

Abstract
Background
Mitochondrial aldehyde dehydrogenase 2 (ALDH2) is a key enzyme in alcohol metabolism. The ALDH2*2 mutations are found in approximately 45% of East Asians, with 40% being heterozygous (HE) ALDH2*1/*2 and 5% homozygous (HO) ALDH2*2/*2. Studies have shown that HO mice lack cardioprotective effects induced by moderate alcohol consumption. However, the impact of moderate alcohol consumption on cardiac function in HE mice is unknown.
Methods
In this study, HO, HE, and wild‐type (WT) mice were subjected to a 6‐week moderate alcohol drinking protocol, following which myocardial tissue and cardiomyocytes of the mice were extracted.
Results
We found that moderate alcohol exposure did not increase mortality, myocardial fibrosis, apoptosis, or inflammation in HE mice, which differs from the effects observed in HO mice. After exposure to the 6‐week alcohol drinking protocol, there was impaired cardiac function, cardiomyocyte contractility, and intracellular Ca2+ homeostasis and mitochondrial function in both HE and HO mice as compared to WT mice. Moreover, these animals showed overt oxidative stress production and increased levels of the activated forms of calmodulin‐dependent protein kinase II (CaMKII) and ryanodine receptor type 2 (RYR2) phosphorylation protein.
Conclusion
We found that moderate alcohol exposure impaired cardiac function in HE mice, possibly by increasing reactive oxygen species (ROS)/CaMKII/RYR2‐mediated Ca2+ handling abnormalities. Hence, we advocate that people with ALDH2*1/*2 genotypes rigorously avoid alcohol consumption to prevent potential cardiovascular harm induced by moderate alcohol consumption.
Keywords: aldehyde dehydrogenase 2, calcium handling, CaMKII, moderate alcohol, reactive oxygen species
This study revealed that moderate alcohol treatment played a detrimental role in cardiomyocytes and myocardial contractility in ALDH2 heterozygous deficiency mice, possibly via the underlying mechanism of increased ROS/CaMKII/RYR2‐mediated Ca2+ handling disturbance. Together with the results of our present study, these findings supply relevant evidence to enhance our understanding of ALDH2 genetic polymorphisms on alcohol consumption‐induced impact on cardiac function. To this end, the initiative that people with ALDH2*1/*2 (GA) genotypes should avoid alcohol consumption is now proposed.
INTRODUCTION
While alcoholism is considered the most common and detrimental lifestyle worldwide, moderate alcohol intake is known to possess protective impacts on the risk of cardiovascular diseases (CVDs), likely by reducing coronary artery‐associated events and heart failure, as opposed to nonalcoholic consumption or heavy drinking (Dorans et al., 2015; Hung et al., 2016; Xie et al., 2012). In the USA, moderate alcohol intake, which is generally considered a maximum of 1 drink per day for women and 2 drinks per day for men (1 drink = 14 g of pure alcohol), is advised following the 2015 to 2020 Dietary Guidelines for Americans. In addition, the phrase “precision medicine” has been used lately to emphasize the urgent need to profoundly comprehend the importance of personal genetic profiles in clinical treatment approaches.
Mitochondrial aldehyde dehydrogenase 2 (ALDH2) has been well known to oxidize toxic acetaldehyde, which is derived from the catalysis of alcohol by alcohol dehydrogenase (ADH), into nontoxic acetic acid. The ALDH2 genetic polymorphism in humans is designated as ALDH2*2 (ALDH2 point mutation), whereas the wild‐type (WT) is designated as ALDH2*1. The combination of WT and variant alleles forms three types of ALDH2 genotypes: WT (ALDH2*1/*1, also known as GG), heterozygous (HE) (ALDH2*1/*2, also known as GA), and variant‐type homozygous (HO) (ALDH2*2/*2, also known as AA; Brooks et al., 2009). ALDH2*2 is the most prevalent variation in humans and represents approximately 8% of the total population of the world, estimated at 560 million East Asians, including Thai, Korean, Japanese, and Han Chinese individuals (Gross et al., 2015). A survey of the population distribution of allelic ALDH2 indicated that ALDH2*1/*2 constitutes 40%, and ALDH2*2/*2 constitutes 5% population among East Asians (Ginsberg et al., 2002).
People with the ALDH2*2 allele exhibit extremely low ALDH2 enzyme activity and cannot effectively detoxify acetaldehyde, leading to a recognizable clinical phenotype that includes the well‐known Asian alcohol facial flushing response and elevated heart rate following alcohol consumption (Brooks et al., 2009). A study investigating Taiwanese men with various alcohol dehydrogenase 2 (ADH2) and ALDH2 genotypes demonstrated that all participants with heterozygous ALDH2*2 were significantly sensitive to moderate EtOH, as reflected by the considerable elevation of heart rate and facial capillary blood flow (Peng et al., 2002). In addition, another study suggested that there is no safe amount of alcohol consumption for CVDs and health (Wood et al., 2018). However, many reports have clarified the beneficial roles of alcohol consumption in the cardiovascular system, and the mechanisms appear to be related to elevated serum high‐density lipoprotein cholesterol (HDL‐C) and antioxidant effects (Krenz & Korthuis, 2012; Providência, 2006). Our previous study demonstrated that moderate alcohol protects the heart via increased HDL‐C and heme oxygenase‐1(HO‐1), while HO mice abolished these cardioprotective benefits (Fan et al., 2014; Shen et al., 2017). Therefore, controversy exists regarding whether those with the ALDH2*1/*2 genotype gain benefits from moderate alcohol consumption or not. Epidemiological research has shown individuals with the ALDH2*1/*2 genotype self‐reported that the rate of alcohol intake and heavy drinking behaviors were lower than actual, because there may exist bias in the accuracy of self‐report (Higuchi et al., 1996; Luczak et al., 2017), so understanding the precise consequences of moderate alcohol consumption on the hearts of people with the ALDH2*1/*2 genotype and the underlying mechanism is of importance for guiding their drinking habits. Intracellular Ca2+ homeostasis ensures the maintenance of normal cardiac function. Disorders of calcium homeostasis in myocardial cells can lead to dysfunction of cardiac contraction (Shattock et al., 2015; Yeung et al., 2007; Zhao et al., 2016). It has been proposed that altered intracellular Ca2+ homeostasis underscores the compromised mechanical function in alcohol‐induced myocardial injury (Ren et al., 1997). However, it is not well understood whether Ca2+ homeostasis mediates the effect of moderate alcohol on HE. The present study explored the precise role of moderate alcohol in HE mice and illustrated the possible mechanism by focusing on the disordered Ca2+‐mediated signaling pathway.
MATERIALS AND METHODS
Animal models and treatment procedures
Male C57BL/6J WT (ALDH2+/+) mice (20 to 25 g, 6 to 8 weeks old) were procured from Shanghai Animal Administration Center (Shanghai, China). The technique described previously was used to create age‐ and weight‐matched C57BL/6J ALDH2 gene HO (ALDH2−/−) mice (Sun et al., 2014). C57BL/6J ALDH2 gene HE (ALDH2+/−) mice were produced by a cross between male HO mice and female WT mice, commissioned and finished by Cavens Biogle Model Animal Research Co., Ltd. The genotyping of WT, HO, and HE mice was performed by PCR analysis of genomic DNA as described, and representative images of ALDH2 genetic identification by nucleic acid gel electrophoresis are shown in Figure S1e (Kitagawa et al., 2000). All animals were kept in temperature‐controlled environments (24°C and relative humidity of 55 ± 5%), with cycles of 12 h of darkness and 12 h of light and free access to water and food. The care of the animals, as well as the evaluators of the outcomes, was blinded. After a one‐week adaptation period, the WT, HE, and HO mice were randomly assigned to the moderate ethanol (EtOH) cohort and control (con) cohort. The following alcohol administration experiment was conducted: 2.5% (v/v) for 1 week, 5% (v/v) for 1 week, 10% (v/v) for 1 week, and 18% (v/v) for the next 3 weeks; meanwhile, mice in the con cohort were provided with untreated water for 6 weeks, as previously described (Fan et al., 2014; Shen et al., 2017; Zhou et al., 2002). The drinking water of the EtOH and con groups was changed every day. EtOH was present until harvesting. At six weeks after alcohol intake, the animals were used for ventricular myocyte separation or tissue analysis. The animal experimentation techniques adhered to the US National Institutes of Health's Guide for the Care and Use of Laboratory Animals (NIH publication no. 85‐23, updated 1996) and were thoroughly vetted by the Animal Ethics Committee of Zhongshan Hospital, Fudan University, China.
Reagents and antibodies
Reagents required for the isolation and culture of adult cardiomyocytes, including collagenase type 2 (LS004176) and collagenase type 4 (LS004188), were obtained from Worthington Biochemical Corporation; medium 199 (12340‐030) and chemically defined lipid concentrations (11905‐031) were obtained from Gibco; protease type XIV (P5147), ITS liquid media supplement (100×) (I3146), laminin (11243217001), and 2,3‐butanedione monoxime (BDM) (B0753) were obtained from Sigma. Anti‐CaMKII antibody (ab134041), anti‐SOD2/MnSOD antibody (ab68155), anti‐heme oxygenase 1 antibody (ab13243), and anti‐Bcl‐2 antibody (ab182858) were obtained from Abcam. Anti‐phospho‐CaMKII (Thr286) antibody (12716) and anti‐Bax antibody (2772) were obtained from Cell Signaling Technology. The anti‐SERCA2 antibody (A1097) was procured from ABclonal. The anti‐CaMKII (oxidized) antibody (GTX36254) was obtained from GeneTex. Anti‐ALDH2 antibody (15310‐1‐AP), anti‐cleaved caspase 3 antibody (19677‐1‐AP), and anti‐NCX1 antibody (55075‐1AP) were obtained from Proteintech. The anti‐phospho‐RYR2 (Ser2808) antibody (PA5‐104444) was obtained from Invitrogen. The anti‐cardiac troponin T antibody (ab8259) was obtained from Servicebio. The anti‐4‐hydroxynonenal antibody (MAB3249) was obtained from R&D Systems.
Echocardiographic and hemodynamic measurements
Following a 6‐week research duration, heart function was measured in minimally anesthetized mice utilizing isoflurane at 2% inducement and 1% maintenance. M‐mode pictures of the left ventricle (LV) were obtained using the Vevo 770 imaging system (VisualSonics) to evaluate LV anatomical and functional characteristics during systolic and diastolic heart contractions. The heart rate (HR), left ventricular posterior wall thickness in diastole (LVPWD), left ventricular anterior wall thickness in diastole (LVAWD), left ventricular end‐systolic diameter (LVESD), left ventricular end‐diastolic diameter (LVEDD), and left ventricular ejection fraction(LVEF) were all measured, and the mean value of five cardiac cycles was utilized. Left ventricular fractional shortening (LVFS) was assessed according to the following formula: LVFS = [(LVEDD − LVESD)/LVEDD] × 100.
