

Abstract
The DNA damage response (DDR) is a complex set of downstream pathways triggered in response to DNA damage to maintain genomic stability. Many tumours exhibit mutations which inactivate components of the DDR, making them prone to the accumulation of DNA defects. These can both facilitate the development of tumours and provide potential targets for novel therapeutic interventions. The inhibition of the DDR has been shown to induce radiosensitivity in certain cancers, rendering them susceptible to treatment with radiotherapy and improving the therapeutic window. Moreover, DDR defects are a strong predictor of patient response to immune checkpoint inhibition (ICI). The ability to target the DDR selectively has the potential to expand the tumour neoantigen repertoire, thus increasing tumour immunogenicity and facilitating a CD8+ T and NK cell response against cancer cells. Combinatorial approaches, which seek to integrate DDR inhibition with radiotherapy and immunotherapy, have shown promise in early trials. Further studies are necessary to understand these synergies and establish reliable biomarkers.
Keywords: cancer, DNA damage response, double‐strand breaks, immunotherapy, radiotherapy
Introduction
To combat threats posed by DNA damage, cells have evolved a complex set of mechanisms – the DNA damage response (DDR) – to detect DNA damage, flag its presence and promote its repair to ensure genomic stability. 1 The DDR is an intricate, highly coordinated signalling network comprising many levels of crosstalk and feedback control between a vast variety of factors. Ultimately, activation of the DDR promotes cell cycle arrest to provide adequate time to repair genotoxic injury or, in the case of excessive DNA damage, to trigger permanent senescence or apoptosis. 2 As a result, the DDR plays an important tumour‐suppressive role. 3 , 4 Many cancers exhibit inactivation of DDR components, allowing for uncontrolled proliferation of cancer cells – highlighted by hereditary conditions such as Lynch syndrome or xeroderma pigmentosum. 5 , 6 , 7 Moreover, not only does DDR inactivation promote oncogenesis, but it also renders cancer cells more dependent on other repair components, especially under increased genotoxic stress induced by radiotherapy and chemotherapy. 8 This can be leveraged therapeutically, as exemplified by poly(ADP‐ribose) polymerase inhibitors (PARPi), which selectively target breast cancer genes 1/2 (BRCA1/2) deficient tumours by synthetic lethality. 9 , 10
Evasion of the immune system is another fundamental hallmark of cancer. Emerging data provides compelling evidence that DDR and the cellular pathological DNA sensing pathways of the innate immune system share effector molecules, demonstrating inextricable links between the DDR and the immune response. 11 Genomic instability resulting from DDR defects can lead to an increase in the generation of DNA‐based neoantigens, upregulate the expression of programmed death ligand 1 (PD‐L1) and engage signalling pathways such as cyclic GMP–AMP synthase‐stimulator of interferon genes (cGAS–STING). 12 Immune checkpoint inhibitors (ICIs) such as anti‐PD1/PD‐L1 and anti‐cytotoxic T‐lymphocyte‐associated protein 4 (anti‐CTLA4) antibodies have led to notable treatment improvements in a limited subset of patients. 8 , 13 Intriguingly, radiotherapy can enhance immunotherapy response, likely through activation of DDR and effector immune molecules. These advances open the door for new possibilities, in which multi‐modal approach targeting different molecular pathways can be utilized to improve cancer outcomes. 14 , 15
This review provides a basic summary of the main DDR pathways, the prevalence of DDR mutations in cancers commonly treated with radiotherapy and immunotherapy, the impact of DDR alterations on the immune response and its potential applications as a therapeutic target in radiotherapy and immunotherapy.
DNA damage response signalling
The DDR can be activated by a variety of insults and is mainly mediated by proteins of the phosphatidylinositol 3‐kinase (PI3K)‐like protein kinase family – ataxia‐telangiectasia mutated (ATM), ATM‐ and RAD3‐related (ATR) and DNA‐dependent protein kinase (DNA‐PK) – and by members of the PARP family. 16 The ATM pathway is initiated primarily by DNA double‐strand breaks (DSB). 17 DSBs are first detected by the MRN (MRE11:RAD50:NBS1) sensor complex, which triggers autophosphorylation of ATM. 18 Once stimulated, ATM phosphorylates serine‐139 residues on histone H2AX, stimulating a conformational change into γ‐H2AX. In turn, γ‐H2AX facilitates activation of many downstream enzymes, most important being checkpoint kinase 2 (CHK2). The ATM‐CHK2 pathway increases intracellular concentration of protein p53 via its direct phosphorylation and inactivation of MDM2, an endogenous inhibitor of p53. 19 Increase in p53 arrests the cell cycle and coordinates an adequate damage response, ranging from DNA repair to cell apoptosis. 17 In contrast, the ATR‐checkpoint kinase 1 (CHK1) response is evoked mostly by stretches of single‐strand DNA (ssDNA) exposed by the uncoupling of the helicase–polymerase complex at stalled replication forks and at resected DSBs. 18 , 20 Presence of an isolated DNA strand leads to avid binding of replication protein A (RPA) and establishment of RPA‐ssDNA platform at the site of injury. 21 This in turn leads to binding of the ATR‐interacting protein (ATRIP), with subsequent recruitment of ATR and several of its regulators. 22 Together, these proteins enable DNA topoisomerase 2‐binding protein 1 to stimulate the kinase activity of ATR‐ATRIP. 23 Aided by mediator proteins, ATR phosphorylates its main downstream effector kinase CHK1, 22 which slows down S phase progression, stabilizes replication forks and promotes DNA repair. 23
DSB repair pathways
Considered the most severe form of DNA injury, DSBs are induced by exogenous factors such as ionising radiation and endogenous sources such as oxidative stress due to generation of reactive oxygen species (ROS). 24 Incorrect DSB repair can lead to mutation and instability of key regulatory genes, resulting in oncogenesis. 25 Two canonical pathways dominate the repair of DSBs: homologous recombination (HR) and non‐homologous end joining (NHEJ) 26 (Fig. 1). Two more error‐prone pathways may also be important in response to radiation therapy‐induced DSBs: microhomology‐mediated end joining (MMEJ) and single strand annealing (SSA).
Fig. 1.
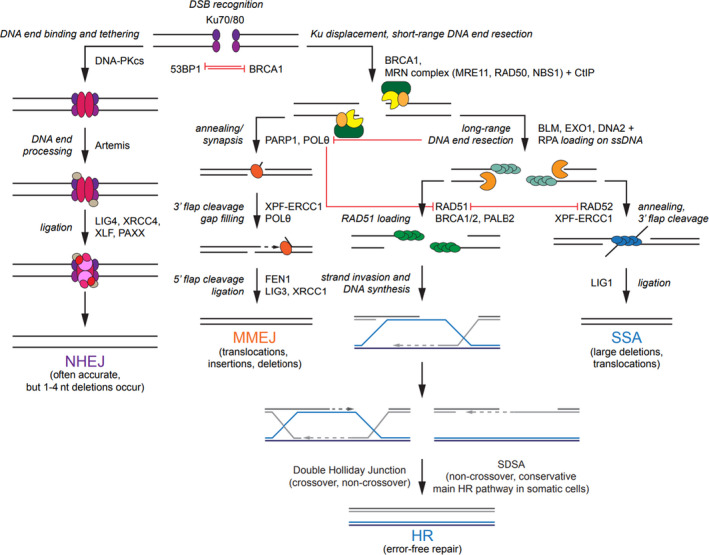
DSB repair pathways. Choice of the pathway is initially determined by 53BP1 and BRCA1, with 53BP1 promoting NHEJ and BRCA1 stimulating HR. NHEJ begins with Ku70‐Ku80 heterodimer binding to the broken DNA ends, followed by trimming via endonuclease Artemis and final ligation step via the LIG4‐XRCC4‐XLF‐PAXX complex. In MMEJ (sometimes also known as aEJ or TMEJ), PARP1 promotes DNA end synapsis and POLθ recruitment. After annealing of the microhomologous sequences (2–20 base pairs), the XPF/ERCC1 complex removes the redundant 3′ flaps. Subsequent ligation is mediated by LIG3/XRCC1. HR is initiated by DSB sensing by the MRN complex. Facilitated by BRCA1 and CtIP, MRN performs a short‐range resection, followed by a more extensive resection by EXO1 and/or BLM with DNA2 nuclease with subsequent coating of the 3’ ssDNA overhangs by RPA. BRCA1‐PALB2‐BRCA2 complex promotes RAD51 filament assembly. At this point, the DNA can be extended in a template‐dependent manner via SDSA, which results in a non‐crossover gene conversion. Alternatively, the formation of the double Holliday junction can resolve either as a crossover or as a non‐crossover. In contrast to MMEJ, SSA requires long regions of homology (20–25 base pairs) between the resected DNA ends. Annealing of the complementary ssDNA is mediated by RAD52, while non‐homologous flaps are digested by the XPF/ERCC1 complex. DSB, double‐strand break; 53BP1, p53 binding protein 1; BRCA1/2, breast cancer gene 1/2; NHEJ, non‐homologous end joining; HR, homologous recombination; LIG3/4, ligase 3/4; XRCC1/4, Xray repair cross‐complementing protein 1/4; XLF ‐ X‐ray repair cross‐complementing protein‐like factor; PAXX, paralogue of X‐ray repair cross‐complementing protein and X‐ray repair cross‐complementing protein‐like factor; MMEJ, microhomology‐mediated end joining; aEJ, alternative end joining; TMEJ, theta‐mediated end joining; PARP1, poly(ADP‐ribose) polymerase 1; POLθ, polymerase θ; XPF, xeroderma pigmentosum complementation group F; ERCC1 ‐ excision repair cross‐complementation group 1; DSB, double‐stranded break; MRN, meiotic recombination 11: radiation sensitive 50: Nijmegen breakage syndrome 1; CtIP, C‐terminal interacting protein; EXO1, exonuclease 1; BLM, Bloom syndrome helicase; DNA2, DNA replication helicase/nuclease 2; ssDNA, single‐stranded DNA; RPA, replication protein A; PALB2, partner and localiser of BRCA2; RAD51/52, radiation sensitive 51/52; SDSA, synthesis‐dependent strand annealing. [Colour figure can be viewed at wileyonlinelibrary.com]
Non‐homologous end joining
Classical NHEJ (known as NHEJ) – so called to distinguish it from its more error‐prone substitute, MMEJ – is a fast, high capacity pathway operating throughout the cell cycle and responsible for the repair of the majority of DSBs 27 (Fig. 2). cNHEJ is initiated by the binding of Ku70‐Ku80 heterodimer, which exhibits strong affinity towards DNA ends that are blunt or possess short, single‐stranded overhangs (Fig. 1). 26 Once bound, Ku70‐Ku80 recruits DNA‐PK, which in turn captures and phosphorylates Artemis, a protein with both 3′ and 5′ endonuclease activity. 28 , 29 Artemis prepares the DNA for ligation by trimming the overhanging strands into blunt ends. 28 Furthermore, DNA ends can be modified by the action of polymerases μ and λ into regions of microhomology (<4 nucleotides), which facilitate repair in certain types of damage. 30 Following trimming, a complex comprising PAXX, XRCC4, XRCC4‐like factor, and DNA ligase IV (LIG4) is assembled, 31 , 32 after which LIG4 completes the process of repair by ligating the broken DNA strand. 27 Unlike HR, NHEJ does not require a template in the form of a sister chromatid or homologous chromosome for DNA repair and does not result in the synthesis of new DNA strands. NHEJ is relatively accurate, although small insertions and deletions sometimes occur.
