard ratios (HRs) and 95% confidence intervals (95% CIs) were calculated as appropriate. Analyses were not adjusted for multiplicity.
RESULTS
Patients
Of the 663 patients in the INBUILD trial (Supplementary Figure 1, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075), 170 (25.6%) had autoimmune disease–related ILDs, of whom 89 had RA‐ILD, 39 had SSc‐ILD, 19 had MCTD‐ILD, and 23 had other autoimmune disease–related ILDs. In subjects with other autoimmune disease–related ILDs, the diagnoses reported on the case report form included Sjögren's disease–related ILD (n = 7), interstitial pneumonia with autoimmune features (n = 5), undifferentiated CTD‐ILD (n = 3), ILD associated with lupus (n = 2), and antineutrophil cytoplasmic antibody–associated ILD, microscopic polyangiitis–associated ILD, polymyositis‐associated ILD, antisynthetase syndrome, CTD‐associated organizing pneumonia, and CTD‐ILD (n = 1 per condition). Among 158 subjects with available data, the autoimmune disease diagnosis was confirmed by a rheumatologist in 144 subjects (91.1%).
The baseline characteristics of the subgroup with autoimmune disease–related ILDs are shown in Table 1. The mean ± SD age was 64.3 ± 10.6 years, 52.9% of subjects were female, and 67.6% were White; the mean ± SD time since ILD diagnosis was 4.2 ± 4.1 years. Baseline characteristics of the subgroups according to ILD diagnosis are shown in Supplementary Table 1 (available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). Based on customized drug groupings, 77.1% of subjects were receiving ≥1 immunomodulatory therapy (any dose). Glucocorticoids were taken by 67.6% of subjects, nonbiologic DMARDs by 35.9%, and biologic DMARDs by 11.8% (Supplementary Table 2, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). There were some differences between the groups of subjects who were and those who were not receiving DMARDs and/or glucocorticoids at baseline (Supplementary Table 3, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). The majority of the subjects receiving DMARDs and/or glucocorticoids at baseline had RA‐ILD.
Table 1.
Baseline characteristics of subjects with autoimmune disease–related ILDs in the INBUILD trial*
| Nintedanib (n = 82) | Placebo (n = 88) | |
|---|---|---|
| Female | 47 (57.3) | 43 (48.9) |
| Age, mean ± SD years | 63.3 ± 10.0 | 65.1 ± 11.1 |
| BMI, mean ± SD kg/m2 | 26.7 ± 5.2 | 28.0 ± 4.9 |
| Current or former smoker | 40 (48.8) | 45 (51.1) |
| Race | ||
| White | 53 (64.6) | 62 (70.5) |
| Asian | 25 (30.5) | 25 (28.4) |
| Black/African American | 3 (3.7) | 1 (1.1) |
| American Indian/Alaska Native/Native Hawaiian/other Pacific Islander | 1 (1.2) | 0 (0.0) |
| Criteria for ILD progression within 24 months before screening | ||
| Relative decline in FVC% ≥10% | 43 (52.4) | 42 (47.7) |
| Relative decline in FVC% ≥5% to <10%, plus worsened respiratory symptoms and/or increased extent of fibrosis on HRCT | 26 (31.7) | 31 (35.2) |
| Worsened respiratory symptoms and increased extent of fibrosis on HRCT only | 13 (15.9) | 15 (17.0) |
| ILD diagnosis | ||
| RA‐associated ILD | 42 (51.2) | 47 (53.4) |
| SSc‐associated ILD | 23 (28.0) | 16 (18.2) |
| MCTD‐associated ILD | 7 (8.5) | 12 (13.6) |
| Other autoimmune ILDs† | 10 (12.2) | 13 (14.8) |
| Time since diagnosis of ILD based on imaging, mean ± SD years | 4.6 ± 4.4 | 4.0 ± 3.9 |
| UIP‐like fibrotic pattern on HRCT‡ | 62 (75.6) | 65 (73.9) |
| FVC, mean ± SD ml | 2,291 ± 722 | 2,366 ± 680 |
| FVC%, mean ± SD | 69.6 ± 15.1 | 72.1 ± 14.6 |
| DLco%, mean ± SD§ | 44.9 ± 13.4 | 50.8 ± 16.0 |
| K‐BILD total score, mean ± SD¶ | 51.3 ± 10.8 | 52.6 ± 9.7 |
Except where indicated otherwise, values are the number (%) of subjects. Not all subjects provided data for all variables. ILDs = interstitial lung diseases; BMI = body mass index; FVC% = forced vital capacity percent predicted; RA = rheumatoid arthritis; SSc = systemic sclerosis; MCTD = mixed connective tissue disease; UIP = usual interstitial pneumonia; DLco% = diffusing capacity of the lungs for carbon monoxide percent predicted; K‐BILD = King's Brief Interstitial Lung Disease questionnaire.