Analysis of serum acetaldehyde and lipid level, and blood alcohol concentration
Following cardiac functional recordings, blood specimens were drawn via retro‐orbital hemorrhage while the mice were anesthetized with pentobarbital sodium (80 mg/kg, intraperitoneal injection, i.p.). Part of the whole blood samples was collected into the tubes containing the K2EDTA anticoagulant. The blood collection tube was gently mixed by inverting 8 to 10 times immediately after collection. A Headspace‐Gas Chromatography (HS‐GC) method was used to assess blood alcohol concentration. The rest whole blood samples were placed at ambient temperatures for 30 min first. The serum was subsequently isolated at 1006 g at a temperature of 4°C for 15 min and kept at a temperature of −80°C until analyzed. An acetaldehyde assay kit (Megazym, Bray) was utilized for the determination of serum acetaldehyde levels. For lipid analysis, commercial kits (Nanjing Jiancheng Bioengineering Institute, China) were utilized to quantify plasma triglyceride (TG), total cholesterol (TC), low‐density lipoprotein cholesterol (LDL‐C), and high‐density lipoprotein cholesterol (HDL‐C).
Isolation of adult mouse cardiomyocytes and measurement of mechanical properties
After echocardiographic evaluation, the ventricles were immediately removed from the hearts of WT, HE, and HO mice under anesthesia and were digested with collagenase buffer. Adult mouse cardiomyocyte isolation was performed by the described methods (Ackers‐Johnson et al., 2016; Jiang et al., 2020). The only cardiomyocytes chosen for usage were those with rod shapes and distinct edges. An inverted microscope (IX‐70, Olympus) was utilized to visualize cardiomyocytes, and a SoftEdge MyoCam system (IonOptix Corporation) was utilized to assess their mechanical characteristics. The following indicators were utilized to quantify cardiomyocyte contraction and relengthening profiles: resting cell length; peak shortening (PS, normalized to the resting cell length); time‐to‐PS (TPS); maximal rates of shortening (+dL/dt) and relengthening (−dL/dt) and time‐to‐90% relengthening (TR90).
Intracellular Ca2+ transients
Fura‐2, a recognized ratio‐metric dye, was utilized to assess free intracellular Ca2+ by recording the emitted fluorescence signals at excitation wavelengths of 360 and 380 nm and presented as 360/380 nm fluorescence. After incubation using fura‐2/AM (0.5 μM; Molecular Probes, Thermo Fisher) for 30 to 60 min, the fluorescence intensity of cardiomyocytes was measured utilizing a dual excitation fluorescence photomultiplier system (IonOptix). The myocytes were subsequently loaded onto an Olympus IX‐70 inverted microscope, and imaging was performed via a Fluor 40× oil objective. Cells were induced to contract at 0.5 Hz while being subjected to light produced by a 75‐W lamp and passing through either a 360‐ or a 380‐nm filter. The FFI ratio of both wavelengths (360/380) was used to detect fluorescence emissions between 480 and 520 nm and for qualitative changes in the FFI. As an indicator of intracellular Ca2+ clearance, the fluorescence degradation time was measured.
Histological staining
Hearts fixed with 4% paraformaldehyde were embedded in paraffin or OCT compound. For routine histological analysis, 5‐μm‐thick segments were subjected to staining using H&E. Sirius red staining and Masson's trichrome staining were utilized to evaluate collagen deposition. The heart's cross‐sectional area was investigated using WGA staining. Frozen specimens incubated with dihydroethidium (DHE) were used to evaluate the excessive production of myocardial reactive oxygen species (ROS).
ALDH2 enzymatic activity and ATP measurement
The activity of ALDH2 was analyzed using a kit for the quantitative colorimetric detection of tissue ALDH2 activity per the guidelines provided by the manufacturer (GMS50300.2, GENMED Scientifics, Inc.). The ATP content was measured utilizing an enhanced ATP test kit (Beyotime Biotechnology). Lysis of the tissues was performed in ATP lysis buffer followed by centrifugation for 10 min at 16099 g to collect the supernatant. The detection working dilution of ATP and the supernatant were combined. The ATP content was assessed utilizing a microplate reader (BioTek), followed by normalization of the amount to the protein concentration.
Mitochondrial membrane potential measurement
JC‐1 spontaneously creates J‐aggregates that produces red fluorescence in the mitochondria of healthy cells with elevated mitochondrial membrane potential (MMP). Nevertheless, the MMP decreases, and JC‐1 is discharged from the mitochondria of unhealthy cells and resides as a green fluorescent monomer in the cytoplasm. A JC‐1 staining kit (Beyotime Biotechnology) was utilized to monitor the MMP according to the instructions stipulated by the manufacturer. Adult mouse cardiomyocytes were stained with JC‐1 (10 μg/ml) for 20 min at 37°C. A fluorescence microscope (Olympus) was utilized to visualize MMP. JC‐1 monomers exhibit green fluorescence, indicative of reduced MMP, while the red fluorescence of JC‐1 aggregates indicates elevated MMP. The data are displayed as the red‐to‐green fluorescence intensity ratio.
Mitochondrial ROS measurement
MitoSOX Red Superoxide Indicator (Invitrogen) was utilized to detect mitochondrial ROS (mtROS) according to the manufacturer's instructions. To produce a 5 mM MitoSOX™ reagent stock solution, the content (50 μg) of a single vial of MitoSOX™ mitochondrial superoxide indicator was dissolved in 13 μl of dimethylsulfoxide (DMSO). Subsequently, preparation of the working solution (5 μM) was done by diluting 5 mM MitoSOX™ reagent stock solution utilizing cell culture medium at 1:1000. Then, the cardiomyocytes of the adult mice were incubated with the working solution for 10 min at a temperature of 37°C in darkness. Finally, a fluorescence microscope (Olympus) was utilized to observe the cells.
Determination of the malondialdehyde content and caspase 3 activity
The malondialdehyde (MDA) content in heart tissue homogenates was determined using a commercial kit by spectrophotometry according to the protocols stipulated by the manufacturer (Beyotime Biotechnology). The Caspase 3 Activity Assay Kit (Beyotime Biotechnology) was utilized to assess the caspase 3 activity. Tissue homogenates were centrifuged at 20,000 × g for 15 min at a temperature of 4°C, followed by lysing of the pellets in lysis buffer for an additional 5 min on ice. After the tissue was lysed, the assay was performed in a 96‐well plate. In each of the wells, 50 μl of tissue lysate were added to assay buffer (40 μl), followed by an additional 10 μl of the caspase‐3 colorimetric substrate (Ac‐DEVD‐pNA). The 96‐well plate was incubated for 1 h at a temperature of 37°C, during which the caspase in the specimen was able to cleave the chromophore p‐NA from the substrate. Subsequently, the specimens were examined at 405 nm utilizing a microplate reader. The activity of caspase 3 is presented as picomoles of pNA produced per microgram of protein per minute.
Protein extraction and western blot analysis
The left ventricular cardiac tissues were homogenized and sonicated in RIPA lysis buffer containing a 1% protease‐inhibitor cocktail. The Bradford assay was employed to quantify the protein content. After being extracted from tissues, proteins (normalized to 30 to 60 μg) were injected into each well, followed by separation using 10% to 15% SDS‐PAGE and electrotransfer to a PVDF membrane (Bio‐Rad). One hour before incubating the membranes with specified primary antibodies at a temperature of 4°C overnight, they were blocked in Tris‐buffered saline containing 0.1% Tween 20 (TBST) and 5% bovine serum albumin (BSA; Sigma). Subsequently, horseradish peroxidase (HRP)‐conjugated secondary antibodies were utilized to incubate the membranes for a duration of 2 h at room temperature (Abcam). The particular immunoreaction bands were identified by Image Lab software, version 3.0, utilizing Pierce ECL western blotting substrate (Thermo Fisher, 32109). Densitometric quantification was performed utilizing ImageJ analysis software, followed by normalization to the loading control β‐actin.
RNA isolation and RT–qPCR analysis
TRIzol™ Reagent (Invitrogen) was utilized to extract total RNA from the myocardial tissue according to the instructions stipulated by the manufacturer. A UV absorption assay (NanoDrop® ND‐1000) was utilized to assess the purity and concentration of RNA. The cDNA was reverse transcribed by purified 1000‐ng RNA with an A260/A280 ratio of 1.8 to 2.0. PCR was performed with RT–PCR detection. Next, PrimeScript™ RT Master Mix (TaKaRa) was utilized to perform RT–PCR. The cDNA was obtained by reverse transcription with purified 1000 ng RNA with an A260/A280 ratio of 1.8 to 2.0. Next, TB Green Premix Ex Taq II (TaKaRa) was utilized to perform qPCR. An aggregate of a 10 μl reaction system was prepared, which included 0.8μl DNA template (cDNA), 3.4 μl ddH2O, 0.4 μl forward and reverse primers respectively, 5 μl TB Green Premix Ex Taq II. Table S3 lists the primers applied in this experiment. The PCR response cycles were configured as indicated below: 30 s at a temperature of 95°C, then 5 s at a temperature of 95°C, and 30 s at a temperature of 60°C for 40 cycles. The method was employed to record fluorescent signals, which were normalized to 18S.
Immunostaining and immunofluorescence analysis
For histological analysis, the myocardial tissues were serially sectioned at a thickness of 5 μm. For 30 min, the cryosections were fixed in 4% paraformaldehyde and then washed three times for 10 min each in phosphate‐buffered saline (PBS). Then, 0.1% Triton X‐100 (Sigma) was applied to permeabilize the plasma membranes for 10 min. Blocking of nonspecific binding sites was performed by incubation with 10% BSA (Sigma) in PBS for 30 min. Incubation of the slides was performed with primary antibodies overnight at a temperature of 4°C. This step was followed by washing the slides and incubating them with secondary antibodies at ambient temperature in the dark for 2 h. Nuclei were stained with DAPI (Thermo Fisher) at room temperature. Finally, the slides were covered with a glass slide. The immunofluorescence staining was viewed and captured using a microscope (Olympus).
Statistical analysis
Prism 8 (GraphPad Software, Inc.) was used to conduct all of the statistical analyses. Continuous variables are presented as the mean ± standard error of the mean (SEM). The Shapiro–Wilk test was employed to determine normal distribution. The distinctions of normal variates were evaluated utilizing Student's t‐test (within 2 cohorts) or one‐way analysis of variance (ANOVA, among 3 cohorts or more), while Tukey's multiple comparisons test was utilized for post hoc comparisons. The Kruskal–Wallis H test or Mann–Whitney U test was used to analyze nonnormal data. Two‐tailed p < 0.05 was recognized as statistically significant.
