

Abstract
Fluorination is a potent method to modulate chemical properties of glycans. Here, we study how C3‐ and C6‐fluorination of glucosyl building blocks influence the structure of the intermediate of the glycosylation reaction, the glycosyl cation. Using a combination of gas‐phase infrared spectroscopy and first‐principles theory, glycosyl cations generated from fluorinated and non‐fluorinated monosaccharides are structurally characterized. The results indicate that neighboring group participation of the C2‐benzoyl protecting group is the dominant structural motif for all building blocks, correlating with the β‐selectivity observed in glycosylation reactions. The infrared signatures indicate that participation of the benzoyl group in enhanced by resonance effects. Participation of remote acyl groups such as Fmoc or benzyl on the other hand is unfavored. The introduction of the less bulky fluorine leads to a change in the conformation of the ring pucker, whereas the structure of the active dioxolenium site remains unchanged.
Keywords: Carbohydrates, Fluorine, Glycosylation, IR Spectroscopy, Mass spectrometry
The influence of fluorination on the gas‐phase structure of the glycosyl cation is probed with cryogenic infrared spectroscopy. It is shown that for building blocks, readily used in automated‐glycan assembly, C2 benzoyl protecting groups participate and shield the α‐side of the anomeric carbon. This effect is enhanced through resonance stabilization of the positive charge of the phenyl ring in the benzoyl group.
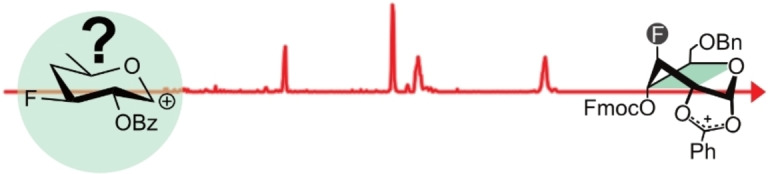
Introduction
Beyond the various roles of glycans in biological processes, [1] they exhibit a great pharmaceutical potential. Fractionated heparin is used as anti‐coagulating agent since the 1940s. Glycans used in biomedical applications are often extracted from natural sources. This approach not only limits the number of available compounds to those occurring in nature, but also requires elaborate separation workflows to produce pure and well‐defined molecules. [2] Furthermore, the short lifetimes of glycan‐based pharmaceuticals and their absorption properties, such as low lipophilicity, in the human body are impeding their usage. [3] An efficient method to modulate glycan properties is the incorporation of fluorine. Fluorinated glycans are more stable, [4] exhibit an increased lipophilicity [5] and are more potent against certain pathogens than their non‐fluorinated counterparts. [6] Moreover, site‐selective introduction of fluorine impacts material properties of carbohydrates as demonstrated for cellulose. [7]
Well‐defined fluorinated glycans can be synthesized by automated glycan assembly (AGA) [8] using fluorinated monosaccharide building blocks. AGA allows to control sequence, branching, and length, up to 100‐mers. [9] A major challenge in the glycosylation reactions is the stereoselective formation of α‐ and β‐glycosidic linkages. However, the underlying reaction mechanism is still not fully understood today, thus rendering the prediction of the stereochemical outcome of a reaction difficult. Generally, it is believed that the reaction is governed by a mechanistic continuum between SN1 and SN2, dependent on various parameters such as the nature of acceptor and donor, temperature, solvent, counter ions, or leaving groups. [10] Recently, a correlation between the stereoselectivity of the SN1 side of the continuum and the structure of the positively charged intermediate that is formed during the reaction, the glycosyl cation, has been determined. [11] To selectively generate 1,2‐trans linkages, participating acyl protecting groups such as benzoyl or acetyl at the C2 position are commonly used. [12] For glucose, it has been postulated that these neighboring protecting groups (PGs) shield the α‐side in glycosyl cations, forcing nucleophiles to attack from the β‐side.