Fig. 2.
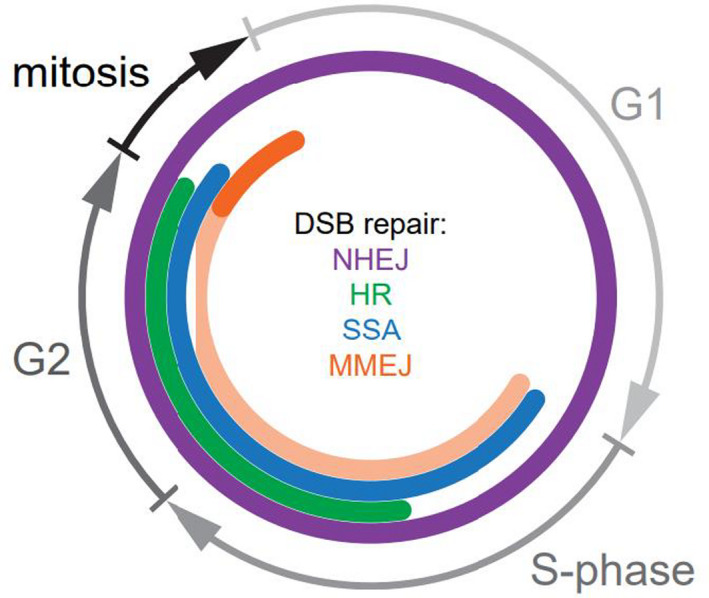
DSB repair throughout the cell cycle. NHEJ is active in all phases of the cell cycle. In contrast, the HR pathway, which requires the presence of a homologous template for strand synthesis, takes place in the late S/G2 phase. MMEJ repair is active from early S phase until mid‐late mitosis (anaphase). However, under normal conditions, its activity is delayed until mitotic onset due to inhibitory effect of BRCA2 and RAD52. SSA is active in both early mitosis and S/G2 phase. DSB, double‐strand break; NHEJ, non‐homologous end joining; HR, homologous recombination; MMEJ, microhomology‐mediated end joining; BRCA2, breast cancer gene 2; RAD52, radiation sensitive 52; SSA, single‐strand annealing. [Colour figure can be viewed at wileyonlinelibrary.com]
Microhomology‐mediated end joining
Microhomology‐mediated end joining pathway is often upregulated in cancer cells deficient in NHEJ and/or HR repair, acting as a back‐up mechanism to deal with extensive DNA damage. 30 , 33 This pathway requires the presence of microhomology regions and is triggered by the binding of PARP1, which provides a scaffold for the assembly of other factors such as MRN, X‐ray repair cross‐complementing protein (XRCC1) and LIG3 (Fig. 1). 34 Through the action of MRN and C‐terminal interacting protein (CtIP), DNA ends are resected to form 2–20 nucleotide‐long microhomologous overhangs. 34 , 35 End‐bridging is accomplished by coordinated activity of PARP1 and polymerase θ (POLθ). 36 Subsequently, LIG3/XRCC1 complex ligates the DNA strand. 34 MMEJ is highly error prone and carries an increased risk of chromosomal translocations 37 and genetic alterations at the site of the repair, particularly deletions, insertions and other complex rearrangements. 35 , 38 While under normal circumstances only a small proportion of DSB repair is thought to be carried out by MMEJ, this proportion is thought to increase significantly after radiotherapy. 39
Homologous recombination
In contrast to NHEJ, HR is a very accurate repair mechanism which requires extensive sequence homology between the broken DNA ends and functions in the late S/G2 phase of the cell cycle due to the availability of sister chromatids or homologous chromosomes for templated DNA synthesis (Fig. 2). 24 , 26 HR begins with the MRN complex, which, aided by BRCA1 and CtIP, performs a short‐range resection of the DNA upstream from the break (Fig. 1). 26 This is followed by DNA unwinding and long‐range resection by the exonuclease 1 (EXO1) or by the Bloom syndrome helicase together with the DNA replication helicase/nuclease 2 40 to create extensive 3′ overhangs approximately 1,000 base pairs in length. 26 Emergent ssDNA is coated with RPA, which is then displaced by BRCA2 and replaced with RAD51 recombinase. 41 Facilitated by BRCA1 – partner and localiser of BRCA2 (PALB2) – BRCA2 complex, 42 RAD51–ssDNA nucleofilament mediates homology search by invading the duplex DNA and facilitating base pairing with complementary sequences. 43 Strand invasion of the 3′ overhang forms a displacement loop (D‐loop), whereupon DNA polymerase δ/ε synthesizes a new DNA strand using the 3′ end as a primer. 41 Following the D‐loop formation, HR can proceed via several mechanisms including the formation of a double Holliday junction or synthesis‐dependent strand annealing. 44 While the latter exclusively produces non‐crossover recombinants, resolution of the double Holliday junction may generate crossover and non‐crossover products. 41
Single‐strand annealing
Single‐strand annealing pathway is unique as it requires only one DNA duplex for repair and does not depend on the formation of HJ. SSA requires the presence of 20–25 bp homology regions between the DNA sequence. 45 The 3′ tails created by DNA end resection are coated with RPA and anneal to repeat sequences on either side of the break via the action of RAD52 (Fig. 1). 46 Next, non‐homologous ssDNA flaps are digested by excision repair cross‐complementation group 1 (ERCC1)/ xeroderma pigmentosum complementation group F complex, with any leftover gaps possibly filled in by polymerases and ligases, thus restoring strand continuity. 46 The factors that promote gap filling and ligation are not fully elucidated. 45 Similar to MMEJ, SSA is a highly mutagenic pathway which can lead to large deletions and chromosomal translocations. 47
Single‐strand break (SSB) repair mechanisms
Base excision repair (BER)
Base excision repair corrects little distortions of the DNA helix caused by damage from oxidation, deamination and alkylation. 48 BER is initiated by over 11 distinct DNA glycosylases, the choice of which depends on the type of damage. Repair begins by localisation of extrahelical nucleotides and their excision by an appropriate DNA glycosylase, which cuts the glycosidic bond, leaving behind an abasic (AP) site. 48 At this point repair proceeds either via short (SP‐BER; single nucleotide damage) or long‐patch repair (LP‐BER; 2–10 nucleotide patches). 48 While the exact enzymology of downstream steps depends on whether the chosen glycosylase is monofunctional or bifunctional with β or β/δ lyase activity, in general the AP‐endonuclease (APE1) cleaves the DNA backbone at the AP site to generate 3′ OH and 5′ deoxyribose phosphate (dRP) terminus. 49 This allows for strand synthesis by DNA polymerase β, followed by ligation by LIG3. 49 LP‐BER occurs mainly in proliferating cells and utilizes DNA replication machinery. Following the initial incision by APE1, a complex consisting of a processivity factor PCNA and replication factor C (RCF) enables binding of polymerase δ/ε (POLδ) to synthesize a new strand, which is then ligated by LIG1. 50
Mismatch repair (MMR)