RESULTS
Analysis of the general parameters of mice
After 6 weeks of moderate EtOH treatment, no mice died in the WT‐EtOH and HE‐EtOH groups, but the survival rate was only 46.67% in HO‐EtOH mice (p < 0.05) (Figure 1A). At the study end, the body weight of HO‐EtOH mice was significantly lower than that of WT‐EtOH and HE‐EtOH mice (18.23 ± 0.5046 vs. 25.06 ± 0.4397, 24.83 ± 0.4993), while the baseline body weight of the mouse was similar among six groups (WT‐con: 22.26 ± 0.2603; WT‐EtOH: 23.04 ± 0.3106; HE‐con: 23.25 ± 0.4032; HE‐EtOH: 22.77 ± 0.2539; HE‐con: 22.49 ± 0.2539; HO‐EtOH: 23 ± 0.3461). The body weight at baseline and the study end demonstrated that the incremental step‐up in EtOH concentration with moderate with six weeks led to a significant decline in body weight of HO‐EtOH mice compared to that of WT‐EtOH mice and HE‐EtOH mice (Figure 1B). The above result was consistent with the previous studies (Fan et al., 2014; Shen et al., 2017; Zhou et al., 2002). The results might be a consequence of providing EtOH in drinking water, which is a major limitation of the study, particularly when the EtOH concentration reached 18% (v/v). With such a high EtOH concentration from week 4 to week 6, the animals in the HO‐EtOH group drank less water and consumed less food than animals in the WT‐EtOH and HE‐EtOH groups (Table S1), which might be responsible for the lower body weight observed in HO‐EtOH mice. It remains unknown why the drinking and eating behavior were only significantly affected in HO mice but not in WT and HE mice, which needs to be clarified in future studies. The heart weight/body weight (HW/BW) ratio after alcohol treatment was similar among six groups (Figure 1C). And the ratios of lung wet weight/body weight (LWW/BW) were considerably elevated in HE‐EtOH and HO‐EtOH mice compared to that in WT‐EtOH mice (Figure 1D). Improved dyslipidemia might be associated with the cardioprotective impacts of moderate alcohol ingestion according to previous studies (Fan et al., 2014; Gardner & Mouton, 2015; Gaziano et al., 1993). Therefore, we measured the levels of lipids in mice after 6 weeks. WT, HE, and HO mice had similar basic values of blood lipids, including TG, TC, and LDL‐C. However, after moderate alcohol intake for 6 weeks, the levels of TG, TC, and LDL‐C in HE‐EtOH mice increased significantly compared to HE‐con mice, and they were analogous to that in WT‐EtOH and HO‐EtOH mice (Figure 1E to G). However, moderate alcohol markedly elevated the levels of HDL‐C in WT‐EtOH mice compared to the WT‐con group, while this effect was not observed in HE‐EtOH and HO‐EtOH mice post 6 weeks EtOH exposure (Figure 1H), similar to our previous study (Shen et al., 2017). The whole blood of the mice was collected on days 7, 14, 28, 35, 42 to measure their blood alcohol concentration, respectively. And the data of blood alcohol concentration per mouse were illustrated in Table S2. Blood alcohol concentrations were significantly higher in the HO‐ETOH group than in the WT‐ETOH and HE‐ETOH group on days 14, 21, 28, 35, and 42. And the acetaldehyde level in mice serum from the WT, HE, and HO groups increased obviously, while the acetaldehyde level in mice serum from HE‐EtOH mice was significantly higher than that of WT‐EtOH mice and was similar to that of HO‐EtOH mice (Figure S1b). In summary, these outcomes illustrated that six weeks of moderate alcohol seemed to have no obvious impact on the survival, body weight, or HW/BW in HE mice. We further detected the heart function of mice with echocardiography.
FIGURE 1.
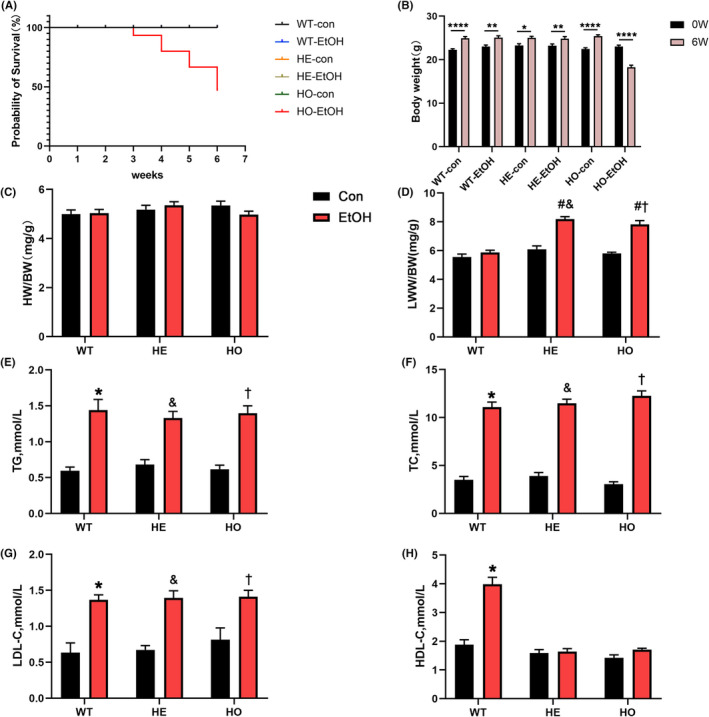
General parameters of WT, HE, and HO mice with or without moderate alcohol exposure. (A) The survival curve of six groups. p < 0.05, HO‐EtOH versus WT‐con, WT‐EtOH, HE‐con, HE‐EtOH, and HO‐con, Log‐Rank test. n = 15. (B) Quantitative analysis of body weight at baseline and the study end. n = 15. (C) Ratio of heart weight (HW) versus body weight (BW). n = 15. (D) Ratio of lung wet weight (LWW) versus body weight (BW). n = 15. Quantitative analysis of plasma (E) triglyceride (TG), (F) total cholesterol (TC), (G) low‐density lipoprotein cholesterol (LDL‐C), and (H) high‐density lipoprotein cholesterol (HDL‐C). n = 6 to 11. The data are presented as the mean ± SEM. Statistical comparisons were carried out by one‐way ANOVA. *p < 0.05, versus WT‐con, # p < 0.05, versus WT‐EtOH, & p < 0.05, versus HE‐con, $ p < 0.05, versus HE‐EtOH, † p < 0.05, versus HO‐con; (b), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Moderate alcohol harmed cardiac function in HE mice
Echocardiography was conducted to monitor the progression of heart structural and functional changes and the representative M‐mode echocardiographic images were shown in Figure 2A. LVEF in the HE‐EtOH group (52.46 ± 1.107, p < 0.0001) and the HO‐EtOH group (52.14 ± 1.219, p < 0.0001) were both decreased significantly compared to that in the WT‐EtOH group (68.01 ± 1.344) (Figure 2B). And LVFS in the HE‐EtOH group (26.63 ± 1.032, p < 0.0001) and HO‐EtOH group (26.28 ± 0.7139, p < 0.0001) were both reduced clearly compared to the WT‐EtOH group (37.36 ± 1.046) (Figure 2C). Moderate alcohol intake overtly increased the LVESD (Figure 2D) but had no serious effect on the LVEDD (Figure 2E) or LVPWD (Figure 2G) in HE‐EtOH and HO‐EtOH mice compared to that in WT‐EtOH mice. In HE‐EtOH and HO‐EtOH groups, the LVAWD was decreased compared to that in WT‐EtOH group, but the difference was not statistically significant (Figure 2F). These echocardiographic measurements illustrated that moderate alcohol ingestion in HE mice resulted in cardiac abnormalities, as shown by the decreased LVEF and LVFS, instead of cardioprotective impacts.
FIGURE 2.
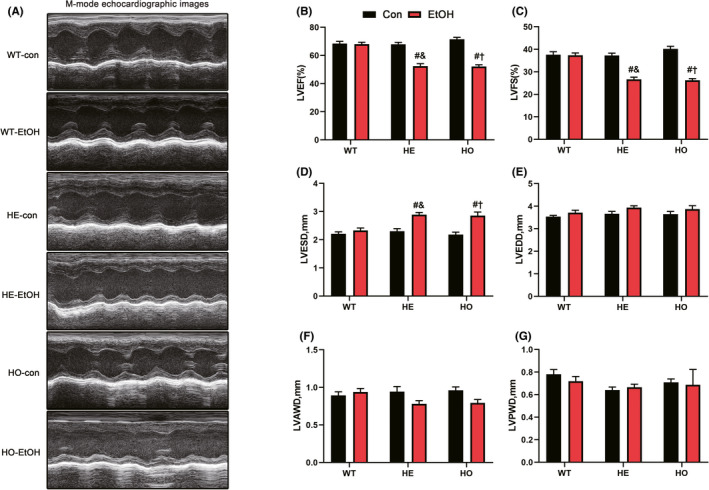
Cardiac function of WT, HE, and HO mice with or without moderate alcohol challenge. (A) Representative M‐mode echocardiographic images of each study group. (B) left ventricular ejection fraction (LVEF). n = 15. (C) Left ventricular fractional shortening (LVFS). n = 15. (D) Left ventricular end‐systolic dimension (LVESD). n = 15. (E) Left ventricular end‐diastolic dimension (LVEDD). n = 15. (F) Left ventricular anterior wall thickness in diastole (LVAWD). n = 15. (G) Left ventricular posterior wall thickness in diastole (LVPWD). n = 15. All measurements were based on similar anesthesia conditions. The data are presented as the mean ± SEM. Statistical comparisons were carried out by one‐way ANOVA. The data are presented as the mean ± SEM. Statistical comparisons were carried out by one‐way ANOVA. *p < 0.05, versus WT‐con, # p < 0.05, versus WT‐EtOH, & p < 0.05, versus HE‐con, $ p < 0.05, versus HE‐EtOH, † p < 0.05, versus HO‐con
Moderate alcohol impaired adult cardiomyocyte contractile function in HE mice
Adult mouse cardiomyocytes were isolated after echocardiographic evaluation and then their contractile functions were measured. After subjecting to moderate alcohol, PS (normalized to the resting cell length), also known as the length of cardiomyocytes reduced by electrical stimuli, in adult mouse cardiomyocytes from HE‐EtOH and HO‐EtOH mice was markedly reduced compared to that in adult mouse cardiomyocytes from WT‐EtOH mice (Figure 3B). In addition, the −dL/dt and +dL/dt in the HE‐EtOH and HE‐EtOH groups decreased sharply compared to those in the WT‐EtOH group (Figure 3C,D). The length of the resting cell (Figure 3A), TPS, also known as the period to peak shortening (Figure 3E), and TR90 which represents the duration of relaxation (Figure 3F) were not altered among the six groups. These conclusions were consistent with the in vivo echocardiographic findings, indicating that moderate alcohol intake markedly suppressed the contractile function of mouse cardiomyocytes isolated from HE mice.
FIGURE 3.