Due to their short lifetimes, it is generally difficult to directly characterize glycosyl cations experimentally. They can be stabilized by super acids and subsequently be probed via NMR spectroscopy. However, the super acids fully protonate the glycosyl cation, leading to a distortion of its structure and properties. [13] Recently, it was shown that bare glycosyl cations can be isolated in the “clean‐room” environment of a mass spectrometer and subsequently characterized by gas‐phase infrared spectroscopy. First experiments demonstrated that acetyl groups in model building blocks show neighboring group participation (I, Scheme 1)[ 11a , 11b , 14 ] and remote participation (II), [15] in which the carbonyl oxygen forms a covalent bond with the anomeric carbon to yield a bicyclic dioxolenium intermediate. The studies also revealed that the gas‐phase structures of the investigated glycosyl cations correlate with the experimental stereoselectivity observed in solution‐phase studies of their precursors.
Scheme 1.
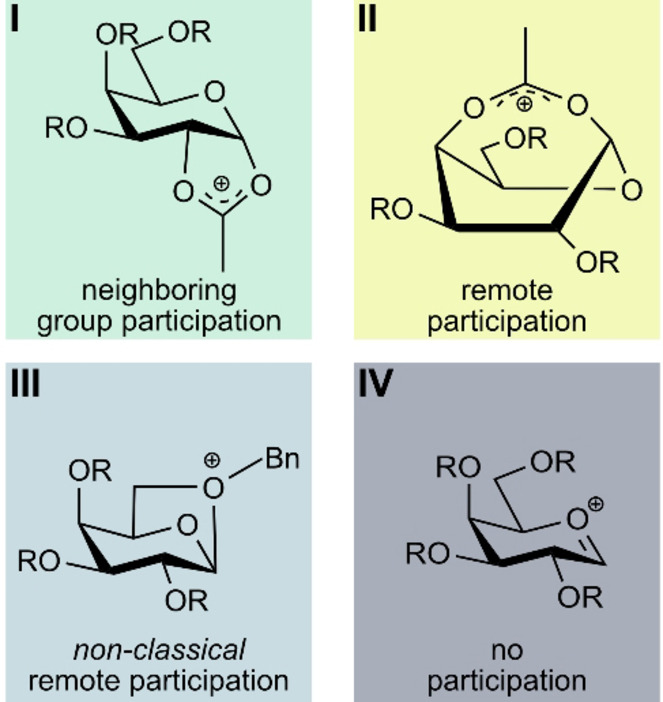
Modes of participation in glycosyl cations.
Interestingly, despite being formally known as non‐participating PGs, benzyl ether oxygens can also stabilize the positive charge at the anomeric carbon, resulting in the formation of oxonium ions (III). [15b]
Here, we combine cryogenic infrared spectroscopy with density functional theory (DFT) to probe glycosyl cations of functionalized glucose building blocks that are commonly used in glycan synthesis. The C2 position is always benzoylated (Bz), while the other hydroxyl groups are either protected with fluorenylmethoxycarbonyl (Fmoc) or benzyl (Bn) groups. In selected building blocks fluorine is introduced at the C3 or C6 position to study its impact on the structure of the glycosyl cation (Scheme 2). Further, the gas‐phase structures are correlated to the experimentally observed β‐stereoselectivity.
Scheme 2.
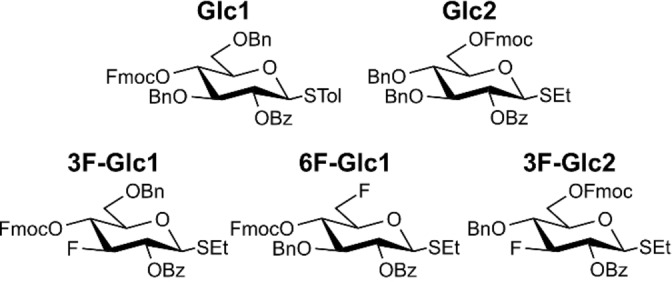
Differentially protected monosaccharide building blocks used in this study to generate glycosyl cations, which are subsequently probed by cryogenic infrared spectroscopy.