The main role of MMR is to correct mispaired nucleotides and small insertion–deletion loops generated during DNA replication. Repair process begins with the assembly of a MSH/MSH6 (MutSα) or MSH2/MSH3 (MutSβ) ATPase heterodimer, forming a sliding clamp tasked with detection and initiation of DNA repair. 51 Once the mismatch is recognized, other factors are recruited to the site. MLH1/PMS2 (MutLα) complex possesses endonuclease activity and creates an initial incision within the helix, followed by wider EXO1‐mediated resection. 51 Subsequently, RFC enables loading of PCNA onto the DNA strand. 52 PCNA serves as a scaffold for the binding of POLδ, which resynthesises the removed DNA, 53 while any remaining nicks are ligated by LIG1. 52
Nucleotide excision repair (NER)
Nucleotide excision repair is an important repair mechanism for removing bulky DNA adducts caused by UV light damage or chemotherapeutics. NER can be initiated by two sub‐pathways: global genome NER (GG‐NER) or transcription coupled NER (TC‐NER). GG‐NER can occur anywhere in the genome, whereas TTC‐NER conducts accelerated repair of any lesions in actively transcribed genes. While each process utilizes different machinery for injury detection, both pathways converge at the lesion excision step, which is mediated by the ERCC1‐xeroderma pigmentosum complementation group G complex. 54 , 55 Excision is then immediately followed by strand resynthesis and ligation by POLδ/ε/κ and LIG1/3‐XRCC1. 54
DDR mutations in common cancers treated with radiotherapy
Mutational landscape of common cancers treated with radiotherapy shows frequent alterations of DDR genes thought to be involved in both cancer development and response to radiotherapy. For example, a study looking at 266 patients with advanced non‐small cell lung cancer (NSCLC) found DDR mutations in 132 of cases. 56 Out of the DDR‐positive patients, 85 had only one mutation, with 47 harbouring ≥2. 56 The most commonly affected gene was ATM, followed by ATR, BRCA2, POLQ and RAD50. 56 In a large study of 3,182 tumour samples, including 1,461 lung cancers, the most common mutations across all cancer types were TP53 (66%) and ATM (28%). 57 Another recent project looking at DDR mutations in small cell lung cancer (SCLC) analysed a cohort of 166 patients, which consisted of 100 cases of extensive stage disease and 66 cases of limited stage disease and found DDR‐related mutations in 96 participants. 58 Based on pre‐defined DDR gene sets, half of patients had DSB and one‐fifth had SSB‐related gene set alterations. 58 TP53 alterations were most commonly observed in both the extensive stage disease and limited stage disease groups, present in approximately 90% of samples. 58
Genomic alterations involving DDR genes are highly prevalent in prostate cancer, the second most common cancer in men. 59 , 60 , 61 , 62 , 63 Molecular analysis of 333 primary prostate tumours published by The Cancer Genome Atlas (stage pT2a – pT4) showed that 19% harboured inactivating mutations in various DDR genes, most commonly BRCA2, BRCA1, ATM, CDK12, RAD51C and FANCD2, all of which are important factors in the HR pathway. 64 Similarly, an investigation of 150 metastatic castrate‐resistant prostate cancer (mCRPC) samples found alterations in DDR genes in 34 cases, most commonly in BRCA2, ATM and MSH2, followed by BRCA1, FANCA, MLH1, RAD51B and RAD51C. 59 Furthermore, a study of 1,033 patients, which looked at the prevalence of MMR deficiency in prostate cancer, identified MMR gene mutations in 32 cases and found MSH2 to be the most commonly altered gene. 65
The importance of intact DDR mechanisms and their contribution to oncogenesis have been highlighted by various heritable conditions associated with cancer development. For example, Lynch syndrome – the commonest cause of familial predisposition to colorectal cancer (CRC), accounting for approx. 3% of newly diagnosed cases 66 – is caused by an autosomal dominant mutation of the MLH1, MSH2, MSH6 and PMS2 genes, all critical factors in the MMR pathway. 67 Dysfunction of the MMR leads to alterations in the repetitive sequence number of microsatellites, defined as microsatellite instability. 68 An investigation based on the data from the Colon Cancer Family Registry found that among 386 probands, approximately one‐third had an MMR gene mutation, and one‐fifth had a MUTYH alteration (gene encoding for MYH glycosylase involved in BER). 69 In the presence of MMR mutations, the cumulative incidence of CRC by the age of 75 ranged from 10% to 48% for females and 10% to 57% for males depending on the affected gene, as reported by a prospective observational study of 6,350 participants. 70 Another investigation looking at the mutational landscape of DDR alterations in a large cohort of 9,321 CRC patients reported that 1,290 carried an alteration in at least one of the 29 investigated DDR genes. 71
DNA damage response defects have also been associated with oncogenesis in breast cancer. Pathogenic mutations in high penetrance genes (BRCA1, BRCA2, TP53, PTEN, SKT11 and CDH1) account for perhaps 25% of familial breast cancer cases. 72 Chief among these are mutations in BRCA1 and BRCA2, key regulators of the HR pathway, accounting for 20% of all breast cancers in total. 72 , 73 , 74 , 75 , 76 A recent study looking at 925 breast cancer patients found BRCA1 and BRCA2 mutations in 171 and 95, respectively. 73 Another investigation of a larger cohort of 1824 patients with triple‐negative breast cancer reported the presence of BRCA1 and BRCA2 mutations in 155 and 49, respectively. 77 Furthermore, mutations in moderate‐penetrance genes CHK2, ATM, PALB2 and BRIP1 confer a twofold to fourfold increased risk of oncogenesis and are responsible for approximately 2–3% of all breast cancers. 77 , 78 , 79 , 80 CHK2 mutations are the most common and can be found in 3–5% of breast cancer patients, 81 , 82 followed by ATM alterations, which are responsible for approximately 1% of cases. 83 PABL2 and BRIP1 mutations are rarer, accounting for <1% of tumours. 78 A large study looking at 1054 BRCA‐negative breast cancer patients found pathogenic variants in moderate‐penetrance genes in 49 of participants, including CHK2, PALB2, ATM and BRIP1. 84 Important to note is the large variability of pathogenic mutations depending on population and ethnicity, making any kinds of generalizations difficult.