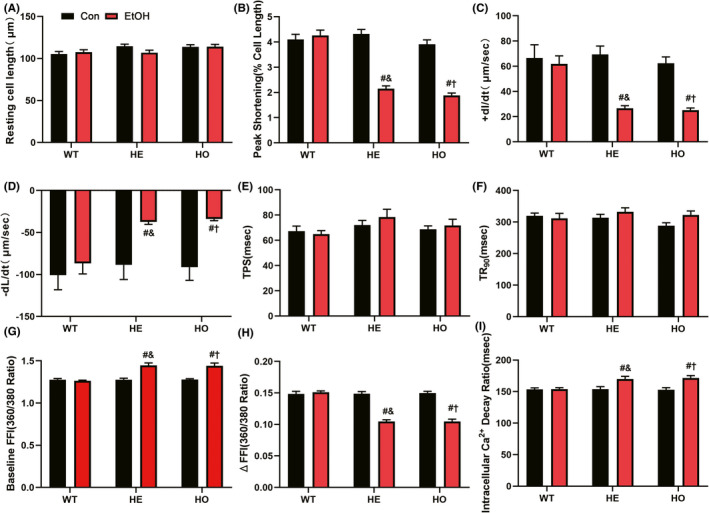
Contractile properties and intracellular Ca2+ properties of cardiomyocytes isolated from WT, HE, and HO mice with or without moderate alcohol challenge. (A to F) showed contractile properties of cardiomyocytes: (A) resting cell length; (B) peak shortening (PS, normalized to resting cell length); (C) maximal velocity of shortening (+ dL/dt); (D) maximal velocity of relengthening (− dL/dt); (E) time‐to‐peak shortening (TPS); (F) time‐to‐90% relengthening (TR90). n = 82 to 86 cells from 6 to 8 mice per group. (G to I) showed intracellular Ca2+ properties of cardiomyocytes: (G) resting Fura‐2 fluorescence intensity (FFI); (H) electrically‐stimulated rise in FFI (ΔFFI); (I) intracellular Ca2+ decay rate. n = 28 to 30 cells from 6 to 8 mice per group. The data are presented as the mean ± SEM. Statistical comparisons were carried out by one‐way ANOVA. *p < 0.05, versus WT‐con, # p < 0.05, versus WT‐EtOH, & p < 0.05, versus HE‐con, $ p < 0.05, versus HE‐EtOH, † p < 0.05, versus HO‐con
Moderate alcohol had no obvious impact on myocardial fibrosis, apoptosis, inflammation, or hypertrophy in HE mice
We further explored why moderate EtOH exposure exerted an adverse effect on cardiac function in HE mice hearts. Cardiac hypertrophy (Hajnóczky et al., 2005), apoptosis (Fernández‐Solà et al., 2006), fibrosis (Piano et al., 2002), and inflammation (Lang & Korzick, 2014; Liu et al., 2011) play important physiopathological roles in the deleterious effects of alcohol on the cardiovascular system. Therefore, we performed hematoxylin and eosin (H&E) staining, Sirius red staining, Masson's trichrome staining, and wheat germ agglutinin (WGA)‐fluorescein isothiocyanate staining of the myocardium, and the representative images were shown in Figure 4A. After 6 weeks drinking, compared to those of WT mice, the H&E‐stained images did not exhibit any pathological changes in tissue arrangement in the hearts of HE‐EtOH mice, while the disordered arrangement was observed in the hearts of HO‐EtOH mice (Figure 4A). Myocardial fibrosis detected by Sirius red staining or Masson's trichrome staining was similar between HE‐EtOH and WT‐EtOH groups (Figure 4A and Figure S1b). And the myocardial mRNA levels of the profibrotic genes collagen type I alpha 1 chain (col1a1), collagen type III alpha 1 chain (col3a1), smooth muscle alpha‐actin (α‐SMA), and transforming growth factor‐beta 1 (TGF‐β1) were also similar between HE‐EtOH and WT‐EtOH groups (Figure 4B). At the same time, they were overtly increased in HO‐EtOH mouse hearts compared to those in WT‐EtOH and HE‐EtOH mice after 6 weeks. Cardiomyocyte hypertrophy by WGA staining (Figure 4A and Figure S1c), and the mRNA levels of the pro‐hypertrophic genes atrial natriuretic peptide (ANP), natriuretic peptide B (BNP), and β‐myosin heavy chain (β‐MyHC) (Figure 4C) were similar among the six groups. The protein and mRNA levels of the antiapoptotic protein B‐cell lymphoma 2 (Bcl2) and the proapoptotic protein Bcl2‐associated X (Bax), as well as the protein levels of apoptotic executive protein cleaved caspase 3, also remained unchanged in the HE‐EtOH group compared to those in the WT‐EtOH group. Nevertheless, the protein and mRNA levels of Bcl2 decreased significantly, while the protein and mRNA levels of Bax and the protein levels of cleaved caspase 3 expression increased significantly in the HO‐EtOH group compared to those in the WT‐EtOH group (Figure 4 and Figure S1d). Consistently, compared to those of WT‐EtOH mice, the activities and mRNA expression of caspase 3 were increased in HO‐EtOH mice but remained unchanged in HE‐EtOH mice after alcohol intake (Figure 4G and Figure S1d). RT–qPCR analysis of inflammatory cytokine levels in myocardial tissue showed that, in the HO‐EtOH group, the pro‐inflammatory factors tumor necrosis factor‐alpha (TNFα), monocyte chemoattractant protein‐1 (MCP‐1), interleukin‐6 (IL‐6), and interleukin‐1β (IL‐1β) increased significantly, while the anti‐inflammatory factor interleukin‐10 (IL‐10) decreased significantly as compared to those in WT‐EtOH group. However, these cytokines remained unchanged in the HE‐EtOH group compared to those in the WT‐EtOH group (Figure 4D). Altogether, there were no distinct variations in myocardial fibrosis, apoptosis, inflammation, or cardiomyocyte hypertrophy in HE mice after moderate alcohol exposure for six weeks.
FIGURE 4.
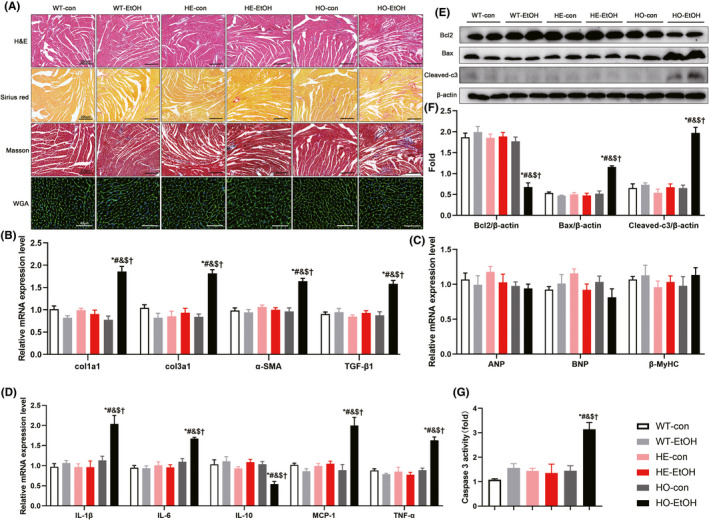
Myocardial fibrosis, cross‐sectional areas, inflammation, and apoptosis in WT, HE, and HO mice with or without moderate alcohol drinking. The data indicated that after 6 weeks of moderate alcohol treatment, there were no differences in cross‐sectional areas among six groups. There were also no significant differences in myocardial fibrosis, inflammation, and apoptosis in HE‐EtOH mice compared to WT‐EtOH mice after 6 weeks of moderate alcohol challenge. (A) Representative images of hematoxylin and eosin (H&E) staining of the entire heart in transverse midsection showing the heart structural deficits (images in the first row, Scale bars = 200 μm); representative images of Sirius red staining (images in the second row, Scale bars = 200 μm) and Masson's trichrome staining (images in the third row, Scale bars = 200 μm) showing the collagen deposition in heart tissues of mice; representative images of wheat germ agglutinin (WGA)‐fluorescein isothiocyanate staining of heart tissues showing cardiomyocytes hypertrophy (images in the fourth row, Scale bars = 40 μm). (B) mRNA expression levels of pro‐fibrosis genes col1a1, col3a1, α‐SMA, and TGF‐β1 in mouse hearts. n = 8. (C) mRNA expression levels of pro‐hypertrophic genes ANP, BNP, β‐MyHC in the hearts of mice. n = 8. (D) mRNA expression levels of inflammatory cytokines IL‐1β, IL‐6, IL‐10, MCP‐1, and TNFα levels in the hearts of mice. n = 8. (E) Representative western blots of apoptosis‐related Bcl2 (26 KDa), Bax (21 KDa), and cleaved caspase 3 (Cleaved‐c3) (17 KDa) with β‐actin (43 KDa) used as loading control in mouse heart tissues; (F) densitometry values (normalized to β‐actin) of Bcl2, Bax, and Cleaved‐c3. n = 6. (G) myocardial apoptosis was measured using Caspase‐3 activity. n = 4. The data are presented as the mean ± SEM. Statistical comparisons were carried out by one‐way ANOVA. *p < 0.05, versus WT‐con, # p < 0.05, versus WT‐EtOH, & p < 0.05, versus HE‐con, $ p < 0.05, versus HE‐EtOH, † p < 0.05, versus HO‐con
Moderate alcohol elicited mitochondrial dysfunction in HE mice
The protein levels of ALDH2 in HE‐con mice were considerably decreased compared to those in WT‐con mice and were significantly higher than those in HO‐con mice, and moderate alcohol treatment did not significantly change their ALDH2 protein expression levels compared to their baseline levels (Figure 5A,B). These changes paralleled the mRNA levels of ALDH2 (Figure 5C). Moreover, the activity of ALDH2 in HE‐con mice was considerably decreased compared to that in WT‐con mice at the basic levels. And the activity of ALDH2 in HE‐con mice was higher than that of HO‐con mice, but the difference did not reach statistically significant (p > 0.05). Meanwhile, moderate EtOH drinking did not alter the ALDH2 activity levels in WT‐EtOH, HE‐EtOH, and HO‐EtOH mice compared to their con group, respectively (Figure 5D). Contraction of the heart is a process that demands a high amount of energy, which is supplied in the form of ATP generated by mitochondria (Page & McCallister, 2007). ATP synthesis, the decrease of which is used as an indicator of mitochondrial damage, is a primary mitochondrial function (Rhoads et al., 2006). Reduced ATP generation was found in the HO‐EtOH cohort and the HE‐EtOH cohort compared to that in the WT‐EtOH cohort (Figure 5E). A decrease in red fluorescence and an increase in green fluorescence in the HE‐EtOH and HO‐EtOH groups were observed by inverted fluorescence microscopy compared to the WT‐EtOH group (Figure 5H). The histograms depicted the ratios of red/green fluorescence, which represented the outcomes of the immunofluorescence analysis, revealing sharp decreases in MMP in the HE‐EtOH and HO‐EtOH cohorts compared to that in the WT‐EtOH cohort (Figure 5I). These data demonstrated that moderate alcohol could induce mitochondrial dysfunction, manifested as decreased ATP generation and MMP in HE mice.
FIGURE 5.