Results and Discussion
First, the IR signature of the non‐fluorinated glycosyl cation Glc1 is shown (Figure 1a). The functional group region (1450–1800 cm−1) shows five resolved absorption bands that clearly match the computed spectrum of the lowest‐energy structure I (Glc1), with an O,3 B ring pucker, exhibiting neighboring group participation (NGP) of the C2‐benzoyl group with a covalent bond (1.51 Å) between the carbonyl oxygen and the anomeric carbon. The signals at 1466 and 1500 cm−1 originate from the symmetric and antisymmetric dioxolenium stretches ν(O−C−O) of the participating Bz PG, while the signal at 1759 cm−1 stems from a carbonyl stretch ν(C=O) within the non‐participating Fmoc PG. Interestingly, the vibrations at 1519 and 1600 cm−1 are due to ν(C=C) stretches connected to resonance stabilization of the positive charge by the phenyl ring of the Bz PG in the dioxolenium motif (Scheme 3). The strong absorption at 1519 cm−1 is caused by the vibration of the formed C=C double bond, while the weak absorption at 1600 cm−1 can be attributed to the ν(C=C) stretches within the phenyl ring. The increased partial double bond character is also visible in the length of the C−C bond that decreases from 1.47 to 1.43 Å compared to the lowest‐energy oxocarbenium structure where the PGs do not participate. Thus, the charge of the glycosyl cation is not only delocalized within the dioxolenium motif, but also within the phenyl ring, leading to further stabilization. A similar stabilization by resonance effects in cations was previously reported for 4‐aminobenzoic acid in gas‐phase IR experiments. [16]
Figure 1.

Infrared spectra of (a) Glc1, (b) 3F−Glc1, and (c) 6F−Glc1 glycosyl cations generated from β‐thiotolyl (a) and β‐thioethyl (b,c) precursors. Experimental IR spectra are shown as light gray traces. Computed spectra of lowest‐energy dioxolenium structures, exhibiting neighboring group (green) and remote participation (yellow), oxonium (blue), and oxocarbenium structures (dark gray) are shown as inverted traces in respective colors. Relative free energies at 90 K are indicated. The lowest‐energy structures are shown in a simplified representation below the spectra, with their ring pucker annotated. For clarity, some protecting groups have been omitted and R used as abbreviation for fluorenylmethyl. 3D‐representation of the structures and xyz‐coordinates can be found in the SI.
Scheme 3.
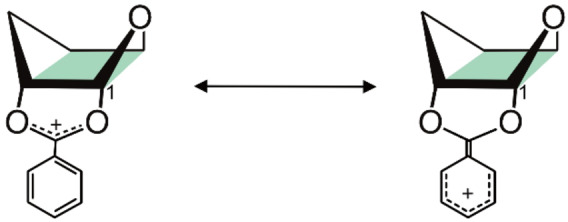
Resonance stabilization of the positive charge by the phenyl ring in benzoyl neighboring group participation. Glycosyl cations with this mode of participation are further stabilized by increased delocalization of the positive charge.
The fingerprint region (1000–1450 cm−1) contains a unique signature for each species, however, it is rather difficult to derive a structural assignment solely based on this region. Computational methods often fail to accurately model the fingerprint region in more complex systems, also due to anharmonicities. [17] The vibrations observed herein are mainly originating from C−C and C−O stretching vibrations (1000–1350 cm−1) as well as C−H bends (1350–1450 cm−1). The spectral signature corresponds the best to the lowest‐energy structure I (Glc1). Other structural motifs, such as remote participation of the Fmoc PG II (Glc1) (+61 kJ mol−1), remote benzyl ether participation III (Glc1) (+57 kJ mol−1) or oxocarbenium structures IV (Glc1) (+80 kJ mol−1), can be clearly ruled out due to two reasons: 1) their free energies at 90 K are significantly higher than those of structures exhibiting NGP; 2) their computed infrared spectra do not agree with the experimental spectrum (Figure 1a).
The IR spectra of the C3‐ and C6‐fluorinated glycosyl cations 3F−Glc1 and 6F−Glc1 are shown in Figure 1b and Figure 1c. Compared to Glc1, the spectral signature of the fluorinated counterparts is less crowded in the fingerprint region. Here, mainly one intense absorption band can be observed at 1234 cm−1 associated with a ν(C−O) stretch within the Fmoc PG. Otherwise, the spectral signature resembles that of Glc1. As a consequence, the glycosyl cations 3F−Glc1 and 6F−Glc1 mainly adopt dioxolenium‐type structures I exhibiting benzoyl NGP. Although all three experimental spectra share some similarities, the absorption bands differ in shape and exact position. Thus, each spectrum is a unique pattern for the probed glycosyl cation. Further evidence for a C2‐dioxolenium motif is provided by the computed spectra of structures exhibiting benzoyl NGP that also possess the lowest free energy of all sampled structures. In both cases, a 3 S 1 pucker is adopted with a bond distance of 1.50 Å between the carbonyl oxygen of the Bz PG and the anomeric carbon. The vibrations associated with the dioxolenium motif and the +M effect within the benzoyl group clearly correspond to the experimental signature. The carbonyl absorption band in I (3F−Glc1) corresponds to the experiment, while the experimental spectrum of 6F−Glc1 exhibits two carbonyl bands, which is diagnostic for a second low‐energy conformer (IB) simultaneously present in the ion trap. Like for Glc1, other structural motifs can be excluded based on their computed spectral signatures and unfavorable free energies.