Subjects with an autoimmune disease noted in the “Other fibrosing ILDs” category of the case report form.
Two subjects with an undetermined high‐resolution computed tomography (HRCT) pattern were randomized and counted as having other fibrotic patterns.
Corrected for hemoglobin level.
Scores range from 0 to 100, with higher scores representing better health status.
Exposure
The mean ± SD exposure over 52 weeks was 10.1 ± 4.0 months in the nintedanib group and 11.1 ± 3.1 months in the placebo group. The mean ± SD exposure over the whole trial was 15.4 ± 7.4 months and 16.9 ± 6.1 months in these groups, respectively.
FVC end points
Among subjects with autoimmune disease–related ILDs, the adjusted annual rate of decline in FVC over 52 weeks was −75.9 ml/year in the nintedanib group compared to −178.6 ml/year in the placebo group (difference 102.7 ml/year [95% CI 23.2, 182.2]; nominal P = 0.0117) (Figure 1). No heterogeneity was detected in the effect of nintedanib versus placebo across the subgroups according to ILD diagnosis (P = 0.91) (Figure 1 and Supplementary Figure 2, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). In subjects with RA‐ILD (89 of the 170 subjects with autoimmune disease–related ILDs), the adjusted annual rate of decline in FVC over 52 weeks was −79.0 ml/year in the nintedanib group versus −196.9 ml/year in the placebo group (difference 117.9 ml/year [95% CI 5.2, 230.7]; nominal P = 0.041).
Figure 1.
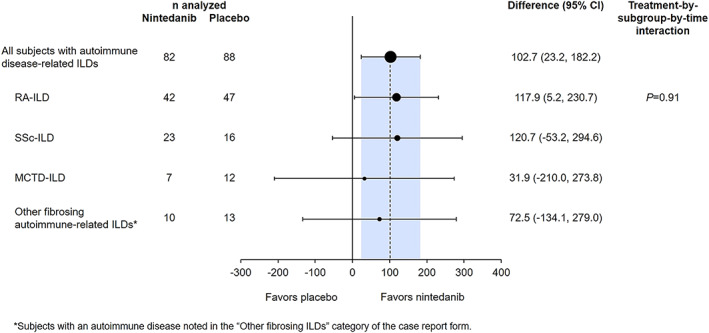
Rate of decline in forced vital capacity (FVC) (ml/year) over 52 weeks in subjects with autoimmune disease–related interstitial lung diseases (ILDs) treated with nintedanib compared to placebo in the INBUILD trial. RA‐ILD = rheumatoid arthritis–associated ILD; SSc‐ILD = systemic sclerosis–associated ILD; MCTD‐ILD = mixed connective tissue disease–associated ILD; 95% CI = 95% confidence interval.
In subjects with a UIP‐like fibrotic pattern on HRCT (n = 127), the adjusted annual rate of decline in FVC over 52 weeks was −58.6 ml/year in the nintedanib group versus −182.8 ml/year in the placebo group (difference 124.2 ml/year [95% CI 31.1, 217.4]) (Figure 2). In subjects with other fibrotic patterns on HRCT (n = 43), the rates of FVC decline in these groups were −126.4 ml/year versus −168.1 ml/year, respectively (difference 41.7 ml/year [95% CI −112.2, 195.5]). The effect of nintedanib versus placebo was numerically greater in subjects with a UIP‐like fibrotic pattern on HRCT than in those with other fibrotic patterns, but the exploratory interaction P value did not indicate heterogeneity in the treatment effect between these subgroups (P = 0.37) (Figure 2).
Figure 2.
