

Abstract
Thermosetting polymeric materials have advantageous properties and are therefore used in numerous applications. In this study, it was hypothesized and ultimately shown that thermosets could be derived from comparably sustainable sub‐components. A two‐step procedure to produce a thermoset comprising of Kraft lignin (KL) and the cross‐linker adipic acid (AdA) was developed. The cross‐linking was activated by means of an acetylating agent comprising isopropenyl acetate (IPA) to form a cross‐linking mixture (CLM). The cross‐linking was confirmed by FTIR and solid‐state NMR spectroscopy, and the esterification reactions were further studied using model compounds. When the KL lignin was mixed with the CLM, partial esterification occurred to yield a homogeneous viscous liquid that could easily be poured into a mold, as the first step in the procedure. Without any additions, the mold was heated and the material transformed into a thermoset by reaction of the two carboxylic acid‐derivatives of AdA and KL in the second step.
Keywords: adipic acid, bioplastics, lignin, renewable materials, thermosets
Renewable thermoset: Kraft lignin, a by‐product from the paper and pulp industry is mixed with a cross‐linking mixture based on adipic acid able to react with lignin under heat. The resulting material is a partially esterified mixture with thermoplastic‐like properties. Such a liquid can undergo further thermal treatment inside a mold under pressure to yield a tunable thermoset without any additives.

Introduction
Plastics are produced in 368 Mtons y−1 and are pivotal for our everyday life in vast applications such as sterile hospital instruments, coatings to prolong the lifetime and increase the hydrophobicity of wood products, molecular electronics, materials in our electronic devices, et cetera. [1] Even though there is a strive to substitute plastic materials for bio‐based ones such as wood, there will still be a demand for plastics in the future. Therefore, there has been an effort to substitute fossil‐based plastics for bio‐based alternatives. However, to avoid food competition and better exploit the existing resources, waste or side streams should preferably be used. Such waste streams are common in the Kraft pulp and paper industry, which has an estimated annual production of 260 Mtons y−1 of pulp and where 130 Mtons y−1 of Kraft lignin (KL), in form of black liquor, is processed. [2] Currently, KL is burnt to a low value for the pulp mill. [3] To make the KL source more valuable, several strategies have been reported to produce lignin‐based plastics by either incorporating KL into a thermoplastic polymer or by producing KL‐based thermosets.[ 4 , 5 ] However, KL is extracted as an amphiphilic solid with scarce affinity with common plastics, and therefore, to achieve blends or composites with non‐polar matrices, further chemical functionalization is required. [6] These functionalization procedures yield lignin derivatives with increased lipophilicity to allow mixing with the polymer. Among the reported methodologies, the esterification of KL is one of the most investigated.[ 7 , 8 , 9 ] However, different chemical strategies have been reported to achieve thermosets based on KL (Table 1). Gioia et al. reported the synthesis of an epoxy resin‐based on fractionated and subsequently epoxidated KL crosslinked with a polyetheramine. [10] Jawerth et al. have investigated the allylation of fractionated KL that allows the reaction with a set of poly‐thiols‐based crosslinkers to yield materials with tunable mechanical properties. [11] Different esterification strategies to yield thermosets have also been investigated in the production of thermosetting polymers. Scarica et al. showed the possibility to use succinylated KL to yield either polyamides or polyesters after curing. [12] Zhang et al. reported the synthesis of a KL based vitrimer by initial ozonation followed by a curing step with sebacic epoxides. [13] The reactivity of poly‐acids as crosslinkers was demonstrated by Xu et al., where PEG was involved as a dispersant. [14] Noteworthy, most of the above‐mentioned protocols involve multi‐step procedures comprising the first functionalization of lignin, followed by a second cross‐linking with different reagents.
Table 1.
State‐of‐the‐art KL thermosets.
|
Entry |
KL pre‐treatment |
Strategy |
Additives |
Ref. |
|---|---|---|---|---|
|
1 |
solvent fractionation and epoxidation |
epoxy resin |
acetonitrile solution of Polyetheramine D2000 |
[10] |
|
2 |
solvent fractionation and allylation |
thiol‐ene |
EtOAc solution of 3MP3 TMP |
[11] |
|
3 |
solvent fractionation and succinylation |
polyamide or polyester |
MEK solutions of either HMMM and PTSA (cat.) or sodium hypophosphite (cat.) |
[12] |
|
4 |
ozonation |
polyester |
concentrated EtOH solution of sebacic acid‐epoxy, Zn(acac)2 (cat.) |
[13] |
|
5 |
– |
polyester |
dioxane solution of citric acid, PEG, and DMAP (cat.) |
[14] |
|
6 |
– |
polyester |
adipic mixed anhydrides (neat) |
this work |
The solid nature of lignin and its poor solubility hinders the direct generation of a prepolymer, that is, an important intermediate for the production of a thermoset. A cross‐linking mixture (CLM) is generally not able to disperse KL sufficiently where usually one or more pretreatment steps are required, for example, KL fractionation, KL pre‐functionalization, use of dispersants such as PEG or solvents.[ 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 ] Consequently, a synthetic strategy that allows the direct mixing of KL and a CLM to yield a uniform liquid prepolymer would be beneficial. The free hydroxyl moieties are the main functional groups on the Kraft lignin chain (approx. 6.5 mmol g‐1) making the synthesis of esters of lignin with fatty acids a valuable strategy for its functionalization. [19] Polyacids act as cross‐linking agents via esterification with free hydroxyl groups.[ 20 , 21 ] However, only a few reports based on citric acid showed the reaction between lignin, as a minor component, and polyacids to yield a thermoset.[ 14 , 22 ] Due to the abovementioned compatibility issues, a high concentration of untreated lignin is usually detrimental to the mechanical properties of the resulting resins, due to the presence of residual lignin particles, reducing their potential application. [23]
Aiming to use untreated and non‐purified KL in a two components resin, we here report a new strategy to use activated dioic acids to generate a prepolymer that can be poured and molded as a thermoset material circumventing the need for any purifications, pretreatment or solvents.