Although the substitution of a benzyl group by fluorine changes the ring pucker from O,3 B in Glc1 to 3 S 1 in 3/6F−Glc1, it does not have an influence on the participation of the neighboring benzoyl group. The changes in ring pucker could be attributed to a decreased steric hindrance of fluorine compared to the bulkier benzyl PG. In all three cases, the α‐side of the glycosyl cation is efficiently shielded, leading to β‐stereoselectivity. This selectivity was observed in the AGA of deoxyfluorinated β(1,4) hexaglucoside analogues (employing building blocks Glc1, 3F−Glc1, and 6F−Glc1, see the Supporting Information).[ 7b , 18 ]
In a second set of glycosyl cations, Glc2 and 3F−Glc2, the C4 and C6 PGs are permuted, compared to Glc1 analogues. The IR spectra are shown in Figure 2. Generally, the spectral signature is slightly more congested than the corresponding Glc1 species, which is attributed to the population of multiple low‐energy conformers enabled by the increased flexibility of the Fmoc PG now located at the C6 position. In the functional group region, the spectra look similar to those previously shown, being diagnostic for C2‐dioxolenium structures exhibiting NGP. For Glc2, the lowest‐energy structure IA exhibits benzoyl NGP, with a 3 S 1 pucker and a bond distance of 1.51 Å between the carbonyl oxygen of the benzoyl group and the anomeric carbon. A second low‐energy conformer IB (+5 kJ mol−1) was sampled, in which the Fmoc, the C4‐Bn and the participating Bz PG are stacked. The differently orientated Fmoc PG leads to a shift of the position of the carbonyl band. The population of these two low‐energy conformers might explain the presence of two carbonyl bands and the wealth of absorption bands in the fingerprint region in the experimental spectrum. For 3F−Glc2, the lowest‐energy conformer IA exhibits a 5 H 4 pucker, however, its IR signature matches the experiment slightly less well than that of a second low‐energy structure IB (+3 kJ mol−1) with a O S 2 pucker and a 1.50 Å bond distance. Again, other structural motifs are unlikely, considering their spectral signature and energetics. Here, fluorine has an influence on the ring pucker, but not on the overall structural motif, strongly correlated to the experimental β‐stereoselectivity. Formation of β‐linkages was observed in the AGA of deoxyfluorinated glucosides (employing building blocks Glc2 and 3F−Glc2). [19]
Figure 2.

Infrared spectra of (a) Glc2 and (b) 3F−Glc2 glycosyl cations generated from β‐thioethyl precursors. Experimental IR spectra are shown as light gray traces. Computed spectra of lowest‐energy dioxolenium structures, exhibiting neighboring group (green) and remote participation (yellow), and oxocarbenium structures (dark gray) are shown as inverted traces in respective colors. Relative free energies at 90 K are indicated. The lowest‐energy structures are shown in a simplified representation below the spectra, with their ring pucker annotated (for Glc2, IA and IB the differences in structures are too subtle to represent them in the simplified representation, therefore, the reader is referred to the 3D‐structure in Figure S12). For clarity, some protecting groups have been omitted and R used as abbreviation for fluorenylmethyl. 3D‐representation of the structures and xyz‐coordinates can be found in the SI.