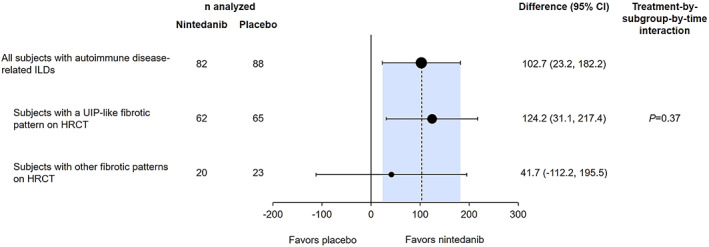
Rate of decline in FVC (ml/year) over 52 weeks in subjects with autoimmune disease–related ILDs treated with nintedanib compared to placebo in the INBUILD trial, according to fibrotic pattern on high‐resolution computed tomography (HRCT). UIP = usual interstitial pneumonia (see Figure 1 for other definitions).
Exploratory interaction P values did not indicate heterogeneity in the effect of nintedanib versus placebo on the annual rate of decline in FVC across subgroups according to use of DMARDs and/or glucocorticoids at baseline (Figure 3 and Supplementary Figure 3, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). The mean ± SE absolute changes from baseline in FVC at week 52 were −92.7 ± 29.1 ml in the nintedanib group and −185.7 ± 27.3 ml in the placebo group (difference 93.0 [95% CI 14.1, 172.0]; nominal P = 0.021). The mean ± SE absolute changes from baseline in FVC% at week 52 were −2.7 ± 0.9 in the nintedanib group and −6.0 ± 0.8 in the placebo group (difference 3.3 [95% CI 0.9, 5.6]; nominal P = 0.0073). The proportions of subjects with absolute declines or increases in FVC% >0% to ≤5%, >5% to ≤10%, >10% to ≤15%, and >15% at week 52 are shown in Supplementary Figure 4 (available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). Over the whole trial, the proportions of subjects with absolute and relative declines in FVC% ≥5% or ≥10% were lower in the nintedanib group than in the placebo group (Table 2).
Figure 3.
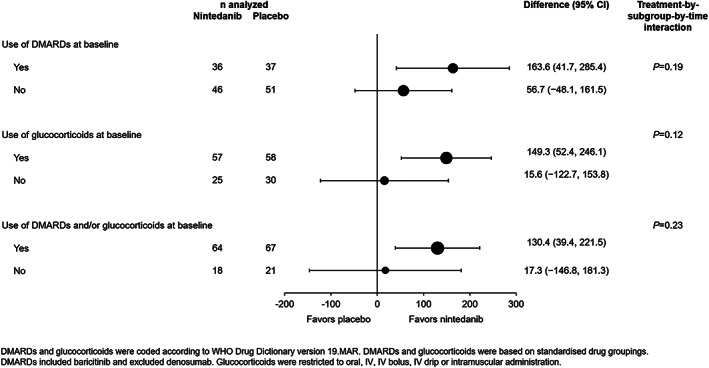
Rate of decline in FVC (ml/year) over 52 weeks in subjects with autoimmune disease–related ILDs treated with nintedanib compared to placebo in the INBUILD trial, according to use of disease‐modifying antirheumatic drugs (DMARDs) and/or glucocorticoids at baseline. WHO = World Health Organization; IV = intravenous (see Figure 1 for other definitions).
Table 2.
Efficacy end points in subjects with autoimmune disease–related ILDs in the INBUILD trial*
| Nintedanib (n = 82) | Placebo (n = 88) | Difference or HR (95% CI) | Nominal P | |
|---|---|---|---|---|
| Annual rate of decline in FVC over 52 weeks | ||||
| Rate of decline in FVC over 52 weeks, adjusted mean ± SE ml/year | −75.9 ± 29.3 | −178.6 ± 27.5 | 102.7 (23.2, 182.2)† | 0.012 |
| Changes in FVC at week 52 | ||||
| Absolute change from baseline in FVC at week 52, adjusted mean ± SE ml | −92.7 ± 29.1 | −185.7 ± 27.3 | 93.0 (14.1, 172.0)† | 0.021 |
| Absolute change from baseline in FVC% at week 52, adjusted mean ± SE | −2.7 ± 0.9 | −6.0 ± 0.8 | 3.3 (0.9, 5.6)† | 0.0073 |
| Change in K‐BILD total score at week 52 | ||||
| Absolute change from baseline in K‐BILD total score at week 52, adjusted mean ± SE | 2.10 ± 1.14 | 1.72 ± 1.08 | 0.38 (−2.71, 3.48)† | 0.81 |
| Time‐to‐event end points assessed over the whole trial, no. (%) | ||||
| Relative decline from baseline in FVC% ≥5% | 60 (73.2) | 73 (83.0) | 0.89 (0.63, 1.26)‡ | 0.50 |
| Relative decline from baseline in FVC% ≥10% | 41 (50.0) | 55 (62.5) | 0.85 (0.57, 1.28)‡ | 0.43 |
| Absolute decline from baseline in FVC% ≥5% | 52 (63.4) | 69 (78.4) | 0.78 (0.54, 1.12)‡ | 0.17 |
| Absolute decline from baseline in FVC% ≥10% | 29 (35.4) | 42 (47.7) | 0.72 (0.45, 1.16)‡ | 0.17 |
| Acute exacerbation of ILD or death | 10 (12.2) | 18 (20.5) | 0.58 (0.27, 1.27)‡ | 0.17 |
| Progression of ILD§ or death | 33 (40.2) | 47 (53.4) | 0.72 (0.46, 1.13)‡ | 0.15 |
| Death | 8 (9.8) | 11 (12.5) | 0.80 (0.32, 1.98)‡ | 0.62 |
Not all subjects provided data for all variables. See Table 1 for other definitions.