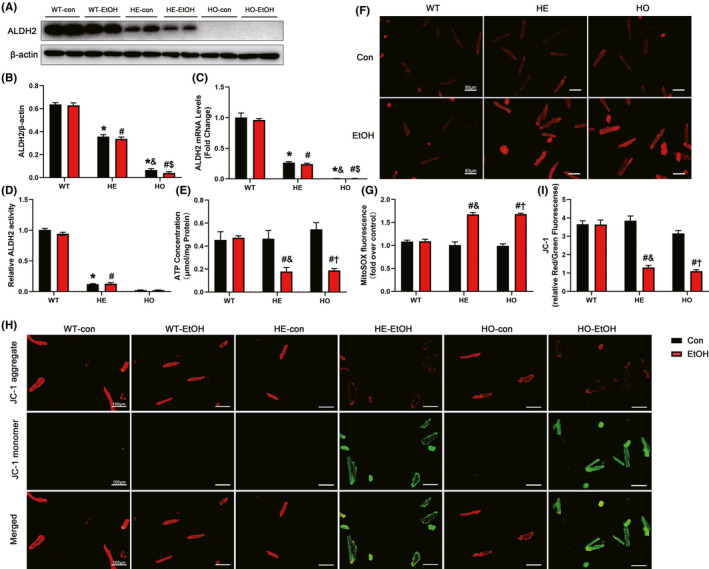
The effect of moderate alcohol on the levels of ATP, mitochondrial membrane potential (MMP), mitochondrial ROS (mtROS), and ALDH2 expression and activity in WT, HE, and HO mice with or without moderate alcohol treatment. (A) Representative western blots of ALDH2(56KDa) with β‐actin(43KDa) used as loading control in mouse heart tissues; (B) densitometry values (normalized to β‐actin) of ALDH2. n = 6. (C) RT‐qPCR analysis of ALDH2 gene in mouse heart. n = 8. (D) ALDH2 activity in mouse heart was quantified and expressed as fold of WT‐con mice. n = 12. (E) ATP generation in mouse heart, normalized to total protein amount and expressed as μmoL/mg protein. n = 4. (F) Representative images of MitoSOX red staining determining the mitochondrial superoxide in isolated adult mouse cardiomyocytes (Scale bars = 80 μm). (G) The image shows quantification of MitoSOX fluorescence intensity from different visual fields of 6 samples in each experimental group. n = 6. (H) Representative images of JC‐1 staining detected the MMP changes in adult mouse cardiomyocytes. The red fluorescence indicates the JC‐1 aggregate, while the green fluorescence indicates the JC‐1 monomer (Scale bars = 100 μm). (I) Quantitative analysis of the ratio of Red/Green fluorescence in each group from different visual fields of 6 samples in each experimental group. n = 6. The data are presented as the mean ± SEM. Statistical comparisons were carried out by one‐way ANOVA. *p < 0.05, versus WT‐con, # p < 0.05, versus WT‐EtOH, & p < 0.05, versus HE‐con, $ p < 0.05, versus HE‐EtOH, † p < 0.05, versus HO‐con
Moderate alcohol increased the generation of oxidative stress in HE mice
Deficiency of ALDH2 activity causes disorders in EtOH metabolism, leading to an increased ratio of nicotinamide adenine dinucleotide hydrogen (NADH)/nicotinamide adenine dinucleotide+ (NAD+), thus promoting the production of ROS (Bailey & Cunningham, 2002). Most endogenous ROS are produced in the mitochondria, damage to which results in subsequent increases in ROS production (Slimen et al., 2014; Suematsu et al., 2003). Here, DHE staining was performed to identify increased intracellular ROS levels in HE‐EtOH and HO‐EtOH mouse hearts post alcohol challenge (Figure 6A,B). Moreover, MitoSOX red staining in isolated adult mouse cardiomyocytes was applied to show elevated levels of mtROS in cardiomyocytes from HE‐EtOH and HO‐EtOH mice compared to that from WT‐EtOH mice post EtOH of 6 weeks (Figure 5F,G). Lipid peroxidation is a well‐recognized process of oxidative degradation induced by a chain response of excess ROS, producing highly endogenous reactive lipid peroxidation aldehydes, such as MDA and 4‐hydroxynonenal (4‐HNE), which are effectively metabolized by ALDH2 (Anderson et al., 2012; Kato & Osawa, 2010; Yoval‐Sánchez & Rodríguez‐Zavala, 2012). Therefore, we inferred that when ALDH2 was heterozygous deficient, the levels of 4‐HNE and MDA were elevated. 4‐HNE immunostaining and MDA concentration assays were performed to verify our speculation. The results showed that the 4‐HNE intensity (Figure 6A,C) and the concentration of MDA (Figure 6D), a marker of lipid peroxidation, were both markedly elevated in the HE‐EtOH and HO‐EtOH cohorts compared to that in the WT‐EtOH cohort. It has been well documented that HO‐1, which plays an essential role against EtOH‐induced protection in cardiomyocytes, is an important antioxidant enzyme (Shen et al., 2017). HO‐1 was found to have overtly increased protein expression in the myocardium of WT‐EtOH mice post alcohol treatment compared to WT‐con mice, while no difference was observed in the HE‐EtOH and HO‐EtOH groups compared to their con groups, respectively (Figure 6E,F). The protein level of superoxide dismutase 2 (SOD2) was strongly reduced in HE‐EtOH and HO‐EtOH mice compared to that in WT‐EtOH mice after EtOH intervention (Figure 6G,H). RT–qPCR analysis also showed a lower mRNA expression level of SOD2 in the HE‐EtOH and HO‐EtOH groups compared to that in the WT‐EtOH group (Figure 6I). As described above, these findings revealed that, on the one hand, moderate alcohol administration increased the protein levels of HO‐1 in WT mice; on the other hand, alcohol intake increased the generation of ROS and reactive lipid peroxidation aldehydes, including 4‐HNE and MDA, while significantly decreasing the expression of the antioxidant enzymes HO‐1 and SOD2 in HE mice.
FIGURE 6.
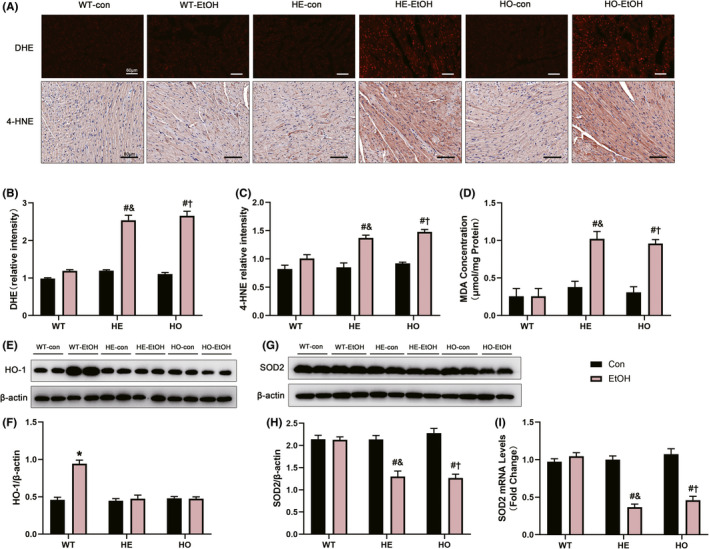
The myocardial oxidative stress generation and the antioxidant protein markers levels in WT, HE, and HO mice with or without moderate alcohol treatment. (A) Representative images of dihydroethidium (DHE) staining were performed to assess ROS levels in mouse heart (Images in the top row, Scale bars = 60μm); representative images of 4‐hydroxynonenal (4‐HNE) immunohistochemistry detection in mouse heart (images in the middle row, Scale bars = 80 μm). (B) Quantification of DHE staining from different visual fields of six samples in each experimental group. n = 6. (C) Measurement of the intensity of 4‐HNE immunostaining from different visual fields of six samples in each experimental group. n = 6. (D) The concentration of MDA in heart tissue homogenates, normalized to total protein amount and expressed as μmoL/mg protein. n = 4. (E) Representative western blots of HO‐1 (32 KDa) with β‐actin (43 KDa) used as loading control in mouse heart tissues. (F) Densitometry values (normalized to β‐actin) of HO‐1. n = 6. (G) Representative western blots of SOD2 (25 KDa) with β‐actin (43 KDa) used as loading control in mouse heart tissues; (H) Densitometry values (normalized to β‐actin) of SOD2. n = 6. (I) RT‐qPCR analysis of SOD2 gene in mouse heart. n = 8. The data are presented as the mean ± SEM. Statistical comparisons were carried out by one‐way ANOVA. *p < 0.05, versus WT‐con, # p < 0.05, versus WT‐EtOH, & p < 0.05, versus HE‐con, $ p < 0.05, versus HE‐EtOH, † p < 0.05, versus HO‐con
Moderate alcohol caused defects in calcium homeostasis and affected Ca2+‐related markers in HE mice
To explore the possible mechanisms underlying moderate alcohol exposure‐stimulated cardiac dysfunction in HE mice, Fura‐2 fluorescence microscopy was applied to evaluate intracellular Ca2+ handling. The cardiomyocytes from the HE‐EtOH and HO‐EtOH cohorts exhibited reduced intracellular Ca2+ production when subjected to electrical stimuli (Δfura‐2 fluorescence intensity (FFI)) (Figure 3H) and prolonged resting intracellular Ca2+ (resting FFI) (Figure 3G), as well as protracted intracellular Ca2+ decay compared to those in the cardiomyocytes from the WT‐EtOH cohort (Figure 3I), demonstrating disturbed Ca2+ homeostasis in adult mouse cardiomyocytes from HE‐EtOH and HO‐EtOH mice after alcohol. Subsequently, Ca2+ handling‐related protein levels were measured. We assessed the activation of CaMKII, a Ca2+‐related marker, in the mouse myocardium using antibodies that detect phosphorylated CaMKII (phosphorylated threonine 286; P‐CaMKII), oxidized CaMKII (M281‐M282 oxidation; Ox‐CaMKII), or total CaMKII (Tot‐CaMKII). The levels of P‐CaMKII/Tot‐CaMKII and Ox‐CaMKII/Tot‐CaMKII were both significantly higher while the levels of Tot‐CaMKII/β‐actin were unchanged in HE‐EtOH and HO‐EtOH mice compared to that in WT‐EtOH mice after a moderate alcohol challenge of six weeks, suggesting greater CaMKII oxidation and phosphorylation in the HE‐EtOH and HO‐EtOH groups (Figure 7A to D). Moreover, other Ca2+‐related markers such as RYR2, NCX1, and SERCA2 were estimated in our present study. Although several research reports have disclosed the involvement of SERCA2 and NCX1 in compromised intracellular Ca2+ handling after alcohol consumption (Li & Ren, 2006; Oba et al., 2008; Zhang et al., 2003), we did not detect a measurable difference in the protein levels of NCX1 and SERCA2 among the six groups after moderate alcohol consumption (Figure 7E to G). RT–qPCR analysis showed that the mRNA expression of the NCX1 and SERCA2 genes in mouse hearts were unchanged among the six groups either (Figure 7H,I). Besides, we measured total RYR2 mRNA levels and found that the influence of moderate alcohol on the total RYR2 mRNA levels was not significantly different among the six groups (Figure7J). However, it has been reported that in CVDs, RYR2 phosphorylation at serine 2814 (RYR2‐pS2814) by CaMKII adversely impacts myocardial outcomes (van Oort et al., 2010; Respress et al., 2012), so we also measured the protein expression levels of phosphorylated S2814 RYR2 (P‐RYR2) using an immunofluorescence staining assay with cardiac troponin T (cTnT), a marker of cardiomyocytes that colocalizes with RYR2 in cardiomyocytes (Figure 7K). Quantification of P‐RYR2 immunofluorescence staining illustrated that P‐RYR2 levels were greatly elevated in HE‐EtOH and HO‐EtOH mice compared to that in WT‐EtOH mice after six weeks of moderate EtOH challenge (Figure 7L). Collectively, these data revealed that moderate EtOH treatment induced Ca2+ defects and elevated levels of the activated forms of CaMKII (Ox‐CaMKII and P‐CaMKII) and RYR2‐S2814 phosphorylation in HE mice, suggesting the possible contribution of CaMKII‐mediated RYR2 to abnormal Ca2+ handling in the HE‐EtOH group.