Conclusion
To conclude, we have shown that it is possible to generate and probe glycosyl cations and their fluorinated analogues from precursors readily used in glycan synthesis. In each case, the underlying structural motif can be clearly identified as neighboring group participation of C2‐benzoyl protecting groups. Interestingly, participation of the Bz protecting groups is connected to resonance effects involving the phenyl ring, which can be directly monitored due to vibrations associated with the delocalized electrons. The permutation of the protecting groups as well as their substitution by the less bulky fluorine leads to a change in the conformation of the ring pucker. However, the structure of the active dioxolenium site remains unchanged and the stereoselectivity observed for these building blocks in glycosylation reactions is therefore not affected. Further experiments are needed to explore the effects of a C2‐ and C4‐fluorination, which are expected to have a much more significant impact on the structure of the reactive glycosyl‐cation intermediate.
Experimental Section
Cryogenic infrared spectroscopy
A detailed description of the experimental setup can be found in the SI (Figure S1) and in previous publications. [20] Briefly, thioglycoside precursors were transferred into the gas phase via nanoelectrospray ionization (nESI). The leaving group is cleaved by in‐source fragmentation leading to glycosyl cations. Mass spectra can be found in the SI (Figures S2–S6). The ions of interest are mass‐to‐charge selected by a quadrupole mass filter and accumulated in a hexapole ion trap, which is cooled to approximately 90 K by liquid nitrogen. Superfluid helium nanodroplets (0.4 K) are generated by an Even‐Lavie valve and traverse the ion trap, picking up ions, and guide them to a detection region, where the embedded ions are excited by IR photons generated by the free‐electron laser of the Fritz Haber Institute (FHI FEL [21] ). Upon absorption of resonant photons, ions are eventually released from the droplets and afterwards detected by a time‐of‐flight detector. Monitoring the ion signal as a function of the IR photon wavenumber leads to a high‐resolution IR signature of the probed ion.
Computational methods
To model the IR spectra of the probed ions, candidate structures were sampled using the genetic algorithm (GA) FAFOOM. [22] The GA allows sampling flexible bonds and ring puckers and sends each sampled geometry to an external software (ORCA 4.1.1) [23] for DFT optimization at the PBE/def2‐SVP [24] level of theory. This conformational search mainly yielded dioxolenium‐type structures I, in which the benzoyl group shields the anomeric carbon from the α‐side, and oxocarbenium‐type structures IV, in which no participation takes place. Furthermore, the algorithm also generated structures in which either the remote Fmoc (C4 and C6) or Bn PGs (C6 only) interact with the anomeric carbon (dioxolenium II and oxonium structures III). A subset of structures of each type was reoptimized and their harmonic frequencies computed at the PBE0+D3/6‐311+G(d,p) [25] level of theory using Gaussian 16. [26] Each computed IR spectrum was normalized and scaled by 0.965. Ring puckers were assigned according to Cremer‐Pople coordinates. [27] The employed DFT functionals were chosen because they showed chemical accuracy in a benchmark study on carbohydrates. [28] Details of the reoptimized structures, such as energetics, ring puckers, and coordinates can be found in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors gratefully acknowledge the expertise of Dr. Wieland Schöllkopf and Sandy Gewinner for running the FHI FEL. K.G. thanks the Fonds National de la Recherche (FNR), Luxembourg, for funding the project GlycoCat (13549747). C.K. is grateful for financial support by Fonds der Chemischen Industrie. R.C. and K.P. thank the Deutsche Forschungsgemeinschaft (DFG) for support under project number 387284271‐SFB 1349. K.P. acknowledges generous funding by the European Research Council, ERC‐2019‐CoG‐863934‐GlycoSpec. M.D., G.F., and P.H.S. thank the MPG‐FhG Cooperation Project Glyco3Dysplay and the German Federal Ministry of Education and Research (BMBF, grant number 13XP5114) for financial support. P.H.S. thanks the Max Planck Society for generous financial support. Open Access funding enabled and organized by Projekt DEAL.
K. Greis, C. Kirschbaum, G. Fittolani, E. Mucha, R. Chang, G. von Helden, G. Meijer, M. Delbianco, P. H. Seeberger, K. Pagel, Eur. J. Org. Chem. 2022, e202200255.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Varki A., Glycobiology 2017, 27, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seeberger P. H., Cummings R. D., in: Essentials of Glycobiology, 3rd ed. (Eds.: A. Varki rd,, Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H.), Cold Spring Harbor (NY), 2015, pp. 729. [PubMed] [Google Scholar]
- 3.