Difference (95% confidence interval [95% CI]).
Hazard ratio (HR) (95% CI).
Defined as absolute decline from baseline in FVC% ≥10%.
K‐BILD questionnaire total score
The adjusted mean absolute changes from baseline in K‐BILD total score at week 52 in the nintedanib and placebo groups, respectively, were 2.10 and 1.72 (difference 0.38 [95% CI −2.71, 3.48]; nominal P = 0.81) (Table 2).
Acute exacerbations, progression of ILD, and death
Over the whole trial, acute exacerbation of ILD or death occurred in 10 subjects (12.2%) in the nintedanib group and 18 (20.5%) in the placebo group (HR 0.58 [95% CI 0.27, 1.27]; nominal P = 0.17) (Table 2 and Supplementary Figure 5, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075). Acute exacerbations of ILD were reported in 4 subjects (4.9%) in the nintedanib group and 8 subjects (9.1%) in the placebo group. The proportions of subjects with progression of ILD or death were 40.2% in the nintedanib group and 53.4% in the placebo group (HR 0.72 [95% CI 0.46, 1.13]; nominal P = 0.15) (Table 2). Deaths occurred in 9.8% of subjects in the nintedanib group and 12.5% in the placebo group (HR 0.80 [95% CI 0.32, 1.98]; nominal P = 0.62) (Table 2; Supplementary Figure 5).
Safety and tolerability
Over 52 weeks, the most frequent AE was diarrhea, reported in 63.4% and 27.3% of subjects treated with nintedanib and placebo, respectively (Table 3). Nausea, vomiting, decreased appetite, constipation, abdominal pain, and weight decrease were also more frequently reported in the nintedanib group than in the placebo group. AEs led to permanent discontinuation of the trial drug in 17.1% of subjects in the nintedanib group and 10.2% of those who received placebo. The most frequent AE leading to permanent discontinuation of nintedanib was diarrhea (Table 3).
Table 3.
AEs in subjects with autoimmune disease–related ILDs in the INBUILD trial*
| Nintedanib (n = 82) | Placebo (n = 88) | |
|---|---|---|
| Any AE | 79 (96.3) | 79 (89.8) |
| Most frequent AEs† | ||
| Diarrhea | 52 (63.4) | 24 (27.3) |
| Nausea | 22 (26.8) | 10 (11.4) |
| Decreased appetite | 15 (18.3) | 1 (1.1) |
| Vomiting | 14 (17.1) | 6 (6.8) |
| Increase in ALT | 14 (17.1) | 3 (3.4) |
| Bronchitis | 13 (15.9) | 13 (14.8) |
| Increase in AST | 11 (13.4) | 4 (4.5) |
| Nasopharyngitis | 10 (12.2) | 13 (14.8) |
| Dyspnea | 6 (7.3) | 10 (11.4) |
| Constipation | 10 (12.2) | 5 (5.7) |
| Upper abdominal pain | 10 (12.2) | 2 (2.3) |
| Decrease in weight | 10 (12.2) | 1 (1.1) |
| Urinary tract infection | 9 (11.0) | 2 (2.3) |
| Severe AE‡ | 13 (15.9) | 16 (18.2) |
| Serious AE§ | 28 (34.1) | 28 (31.8) |
| Fatal AE | 3 (3.7) | 4 (4.5) |
| AE leading to treatment discontinuation | 14 (17.1) | 9 (10.2) |
| Most frequent AEs leading to treatment discontinuation¶ | ||
| Diarrhea | 4 (4.9) | 1 (1.1) |
| Increase in ALT | 3 (3.7) | 0 (0.0) |
| Increase in AST | 2 (2.4) | 0 (0.0) |
| Dyspnea | 0 (0.0) | 2 (2.3) |
Values are the number (%) of subjects with ≥1 such adverse event (AE) reported over 52 weeks (or until 28 days after last trial drug intake for subjects who discontinued the trial drug before week 52). ILDs = interstitial lung diseases; ALT = alanine aminotransferase; AST = aspartate aminotransferase.