Results and Discussion
Synthesis of the cross‐linking mixture
To perform the cross‐linking of KL, we envisioned exploiting a diester of adipic acid (AdA) between two separate parts of KL. Acid catalysis can promote AdA esterification.[ 24 , 25 ] However, the direct condensation of KL with carboxylic groups would require high temperatures associated with undesired reactivity. [8] Thus, an activated version of AdA was envisioned. The activation of AdA was performed by reaction with an excess of isopropenyl acetate (IPA) using catalytic amounts of H2SO4 (Scheme 1). IPA was chosen as the preferred activating agent as it is a less toxic and commercially available alternative to the strictly regulated Ac2O, and its reaction with carboxylic acids yields comparable products, giving acetone and AcOH as by‐products. After completion of the reaction, the excess IPA was easily removed by distillation and both acetone and acetic acid were recovered as by‐products, that could in principle be reused to produce a new IPA. [26] The resulting mixture was characterized by the means of nuclear magnetic resonance (NMR) spectroscopy and high‐resolution mass spectrometry (HR‐MS). The primary advantage of IPA is the low associated boiling points, that is, IPA 97 °C and acetone 56 °C.[ 27 , 28 ] Moreover, IPA is a less toxic reagent and not as strictly regulated as Ac2O since it is not included in the drug precursor list. While performing the optimization to generate the activated AdA by IPA using catalytic amounts of H2SO4, the reaction temperature, and the reaction time were evaluated and the optimal parameters were found to be AdA (5 g, 34.2 mmol), IPA (4 equiv.), H2SO4 (0.8 mol %) and 4 h at 97 °C. Under the optimized reaction conditions, a product mixture (6.5 g) comprising isopropenyl functional groups (2.4 equiv. g‐1) and anhydrides moieties (8.0 equiv. g−1) was formed. Both acetyl anhydrides (6.2 equiv. g‐1) and adipoyl anhydrides (1.8 equiv. g−1) were generated (Scheme 1). The HR‐MS analysis showed up to trimeric poly‐adipic anhydride with all the combinations for the terminal groups, either isopropenyl esters or acetyl anhydrides. Noteworthy, the reaction could easily be scaled up to 500 g of AdA giving identical yields and compositions of the resulting CLM.
Scheme 1.

Optimized synthesis of the crosslinking mixture (CLM).
Synthesis of lignoboost Kraft lignin poly‐adipate (KLPA)
Among the polyacids, AdA is the most common within the polymer industry since it is involved in the production of several polyamides. [29] Recent studies have disclosed synthetic strategies to prepare AdA directly from lignin to assess the environmental concerns raised by that AdA currently is produced from fossil feedstocks. This, in turn, would potentially allow a thermoset fully based on lignin.[ 30 , 31 , 32 , 33 ] Reacting KL with the CLM resulted in a viscous prepolymer which was easily pourable into a mold. The consequent thermal treatment of the prepolymer led to the final KL thermoset. The two components, dry Lignoboost KL (30 g) and CLM (19.5 g) were mixed in an equimolar amount to lignin hydroxyl groups. A solid primer was obtained by letting CLM and KL mix. The resulting dark brown powder was then heated swiftly to 100 °C for 10 min. When the KL started to react with the CLM, a viscous homogenous liquid, a “masterbatch”, was obtained. The masterbatch was poured, while still warm, into a PTFE rectangular mold placed between two flat surfaces and heated at 100 °C for 16 h under pressure to achieve the cross‐linking while maintaining the shape. After completion of the reaction, a Kraft lignin poly‐adipate in a form of a dark brown rigid solid was obtained. The overall synthetic procedure is illustrated in Figure 1. During the curing process, AcOH is released upon heating as a side product of the cross‐linking, which can be recovered and merged with the fraction produced during the synthesis of the CLM and undergo full recycling. Both primer and masterbatch shelf life was found longer than 24 h (Supporting Information). Shorter curing times led to incomplete cross‐linking and worsening of both mechanical properties and reproducibility of the resulting material (Supporting Information).
Figure 1.
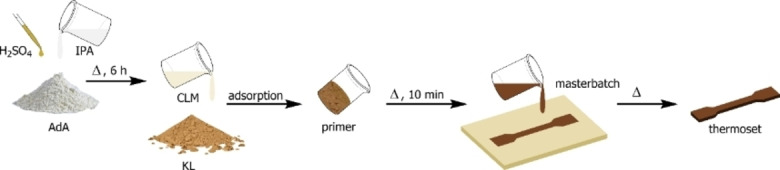
Overall synthetic process of KLPA.