FIGURE 7.
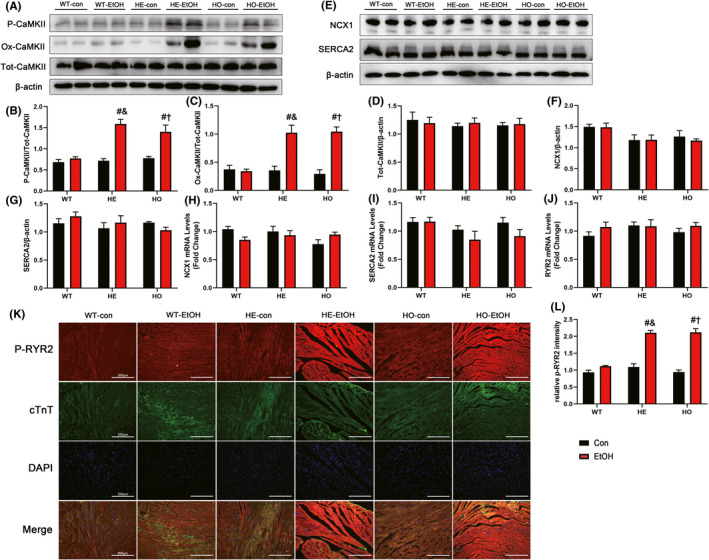
Ca2+‐related protein markers in WT, HE, and HO mice with or without moderate alcohol treatment. (A) Representative western blots of P‐CaMKII (phosphorylated‐Thr286 CaMKII) (60 KDa), Ox‐CaMKII (oxidized CaMKII) (55 KDa), and Tot‐CaMKII (total CaMKII) (54 KDa) with β‐actin (43 KDa) used as loading control in mouse heart tissues; densitometry values (normalized to β‐actin) of (B) P‐CaMKII, (C) Ox‐CaMKII, and (D) Tot‐CaMKII. n = 6. (E) Representative western blots of NCX1 (120 KDa) and SERCA2 (115 KDa) with β‐actin (43 KDa) used as loading control in mouse heart tissues; densitometry values (normalized to β‐actin) of (F) NCX1 and (G) SERCA2. n = 6. RT‐qPCR analysis of (H) NCX1, (I) SERCA2, and (J) RYR2 (ryanodine receptor type 2) gene in mouse heart. n = 8. (K) Immunofluorescence staining images of P‐RYR2 (phosphorylated‐S2814 RYR2) in mouse heart (Scale bars = 200 μm). (L) Quantification of P‐RYR2 immunofluorescence staining from different visual fields of six samples in each experimental group. n = 6. The data are presented as the mean ± SEM. Statistical comparisons were carried out by one‐way ANOVA. *p < 0.05, versus WT‐con, # p < 0.05, versus WT‐EtOH, & p < 0.05, versus HE‐con, $ p < 0.05, versus HE‐EtOH, † p < 0.05, versus HO‐con
DISCUSSION
The key findings from this study are that chronic EtOH administration at the moderate dose leads to cardiac dysfunction in vivo and calcium handling abnormalities in adult cardiomyocytes in vitro in HE mice, possibly via the underlying mechanism of increased ROS/CaMKII/RYR2. The experimental animal research here elucidates the harmful effects of chronic moderate EtOH on cardiac function in HE mice. These findings indicate that people with ALDH2*1/*2 genotypes, like people with ALDH2*2/*2 genotypes, should thus avoid alcohol, even in moderate amounts, in their daily lives.
Previous studies have reported that the levels of apolipoprotein A1, adiponectin, and HDL‐C increased, but the levels of plasminogen decreased when using moderate alcohol (Brien et al., 2011). The most substantial cardioprotective impact of moderate alcohol is the influence of alcohol usage on blood lipoproteins, particularly HDL. Our research revealed pronounced upregulation of serum HDL‐C concentrations and the myocardial antioxidant enzyme HO‐1 protein expression, in line with our previous findings of elevated HDL‐C and HO‐1 levels with moderate doses of EtOH in HO mice (Fan et al., 2014; Shen et al., 2017). Although we did not observe a significantly decreased probability of survival, body weight, LVAWD, or LVPWD, or increased myocardial fibrosis, apoptosis, and inflammation in HE mice, which were exhibited in HO mice after six weeks of alcohol, these findings could be explained by the higher activity of ALDH2 in HE mice than in HO mice, as our present study demonstrated.
Our data suggested that in HE mice, the levels of acetaldehyde increased significantly after moderate alcohol treatment, mainly resulting from the decreased activities of ALDH2. When ALDH2 is deficient, the oxidative metabolism of EtOH cannot proceed normally, and thus causing the accumulation of acetaldehyde, finally resulting in the generation of ROS (Kumar et al., 2013). Similarly, in our study, significantly increased levels of intracellular ROS and mtROS were found in the HE‐EtOH and HO‐EtOH groups. And the decreased expression of the antioxidant enzyme SOD2 was found in the HE‐EtOH and HO‐EtOH groups. Excessive generation of ROS and failure of antioxidative mechanisms (including SOD2) to remove redundant ROS produces oxidative stress. ROS generation promotes the formation of toxic aldehydes through lipid peroxidation, and the buildup of 4‐HNE‐like aldehydes might induce the generation of ROS, resulting in a negative cycle (Peng et al., 2014). Increased ROS accumulation in mitochondria results in mitochondria dysfunction (Slimen et al., 2014; Suematsu et al., 2003). Moreover, it has been well established that, in CVDs, abnormal mitochondrial function is often accompanied by an increased generation of ROS (Brown et al., 2017). In other words, ROS production and mitochondrial dysfunction produce a negative cycle. It is worth noting that our current findings demonstrated that increased endogenous reactive lipid peroxidation aldehydes represented by 4‐HNE and MDA, and apparent mitochondrial dysfunction represented by decreased ATP generation and MMP disruption, were found in HE and HO mice after six weeks of moderate alcohol challenge. All in all, chronic moderate alcohol drinking increased the production of ROS in HE and HO mice.
Our present observation favored significantly disturbed Ca2+ homeostasis in adult cardiomyocytes from HE and HO mice after moderate alcohol exposure, as shown by the decreased intracellular Ca2+ release in response to electrical stimuli and prolonged intracellular Ca2+ decay, as well as elevated resting intracellular Ca2+. Similarly, the research performed by Beuckelmann et al. (1992) showed unbalanced calcium homeostasis in human ventricular myocytes with heart failure. CaMKII is a vital kinase in the modulation of the activity of multiple Ca2+‐handling proteins, including RYR2 (Erickson et al., 2011; Wehrens et al., 2004). The production of ROS has been proposed to be an essential process for the elevation of CaMKII activity, manifesting as phosphorylation and oxidation of CaMKII (Erickson et al., 2011; Wang et al., 2021). A very recent study found that ROS‐CaMKII–RYR2 pathway contributed to disordered calcium handling and damaged cardiac muscle contraction in Barth syndrome (Liu et al., 2021). In both animal models of heart failure and failing human hearts, the levels of CaMKII activity and expression are considerably elevated (Zhang, 2017). Correspondingly, our present findings revealed that the generation of ROS were increased significantly, as mentioned before, and the protein expression levels of activated forms of CaMKII (Ox‐CaMKII and P‐CaMKII) were both elevated in the HE‐EtOH and HO‐EtOH cohorts. RYR2, which has been identified as being affiliated with the RYR family, represents the cardiac RYR isoform and is considered to be the CaMKII substrate (Ho et al., 2016). CaMKII phosphorylation is crucial to modulating RYR2 activity, and CaMKII phosphorylates RYR2 at Ser2808 and Ser2814 (Ho et al., 2016; Respress et al., 2012). The present data suggested notably elevated RYR2 phosphorylation at Ser2814, as shown by P‐RYR2 immunofluorescence staining in HE and HO mice after moderate alcohol treatment. The quantity of Ca2+ produced from the SR through RYR2 essentially influences the Ca2+‐transient amplitude, which correlates with systolic heart contraction intensity. The general effects of these variations in RYR2 phosphorylation are a decrease in Ca2+‐transient amplitude, leading to disordered Ca2+ handling (Nikolaienko et al., 2018). Redox modification of RYR2 contributes to abnormal function in heart failure, as reported by Terentyev et al. (2008). The present evidence showed that HE‐EtOH and HO‐EtOH mice cardiomyocytes exhibited dampened intracellular Ca2+ transient maximal upstroke velocity. Given that CaMKII is highly concentrated near the t‐tubules of cardiomyocytes, close to L‐type calcium channel (LTCC) and RYR2 channels of SR (Zhang, 2017), we speculated that activated CaMKII‐mediated RYR2 phosphorylation and thus contributing to dysregulated intracellular Ca2+ homeostasis. Although SERCA2 and NCX1 ensure that the concentration of Ca2+ in the cytoplasm of cardiomyocytes is maintained at a normal, low level (Dobrev & Wehrens, 2014), the mRNA and protein expression levels of SERCA2 and NCX1 were not affected in HE and HO mice following moderate alcohol in our study. However, CaMKII promotes RYR2 diastolic Ca2+ leak, promotes arrhythmia, and adversely impacts myocardial outcome (van Oort et al., 2010; Respress et al., 2012). So, it should be noted that in this study, we have evaluated only increased resting intracellular Ca2+, but SR Ca2+ leak could be measured in future research to illustrate whether elevated resting intracellular Ca2+ was associated with diastolic Ca2+ leak through RYR2. Besides, CaMKII was recently found in mitochondria, whose CaMKII inhibition has been reported to be beneficial to cardiac function. And it is well konwn that ALDH2 mainly exists in mitochondria where is the main place for alcohol metabolism. (Luczak et al., 2020; Swaminathan et al., 2012). Therefore, further studies involved in illuminating the roles of mitochondrial CaMKII in moderate alcohol‐mediated damaging effects on mice with ALDH2 deficiency will make sense.