AEs reported in >10% of subjects in either treatment group, coded using preferred terms in the Medical Dictionary for Regulatory Activities.
AE that was incapacitating or that caused an inability to work or to perform usual activities.
AE that resulted in death, was life‐threatening, resulted in hospitalization or prolongation of hospitalization, resulted in persistent or clinically significant disability or incapacity, was a congenital anomaly or birth defect, or was deemed to be serious for any other reason.
AEs that led to treatment discontinuation reported in >2% of subjects in either treatment group.
Increases in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) were more common in subjects treated with nintedanib compared to those who received placebo (Table 3). Based on laboratory tests, elevations in ALT and/or AST up to ≥3 times the upper limit of the normal range were observed in 12 subjects (14.6%) in the nintedanib group and 1 subject (1.1%) in the placebo group. Liver enzyme levels normalized or showed a trend toward normalization after dose adjustment or discontinuation (or spontaneously). No subjects met the criteria for Hy's law. AEs among subjects receiving versus those not receiving DMARDs and/or glucocorticoids at baseline are presented in Supplementary Table 4 (available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075).
DISCUSSION
These analyses of data from the INBUILD trial support the clinical observation that there is a group of patients with autoimmune disease–related ILDs who develop a progressive fibrosing phenotype with a natural history similar to IPF. In the INBUILD trial, such patients were identified using inclusion criteria based on worsening fibrosis on HRCT, lung function decline, and worsening symptoms, which are similar to the criteria used in clinical practice to identify patients with progressive ILD. Compared to placebo, nintedanib reduced the rate of decline in FVC over 52 weeks by 58% in subjects with progressive fibrosing autoimmune ILDs, a similar relative reduction to that observed in the overall population of the INBUILD trial (57%) (21), as well as in patients with SSc‐ILD in the SENSCIS trial (44%) (20) and patients with IPF in the INPULSIS trials (49%) (19). The INBUILD trial was not designed or powered to study patients with individual diseases. However, in subgroup analyses, no heterogeneity was detected in the effect of nintedanib versus placebo among patients with RA‐ILD, SSc‐ILD, MCTD‐ILD, or other fibrosing autoimmune ILDs.
Previous studies have suggested that in patients with autoimmune‐related ILDs, a UIP‐like fibrotic pattern on HRCT is associated with faster decline in lung function than other fibrotic patterns (5, 24, 25, 26), although this has not been demonstrated in patients with SSc‐ILD. In the overall population of the INBUILD trial, patients with a UIP‐like fibrotic pattern on HRCT showed a greater rate of decline in FVC over 52 weeks than patients with other fibrotic patterns on HRCT imaging (8,21). In patients with progressive autoimmune disease–related ILDs, the rate of decline in FVC over 52 weeks in the placebo group was similar between subjects with a UIP‐like pattern and those with other fibrotic patterns on HRCT. Although numerical differences were observed, based on statistical testing, no heterogeneity was detected in the effect of nintedanib versus placebo across subgroups according to fibrotic pattern on HRCT, either in the overall population (21) or in patients with progressive autoimmune disease–related ILDs. These subgroup analyses should be interpreted with caution due to the fact that the INBUILD trial was not powered for these analyses.
Patients who were receiving low‐dose glucocorticoids or DMARDs to treat their autoimmune disease were allowed to participate in the INBUILD trial. Among patients with autoimmune disease–related ILDs, glucocorticoids (mostly prednisone or prednisolone <20 mg/day) were taken at baseline by 68% of patients, nonbiologic DMARDs by 36%, and biologic DMARDs by 12%. More than half of the overall trial population were receiving glucocorticoids at baseline. Previous analyses of data from the overall trial population showed that the effect of nintedanib on reducing the rate of decline in FVC, and its AE profile, were consistent between subgroups according to use of glucocorticoids at baseline and between patients who received immunomodulatory medications at baseline or during the 52 weeks of the trial and those who did not (27).