KL polyesters tensile properties
The tensile properties of the thermoset materials with different compositions and process parameters were evaluated by performing stress‐strain analysis on rectangular samples (70 mm×15 mm×1.2 mm, clamp distance 40 mm, and elongation rate 5 mm min−1). A negative trend was registered in the elastic behavior of the KLPAs produced with 1, 1.5, and 2 equiv. of CLM. A higher CLM:KL ratio led to materials with lower tensile strength and Young's modulus (Table 2, entries 1–3), disclosing the plasticizing effect of the extra CLM. The same trend was observed when adding glycerol (10 and 20 wt %) (Table 2, entries 4 and 5). As a control, KL reacted with a CLM based on the corresponding monoacid, that is, hexanoic acid, which gave a viscous material not suitable for stress/strain analysis, although it was fully soluble in THF suggesting no degree of cross‐linking (Table 2, entry 6). The consequent GPC showed a KL with increased molecular weight compatible with a lignin ester (Supporting Information). This increased molecular weight clearly shows that the mechanical properties can easily be tuned based on either ratio of CLM and KL and the addition of glycerol.
Table 2.
KL‐based polyester tensile properties.[a]
|
Entry |
CLM [equiv.] |
Tensile strength [MPa] |
Tensile strain [%] |
E (σ/ϵ) [MPa] |
Gel content [wt %] |
|---|---|---|---|---|---|
|
1 |
1 |
15±3 |
10±3 |
1400±254 |
97 |
|
2 |
1.5 |
7±1 |
90±30 |
300±16 |
95 |
|
3 |
2 |
1.3±0.1 |
120±14 |
11±1 |
98 |
|
4[b] |
1 |
1.9±0.3 |
6±2 |
125±7 |
|
|
5[c] |
1 |
0.33±0.01 |
145±10 |
14±5 |
|
|
6[d] |
1 |
– |
– |
– |
– |
[a] Reaction conditions: Lignoboost Kraft lignin (30 g), 100 °C, 16 h. [b] Glycerol (10 wt %) added on top. [c] Glycerol (20 wt %) added on top. [d] CLM based on caprylic acid, the resulting material was not analyzable by a tensile stress machine. Mean values of 3 repetitions associated with standard deviation.
The mechanical properties shown in Table 2 fall in the same range of most thermoset materials based on KL already published,[ 10 , 11 , 14 , 34 ] and the ones meant to be used as coatings.[ 12 , 13 ] More specifically the tensile strength range reported in this study, that is, 15–0.33 MPa, overlaps with the previously reported: 5–1.2 MPa, [10] 34.3–1.2 MPa, [14] and 12.9–5.1 MPa. [13] However, in the presented methodology moldable materials with no pre‐functionalization and/or dispersing agents were achieved directly with KL. Additional DMA studies and gel content analysis were carried out on the samples containing only KL and CLM to evaluate the cross‐linking. The DMA was performed in tensile mode in the temperature range of 25–150 °C. The study shows that at room temperature the materials are in the glassy state, confirmed by the magnitude of storage modulus. The increase in temperature induced a relaxation process. The relaxation temperature and the tan Δ peak were observed to be dependent on the composition of the studied compound (Supporting Information). The gel content was evaluated by the means of a Soxhlet extraction using THF for 24 h. The analysis showed high gel content (>95 wt %) and independent by the composition of the crosslinked lignin materials.
KLPA thermal analysis
The materials resulting from the reaction of KL and an equimolar amount of CLM were further tested by means of differential scanning calorimetry and thermo‐gravimetric analysis. The thermograms of the KL‐based materials and intermediates are depicted in Figure 2. The thermogram of CLM alone shows an endo peak at 155 °C, corresponding to the melting point of the residual AdA. [35] Interestingly, both the primer and masterbatch behaved similarly, both showing an exothermic peak at around 150 °C indicating the reaction of the CLM with KL. Indeed, the thermogram of the corresponding polyester, which only contains fully reacted CLM did not show any exo‐peak, not even any endo‐peak in the region around 100 °C which would be produced by the evaporation of the residual acetic acid or water. The TGA analysis confirmed this by showing the thermal stability of the polyester up to 125 °C with a negligible mass loss of 1.0 wt % (Supporting Information) and confirming the absence of any free byproduct in the matrix.
Figure 2.
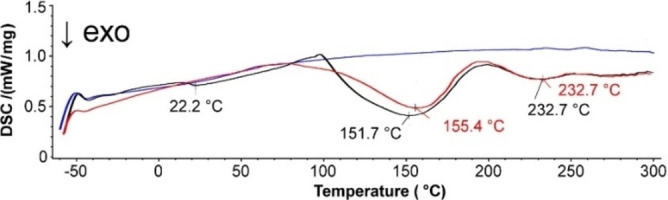
Thermograms of KL with equimolar CLM, primer (black), masterbatch (red), and KLPA (blue).