There are many other examples of mutations in DDR in all types of cancer, beyond the scope of this article, and readers are invited to read further. 4 , 6 , 57
DDR and sensitisation to radiation therapy
Radiation therapy (RT) is a cornerstone of cancer care, with almost 50% of patients with solid tumours receiving RT at some stage of their management. 85 RT induces DNA lesions – approximately 10,000 damaged bases, 1,000 SSBs and 20–40 DSBs are produced per Grey per cell. 86 , 87 Despite their low proportion, DSBs are the most lethal type of injury 88 and can be caused by RT both directly and indirectly. 89 Indirect damage occurs via production of free radicals 89 or by conversion of SSBs to DSBs at blocked replication forks. 90 , 91 , 92 Detection of these lesions activates the DDR, leading to cell cycle arrest and repair, or cell death. 89 Unsurprisingly, tumours with efficient DDR mechanisms are highly radioresistant, 93 while deficiencies in repair pathways are detrimental to the cell. 5 Many cancers exhibit characteristics which make them a valid target for DDR inhibition: dysregulated DNA repair, 3 , 94 failure to halt the cell cycle and provide adequate time to repair DNA damage induced by RT, 94 increased frequency of endogenous DNA damage 95 and overreliance on compensatory repair mechanisms. 96 As a result, therapies aiming to inhibit key enzymes involved in the DDR have the potential to enhance RT efficacy. 97 For example, Higgins et al. have shown that POLθ gene knockout led to tumour cell radiosensitisation in vitro. 98 Conversely, high levels of MRE11 expression were associated with better outcomes following adjuvant radiotherapy and improved cause‐specific survival. 99
Given the lethality of DSBs, disruption of DSB repair pathway has been investigated to improve the therapeutic window with radiotherapy. The use of selective inhibitors of DNA‐PK, a key enzyme involved in NHEJ, showed promising results in preclinical studies. 100 , 101 , 102 , 103 For example, NU7441 produced a radiosensitising effect in nasopharyngeal and liver cancer, 100 , 101 as well as enhanced the radiosensitivity of lung cancer cells at lower concentrations. 104 VX‐984 radiosensitises glioblastoma cells in vitro and in orthotopic tumours. 102 Another DNA‐PK inhibitor NU5455 increased the efficacy of radiotherapy treatment of lung cancer xenografts. 103 While considered promising by some, 3 such approaches have drawn criticism from others due to their low specificity and unwanted side effects on healthy cells. 105 In contrast, HR is a less popular choice for radiosensitisation efforts, as it plays a critical role in the maintenance of genome stability in healthy tissues, preventing the development of secondary cancers. 106 A potential target in the HR pathway is the RAD51 nucleoprotein. 106 A small molecule inhibitor RI‐1 has been shown to block the binding of RAD51 to ssDNA and radiosensitise glioblastoma and glioma cells. 107 , 108 BRCA 1/2 deficiency or other common mutations such as ATM, CHEK2 and PALB2 alone do not appear to affect radiosensitivity or outcomes for patients with breast cancer. 109
In another clinically tested strategy, synthetic lethality for cells deficient in HR has been exploited by inhibiting BER with PARPi – BER being the major repair pathway activated in response to SSBs produced by RT‐induced oxidative damage. 110 Blockage of BER results in unrepaired SSBs, which are converted to DSBs upon encountering a replication fork. 92 Inhibition of BER by PARPi in cells already defective in HR, such as BRCA‐negative breast cancer, leads to unrepaired DSBs and cell death. 3 , 111 Inhibition of BER has the potential to induce radiosensitivity even in HR‐intact cells by delaying SSB repair. 112 The ability of PARPi to enhance RT has been demonstrated both in vitro and in vivo. 113 , 114 , 115 Alternatively, BER blockade can be achieved via the use of APE1 inhibitors. 110 APE1, a crucial factor in the BER pathway, is commonly overexpressed in several cancer types, such as NSCLC. 116 , 117 APE1 inhibitors have shown efficacy in combination with RT in several studies. 118 , 119
Another approach is to target upstream enzymes involved in multiple repair pathways. 106 , 110 ATM inhibitors increase radiosensitisation in glioblastoma and squamous cell carcinoma cells in preclinical studies 120 , 121 by impairing both the HR and NHEJ. 22 Deletion of ATM results in suppression of tumour growth in lung adenocarcinoma after a single 15Gy RT dose. 122 Furthermore, cancer cells are often more reliant on ATR signalling for survival due to high levels of replication stress, 123 making ATR a potential therapeutic target. ATR inhibition with M6620 has been shown to improve RT response in triple‐negative breast cancer xenograft models, 124 while another study found that ATR inhibitor AZD6738 lead to radiosensitisation in a panel of human cancer cell lines. 125 Therapies targeting the MRN complex have also generated interest. Overexpression of MRN has been associated with radioresistance and poor treatment outcomes in certain cancers. 126 Conversely, therapies aimed at inhibition of the MRN subunits RAD50, NBS1 and MRE11 reported synergistic effects with RT. 127 , 128 , 129
DDR and response to immunotherapy
DNA damage response gene alterations and resultant genomic instability are important factors determining tumour antigenicity through neoantigen‐dependent and neoantigen‐independent mechanisms. 130 , 131 As such, attention has been drawn to the use of DDR mutational status as a predictive biomarker for the response to immune checkpoint blockade to improve selection of patients and guide therapeutic choices. 132 , 133 , 134 Just like RT, DDR deficiency leads to an increase in DNA damage and tumour mutational burden (TMB) via accumulation of point mutations and indels, a hallmark of cancer. 130 , 135 , 136 Higher number of nonsynonymous mutations has been shown to influence the efficacy of immunotherapy. 137 , 138 Such genomic alterations are associated with an increase in the neoantigen load, which enhances tumour antigenicity and raises the chances for the immune system to recognize the tumour as non‐self and elicit an anti‐tumour response. 135 , 139 , 140 NGS analysis of 240 advanced NSCLC patients treated with ICIs reported a significantly better response in those with a higher TMB. 141 In a phase 2 study of 41 patients with progressive metastatic carcinoma, MMR status was reported to predict the response to PD‐1 inhibitor pembrolizumab. 142 , 143 Another series found that deleterious mutations in several DDR genes correlated with pembrolizumab efficacy in NSCLC. 144 Also, loss of BRCA1 and defects in MMR in cancer cells were shown to result in increased mutational burden and continuous neoantigen renewal, as well as enhanced immune response and surveillance. 142 , 145 , 146 , 147 Analysis of 22 DDR genes in prostate adenocarcinoma found significant inverse association between gene expression (as measured by mRNA levels) and cytotoxic T‐cell infiltration in 19 of them, 11 implying that inhibition of the DDR pathways may be utilized to enhance the innate immune response. 148
In addition to expanding the neoantigen repertoire, DNA damage induced by DDR deficiency can increase tumour immunogenicity via neoantigen‐independent mechanisms (Fig. 3). 148 , 149 DNA damage leads to the accumulation of DNA fragments in the cell cytoplasm, which can be detected by the cyclic GMP‐AMP synthase (cGAS). 150 cGAS binds dsDNA sequences and initiates downstream signalling via the STING pathway, 11 , 131 a protein with a modulatory effect on the immune system. 150 Subsequently, STING promotes gene transcription through the interferon regulatory factor 3 (IRF3), increasing the expression of type I interferon (IFN‐1) and other inflammatory cytokines. 150 Additionally, STING can activate a transcriptional response through the canonical and noncanonical nuclear factor kappa‐light‐chain enhancer of activated B cells pathways. 151 As a consequence, tumour antigen presentation by dendritic cells is increased, in turn potentiating the CD8+ T‐cell response. 151 In animal models, cGAS/STING‐deficient mice failed to reject cancer growth spontaneously and following local RT. 152 , 153 , 154 Furthermore, prostate adenocarcinoma cells were found to exhibit elevated levels of DNA fragments, STING‐dependent signalling, IFN‐1 production and tumour rejection in mice. 155 In conditions of DDR deficiency, such as BRCA1/2 inactivation, the cGAS‐STING pathway can stimulate anti‐tumour immunity in response to the formation of micronuclei derived from chromosomal missegregation. 135 Several studies report upregulated cGAS‐STING activity in ATM and BRCA1/2‐mutated tumour cell lines, with a durable response to ICIs. 156 , 157 , 158 It is important to note that cGAS signalling is dispensable in some human cell lines. 159 Another pathway called the retinoic acid‐inducible gene I/melanoma differentiation‐associated gene 5/mitochondrial antiviral‐signalling protein can be initiated by the conversion of cytosolic dsDNA fragments into RNA transcript by the action RNA polymerase III, leading to downstream IRF3 dimerisation and IFN‐1 production (Fig. 3). 159 , 160 In addition, the DDR appears to be involved in the upregulation of PD‐L1 on tumour cells both through ATM‐ and ATR‐dependent mechanisms, 161 , 162 a phenomenon which has been associated with immune exhaustion.
Fig. 3.