- 3a. Hevey R., Chem. Eur. J. 2021, 27, 2240; [DOI] [PubMed] [Google Scholar]
- 3b. Linclau B., Ardá A., Reichardt N. C., Sollogoub M., Unione L., Vincent S. P., Jiménez-Barbero J., Chem. Soc. Rev. 2020, 49, 3863. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Geissner A., Baumann L., Morley T. J., Wong A. K. O., Sim L., Rich J. R., So P. P. L., Dullaghan E. M., Lessard E., Iqbal U., Moreno M., Wakarchuk W. W., Withers S. G., ACS Cent. Sci. 2021, 7, 345; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Axer A., Jumde R. P., Adam S., Faust A., Schäfers M., Fobker M., Koehnke J., Hirsch A. K. H., Gilmour R., Chem. Sci. 2021, 12, 1286; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Lo H. J., Krasnova L., Dey S., Cheng T., Liu H., Tsai T. I., Wu K. B., Wu C. Y., Wong C. H., J. Am. Chem. Soc. 2019, 141, 6484. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. St-Gelais J., Côté E., Lainé D., Johnson P. A., Giguère D., Chem. Eur. J. 2020, 26, 13499; [DOI] [PubMed] [Google Scholar]
- 5b. St-Gelais J., Bouchard M., Denavit V., Giguère D., J. Org. Chem. 2019, 84, 8509; [DOI] [PubMed] [Google Scholar]
- 5c. Lainé D., Lessard O., St-Gelais J., Giguère D., Chem. Eur. J. 2021, 27, 3799. [DOI] [PubMed] [Google Scholar]
- 6. Vaugenot J., El Harras A., Tasseau O., Marchal R., Legentil L., Le Guennic B., Benvegnu T., Ferrières V., Org. Biomol. Chem. 2020, 18, 1462. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Delbianco M., Seeberger P. H., Mater. Horiz. 2020, 7, 963; [Google Scholar]
- 7b. Yu Y., Tyrikos-Ergas T., Zhu Y., Fittolani G., Bordoni V., Singhal A., Fair R. J., Grafmuller A., Seeberger P. H., Delbianco M., Angew. Chem. Int. Ed. 2019, 58, 13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Plante O. J., Palmacci E. R., Seeberger P. H., Science 2001, 291, 1523. [DOI] [PubMed] [Google Scholar]
- 9. Joseph A. A., Pardo-Vargas A., Seeberger P. H., J. Am. Chem. Soc. 2020, 142, 8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Adero P. O., Amarasekara H., Wen P., Bohe L., Crich D., Chem. Rev. 2018, 118, 8242; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Chatterjee S., Moon S., Hentschel F., Gilmore K., Seeberger P. H., J. Am. Chem. Soc. 2018, 140, 11942. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Mucha E., Marianski M., Xu F.-F., Thomas D. A., Meijer G., von Helden G., Seeberger P. H., Pagel K., Nat. Commun. 2018, 9, 4174; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Elferink H., Severijnen M. E., Martens J., Mensink R. A., Berden G., Oomens J., Rutjes F., Rijs A. M., Boltje T. J., J. Am. Chem. Soc. 2018, 140, 6034; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Hettikankanamalage A. A., Lassfolk R., Ekholm F. S., Leino R., Crich D., Chem. Rev. 2020, 120, 7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hahm H. S., Hurevich M., Seeberger P. H., Nat. Commun. 2016, 7, 12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lebedel L., Ardá A., Martin A., Désiré J., Mingot A., Aufiero M., Aiguabella Font N., Gilmour R., Jiménez-Barbero J., Blériot Y., Thibaudeau S., Angew. Chem. Int. Ed. 2019, 58, 13758. [DOI] [PubMed] [Google Scholar]
- 14. Greis K., Kirschbaum C., Leichnitz S., Gewinner S., Schöllkopf W., von Helden G., Meijer G., Seeberger P. H., Pagel K., Org. Lett. 2020, 22, 8916. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Elferink H., Mensink R. A., Castelijns W. W. A., Jansen O., Bruekers J. P. J., Martens J., Oomens J., Rijs A. M., Boltje T. J., Angew. Chem. Int. Ed. 2019, 58, 8746; [DOI] [PubMed] [Google Scholar]