Mechanistic study
We were curious about the chemistry that occur at the masterbatch stage and also during cross‐linking to produce the thermoset. The infrared band at 3370 cm−1, which corresponds to the free hydroxyl groups of KL, was reduced at the masterbatch stage as can be seen in the IR spectra in Figure 3. The same reductions in the band intensities were observed for the bands at 1744 cm‐1 and 1816 cm−1, assigned to the C−H stretching in the anhydrides. [36] At the same time as the intensity of these bands decreased, the band intensities corresponding to lignin esters at 1708 cm‐1, increased, and at the KLPA stage were dominating. (Figure 3). In addition, the reaction of KL with 2 equiv. of CLM resulted in full conversion of the free hydroxy as shown by the relative IR spectrum (Supporting Information).
Figure 3.
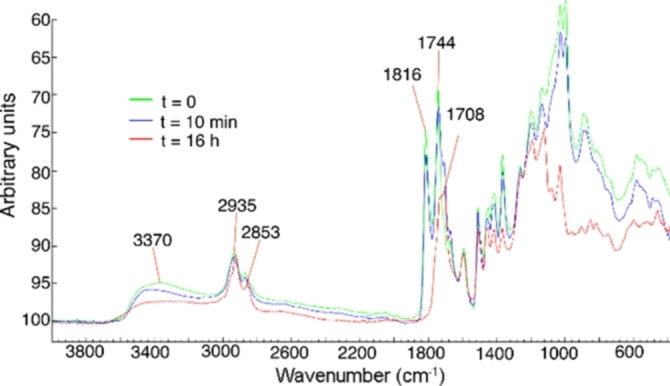
ATR‐IR spectra of KL and equimolar amount of CLM at primer (t= 0), masterbatch (t=10 min) and KLPA (t=16 h) stages.
To gain more insight into the chemistry occurring at the cross‐linking stage to yield the KLPA, solid‐state 13C MAS NMR spectra were recorded. A broad peak at 172 ppm was observed only in the case of KLPA and corresponded to the esters formed between KL and AdA (Figure 4). Signals corresponding to carboxylic acids were observed in the KLPA. However, since it was not possible to extract any residual AdA from the material we assign those signals to the terminal carboxylic acids of KL adipate that were not reacted. Moreover, its insolubility in common organic solvents confirmed the cross‐linking. For instance, the masterbatch was fully soluble in THF, as well as the KL esterified by hexanoic acid.
Figure 4.
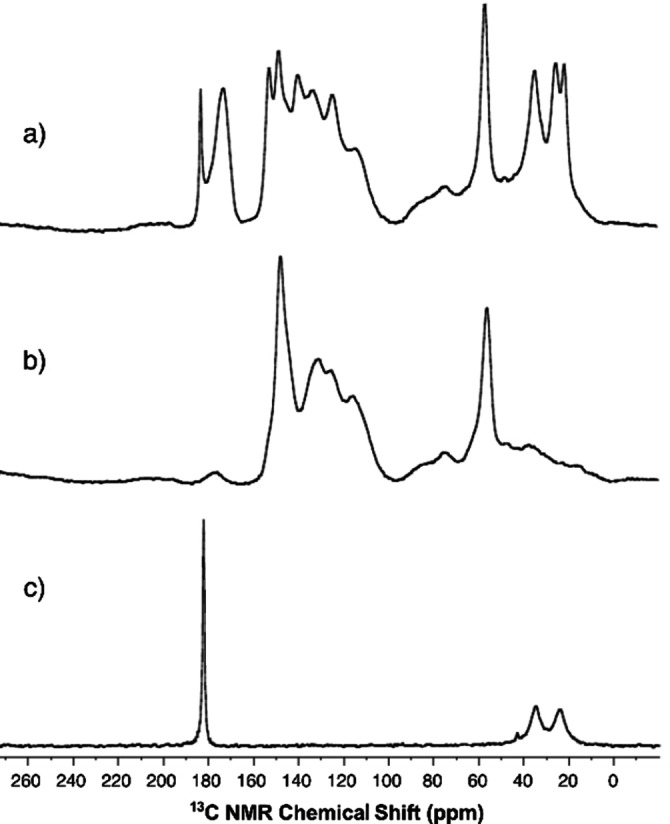
13C MAS NMR spectra of KLPA (a), KL (b), and AdA (c).
To ascertain which of the two moieties: adipic derivatives of either isopropenyl or acetate reacted faster, the two compounds were synthesized and isolated in pure forms that is, bis(isopropenyl) adipate (BIPA) and bis(acetyl) adipate (BACA) (Scheme 2, Supporting Information). The esterification reaction of KL was then independently evaluated. The reactions were performed by mixing KL with an equimolar amount of either BIPA or BACA at 100 °C for 16 h following the same procedure as for the CLM. As for the CLM, BIPA and BACA were first blended with KL at room temperature to obtain light brown primers. However, the preheating step (100 °C, 10 min) did not give any effect with BIPA, while BACA produced the usual dark brown mixture soluble in THF. GPC analysis demonstrated that the molecular weight of lignin increased comparably with the one made with CLM when reacted with BACA, while no significant modification of KL was achieved with BIPA (Supporting Information). By further reacting the masterbatches at 100 °C for 16 h produced an insoluble material in the case of BACA, confirming its role as the cross‐linking agent, while an only slight increase in the molecular weight was obtained for BIPA suggesting a slower reactivity (Supporting Information). Thus, the anhydride synthon/group in the CLM is responsible for esterification at the masterbatch step, where both of the moieties react during the cross‐linking as proved by the disappearance of the isopropenyl signals after curing.