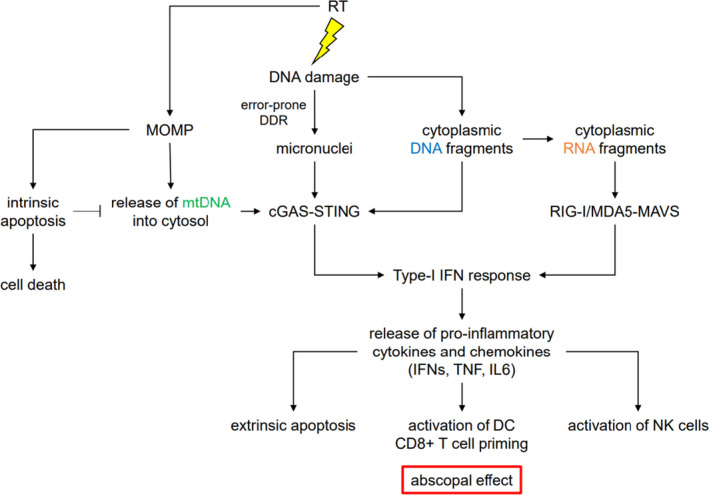
RT induces MOMP, which triggers activation of Caspase 9‐mediated intrinsic apoptosis. If the intrinsic apoptosis is inhibited, MOMP results in release of mtDNA into a cytosol. RT also directly damages DNA leading to accumulation of cytoplasmic DNA fragments. Moreover, incorrect damage repair, drives the formation of micronuclei. mtDNA, cytoplasmic DNA and micronuclei are recognized by cGAS, which stimulates production of type‐1 IFNs in a STING‐IRF3‐dependent manner. Additionally, cytoplasmic DNA fragments can be transcribed into RNAs by the RNA polymerase III. Through activation of RIG‐I/MDA5‐MAVS‐IRF3 pathway cytoplasmic RNA species can also promote IFN‐1 signalling. Subsequent release of pro‐inflammatory cytokines and chemokines triggers Caspase 8‐mediated extrinsic apoptosis, and anti‐tumour immunity by CD8+ T and NK cells. Importantly, the anti‐tumour immunity is directed against distal lesions as well as the irradiated site in a process called the abscopal effect. RT, radiotherapy; MOMP, mitochondrial outer membrane permeabilisation; mtDNA, mitochondrial DNA; cGAS, cyclic GMP–AMP synthase; IFN, interferon; STING, stimulator of interferon genes; IRF3, interferon regulatory factor 3; RIG‐I/MDA5‐MAVS, retinoic acid‐inducible gene I/melanoma differentiation‐associated gene 5‐mitochondrial antiviral‐signalling protein. [Colour figure can be viewed at wileyonlinelibrary.com]
Given the link between DDR deficiency and immune response, combining DDR‐inhibiting agents with ICI‐based treatments is under ongoing investigation. 11 Currently, ICIs are approved for treatment across several cancer types; however, the durable response rate is only 10–20%. 163 The most studied DDR inhibitors in cancer immunotherapy are PARPi, comprising almost 85% of all clinical trials. 164 Combination of PARPi with anti‐PD‐(L)1 inhibitors in ERCC1‐deficient NSCLC patients resulted in cell‐autonomous and constitutive increase in STING expression and IFN‐1 signalling in vitro, as well as increased intratumoural lymphocytic infiltration in vivo. 165 Another series reported that coupling of PD‐L1 blockade with PARP or CHK1 inhibitors resulted in remarkable improvements in cGAS‐STING activation, tumour T‐cell infiltration and treatment response in SCLC patients. 166 Similarly, PD‐1/ CTLA‐4 blockade was found to synergise with PARPi in BRCA1‐deficient ovarian cancer to improve immune‐mediated tumour clearance and long‐term survival in animal models. 147 , 167 Interestingly, combination of PARPi and PD‐1 inhibitors can elicit a strong therapeutic response regardless of BRCA1/2 status 168 , 169 , 170 ; however, the mechanisms behind this have not been fully elucidated. Other DDR targets under investigation for potential synergism between DDR inhibitors and ICIs include cyclin‐dependent kinase 4/6, 171 ATR, 172 CHK1, 166 ATM 173 , 174 and DNA‐PK. 164
Exploiting DDR for more effective combination of immunotherapy and radiotherapy
As discussed elsewhere in this series of review articles, the addition of radiotherapy to immunotherapy enhances clinical effectiveness. For example, subset analysis of the well‐known PACIFIC trial adding immunotherapy to chemo‐radiotherapy for stage III NSCLC has suggested efficacy only if immunotherapy is given within 14 days of RT (HR 0.43, CI 0.28–0.66), 175 although this may be confounded by the improved performance status of patients able to commence adjuvant durvalumab earlier. The addition of stereotactic body radiation therapy to immunotherapy in Stage IV NSCLC doubled the response rate to ICI. 176 However, the most immunogenic dose, fractionation and sequence of radiotherapy remain elusive. Enhanced understanding of DDR would enable more effective combination by eliciting more immunogenic DNA and RNA species and enlarging the therapeutic window between normal and tumour tissue.
Landmark pre‐clinical studies have shown that the type of DNA damage induced by radiotherapy is particularly effective in generating an immune response due to the generation of micronuclei (cytoplasmic dsDNA) which is sensed by cGAS. 177 , 178 Moreover, clinically relevant doses of RT result in mitochondrial outer membrane permeabilisation in cancer cells and exposure of the mtDNA to the cytosol, which also acts as a potent immunogenic signal stimulating the production of IFN‐1. 179 A study by Dillon and colleagues in an immunocompetent mouse model of HPV‐driven cancers showed that an ATR inhibitor given with radiotherapy enhanced antigen presentation and innate immune cell infiltration, with results expected from a phase 1 clinical trial of ATR inhibitor in combination with radiotherapy. 180 It is also possible that by inducing DNA lesions which are difficult to repair (either due to complexity or due to large burden), RT can force cells to use more mutagenic pathways such as SSA or MMEJ generating more immunogenic DNA and RNA strands which in turn enhance response to immunotherapy (Fig. 3). As DDR is frequently defective in cancer cells this may provide a difference between tumour and normal tissue to enlarge the therapeutic window. For example, POLθ, key in MMEJ, is frequently upregulated in cancers, suggesting dependence on this pathway. 45 As a cautionary note, however, chronic interferon signalling can unfavourably modulate PD‐L1 pathways. 6 Further studies are needed to understand synergies and develop robust biomarkers of response.
Summary and conclusions
Recent advances in understanding of the DDR and immunotherapy have led to improvements in patient care. Defect in the DNA repair mechanisms are emerging as one of the strongest predictors of response to ICI. Moreover, available data show that DNA damage induced by RT plays a significant role in stimulating an immune response to control tumour growth. A better understanding of the interplay between the DDR and tumour immunity will provide us with a better insight into optimal ways of combining radiotherapy with immunotherapy, as well as facilitate the implementation of novel therapies, including those that target the DDR directly, to improve cancer outcomes.
Acknowledgements
Open access publishing facilitated by The University of Sydney, as part of the Wiley ‐ The University of Sydney agreement via the Council of Australian University Librarians.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
D Czajkowski BSc (Hons); R Szmyd PhD; HE Gee MBBS (Hons), PhD, FRANZCR.
Conflict of interest: None.
References
- 1. Chabanon RM, Rouanne M, Lord CJ, Soria JC, Pasero P, Postel‐Vinay S. Targeting the DNA damage response in immuno‐oncology: developments and opportunities. Nat Rev Cancer 2021; 21: 701–17. [DOI] [PubMed] [Google Scholar]
- 2. Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013; 5: a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pilié PG, Tang C, Mills GB, Yap TA. State‐of‐the‐art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 2019; 16: 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knijnenburg TA, Wang L, Zimmermann MT et al. Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep 2018; 23: 239–54.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer 2016; 16: 35–42. [DOI] [PubMed] [Google Scholar]
- 6. Samstein RM, Riaz N. The DNA damage response in immunotherapy and radiation. Adv Radiat Oncol 2018; 3: 527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campbell BB, Light N, Fabrizio D et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017; 171: 1042–56.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J, Shih DJH, Lin S‐Y. Role of DNA repair defects in predicting immunotherapy response. Biomark Res 2020; 8: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nizialek E, Antonarakis ES. PARP inhibitors in metastatic prostate cancer: evidence to date. Cancer Manag Res 2020; 12: 8105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rose M, Burgess JT, O'Byrne K, Richard DJ, Bolderson E. PARP inhibitors: clinical relevance, mechanisms of action and tumor resistance. Front Cell Dev Biol 2020; 8: 564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ye Z, Shi Y, Lees‐Miller SP, Tainer JA. Function and molecular mechanism of the DNA damage response in immunity and cancer immunotherapy. Front Immunol 2021; 12: 797880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang M, Jia K, Wang L et al. Alterations of DNA damage response pathway: biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm Sin B 2021; 11: 2983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018; 8: 1069–86. [DOI] [PubMed] [Google Scholar]
- 14. Han C, Liu Z, Zhang Y et al. Tumor cells suppress radiation‐induced immunity by hijacking caspase 9 signaling. Nat Immunol 2020; 21: 546–54. [DOI] [PubMed] [Google Scholar]
- 15. Li P, Zhou L, Zhao T et al. Caspase‐9: structure, mechanisms and clinical application. Oncotarget 2017; 8: 23996–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol 2014; 6: 442–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith J, Mun Tho L, Xu N, Gillespie DA. Chapter 3 – The ATM–Chk2 and ATR–Chk1 pathways in DNA damage signaling and cancer. In: Vande Woude GF, Klein G (eds). Advances in Cancer Research, Vol. 108. Academic Press, Cambridge, 2010; 73–112. [DOI] [PubMed] [Google Scholar]
- 19. Chen L, Gilkes D, Pan Y, Lane W, Chen J. ATM and Chk2‐dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J 2005; 24: 3411–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science 2003; 300: 1542–8. [DOI] [PubMed] [Google Scholar]
- 21. Maréchal A, Zou L. RPA‐coated single‐stranded DNA as a platform for post‐translational modifications in the DNA damage response. Cell Res 2015; 25: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Awasthi P, Foiani M, Kumar A. ATM and ATR signaling at a glance. J Cell Sci 2015; 128: 4255–62. [DOI] [PubMed] [Google Scholar]