Acute exacerbations in patients with fibrosing autoimmune‐related ILDs are rare events but are associated with high mortality (28, 29, 30). While the INBUILD trial was not powered to show an effect of nintedanib on this end point, throughout the whole trial, nintedanib was associated with a numerically reduced risk of the composite of acute exacerbation of ILD or death (HR 0.58). In the overall trial population, the HR for this end point was 0.67, and statistical significance was reached (P = 0.04) (31). Further research is needed to examine the effect of nintedanib on acute exacerbations of autoimmune‐related ILDs.
The AEs associated with nintedanib in patients with autoimmune ILDs in the INBUILD trial were consistent with those observed in the overall trial population (21,31) and in patients with SSc‐ILD (20, 32) and IPF (19, 33). Gastrointestinal AEs, particularly diarrhea, were the most common AEs. Elevations in liver enzymes were more common in patients treated with nintedanib than placebo.
Consistent with observations in the overall trial population (21), there was no meaningful change in health‐related quality of life assessed using the K‐BILD questionnaire in patients with autoimmune disease–related ILDs in either treatment group. This likely reflects the challenges of measuring changes in health‐related quality of life in patients with autoimmune diseases over relatively short timeframes and the weak association observed between small changes in FVC and changes in quality of life in patients with ILD and moderately impaired FVC at baseline (19, 20, 34).
Strengths of our analyses include the large cohort of patients with autoimmune disease–related ILDs (n = 170), the identification of ILD progression based on criteria that may be used in clinical practice, the randomized placebo‐controlled trial design, and the robust collection of FVC measurements and data on AEs throughout the trial. Limitations of our analyses include that the INBUILD trial was not designed or powered to show a benefit of nintedanib in the subgroup of patients with autoimmune disease–related ILDs, and the trial protocol excluded patients receiving some commonly used immunomodulatory medications. Data were not collected on medications that patients received prior to being enrolled in the INBUILD trial. The trial was not designed or powered to enable analyses based on individual clinical diagnoses or use of background therapies. In interpreting the subgroup analyses, it is important not to overinterpret the point estimates and confidence intervals for individual subgroups, but rather to look at the interaction P value that assessed whether there was evidence of heterogeneity across subgroups.
In conclusion, data from the INBUILD trial suggest that nintedanib slows the rate of decline in FVC in patients with progressive fibrosing ILDs, including autoimmune disease–related ILDs, with AEs that can be tolerated by most patients. Rheumatologists need to be aware of risk factors for ILD, incorporate judicious assessment strategies that can aid in identifying patients with ILD, monitor for progression of ILD, and formulate an appropriate treatment plan for patients with ILD in cooperation with colleagues in pulmonology and other relevant providers.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Matteson had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Seibold, Dellaripa, James, Schlenker‐Herceg, Stowasser, Quaresma, Flaherty.
Acquisition of data
Flaherty.
Analysis and interpretation of data
Matteson, Kelly, J. Distler, Hoffmann‐Vold, Seibold, Mittoo, Dellaripa, Aringer, Pope, O. Distler, James, Schlenker‐Herceg, Stowasser, Quaresma, Flaherty.
ROLE OF THE STUDY SPONSOR
Boehringer Ingelheim International GmbH (BI) participated in the study design, data collection, statistical analyses, data interpretation, and the writing of the report. The corresponding author had full access to all the data presented in this manuscript and had final responsibility for the decision to submit for publication. Writing assistance was provided by Elizabeth Ng, BSc and Wendy Morris, MSc of FleishmanHillard, London, UK, which was contracted and funded by BI. BI was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations. BI did not control the interpretation of the study results. Publication of this article was not contingent upon approval by BI. Data are available upon reasonable request (see Supplementary Materials, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42075).
ADDITIONAL DISCLOSURES
Author James is an employee of elderbrook solutions, GmbH, which was contracted by BI to conduct some of the analyses in this manuscript. Author Schlenker‐Herceg was an employee of BI at the time this manuscript was developed and is now an employee of CSL Behring. Authors Stowasser and Quaresma are employees of BI.