Scheme 2.

Cross‐linking mixture synthesis.
Lignin model compounds
To better understand the reactivity of the abovementioned CLM, we investigated the reaction between AdA‐based CLM and several lignin model compounds bearing different primary and secondary aliphatic, and phenolic hydroxyl moieties found on KL.[ 37 , 38 ] For each set of reactions, four main products were detected by high‐resolution mass spectrometry: acetates, mono adipates, mixed adipates, and symmetrical diesters (Scheme 3). The products were quantified by 1H NMR using 1,4‐dioxane as an internal standard. Due to overlapping signals, it was not possible to quantify independently mono‐ and diesters, and the total amount of adipate was reported as yield of esters. The reactions were performed by mixing each lignin model (1 mmol) with the CLM (1.1 equiv.) at 100 °C for 16 h. Similar reactivities among the tested compounds were observed. All the substrates bearing hydroxyl moieties were transformed within the experimental error into acetate and adipate in an approximate ratio of 1 : 2 (Table 3, entries 1–5). This was also confirmed by GPC analysis (Supporting Information). 1‐Phenyletanol underwent an elimination reaction, probably catalyzed by the residual H2SO4 present in the CLM, yielding styrene in 20 % amount among the products (Table 3, entry 5).
Scheme 3.
Lignin model compounds product distribution.
Table 3.
Lignin model compound reactivity with CLM.[a]
|
Entry |
Substrate |
Conv. [%] |
Yieldacetate [%] |
Yieldesters [%] |
|---|---|---|---|---|
|
1 |
|
96 |
28 |
68 |
|
2 |
|
97 |
31 |
66 |
|
3 |
|
99 |
35 |
64 |
|
4 |
|
99 |
38 |
61 |
|
5[b] |
|
99 |
24 |
56 |
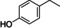
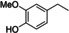
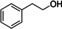
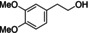
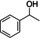
[a] Reaction conditions: substrate (1 mmol), CLM (1.1 equiv.), 100 °C, 16 h, neat. Conversions and yield by 1H NMR. [b] Styrene was observed among the products.
Three models bearing two hydroxyl groups were reacted with CLM, in order to confirm the cross‐linking reactivity, that is, 4‐(2‐hydroxyethyl)phenol, 4‐(2‐hydroxyethyl)‐2‐methoxyphenol, and 4‐(1‐hydroxy‐ethyl)‐2‐methoxyphenol. To assure the stability of the substrates, they were reacted under the same reaction conditions in absence of the CLM, the results from these reactions were used as references for the GPC analysis (Supporting Information). 1H NMR spectroscopy together with high‐resolution mass spectrometry proved the formation of acetylated and esterified products. Gel permeation chromatography (GPC) demonstrated the effectiveness of the CLM in the esterification of the tested diols to give oligomers (Figure 5).
Figure 5.
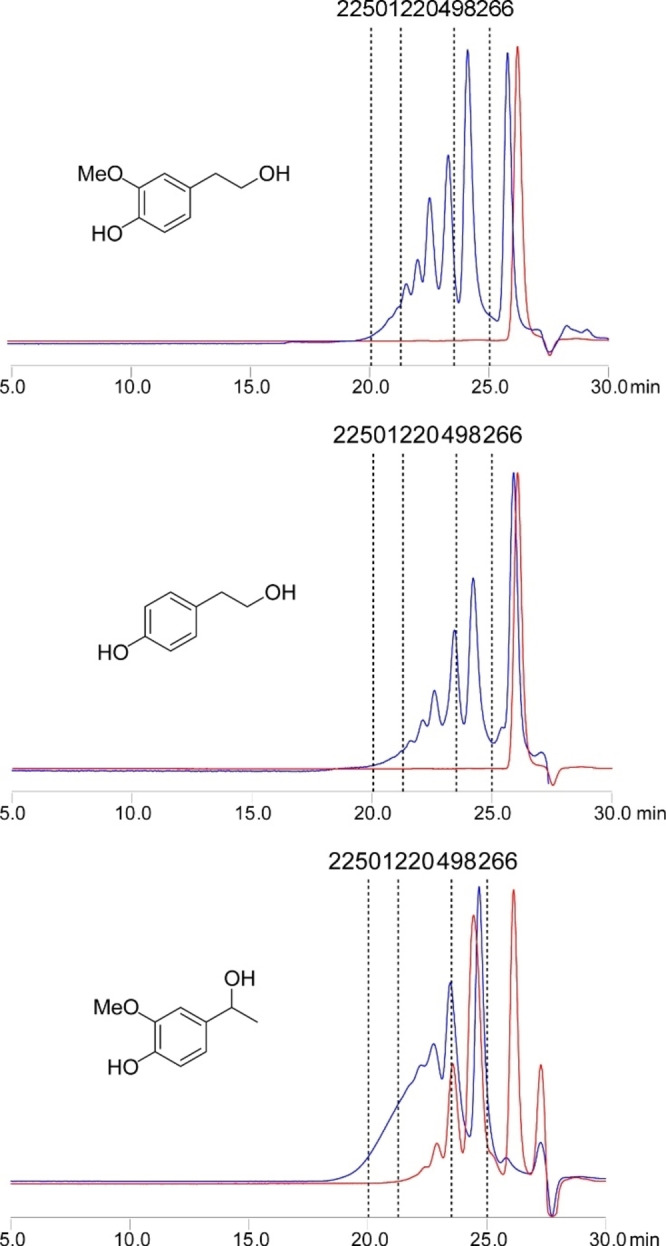
GPC chromatograms of lignin model compounds, as blank (red) and reacted with CLM (blue). Normalized by height. Dashed lines report apparent molecular weight (Da) based on polystyrene calibrant.