- 23. Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res 2015; 21: 4780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burgess JT, Rose M, Boucher D et al. The therapeutic potential of DNA damage repair pathways and genomic stability in lung cancer. Front Oncol 2020; 10: 1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lieber MR. The mechanism of double‐strand DNA break repair by the nonhomologous DNA end‐joining pathway. Annu Rev Biochem 2010; 79: 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scully R, Panday A, Elango R, Willis NA. DNA double‐strand break repair‐pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 2019; 20: 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deriano L, Roth DB. Modernizing the nonhomologous end‐joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet 2013; 47: 433–55. [DOI] [PubMed] [Google Scholar]
- 28. Chang H, Lieber M. Structure‐specific nuclease activities of artemis and the artemis: DNA‐PKcs complex. Nucleic Acids Res 2016; 44: gkw456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X, Heyer W‐D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 2008; 18: 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nat Rev Mol Cell Biol 2017; 18: 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu PY, Frit P, Malivert L et al. Interplay between Cernunnos‐XLF and nonhomologous end‐joining proteins at DNA ends in the cell. J Biol Chem 2007; 282: 31937–43. [DOI] [PubMed] [Google Scholar]
- 32. Craxton A, Somers J, Munnur D, Jukes‐Jones R, Cain K, Malewicz M. XLS (c9orf142) is a new component of mammalian DNA double‐stranded break repair. Cell Death Differ 2015; 22: 890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strickland KC, Howitt BE, Shukla SA et al. Association and prognostic significance of BRCA1/2‐mutation status with neoantigen load, number of tumor‐infiltrating lymphocytes and expression of PD‐1/PD‐L1 in high grade serous ovarian cancer. Oncotarget 2016; 7: 13587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frit P, Barboule N, Yuan Y, Gomez D, Calsou P. Alternative end‐joining pathway(s): bricolage at DNA breaks. DNA Repair (Amst) 2014; 17: 81–97. [DOI] [PubMed] [Google Scholar]
- 35. Iliakis G, Murmann T, Soni A. Alternative end‐joining repair pathways are the ultimate backup for abrogated classical non‐homologous end‐joining and homologous recombination repair: implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen 2015; 793: 166–75. [DOI] [PubMed] [Google Scholar]
- 36. Sallmyr A, Tomkinson AE. Repair of DNA double‐strand breaks by mammalian alternative end‐joining pathways. J Biol Chem 2018; 293: 10536–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iliakis G, Wang H, Perrault AR et al. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet Genome Res 2004; 104: 14–20. [DOI] [PubMed] [Google Scholar]
- 38. Simsek D, Jasin M. Alternative end‐joining is suppressed by the canonical NHEJ component Xrcc4‐ligase IV during chromosomal translocation formation. Nat Struct Mol Biol 2010; 17: 410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dutta A, Eckelmann B, Adhikari S et al. Microhomology‐mediated end joining is activated in irradiated human cells due to phosphorylation‐dependent formation of the XRCC1 repair complex. Nucleic Acids Res 2017; 45: 2585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qin Z, Bi L, Hou X‐M et al. Human RPA activates BLM's bidirectional DNA unwinding from a nick. Elife 2020; 9: e54098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mehta A, Haber JE. Sources of DNA double‐strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 2014; 6: a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu J, Lu L‐Y, Yu X. The role of BRCA1 in DNA damage response. Protein Cell 2010; 1: 117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Špírek M, Mlcoušková J, Belán O et al. Human RAD51 rapidly forms intrinsically dynamic nucleoprotein filaments modulated by nucleotide binding state. Nucleic Acids Res 2018; 46: 3967–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shah Punatar R, Martin MJ, Wyatt HDM, Chan YW, West SC. Resolution of single and double Holliday junction recombination intermediates by GEN1. Proc Natl Acad Sci USA 2017; 114: 443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trenner A, Sartori AA. Harnessing DNA double‐strand break repair for cancer treatment. Front Oncol 2019; 9: 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bhargava R, Onyango DO, Stark JM. Regulation of single‐strand annealing and its role in genome maintenance. Trends Genet 2016; 32: 566–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic Alu elements in mammalian cells. Mol Cell 2005; 17: 885–94. [DOI] [PubMed] [Google Scholar]
- 48. Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol 2013; 5: a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single‐strand interruption repair pathway in mammalian cells. Cell Res 2008; 18: 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Woodrick J, Gupta S, Camacho S et al. A new sub‐pathway of long‐patch base excision repair involving 5′ gap formation. EMBO J 2017; 36: 1605–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pećina‐Šlaus N, Kafka A, Salamon I, Bukovac A. Mismatch repair pathway, genome stability and cancer. Front Mol Biosci 2020; 7: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li G‐M. Mechanisms and functions of DNA mismatch repair. Cell Res 2008; 18: 85–98. [DOI] [PubMed] [Google Scholar]
- 53. Strzalka W, Ziemienowicz A. Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann Bot 2011; 107: 1127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Borszéková Pulzová L, Ward TA, Chovanec M. XPA: DNA repair protein of significant clinical importance. Int J Mol Sci 2020; 21: 2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schärer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 2013; 5: a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ricciuti B, Recondo G, Spurr LF et al. Impact of DNA damage response and repair (DDR) gene mutations on efficacy of PD‐(L)1 immune checkpoint inhibition in non–small cell lung cancer. Clin Cancer Res 2020; 26: 4135–42. [DOI] [PubMed] [Google Scholar]
- 57. Poonnen PJ, Duffy JE, Hintze B et al. Genomic analysis of metastatic solid tumors in veterans: findings from the VHA National Precision Oncology Program. JCO Precis Oncol 2019; 3: PO.19.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park S, Lee H, Lee B et al. DNA damage response and repair pathway alteration and its association with tumor mutation burden and platinum‐based chemotherapy in SCLC. J Thorac Oncol 2019; 14: 1640–50. [DOI] [PubMed] [Google Scholar]
- 59. Robinson D, Van Allen EM, Wu YM et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015; 161: 1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pritchard CC, Mateo J, Walsh MF et al. Inherited DNA‐repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016; 375: 443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abida W, Armenia J, Gopalan A et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol 2017; 1: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Castro E, Romero‐Laorden N, Del Pozo A et al. PROREPAIR‐B: a prospective cohort study of the impact of Germline DNA repair mutations on the outcomes of patients with metastatic castration‐resistant prostate cancer. J Clin Oncol 2019; 37: 490–503. [DOI] [PubMed] [Google Scholar]
- 63. Armenia J, Wankowicz SAM, Liu D et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet 2018; 50: 645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cancer Genome Atlas Research Network . The molecular taxonomy of primary prostate cancer. Cell 2015; 163: 1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Abida W, Cheng ML, Armenia J et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol 2019; 5: 471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Moreira L, Balaguer F, Lindor N et al. Identification of lynch syndrome among patients with colorectal cancer. JAMA 2012; 308: 1555–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Carethers JM, Stoffel EM. Lynch syndrome and lynch syndrome mimics: the growing complex landscape of hereditary colon cancer. World J Gastroenterol 2015; 21: 9253–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li K, Luo H, Huang L, Luo H, Zhu X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int 2020; 20: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Win AK, Jenkins MA, Dowty JG et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol Biomarkers Prev 2017; 26: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dominguez‐Valentin M, Sampson JR, Seppälä TT et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the prospective lynch syndrome database. Genet Med 2020; 22: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Arai H, Elliott A, Xiu J et al. The landscape of alterations in DNA damage response pathways in colorectal cancer. Clin Cancer Res 2021; 27: 3234–42. [DOI] [PubMed] [Google Scholar]
- 72. Kleibl Z, Kristensen VN. Women at high risk of breast cancer: molecular characteristics, clinical presentation and management. Breast 2016; 28: 136–44. [DOI] [PubMed] [Google Scholar]
- 73. De Talhouet S, Peron J, Vuilleumier A et al. Clinical outcome of breast cancer in carriers of BRCA1 and BRCA2 mutations according to molecular subtypes. Sci Rep 2020; 10: 7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rebbeck TR, Mitra N, Wan F et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015; 313: 1347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mehrgou A, Akouchekian M. The importance of BRCA1 and BRCA2 genes mutations in breast cancer development. Med J Islam Repub Iran 2016; 30: 369. [PMC free article] [PubMed] [Google Scholar]
- 76. Woodward ER, van Veen EM, Evans DG. From BRCA1 to polygenic risk scores: mutation‐associated risks in breast cancer‐related genes. Breast Care 2021; 16: 202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Couch FJ, Hart SN, Sharma P et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple‐negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol 2015; 33: 304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shiovitz S, Korde LA. Genetics of breast cancer: a topic in evolution. Ann Oncol 2015; 26: 1291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lincoln SE, Kobayashi Y, Anderson MJ et al. A systematic comparison of traditional and multigene panel testing for hereditary breast and ovarian cancer genes in more than 1000 patients. J Mol Diagn 2015; 17: 533–44. [DOI] [PubMed] [Google Scholar]