Compared to the model of chronic alcohol consumption which causes alcoholic cardiomyopathy (George & Figueredo, 2011; Kryzhanovskii et al., 2017), in this experiment, moderate EtOH‐induced myocardial injury in ALDH2 heterozygous mice did not have cardiomyocyte hypertrophy, fibrosis, or apoptosis. The reasons for the above results can be explained by the following three points. Firstly, in terms of the dose of alcohol used, we use moderate alcohol in this study, but excessive alcohol was used in the chronic alcohol consumption model. Secondly, in terms of the duration of alcohol use, the present experimental model is a short‐time (6 weeks) alcohol exposure compared to long‐time (usually ≥15 years in humans or ≥12 weeks in mice) necessary to develop chronic alcoholic cardiomyopathy. Thirdly, previous studies have shown that chronic alcohol abuse causes chronic alcohol cardiomyopathy in WT mice, whose mechanisms are implicated in the generation of fibrosis, apoptotic cell death, and adverse cardiac hypertrophy (Gardner & Mouton, 2015). However, we use the mice of ALDH2 heterozygous deficiency in this study, while there is no study about the effect of excessive alcohol consumption on cardiac function of ALDH2 heterozygous mice. Moreover, further studies are needed to determine the impact of high dose or prolonged moderate alcohol consumption on cardiac function of mice with various ALDH2 genotypes.
In conclusion, this study revealed that moderate alcohol treatment played a detrimental role in cardiomyocytes and myocardial contractility, possibly as a result of disordered cardiomyocytes intracellular Ca2+ homeostasis triggered by upregulation of the activated forms of CaMKII (Ox‐CaMKII and P‐CaMKII) and RYR2‐S2814 phosphorylation, likely caused by excessive ROS due to ALDH2 heterozygous deficiency in HE mice (Figure 8). Together with the results of our present study, these findings supply relevant evidence to enhance our understanding of the relationship between ALDH2 genetic polymorphisms and alcohol consumption‐induced impact on cardiac function. To this end, the initiative that people with ALDH2 GA (ALDH2*1/*2) genotypes should avoid alcohol consumption is now proposed.
FIGURE 8.
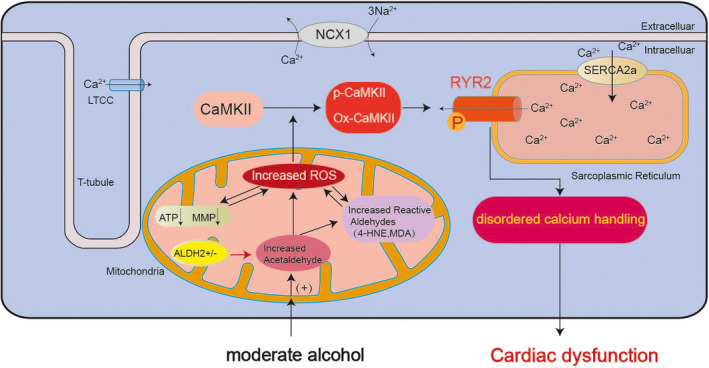
Schematic diagram depicting the mechanism of cardiac dysfunction caused by moderate alcohol consumption in HE mice. Schematic representation showing that after a moderate alcohol challenge of 6 weeks in HE mice, the levels of acetaldehyde in mouse serum were increased, resulting from the insufficient activity of ALDH2 in EtOH metabolism. Then the elevated acetaldehyde accelerated the generation of reactive aldehydes, including 4‐HNE and MDA. Moreover, the increased ROS could produce reactive aldehydes, which in turn promote the production of ROS. And the accumulation of ROS in mitochondria can cause mitochondrial dysfunction manifested as significantly reduced MMP and decreased ATP production. Meanwhile, the ROS would be increased further when the mitochondrial function was impaired. The excessive ROS generated in the above process led to CaMKII being activated to P‐CaMKII and Ox‐CaMKII, which subsequently phosphorylated PYR2, then triggering calcium handling abnormalities, eventually aggravating cardiac malfunction.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Qinfeng Hu, Hang Chen performed experiments and processed the data. Qinfeng Hu wrote the manuscript. Cheng Shen, Beijian Zhang, Xinyu Weng, Xiaolei Sun, Jin Liu, and Zhen Dong contributed reagents/materials/analysis tools and provided critical suggestions. Kai Hu, Junbo Ge, and Aijun Sun designed the experimental and supervised the study. Aijun Sun and Kai Hu revised the manuscript. Aijun Sun administrated the project funding. All authors have read and agreed to the published version of the manuscript.
INSTITUTIONAL REVIEW BOARD STATEMENT
Animal experiments were conducted following the regulations of the US National Institutes of Health's Guide for the Care and Use of Laboratory Animals (NIH publication no. 85‐23, updated 1996) and were approved by the Animal Ethics Committee of Zhongshan Hospital, Fudan University, China.
Supporting information
Fig S1
Table S1
Table S2
Table S3
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for the help of Wenlong Yang, Chaofan Liu, Lihong Pan, and Xiurui Ma in technical support. We thank Xiang Wang and Haixia Xu for their support in detecting the intracellular Ca2+ transients. We would like to thank Lei Yin and Yang Zhang for their scientific ideas and discussions. We thank Yun Cai for the support in the isolation of adult mouse cardiomyocytes.
Hu, Q. , Chen, H. , Shen, C. , Zhang, B. , Weng, X. , Sun, X. , et al (2022) Impact and potential mechanism of effects of chronic moderate alcohol consumption on cardiac function in aldehyde dehydrogenase 2 gene heterozygous mice. Alcoholism: Clinical and Experimental Research, 46, 707–723. Available from: 10.1111/acer.14811
Qinfeng Hu and Hang Chen contributed equally to this work.
Funding information
This research was funded by grant 81725002 from the National Science Fund for Distinguished Young Scholars of China.
REFERENCES
- Ackers‐Johnson, M. , Li, P.Y. , Holmes, A.P. , O'Brien, S.M. , Pavlovic, D. & Foo, R.S. (2016) A simplified, langendorff‐free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circulation Research, 119, 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, E.J. , Katunga, L.A. & Willis, M.S. (2012) Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clinical and Experimental Pharmacology and Physiology, 39, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, S.M. & Cunningham, C.C. (2002) Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radical Biology and Medicine, 32, 11–16. [DOI] [PubMed] [Google Scholar]
- Beuckelmann, D.J. , Näbauer, M. & Erdmann, E. (1992) Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation, 85, 1046–1055. [DOI] [PubMed] [Google Scholar]
- Brien, S.E. , Ronksley, P.E. , Turner, B.J. , Mukamal, K.J. & Ghali, W.A. (2011) Effect of alcohol consumption on biological markers associated with risk of coronary heart disease: systematic review and meta‐analysis of interventional studies. BMJ, 342, d636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, P.J. , Enoch, M.A. , Goldman, D. , Li, T.K. & Yokoyama, A. (2009) The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Medicine, 6, e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, D.A. , Perry, J.B. , Allen, M.E. , Sabbah, H.N. , Stauffer, B.L. , Shaikh, S.R. et al. (2017) Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nature Reviews Cardiology, 14, 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev, D. & Wehrens, X.H. (2014) Role of RyR2 phosphorylation in heart failure and arrhythmias: controversies around ryanodine receptor phosphorylation in cardiac disease. Circulation Research, 114, 1311–1319; discussion 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorans, K.S. , Mostofsky, E. , Levitan, E.B. , Hakansson, N. , Wolk, A. & Mittleman, M.A. (2015) Alcohol and incident heart failure among middle‐aged and elderly men: cohort of Swedish men. Circulation: Heart Failure, 8, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, J.R. , He, B.J. , Grumbach, I.M. & Anderson, M.E. (2011) CaMKII in the cardiovascular system: sensing redox states. Physiological Reviews, 91, 889–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, F. , Cao, Q. , Wang, C. , Ma, X. , Shen, C. , Liu, X.W. et al. (2014) Impact of chronic low to moderate alcohol consumption on blood lipid and heart energy profile in acetaldehyde dehydrogenase 2‐deficient mice. Acta Pharmacologica Sinica, 35, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Solà, J. , Fatjó, F. , Sacanella, E. , Estruch, R. , Bosch, X. , Urbano‐Márquez, A. et al. (2006) Evidence of apoptosis in alcoholic cardiomyopathy. Human Pathology, 37, 1100–1110. [DOI] [PubMed] [Google Scholar]
- Gardner, J.D. & Mouton, A.J. (2015) Alcohol effects on cardiac function. Comprehensive Physiology, 5, 791–802. [DOI] [PubMed] [Google Scholar]
- Gaziano, J.M. , Buring, J.E. , Breslow, J.L. , Goldhaber, S.Z. , Rosner, B. , Vandenburgh, M. et al. (1993) Moderate alcohol intake, increased levels of high‐density lipoprotein and its subfractions, and decreased risk of myocardial infarction. New England Journal of Medicine, 329, 1829–1834. [DOI] [PubMed] [Google Scholar]
- George, A. & Figueredo, V.M. (2011) Alcoholic cardiomyopathy: a review. Journal of Cardiac Failure, 17, 844–849. [DOI] [PubMed] [Google Scholar]
- Ginsberg, G. , Smolenski, S. , Hattis, D. & Sonawane, B. (2002) Population distribution of aldehyde dehydrogenase‐2 genetic polymorphism: implications for risk assessment. Regulatory Toxicology and Pharmacology, 36, 297–309. [DOI] [PubMed] [Google Scholar]
- Gross, E.R. , Zambelli, V.O. , Small, B.A. , Ferreira, J.C. , Chen, C.H. & Mochly‐Rosen, D. (2015) A personalized medicine approach for Asian Americans with the aldehyde dehydrogenase 2*2 variant. Annual Review of Pharmacology and Toxicology, 55, 107–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnóczky, G. , Buzas, C.J. , Pacher, P. , Hoek, J.B. & Rubin, E. (2005) Alcohol and mitochondria in cardiac apoptosis: mechanisms and visualization. Alcoholism: Clinical and Experimental Research, 29, 693–701. [DOI] [PubMed] [Google Scholar]
- Higuchi, S. , Matsushita, S. , Muramatsu, T. , Murayama, M. & Hayashida, M. (1996) Alcohol and aldehyde dehydrogenase genotypes and drinking behavior in Japanese. Alcoholism: Clinical and Experimental Research, 20, 493–497. [DOI] [PubMed] [Google Scholar]
- Ho, H.T. , Belevych, A.E. , Liu, B. , Bonilla, I.M. , Radwański, P.B. , Kubasov, I.V. et al. (2016) Muscarinic stimulation facilitates sarcoplasmic reticulum Ca release by modulating ryanodine receptor 2 phosphorylation through protein kinase G and Ca/Calmodulin‐dependent protein kinase II. Hypertension, 68, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung, C.‐L. , Gonçalves, A. , Lai, Y.‐J. , Lai, Y.‐H. , Sung, K.‐T. , Lo, C.‐I. et al. (2016) Light to moderate habitual alcohol consumption is associated with subclinical ventricular and left atrial mechanical dysfunction in an asymptomatic population: dose‐response and propensity analysis. Journal of the American Society of Echocardiography, 29, 1043–1051.e4. [DOI] [PubMed] [Google Scholar]