To establish the reactivity of the system, a real‐time study was performed by reacting the simplest lignin models bearing phenolic, α‐ and γ‐free hydroxyl groups, for example, 2‐phenylethanol, 4‐ethylphenol, and 1‐phenylethanol, with CLM in neat conditions (Figure 6). The reactions were performed at 50 °C and followed by 1H NMR. The same products as for the reaction performed at 100 °C were observed. In the case of 1‐phenylethanol, the formation of 1‐(phenyl)ethyl ether was also detected (Figure 6a). Each tested substrate reached >95 % conversion within 16 h, and this further supports the reactivity of CLM. Interestingly, the efficiency of acetylation and esterification on model compounds were found to be almost independent of the type of hydroxyl functionality in the substrate generating in all of the studied cases an equimolar amount of acetates and adipates, suggesting a similar reactivity while using KL as substrate.
Figure 6.
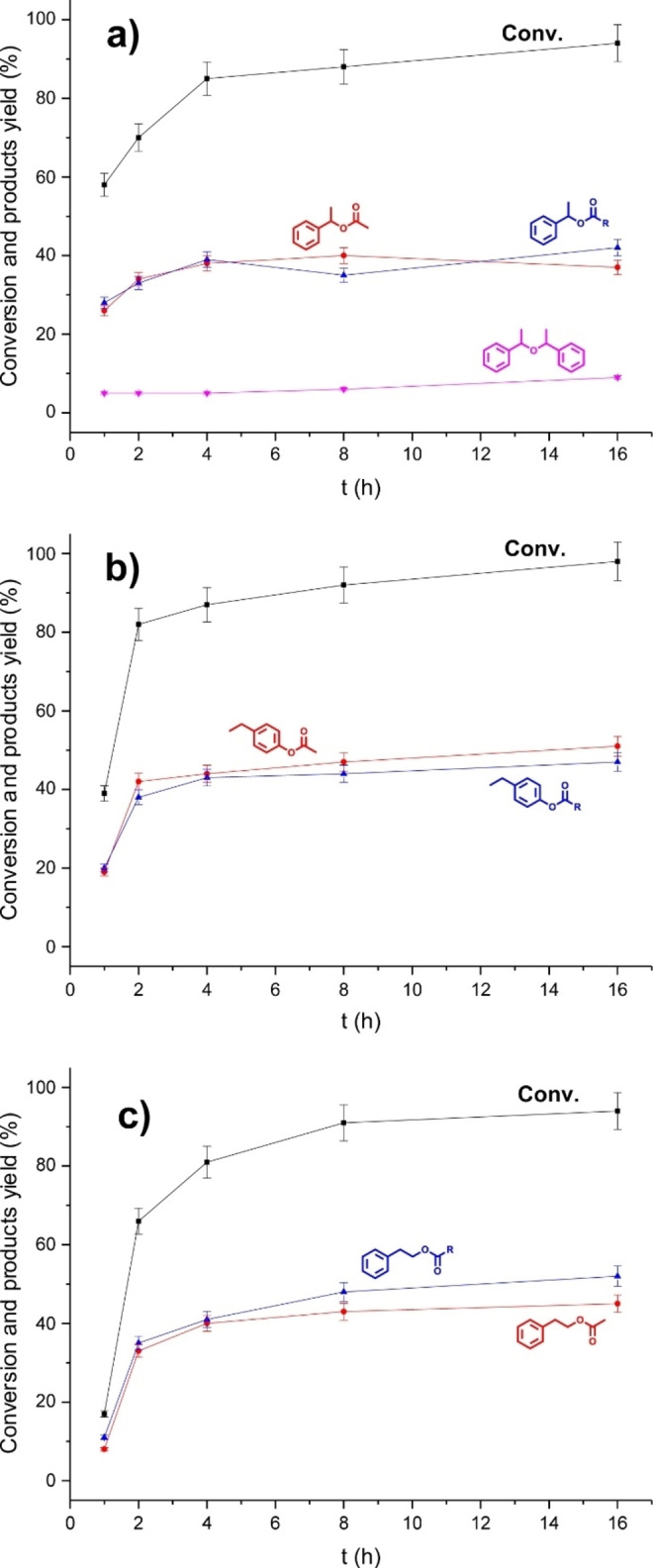
Real‐time analysis of the reactions of Ada‐based CLM with 1‐phenylethanol (a), 4‐ethylphenol (b), and 2‐phenylethanol (c). Reaction conditions: substrate (1 mmol), CLM (1.1 equiv.), 50 °C, neat.