- 80. Maxwell KN, Wubbenhorst B, D'Andrea K et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2‐negative patients with early‐onset breast cancer. Genet Med 2015; 17: 630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Meijers‐Heijboer H, van den Ouweland A, Klijn J et al. Low‐penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 2002; 31: 55–9. [DOI] [PubMed] [Google Scholar]
- 82. Cybulski C, Wokołorczyk D, Jakubowska A et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol 2011; 29: 3747–52. [DOI] [PubMed] [Google Scholar]
- 83. Renwick A, Thompson D, Seal S et al. ATM mutations that cause ataxia‐telangiectasia are breast cancer susceptibility alleles. Nat Genet 2006; 38: 873–5. [DOI] [PubMed] [Google Scholar]
- 84. Weitzel JN, Neuhausen SL, Adamson A et al. Pathogenic and likely pathogenic variants in PALB2, CHEK2, and other known breast cancer susceptibility genes among 1054 BRCA‐negative Hispanics with breast cancer. Cancer 2019; 125: 2829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment. Cancer 2005; 104: 1129–37. [DOI] [PubMed] [Google Scholar]
- 86. Burkart W, Jung T, Frasch G. Damage pattern as a function of radiation quality and other factors. C R Acad Sci III 1999; 322: 89–101. [DOI] [PubMed] [Google Scholar]
- 87. Ward JF. DNA damage and repair. Basic Life Sci 1991; 58: 403–15; discussion 15–21. [DOI] [PubMed] [Google Scholar]
- 88. Radford IR. The level of induced DNA double‐strand breakage correlates with cell killing after X‐irradiation. Int J Radiat Biol Relat Stud Phys Chem Med 1985; 48: 45–54. [DOI] [PubMed] [Google Scholar]
- 89. Baskar R, Lee KA, Yeo R, Yeoh K‐W. Cancer and radiation therapy: current advances and future directions. Int J Med Sci 2012; 9: 193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Biau J, Devun F, Jdey W et al. A preclinical study combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of melanoma. Neoplasia 2014; 16: 835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8: 193–204. [DOI] [PubMed] [Google Scholar]
- 92. Kuzminov A. Single‐strand interruptions in replicating chromosomes cause double‐strand breaks. Proc Natl Acad Sci USA 2001; 98: 8241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jeggo P, Lavin MF. Cellular radiosensitivity: how much better do we understand it? Int J Radiat Biol 2009; 85: 1061–81. [DOI] [PubMed] [Google Scholar]
- 94. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer 2011; 11: 239–53. [DOI] [PubMed] [Google Scholar]
- 95. Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res 2011; 711: 193–201. [DOI] [PubMed] [Google Scholar]
- 96. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 2012; 12: 801–17. [DOI] [PubMed] [Google Scholar]
- 97. Stover EH, Konstantinopoulos PA, Matulonis UA, Swisher EM. Biomarkers of response and resistance to DNA repair targeted therapies. Clin Cancer Res 2016; 22: 5651–60. [DOI] [PubMed] [Google Scholar]
- 98. Higgins GS, Prevo R, Lee YF et al. A small interfering RNA screen of genes involved in DNA repair identifies tumor‐specific radiosensitization by POLQ knockdown. Cancer Res 2010; 70: 2984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Choudhury A, Nelson LD, Teo MT et al. MRE11 expression is predictive of cause‐specific survival following radical radiotherapy for muscle‐invasive bladder cancer. Cancer Res 2010; 70: 7017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dong J, Ren Y, Zhang T et al. Inactivation of DNA‐PK by knockdown DNA‐PKcs or NU7441 impairs non‐homologous end‐joining of radiation‐induced double strand break repair. Oncol Rep 2018; 39: 912–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yang C, Wang Q, Liu X et al. NU7441 enhances the radiosensitivity of liver cancer cells. Cell Physiol Biochem 2016; 38: 1897–905. [DOI] [PubMed] [Google Scholar]
- 102. Timme CR, Rath BH, O'Neill JW, Camphausen K, Tofilon PJ. The DNA‐PK inhibitor VX‐984 enhances the radiosensitivity of glioblastoma cells grown in vitro and as orthotopic xenografts. Mol Cancer Ther 2018; 17: 1207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Willoughby CE, Jiang Y, Thomas HD et al. Selective DNA‐PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J Clin Invest 2020; 130: 258–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sunada S, Kanai H, Lee Y et al. Nontoxic concentration of DNA‐PK inhibitor NU7441 radio‐sensitizes lung tumor cells with little effect on double strand break repair. Cancer Sci 2016; 107: 1250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Brown JM. Beware of clinical trials of DNA repair inhibitors. Int J Radiat Oncol Biol Phys 2019; 103: 1182–3. [DOI] [PubMed] [Google Scholar]
- 106. Nickoloff JA, Taylor L, Sharma N, Kato TA. Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resist 2021; 4: 244–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Balbous A, Cortes U, Guilloteau K et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem‐like cells. BMC Cancer 2016; 16: 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. King HO, Brend T, Payne HL et al. RAD51 is a selective DNA repair target to radiosensitize glioma stem cells. Stem Cell Rep 2017; 8: 125–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Chapman BV, Liu D, Shen Y et al. Breast radiation therapy‐related treatment outcomes in patients with or without germline mutations on multigene panel testing. Int J Radiat Oncol Biol Phys 2022; 112: 437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Biau J, Chautard E, Verrelle P, Dutreix M. Altering DNA repair to improve radiation therapy: specific and multiple pathway targeting. Front Oncol 2019; 9: 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Jannetti SA, Zeglis BM, Zalutsky MR, Reiner T. Poly(ADP‐ribose)polymerase (PARP) inhibitors and radiation therapy. Front Pharmacol 2020; 11: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Vens C, Begg AC. Targeting base excision repair as a sensitization strategy in radiotherapy. Semin Radiat Oncol 2010; 20: 241–9. [DOI] [PubMed] [Google Scholar]
- 113. Dungey FA, Löser DA, Chalmers AJ. Replication‐dependent radiosensitization of human glioma cells by inhibition of poly(ADP‐ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys 2008; 72: 1188–97. [DOI] [PubMed] [Google Scholar]
- 114. Reiss KA, Herman JM, Armstrong D et al. A final report of a phase I study of veliparib (ABT‐888) in combination with low‐dose fractionated whole abdominal radiation therapy (LDFWAR) in patients with advanced solid malignancies and peritoneal carcinomatosis with a dose escalation in ovarian and fallopian tube cancers. Gynecol Oncol 2017; 144: 486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Donawho CK, Luo Y, Luo Y et al. ABT‐888, an orally active poly(ADP‐ribose) polymerase inhibitor that potentiates DNA‐damaging agents in preclinical tumor models. Clin Cancer Res 2007; 13: 2728–37. [DOI] [PubMed] [Google Scholar]
- 116. Tell G, Fantini D, Quadrifoglio F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell Mol Life Sci 2010; 67: 3589–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Long K, Gu L, Li L et al. Small‐molecule inhibition of APE1 induces apoptosis, pyroptosis, and necroptosis in non‐small cell lung cancer. Cell Death Dis 2021; 12: 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Xiang DB, Chen ZT, Wang D et al. Chimeric adenoviral vector Ad5/F35‐mediated APE1 siRNA enhances sensitivity of human colorectal cancer cells to radiotherapy in vitro and in vivo. Cancer Gene Ther 2008; 15: 625–35. [DOI] [PubMed] [Google Scholar]
- 119. Cun Y, Dai N, Xiong C et al. Silencing of APE1 enhances sensitivity of human hepatocellular carcinoma cells to radiotherapy in vitro and in a xenograft model. PLoS One 2013; 8: e55313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 120. Dohmen AJC, Qiao X, Duursma A et al. Identification of a novel ATM inhibitor with cancer cell specific radiosensitization activity. Oncotarget 2017; 8: 73925–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kahn J, Allen J, Karlin JD et al. Next‐generation ATM kinase inhibitors under development radiosensitize glioblastoma with conformal radiation in a mouse orthotopic model. Int J Radiat Oncol Biol Phys 2017; 99: E600–1. [Google Scholar]
- 122. Torok JA, Oh P, Castle KD et al. Deletion of Atm in tumor but not endothelial cells improves radiation response in a primary mouse model of lung adenocarcinoma. Cancer Res 2019; 79: 773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR‐CHK1 Axis in cancer therapy. Cancers 2017; 9: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Tu X, Kahila MM, Zhou Q et al. ATR inhibition is a promising radiosensitizing strategy for triple‐negative breast cancer. Mol Cancer Ther 2018; 17: 2462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dillon MT, Barker HE, Pedersen M et al. Radiosensitization by the ATR inhibitor AZD6738 through generation of acentric micronuclei. Mol Cancer Ther 2017; 16: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ho V, Chung L, Singh A et al. Overexpression of the MRE11‐RAD50‐NBS1 (MRN) complex in rectal cancer correlates with poor response to neoadjuvant radiotherapy and prognosis. BMC Cancer 2018; 18: 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wang Y, Gudikote J, Giri U et al. RAD50 expression is associated with poor clinical outcomes after radiotherapy for resected non–small cell lung cancer. Clin Cancer Res 2018; 24: 341–50. [DOI] [PubMed] [Google Scholar]