- Jiang, H. , Jia, D. , Zhang, B. , Yang, W. , Dong, Z. , Sun, X. et al. (2020) Exercise improves cardiac function and glucose metabolism in mice with experimental myocardial infarction through inhibiting HDAC4 and upregulating GLUT1 expression. Basic Research in Cardiology, 115, 28. [DOI] [PubMed] [Google Scholar]
- Kato, Y. & Osawa, T. (2010) Detection of lipid‐lysine amide‐type adduct as a marker of PUFA oxidation and its applications. Archives of Biochemistry and Biophysics, 501, 182–187. [DOI] [PubMed] [Google Scholar]
- Kitagawa, K. , Kawamoto, T. , Kunugita, N. , Tsukiyama, T. , Okamoto, K. , Yoshida, A. et al. (2000) Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Letters, 476, 306–311. [DOI] [PubMed] [Google Scholar]
- Krenz, M. & Korthuis, R.J. (2012) Moderate ethanol ingestion and cardiovascular protection: from epidemiologic associations to cellular mechanisms. Journal of Molecular and Cellular Cardiology, 52, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryzhanovskii, S.A. , Kolik, L.G. , Tsorin, I.B. , Stolyaruk, V.N. , Vititnova, M.B. , Ionova, E.O. et al. (2017) Alcoholic cardiomyopathy: translation model. Bulletin of Experimental Biology and Medicine, 163, 627–631. [DOI] [PubMed] [Google Scholar]
- Kumar, A. , Lavoie, H.A. , Dipette, D.J. & Singh, U.S. (2013) Ethanol neurotoxicity in the developing cerebellum: underlying mechanisms and implications. Brain Sciences, 3, 941–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, C.H. & Korzick, D.H. (2014) Chronic alcohol consumption disrupts myocardial protein balance and function in aged, but not adult, female F344 rats. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology, 306, R23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. & Ren, J. (2006) Cardiac overexpression of metallothionein rescues chronic alcohol intake‐induced cardiomyocyte dysfunction: role of Akt, mammalian target of rapamycin and ribosomal p70s6 kinase. Alcohol and Alcoholism, 41, 585–592. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Li, J. , Tian, W. , Xu, T. & Zhang, Z. (2011) Chronic alcohol consumption induces cardiac remodeling in mice from Th1 or Th2 background. Experimental and Molecular Pathology, 91, 761–767. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Wang, S. , Guo, X. , Li, Y. , Ogurlu, R. , Lu, F. et al. (2021) Increased reactive oxygen species‐mediated Ca(2+)/Calmodulin‐dependent protein kinase II activation contributes to calcium handling abnormalities and impaired contraction in Barth syndrome. Circulation, 143, 1894–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luczak, E.D. , Wu, Y. , Granger, J.M. , Joiner, M.A. , Wilson, N.R. , Gupta, A. et al. (2020) Mitochondrial CaMKII causes adverse metabolic reprogramming and dilated cardiomyopathy. Nature Communications, 11, 4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luczak, S.E. , Liang, T. & Wall, T.L. (2017) Age of drinking initiation as a risk factor for alcohol use disorder symptoms is moderated by ALDH2*2 and ethnicity. Alcoholism: Clinical and Experimental Research, 41, 1738–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maack, C. & O'Rourke, B. (2007) Excitation‐contraction coupling and mitochondrial energetics.. Basic Research in Cardiology, 102, 369–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaienko, R. , Bovo, E. & Zima, A.V. (2018) Redox dependent modifications of ryanodine receptor: basic mechanisms and implications in heart diseases. Frontiers in Physiology, 9, 1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oba, T. , Maeno, Y. , Nagao, M. , Sakuma, N. & Murayama, T. (2008) Cellular redox state protects acetaldehyde‐induced alteration in cardiomyocyte function by modifying Ca2+ release from sarcoplasmic reticulum. American Journal of Physiology. Heart and Circulatory Physiology, 294, H121–H133. [DOI] [PubMed] [Google Scholar]
- van Oort, R.J. , McCauley, M.D. , Dixit, S.S. , Pereira, L. , Yang, Y. , Respress, J.L. et al. (2010) Ryanodine receptor phosphorylation by calcium/calmodulin‐dependent protein kinase II promotes life‐threatening ventricular arrhythmias in mice with heart failure. Circulation, 122, 2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, G.S. , Chen, Y.C. , Wang, M.F. , Lai, C.L. & Yin, S.J. (2014) ALDH2*2 but not ADH1B*2 is a causative variant gene allele for Asian alcohol flushing after a low‐dose challenge: correlation of the pharmacokinetic and pharmacodynamic findings. Pharmacogenetics and Genomics, 24, 607–617. [DOI] [PubMed] [Google Scholar]
- Peng, G.S. , Yin, J.H. , Wang, M.F. , Lee, J.T. , Hsu, Y.D. & Yin, S.J. (2002) Alcohol sensitivity in Taiwanese men with different alcohol and aldehyde dehydrogenase genotypes. Journal of the Formosan Medical Association, 101, 769–774. [PubMed] [Google Scholar]
- Piano, M.R. (2002) Alcoholic cardiomyopathy: incidence, clinical characteristics, and pathophysiology. Chest, 121, 1638–1650. [DOI] [PubMed] [Google Scholar]
- Providência, R. (2006) Cardiovascular protection from alcoholic drinks: scientific basis of the French Paradox. Revista Portuguesa De Cardiologia, 25, 1043–1058. [PubMed] [Google Scholar]
- Ren, J. , Davidoff, A.J. & Brown, R.A. (1997) Acetaldehyde depresses shortening and intracellular Ca2+ transients in adult rat ventricular myocytes. Cellular and Molecular Biology, 43, 825–834. [PubMed] [Google Scholar]
- Respress, J.L. , van Oort, R.J. , Li, N. , Rolim, N. , Dixit, S.S. , Dealmeida, A. et al. (2012) Role of RyR2 phosphorylation at S2814 during heart failure progression. Circulation Research, 110, 1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads, D.M. , Umbach, A.L. , Subbaiah, C.C. & Siedow, J.N. (2006) Mitochondrial reactive oxygen species. Contribution to oxidative stress and interorganellar signaling. Plant Physiology, 141, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattock, M.J. , Ottolia, M. , Bers, D.M. , Blaustein, M.P. , Boguslavskyi, A. , Bossuyt, J. et al. (2015) Na+/Ca2+ exchange and Na+/K+‐ATPase in the heart. Journal of Physiology, 593, 1361–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, C. , Wang, C. , Han, S. , Wang, Z. , Dong, Z. , Zhao, X. et al. (2017) Aldehyde dehydrogenase 2 deficiency negates chronic low‐to‐moderate alcohol consumption‐induced cardioprotecion possibly via ROS‐dependent apoptosis and RIP1/RIP3/MLKL‐mediated necroptosis. Biochimica Et Biophysica Acta (BBA)‐Molecular Basis of Disease, 1863, 1912–1918. [DOI] [PubMed] [Google Scholar]
- Slimen, I.B. , Najar, T. , Ghram, A. , Dabbebi, H. , Ben Mrad, M. & Abdrabbah, M. (2014) Reactive oxygen species, heat stress and oxidative‐induced mitochondrial damage. A review. International Journal of Hyperthermia, 30, 513–523. [DOI] [PubMed] [Google Scholar]
- Suematsu, N. , Tsutsui, H. , Wen, J. , Kang, D. , Ikeuchi, M. , Ide, T. et al. (2003) Oxidative stress mediates tumor necrosis factor‐alpha‐induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation, 107, 1418–1423. [DOI] [PubMed] [Google Scholar]
- Sun, A. , Zou, Y. , Wang, P. , Xu, D. , Gong, H. , Wang, S. et al. (2014) Mitochondrial aldehyde dehydrogenase 2 plays protective roles in heart failure after myocardial infarction via suppression of the cytosolic JNK/p53 pathway in mice. Journal of the American Heart Association, 3, e000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan, P.D. , Purohit, A. , Hund, T.J. & Anderson, M.E. (2012) Calmodulin‐dependent protein kinase II: linking heart failure and arrhythmias. Circulation Research, 110, 1661–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev, D. , Györke, I. , Belevych, A.E. , Terentyeva, R. , Sridhar, A. , Nishijima, Y. et al. (2008) Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circulation Research, 103, 1466–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Ginnan, R.G. , Wang, Y.X. & Zheng, Y.M. (2021) Interactive roles of CaMKII/ryanodine receptor signaling and inflammation in lung diseases. Advances in Experimental Medicine and Biology, 1303, 305–317. [DOI] [PubMed] [Google Scholar]
- Wehrens, X.H. , Lehnart, S.E. , Reiken, S.R. & Marks, A.R. (2004) Ca2+/calmodulin‐dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circulation Research, 94, e61–70. [DOI] [PubMed] [Google Scholar]
- Wood, A.M. , Kaptoge, S. , Butterworth, A.S. , Willeit, P. , Warnakula, S. , Bolton, T. et al. (2018) Risk thresholds for alcohol consumption: combined analysis of individual‐participant data for 599 912 current drinkers in 83 prospective studies. Lancet (London, England), 391, 1513–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, X. , Ma, Y.T. , Yang, Y.N. , Fu, Z.Y. , Ma, X. , Huang, D. et al. (2012) Alcohol consumption and carotid atherosclerosis in China: the Cardiovascular Risk Survey. European Journal of Preventive Cardiology, 19, 314–321. [DOI] [PubMed] [Google Scholar]
- Yeung, H.M. , Kravtsov, G.M. , Ng, K.M. , Wong, T.M. & Fung, M.L. (2007) Chronic intermittent hypoxia alters Ca2+ handling in rat cardiomyocytes by augmented Na+/Ca2+ exchange and ryanodine receptor activities in ischemia‐reperfusion. American Journal of Physiology. Cell Physiology, 292, C2046–C2056. [DOI] [PubMed] [Google Scholar]
- Yoval‐Sánchez, B. & Rodríguez‐Zavala, J.S. (2012) Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chemical Research in Toxicology, 25, 722–729. [DOI] [PubMed] [Google Scholar]
- Zhang, P. (2017) CaMKII: the molecular villain that aggravates cardiovascular disease. Experimental and Therapeutic Medicine, 13, 815–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Klein, A.L. , Alberle, N.S. , Norby, F.L. , Ren, B.H. , Duan, J. et al. (2003) Cardiac‐specific overexpression of catalase rescues ventricular myocytes from ethanol‐induced cardiac contractile defect. Journal of Molecular and Cellular Cardiology, 35, 645–652. [DOI] [PubMed] [Google Scholar]
- Zhao, F. , Fu, L. , Yang, W. , Dong, Y. , Yang, J. , Sun, S. et al. (2016) Cardioprotective effects of baicalein on heart failure via modulation of Ca(2+) handling proteins in vivo and in vitro. Life Sciences, 145, 213–223. [DOI] [PubMed] [Google Scholar]
- Zhou, H.Z. , Karliner, J.S. & Gray, M.O. (2002) Moderate alcohol consumption induces sustained cardiac protection by activating PKC‐epsilon and Akt. American Journal of Physiology. Heart and Circulatory Physiology, 283, H165–H174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1
Table S2
Table S3
Supplementary Material