Supporting information
Disclosure Form
Appendix S1: Supporting Information
Appendix S2: Supporting Information
Video S1
ACKNOWLEDGMENTS
We thank the patients who participated in the INBUILD trial, and we thank Elizabeth Ng, BSc and Wendy Morris, MSc of FleishmanHillard, London, UK, for providing writing assistance.
A video abstract of this article can be found at https://players.brightcove.net/3806881048001/default_default/index.html?videoId=6295457676001
The INBUILD Trial was funded by Boehringer Ingelheim International GmbH.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42075&file=art42075‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Bongartz T, Nannini C, Medina‐Velasquez YF, Achenbach SJ, Crowson CS, Ryu JH, et al. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population based study. Arthritis Rheum 2010;62:1583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoffmann‐Vold AM, Fretheim H, Halse AK, Seip M, Bitter H, Wallenius M, et al. Tracking impact of interstitial lung disease in systemic sclerosis in a complete nationwide cohort. Am J Respir Crit Care Med 2019;200:1258–66. [DOI] [PubMed] [Google Scholar]
- 3. Reiseter S, Gunnarsson R, Aaløkken TM, Lund MB, Mynarek G, Corander J, et al. Progression and mortality of interstitial lung disease in mixed connective tissue disease: a long‐term observational nationwide cohort study. Rheumatology (Oxford) 2018;57:255–62. [DOI] [PubMed] [Google Scholar]
- 4. Gunnarsson R, Aaløkken TM, Molberg Ø, Lund MB, Mynarek GK, Lexberg AS, et al. Prevalence and severity of interstitial lung disease in mixed connective tissue disease: a nationwide, cross‐sectional study. Ann Rheum Dis 2012;71:1966–72. [DOI] [PubMed] [Google Scholar]
- 5. Zamora‐Legoff JA, Krause ML, Crowson CS, Ryu JH, Matteson EL. Progressive decline of lung function in rheumatoid arthritis‐associated interstitial lung disease. Arthritis Rheumatol 2017;69:542–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adegunsoye A, Oldham JM, Chung JH, Montner SM, Lee C, Witt LJ, et al. Phenotypic clusters predict outcomes in a longitudinal interstitial lung disease cohort. Chest 2018;153:349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jacob J, Hirani N, van Moorsel CH, Rajagopalan S, Murchison JT, van Es HW, et al. Predicting outcomes in rheumatoid arthritis related interstitial lung disease. Eur Respir J 2019;53:1800869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown KK, Martinez FJ, Walsh SL, Thannickal VJ, Prasse A, Schlenker‐Herceg R, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J 2020;55:2000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoffmann‐Vold AM, Allanore Y, Alves M, Brunborg C, Airó P, Ananieva LP, et al. Progressive interstitial lung disease in patients with systemic sclerosis‐associated interstitial lung disease in the EUSTAR database. Ann Rheum Dis 2020;80:219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Solomon JJ, Chung JH, Cosgrove GP, Demoruelle MK, Fernandez‐Perez ER, Fischer A, et al. Predictors of mortality in rheumatoid arthritis‐associated interstitial lung disease. Eur Respir J 2016;47:588–96. [DOI] [PubMed] [Google Scholar]
- 11. Kamiya Y, Fujisawa T, Kono M, Nakamura H, Yokomura K, Koshimizu N, et al. Prognostic factors for primary Sjögren's syndrome‐associated interstitial lung diseases. Respir Med 2019;159:105811. [DOI] [PubMed] [Google Scholar]
- 12. Roofeh D, Lin CJ, Goldin J, Kim GH, Furst DE, Denton CP, et al. Tocilizumab prevents progression of early systemic sclerosis associated interstitial lung disease. Arthritis Rheumatol 2021;73:1301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 2015;45:1434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang J, Beyer C, Palumbo‐Zerr K, Zhang Y, Ramming A, Distler A, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis 2016;75:883–90. [DOI] [PubMed] [Google Scholar]
- 15. Huang J, Maier C, Zhang Y, Soare A, Dees C, Beyer C, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis 2017;76:1941–8. [DOI] [PubMed] [Google Scholar]