- 128. Chang L, Huang J, Wang K et al. Targeting Rad50 sensitizes human nasopharyngeal carcinoma cells to radiotherapy. BMC Cancer 2016; 16: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Flores‐Pérez A, Rafaelli LE, Ramírez‐Torres N et al. RAD50 targeting impairs DNA damage response and sensitizes human breast cancer cells to cisplatin therapy. Cancer Biol Ther 2014; 15: 777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mouw KW, Goldberg MS, Konstantinopoulos PA, D'Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov 2017; 7: 675–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Li T, Chen ZJ. The cGAS‐cGAMP‐STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 2018; 215: 1287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Aiello MM, Vigneri PG, Bruzzi P et al. Excision repair cross complementation group 1 (ERCC‐1) gene polymorphisms and response to nivolumab in advanced non‐small cell lung cancer (NSCLC). J Clin Oncol 2017; 35 (15 Suppl): 3032. [Google Scholar]
- 133. Hauschild A, Eichstaedt J, Möbus L et al. Regression of melanoma metastases and multiple non‐melanoma skin cancers in xeroderma pigmentosum by the PD1‐antibody pembrolizumab. Eur J Cancer 2017; 77: 84–7. [DOI] [PubMed] [Google Scholar]
- 134. Kraemer KH, Tamura D, Khan SG. Pembrolizumab treatment of a patient with xeroderma pigmentosum with disseminated melanoma and multiple nonmelanoma skin cancers. Br J Dermatol 2018; 178: 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ragu S, Matos‐Rodrigues G, Lopez BS. Replication stress, DNA damage, inflammatory cytokines and innate immune response. Genes 2020; 11: 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mavragani IV, Nikitaki Z, Kalospyros SA, Georgakilas AG. Ionizing radiation and complex DNA damage: from prediction to detection challenges and biological significance. Cancers 2019; 11: 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Rizvi NA, Hellmann MD, Snyder A et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015; 348: 124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Balachandran VP, Łuksza M, Zhao JN et al. Identification of unique neoantigen qualities in long‐term survivors of pancreatic cancer. Nature 2017; 551: 512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Riaz N, Morris L, Havel JJ, Makarov V, Desrichard A, Chan TA. The role of neoantigens in response to immune checkpoint blockade. Int Immunol 2016; 28: 411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. McLaughlin M, Patin EC, Pedersen M et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer 2020; 20: 203–17. [DOI] [PubMed] [Google Scholar]
- 141. Rizvi H, Sanchez‐Vega F, La K et al. Molecular determinants of response to anti–programmed cell death (PD)‐1 and anti–programmed death‐ligand 1 (PD‐L1) blockade in patients with non–small‐cell lung cancer profiled with targeted next‐generation sequencing. J Clin Oncol 2018; 36: 633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Le DT, Uram JN, Wang H et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015; 372: 2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Le DT, Durham JN, Smith KN et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017; 357: 409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Rizvi NA, Hellmann MD, Snyder A et al. Mutational landscape determines sensitivity to PD‐1 blockade in non–small cell lung cancer. Science 2015; 348: 124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lu C, Guan J, Lu S et al. DNA sensing in mismatch repair‐deficient tumor cells is essential for anti‐tumor immunity. Cancer Cell 2021; 39: 96–108.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Germano G, Lamba S, Rospo G et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017; 552: 116–20. [DOI] [PubMed] [Google Scholar]
- 147. Higuchi T, Flies DB, Marjon NA et al. CTLA‐4 blockade synergizes therapeutically with PARP inhibition in BRCA1‐deficient ovarian cancer. Cancer Immunol Res 2015; 3: 1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lamberti G, Andrini E, Sisi M, Federico AD, Ricciuti B. Targeting DNA damage response and repair genes to enhance anticancer immunotherapy: rationale and clinical implication. Future Oncol 2020; 16: 1751–66. [DOI] [PubMed] [Google Scholar]
- 149. Kwon J, Bakhoum SF. The cytosolic DNA‐sensing cGAS‐STING pathway in cancer. Cancer Discov 2020; 10: 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS‐STING pathway of cytosolic DNA sensing. Nat Immunol 2016; 17: 1142–9. [DOI] [PubMed] [Google Scholar]
- 151. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS–STING pathway in health and disease. Nat Rev Genetics 2019; 20: 657–74. [DOI] [PubMed] [Google Scholar]
- 152. Deng L, Liang H, Xu M et al. STING‐dependent cytosolic DNA sensing promotes radiation‐induced type I interferon‐dependent antitumor immunity in immunogenic tumors. Immunity 2014; 41: 843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Woo S‐R, Fuertes Mercedes B, Corrales L et al. STING‐dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014; 41: 830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wang H, Hu S, Chen X et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA 2017; 114: 1637–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ho Samantha SW, Zhang Wendy YL, Tan Nikki Yi J et al. The DNA structure‐specific endonuclease MUS81 mediates DNA sensor STING‐dependent host rejection of prostate cancer cells. Immunity 2016; 44: 1177–89. [DOI] [PubMed] [Google Scholar]
- 156. Härtlova A, Erttmann SF, Raffi FAM et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti‐microbial innate immunity. Immunity 2015; 42: 332–43. [DOI] [PubMed] [Google Scholar]
- 157. Parkes EE, Walker SM, Taggart LE et al. Activation of STING‐dependent innate immune signaling by S‐phase‐specific DNA damage in breast cancer. J Natl Cancer Inst 2016; 109: djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Li X, Liu G, Wu W. Recent advances in lynch syndrome. Exp Hematol Oncol 2021; 10: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Feng X, Tubbs A, Zhang C et al. ATR inhibition potentiates ionizing radiation‐induced interferon response via cytosolic nucleic acid‐sensing pathways. EMBO J 2020; 39: e104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG‐I‐dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate. Nat Immunol 2009; 10: 1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Sato H, Niimi A, Yasuhara T et al. DNA double‐strand break repair pathway regulates PD‐L1 expression in cancer cells. Nat Commun 2017; 8: 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Vendetti FP, Karukonda P, Clump DA et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell‐dependent antitumor activity following radiation. J Clin Invest 2018; 128: 3926–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Dempke WCM, Fenchel K, Dale SP. Programmed cell death ligand‐1 (PD‐L1) as a biomarker for non‐small cell lung cancer (NSCLC) treatment‐are we barking up the wrong tree? Transl Lung Cancer Res 2018; 7 (Suppl 3): S275–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Sun W, Zhang Q, Wang R, Li Y, Sun Y, Yang L. Targeting DNA damage repair for immune checkpoint inhibition: mechanisms and potential clinical applications. Front Oncol 2021; 11: 648687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Chabanon RM, Muirhead G, Krastev DB et al. PARP inhibition enhances tumor cell‐intrinsic immunity in ERCC1‐deficient non‐small cell lung cancer. J Clin Invest 2019; 129: 1211–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Sen T, Rodriguez BL, Chen L et al. Targeting DNA damage response promotes antitumor immunity through STING‐mediated T‐cell activation in small cell lung cancer. Cancer Discov 2019; 9: 646–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ding L, Kim H‐J, Wang Q et al. PARP inhibition elicits STING‐dependent antitumor immunity in Brca1‐deficient ovarian cancer. Cell Rep 2018; 25: 2972–80.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kim C, Wang XD, Yu Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping‐induced DNA damage response. Elife 2020; 9: e60637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Wang Z, Sun K, Xiao Y et al. Niraparib activates interferon signaling and potentiates anti‐PD‐1 antibody efficacy in tumor models. Sci Rep 2019; 9: 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Pham MM, Ngoi NYL, Peng G, Tan DSP, Yap TA. Development of poly(ADP‐ribose) polymerase inhibitor and immunotherapy combinations: progress, pitfalls, and promises. Trends Cancer 2021; 7: 958–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Goel S, DeCristo MJ, Watt AC et al. CDK4/6 inhibition triggers anti‐tumour immunity. Nature 2017; 548: 471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Carter L. Phase I modular study of AZD6738, a novel oral, potent and selective ataxia telangiectasia Rad3‐related (ATR) inhibitor in combination (combo) with carboplatin, olaparib or durvalumab in patients (pts) with advanced cancers. Eur J Cancer 2016; 69: S2. [Google Scholar]
- 173. Zhang Q, Green MD, Lang X et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res 2019; 79: 3940–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Wang L, Yang L, Wang C et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A‐deficient tumors. J Clin Invest 2020; 130: 5951–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Gray JE, Villegas A, Daniel D et al. Three‐year overall survival with Durvalumab after chemoradiotherapy in stage III NSCLC—update from PACIFIC. J Thorac Oncol 2020; 15: 288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Theelen WSME, Chen D, Verma V et al. Pembrolizumab with or without radiotherapy for metastatic non‐small‐cell lung cancer: a pooled analysis of two randomised trials. Lancet Respir Med 2021; 9: 467–75. [DOI] [PubMed] [Google Scholar]
- 177. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017; 548: 466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Mackenzie KJ, Carroll P, Martin C‐A et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017; 548: 461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Yamazaki T, Galluzzi L. Mitochondrial control of innate immune signaling by irradiated cancer cells. Oncoimmunology 2020; 9: 1797292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Dillon MT, Boylan Z, Smith D et al. PATRIOT: a phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3‐related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours. Clin Transl Radiat Oncol 2018; 12: 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.