Conclusions
In this study, a novel procedure to obtain thermoset plastic materials from KL was disclosed. The synthetic method involved the reaction of dry KL powder with a cross‐linking liquid based on activated dicarboxylic acid. All the components were relatively inexpensive, and widely available for example, KL, AdA, IPA, and H2SO4. We envisioned and showed that the diester of AdA could undergo further condensation reactions with the KL, and to achieve a prepolymer only heat is required. The compatibilization of KL into the CLM was achieved by the partial esterification of lignin already occurring after 10 min at 100 °C. The resulting viscous liquid became easily pourable and moldable under pressure to yield a plastic solid. DMA and gel content studies clearly indicated an efficient cross‐linking of the lignin materials. The reactivity study performed on model compounds revealed how both the aliphatic and aromatic hydroxyl groups were reactive towards the desired transformation. The mechanical properties of the material could be modulated over time by changing the ratio KL/CLM or by adding glycerol. The proposed strategy overcomes some issues relative to lignin‐based materials, for example, pre‐functionalization, fractionation, and dispersing agents, creating a novel approach for KL valorization into bioplastics. [9] In addition, in the case of the utilization of AdA produced from lignin, up to 90 wt % of the material would be, directly and indirectly, derived from a side‐stream of an existing biorefinery industry not competing with food cultivation. [39] Our vision is that these thermoset plastic materials could be developed further and possibly be an alternative to thermosets used today.
Experimental Section
Synthesis of the adipic‐cross‐linking mixture
A mixture of adipic acid (5 g, 34.2 mmol), IPA (10 equiv., 34.25 g), and H2SO4 (0.8 mol %, 26.7 mg, 0.27 mmol) for 6 h at 97 °C (b.p. of IPA). Once the reaction was complete, the excess of IPA and other volatile co‐products were removed by distillation under vacuum (40 °C, 5 mbar).
Synthesis of lignoboost Kraft lignin poly‐adipate (KLPA)
Softwood Lignoboost Kraft lignin (KL) bearing 6.5 mmol g‐1 of free hydroxyl groups1 was dried at 60 °C overnight before utilization and stored under vacuum. KL (30 g) and Adipic‐CLM (19.5 g) were mixed in an equimolar amount to lignin hydroxyl groups. A homogeneous solid primer was obtained by letting the CLM soak onto the KL. The resulting dark brown powder was then heated to 100 °C for 10 min when the KL started reacting with the CLM to yield a viscous homogenous liquid, the masterbatch. The masterbatch was poured, while still warm, into a PTFE rectangular mold between two flat surfaces and heated at 100 °C for 16 h under pressure to achieve the cross‐linking while maintaining the shape. After the reaction completion, a Kraft lignin poly‐adipate in a form of a dark brown rigid solid was obtained.
Conflict of interest
JS is a founder of RenFuel, a company valorizing lignin. No other authors have any conflicts of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors DDF, KRB, JSMS, APM thank Swedish Foundation for Strategic Environmental Research (Mistra: project Mistra SafeChem, project number 2018/11) and DDF, KRB and JSMS also thank Energimyndigheten for financial support; DR acknowledges the support for his mobility at the University of Stockholm, from the PhD course in Chemistry jointly held by Universities of Ca’ Foscari and Trieste; NH thanks The Swedish Research Council and the grant 2016‐03568; We thank Prof. Claudia Crestini for the supply of lignin model compounds and the fruitful discussions.
D. Di Francesco, D. Rigo, K. Reddy Baddigam, A. P. Mathew, N. Hedin, M. Selva, J. S. M. Samec, ChemSusChem 2022, 15, e202200326.
Contributor Information
Prof. Aji P. Mathew, Email: aji.mathew@mmk.su.se.
Prof. Niklas Hedin, Email: niklas.hedin@mmk.su.se.
Prof. Maurizio Selva, Email: selva@unive.it.
Prof. Joseph S. M. Samec, Email: joseph.samec@su.se.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.“Global plastic production 1950–2020,” can be found under https://www.statista.com/statistics/282732/global-production-of-plastics-since-1950/.
- 2.“Forest products statistics,” can be found under.
- 3. https://www.fao.org/forestry/statistics/en/.