- 16. Hoffmann‐Vold AM, Weigt SS, Saggar R, Palchevskiy V, Volkmann ER, Liang LL, et al. Endotype‐phenotyping may predict a treatment response in progressive fibrosing interstitial lung disease. EBioMedicine 2019;50:379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wollin L, Distler JH, Redente EF, Riches DW, Stowasser S, Schlenker‐Herceg R, et al. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur Respir J 2019;54:1900161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wollin L, Distler JH, Denton CP, Gahlemann M. Rationale for the evaluation of nintedanib as a treatment for systemic sclerosis‐associated interstitial lung disease. J Scleroderma Relat Disord 2019;4:212–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071–82. [DOI] [PubMed] [Google Scholar]
- 20. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for systemic sclerosis‐associated interstitial lung disease. N Engl J Med 2019;380:2518–28. [DOI] [PubMed] [Google Scholar]
- 21. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SL, Inoue Y, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019;381:1718–27. [DOI] [PubMed] [Google Scholar]
- 22. Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases: subgroup analyses by interstitial lung disease diagnosis in the randomised, placebo‐controlled INBUILD trial. Lancet Respir Med 2020;8:453–60. [DOI] [PubMed] [Google Scholar]
- 23. Patel AS, Siegert RJ, Brignall K, Gordon P, Steer S, Desai SR, et al. The development and validation of the King's Brief Interstitial Lung Disease (K‐BILD) health status questionnaire. Thorax 2012;67:804–10. [DOI] [PubMed] [Google Scholar]
- 24. Walsh SL, Sverzellati N, Devaraj A, Keir GJ, Wells AU, Hansell DM. Connective tissue disease related fibrotic lung disease: high resolution computed tomographic and pulmonary function indices as prognostic determinants. Thorax 2014;69:216–22. [DOI] [PubMed] [Google Scholar]
- 25. Adegunsoye A, Oldham JM, Bellam SK, Montner S, Churpek MM, Noth I, et al. Computed tomography honeycombing identifies a progressive fibrotic phenotype with increased mortality across diverse interstitial lung diseases. Ann Am Thorac Soc 2019;16:580–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh N, Varghese J, England BR, Solomon JJ, Michaud K, Mikuls TR, et al. Impact of the pattern of interstitial lung disease on mortality in rheumatoid arthritis: a systematic literature review and meta‐analysis. Semin Arthritis Rheum 2019;49:358–65. [DOI] [PubMed] [Google Scholar]
- 27. Cottin V, Richeldi L, Rosas I, Otaola M, Song JW, Tomassetti S, et al. Nintedanib and immunomodulatory therapies in progressive fibrosing interstitial lung diseases. Respir Res 2021;22:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hozumi H, Nakamura Y, Johkoh T, Sumikawa H, Colby TV, Kono M, et al. Acute exacerbation in rheumatoid arthritis‐associated interstitial lung disease: a retrospective case control study. BMJ Open 2013;3:e003132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manfredi A, Sebastiani M, Cerri S, Vacchi C, Tonelli R, Della Casa G, et al. Acute exacerbation of interstitial lung diseases secondary to systemic rheumatic diseases: a prospective study and review of the literature. J Thorac Dis 2019;11:1621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Salonen J, Purokivi M, Bloigu R, Kaarteenaho R. Prognosis and causes of death of patients with acute exacerbation of fibrosing interstitial lung diseases. BMJ Open Respir Res 2020;7:e000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Flaherty KR, Wells AU, Cottin V, Devaraj A, Inoue Y, Richeldi L, et al. Nintedanib in progressive interstitial lung diseases: data from the whole INBUILD trial. Eur Respir J 2021;2004538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seibold JR, Maher TM, Highland KB, Assassi S, Azuma A, Hummers LK, et al. Safety and tolerability of nintedanib in patients with systemic sclerosis‐associated interstitial lung disease: data from the SENSCIS trial. Ann Rheum Dis 2020;79:1478–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Corte T, Bonella F, Crestani B, Demedts MG, Richeldi L, Coeck C, et al. Safety, tolerability and appropriate use of nintedanib in patients with idiopathic pulmonary fibrosis. Respir Res 2015;16:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Volkmann ER, Tashkin DP, LeClair H, Roth MD, Kim G, Goldin J, et al. Treatment with mycophenolate and cyclophosphamide leads to clinically meaningful improvements in patient‐reported outcomes in scleroderma lung disease: results of Scleroderma Lung Study II. ACR Open Rheumatol 2020;2:362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Appendix S1: Supporting Information
Appendix S2: Supporting Information
Video S1