- 15b. Marianski M., Mucha E., Greis K., Moon S., Pardo A., Kirschbaum C., Thomas D. A., Meijer G., von Helden G., Gilmore K., Seeberger P. H., Pagel K., Angew. Chem. Int. Ed. 2020, 59, 6166; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Hansen T., Elferink H., van Hengst J. M. A., Houthuijs K. J., Remmerswaal W. A., Kromm A., Berden G., van der Vorm S., Rijs A. M., Overkleeft H. S., Filippov D. V., Rutjes F., van der Marel G. A., Martens J., Oomens J., Codee J. D. C., Boltje T. J., Nat. Commun. 2020, 11, 2664; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Greis K., Mucha E., Lettow M., Thomas D. A., Kirschbaum C., Moon S., Pardo-Vargas A., von Helden G., Meijer G., Gilmore K., Seeberger P. H., Pagel K., ChemPhysChem 2020, 21, 1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Seo J., Warnke S., Gewinner S., Schollkopf W., Bowers M. T., Pagel K., von Helden G., Phys. Chem. Chem. Phys. 2016, 18, 25474; [DOI] [PubMed] [Google Scholar]
- 16b. Khuu T., Yang N., Johnson M. A., Int. J. Mass Spectrom. 2020, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Brauer B., Pincu M., Buch V., Bar I., Simons J. P., Gerber R. B., J. Phys. Chem. A 2011, 115, 5859; [DOI] [PubMed] [Google Scholar]
- 17b. Mucha E., Stuckmann A., Marianski M., Struwe W. B., Meijer G., Pagel K., Chem. Sci. 2019, 10, 1272; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Grabarics M., Lettow M., Kirschbaum C., Greis K., Manz C., Pagel K., Chem. Rev. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fittolani G., Shanina E., Guberman M., Seeberger P. H., Rademacher C., Delbianco M., Angew. Chem. Int. Ed. 2021, 60, 13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Delbianco M., Kononov A., Poveda A., Yu Y., Diercks T., Jiménez-Barbero J., Seeberger P. H., J. Am. Chem. Soc. 2018, 140, 5421; [DOI] [PubMed] [Google Scholar]
- 19b. Gim S., Fittolani G., Yu Y., Zhu Y., Seeberger P. H., Ogawa Y., Delbianco M., Chem. Eur. J. 2021, 27, 13139; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Poveda A., Fittolani G., Seeberger P. H., Delbianco M., Jiménez-Barbero J., Front. Mol. Biosci. 2021, 8, 784318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Thomas D. A., Mucha E., Lettow M., Meijer G., Rossi M., von Helden G., J. Am. Chem. Soc. 2019, 141, 5815; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Thomas D. A., Chang R., Mucha E., Lettow M., Greis K., Gewinner S., Schöllkopf W., Meijer G., von Helden G., Phys. Chem. Chem. Phys. 2020, 22, 18400; [DOI] [PubMed] [Google Scholar]
- 20c. Lettow M., Grabarics M., Greis K., Mucha E., Thomas D. A., Chopra P., Boons G. J., Karlsson R., Turnbull J. E., Meijer G., Miller R. L., von Helden G., Pagel K., Anal. Chem. 2020, 92, 10228. [DOI] [PubMed] [Google Scholar]
- 21. Schöllkopf W., Gewinner S., Junkes H., Paarmann A., von Helden G., Bluem H. P., Todd A. M. M., Proc. SPIE-Int. Soc. Opt. Eng. 2015, 9512, 95121 L. [Google Scholar]
- 22. Supady A., Blum V., Baldauf C., J. Chem. Inf. Model. 2015, 55, 2338. [DOI] [PubMed] [Google Scholar]
- 23. Neese F., WIREs Comput. Mol. Sci. 2012, 2, 73. [Google Scholar]
- 24.
- 24a. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865; [DOI] [PubMed] [Google Scholar]
- 24b. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Adamo C., Barone V., J. Chem. Phys. 1999, 110, 6158; [Google Scholar]
- 25b. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 26.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Wallingford, CT, 2016.
- 27. Cremer D., Pople J. A., J. Am. Chem. Soc. 1975, 97, 1354. [Google Scholar]
- 28. Marianski M., Supady A., Ingram T., Schneider M., Baldauf C., J. Chem. Theory Comput. 2016, 12, 6157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.