- 4. Demuner I. F., Colodette J. L., Demuner A. J., Jardim C. M., BioResources 2019, 14, 7543–7581. [Google Scholar]
- 5. Jawerth M., Lawoko M., Lundmark S., Perez-Berumen C., Johansson M., RSC Adv. 2016, 6, 96281–96288. [Google Scholar]
- 6. Li Y., Sarkanen S., Macromolecules 2002, 35, 9707–9715. [Google Scholar]
- 7. Kun D., Pukánszky B., Eur. Polym. J. 2017, 93, 618–641. [Google Scholar]
- 8. Koivu K. A. Y., Sadeghifar H., Nousiainen P. A., Argyropoulos D. S., Sipilä J., ACS Sustainable Chem. Eng. 2016, 4, 5238–5247. [Google Scholar]
- 9. Di Francesco D., Dahlstrand C., Löfstedt J., Orebom A., Verendel J., Carrick C., Håkansson Å., Eriksson S., Rådberg H., Wallmo H., Wimby M., Huber F., Federsel C., Backmark M., Samec J. S. M., ChemSusChem 2021, 14, 2414–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Orebom A., Di Francesco D., Shakari P., Samec J. S. M., Pierrou C., Molecules 2021, 26, 3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gioia C., Lo Re G., Lawoko M., Berglund L., J. Am. Chem. Soc. 2018, 140, 4054–4061. [DOI] [PubMed] [Google Scholar]
- 12. Jawerth M. E., Brett C. J., Terrier C., Larsson P. T., Lawoko M., Roth S. V., Lundmark S., Johansson M., ACS Appl. Polym. Mater. 2020, 2, 668–676. [Google Scholar]
- 13. Scarica C., Suriano R., Levi M., Turri S., Griffini G., ACS Sustainable Chem. Eng. 2018, 6, 3392–3401. [Google Scholar]
- 14. Zhang S., Liu T., Hao C., Wang L., Han J., Liu H., Zhang J., Green Chem. 2018, 20, 2995–3000. [Google Scholar]
- 15. Xu Y., Odelius K., Hakkarainen M., ACS Sustainable Chem. Eng. 2019, 7, 13456–13463. [Google Scholar]
- 16. Olsén P., Jawerth M., Lawoko M., Johansson M., Berglund L. A., Green Chem. 2019, 21, 2478–2486. [Google Scholar]
- 17. Cao Y., Liu Z., Zheng B., Ou R., Fan Q., Li L., Guo C., Liu T., Wang Q., Composites Part B 2020, 200, 108295. [Google Scholar]
- 18. Ganewatta M. S., Lokupitiya H. N., Tang C., Polymer 2019, 11, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu H., Mohsin N., Kim S., Chung H., J. Polym. Sci. Part Polym. Chem. 2019, 57, 2121–2130. [Google Scholar]
- 20. Thielemans W., Wool R. P., Biomacromolecules 2005, 6, 1895–1905. [DOI] [PubMed] [Google Scholar]
- 21. Martel B., Weltrowski M., Ruffin D., Morcellet M., J. Appl. Polym. Sci. 2002, 83, 1449–1456. [Google Scholar]
- 22. Shen L., Xu H., Kong L., Yang Y., J. Polym. Environ. 2015, 23, 588–594. [Google Scholar]
- 23. He X., Luzi F., Yang W., Xiao Z., Torre L., Xie Y., Puglia D., ACS Sustainable Chem. Eng. 2018, 6, 9966–9978. [Google Scholar]
- 24. Mariana M., Alfatah T., Yahya E. B., Olaiya N. G., Nuryawan A., Mistar E. M., Abdullah C. K., Abdulmadjid S. N., Ismail H., J. Mater. Res. Technol. 2021, 15, 2287–2316. [Google Scholar]
- 25. Santacroce V., Bigi F., Casnati A., Maggi R., Storaro L., Moretti E., Vaccaro L., Maestri G., Green Chem. 2016, 18, 5764–5768. [Google Scholar]
- 26. Morgan G. T., Walton E., J. Chem. Soc. Resumed 1933, 91–93. [Google Scholar]
- 27. Rigo D., Fiorani G., Perosa A., Selva M., ACS Sustainable Chem. Eng. 2019, 7, 18810–18818. [Google Scholar]
- 28. Rigo D., Calmanti R., Perosa A., Selva M., Green Chem. 2020, 22, 5487–5496. [Google Scholar]
- 29. Hagemeyer H. J., Hull D. C., Ind. Eng. Chem. 1949, 41, 2920–2924. [Google Scholar]
- 30.B. Herzog, M. I. Kohan, S. A. Mestemacher, R. U. Pagilagan, K. Redmond, R. Sarbandi, in Ullmanns Encycl. Ind. Chem., Wiley, 2020, p. 1–47.
- 31. Yan W., Zhang G., Wang J., Liu M., Sun Y., Zhou Z., Zhang W., Zhang S., Xu X., Shen J., Jin X., Front. Chem. 2020, 8, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rios J., Lebeau J., Yang T., Li S., Lynch M. D., Green Chem. 2021, 23, 3172–3190. [Google Scholar]
- 33. Corona A., Biddy M. J., Vardon D. R., Birkved M., Hauschild M. Z., Beckham G. T., Green Chem. 2018, 20, 3857–3866. [Google Scholar]
- 34. Vardon D. R., Franden M. A., Johnson C. W., Karp E. M., Guarnieri M. T., Linger J. G., Salm M. J., Strathmann T. J., Beckham G. T., Energy Environ. Sci. 2015, 8, 617–628. [Google Scholar]
- 35. Ribca I., Jawerth M. E., Brett C. J., Lawoko M., Schwartzkopf M., Chumakov A., Roth S. V., Johansson M., ACS Sustainable Chem. Eng. 2021, 9, 1692–1702. [Google Scholar]
- 36. Ng W. K., Kwek J. W., Yuen A., Tan C. L., Tan R., AAPS PharmSciTech 2010, 11, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McEvoy J. P., J. Chem. Educ. 2014, 91, 726–729. [Google Scholar]
- 38. Crestini C., Lange H., Sette M., Argyropoulos D. S., Green Chem. 2017, 19, 4104–4121. [Google Scholar]
- 39. Lancefield C. S., Constant S., de Peinder P., Bruijnincx P. C. A., ChemSusChem 2019, 12, 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y., Meng H., Cai D., Wang B., Qin P., Wang Z., Tan T., Bioresour. Technol. 2016, 211, 291–297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.