

Abstract
Objective
Proteasome‐associated autoinflammatory syndrome (PRAAS) is caused by mutations affecting components of the proteasome and activation of the type I interferon (IFN) pathway. This study was undertaken to investigate the pathogenic mechanisms of a newly recognized type of PRAAS caused by PSMD12 haploinsufficiency.
Methods
Whole‐exome sequencing was performed in members of a family with skin rash, congenital uveitis, and developmental delay. We performed functional studies to assess proteasome dysfunction and inflammatory signatures in patients, and single‐cell RNA sequencing to further explore the spectrum of immune cell activation.
Results
A novel truncated variant in PSMD12 (c.865C>T, p.Arg289*) was identified in 2 family members. The impairment of proteasome function was found in peripheral blood mononuclear cells (PBMCs), as well as in PSMD12‐knockdown HEK 293T cell lines. Moreover, we defined the inflammatory signatures in patient PBMCs and found elevated IFN signals, especially in monocytes, by single‐cell RNA sequencing.
Conclusion
These findings indicate that PSMD12 haploinsufficiency causes a set of inflammation signatures in addition to neurodevelopmental disorders. Our work expands the genotype and phenotype spectrum of PRAAS and suggests a bridge between the almost exclusively inflammatory phenotypes in the majority of PRAAS patients and the almost exclusively neurodevelopmental phenotypes in the previously reported Stankiewicz‐Isidor syndrome.
INTRODUCTION
Autoinflammatory diseases are characterized by spontaneous activation of inflammatory pathways primarily driven by innate immune cells (1). Understanding of autoinflammation has expanded drastically with rapid advances in recent decades.
Defects in the constitutive proteasome or immunoproteasome result in proteasome‐associated autoinflammatory syndrome (PRAAS), also known as CANDLE/NNS/JMP/JASL (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature/Nakajo‐Nishimura syndrome/joint contractures, muscle atrophy, microcytic anemia, and panniculitis‐induced lipodystrophy syndrome/Japanese autoinflammatory syndrome with lipodystrophy) (2). PRAAS is clinically characterized by recurrent fever, neutrophilic dermatosis, lipodystrophy, joint contractures, and failure to thrive (2). Several genes encoding immunoproteasome or proteasome 20S subunits, such as PSMB8, PBMB9, PSMB10, PSMB4, PSMA3, and the chaperones essential for proteasome assembly, such as POMP and PSMG2, have been identified as causative genes in PRAAS (2, 3, 4, 5, 6, 7). Biallelic mutations in the proteasome subunits can cause defective proteasome function by disrupting proteasome assembly and/or catalytic activity, ultimately leading to the accumulation of ubiquitin protein conjugates as a consequence of impaired capacity to remove damaged proteins, while deficiency of other proteasome‐associated genes, such as PSMC3 and PSMB1, may cause only neurodevelopmental disorders (8, 9).
Rpn5, a 52.9‐kd protein in the 19S regulatory subunit of the proteasome encoded by PSMD12, facilitates deubiquitination of captured substrates. Previous studies have demonstrated the association between loss‐of‐function variants in PSMD12 and a syndromic neurodevelopmental disorder named Stankiewicz‐Isidor syndrome (OMIM no. 604450). Its symptoms include brain abnormalities, dysmorphic features, ophthalmologic abnormalities, genital anomalies, and skeletal defects (10, 11, 12, 13), without typical autoinflammatory features. In the current study, we investigated a novel PSMD12 nonsense mutation found in 2 generations of family members who presented with skin rash and congenital uveitis in addition to developmental delay. Mechanistic studies revealed that individuals carrying the PSMD12 truncated variant exhibit a variety of inflammatory signatures similar to those found in patients with PRAAS.
PATIENTS AND METHODS
Ethical considerations
The study was approved by the Institutional Review Board of Women's Hospital, Zhejiang University, where the patients were evaluated. The patients and control subjects (or their legal guardians if minors) provided written informed consent.
Cell collection, culture, and stimulation
Peripheral blood mononuclear cells (PBMCs) were separated from peripheral blood using lymphocyte separation medium and SepMate tubes (StemCell Technologies). PBMCs and HEK 293T cells were cultured in RPMI 1640 or Dulbecco's modified Eagle's medium (Gibco) with 10% fetal bovine serum (ExCell Bio) and 10% penicillin/streptomycin (HyClone). Lipopolysaccharide (LPS) (1 μg/ml; Sigma) poly(I‐C) (50 μg/ml; Invitrogen), interferon‐β (IFNβ) (10 ng/ml; PeproTech), and baricitinib (0.5 μM; Selleck) were used to treat PBMCs for 6 hours (LPS and poly[I‐C]) or 4 hours (IFNβ and baricitinib).
Whole‐exome sequencing
Peripheral blood DNA was sequenced using a Roche SeqCap EZ MedExome Enrichment Kit and the Illumina HiSeq X Ten platform. A set of 150‐bp paired‐end reads was mapped to hg38 reference genome using BWA, version 0.7.17 and the bam files were sorted and indexed using Picard followed by calling variants with GATK, version 4.1.4.1.
RNA sequencing
Total RNA was extracted from PBMCs with an RNeasy Mini kit (Qiagen). Libraries were generated from 1 μg RNA with a NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB) followed by sequencing on Illumina HiSeq X Ten.
Single‐cell RNA sequencing
A total of 8,000–10,000 single cells for each sample were captured and barcoded with a 10X Genomics Chromium console. The barcoded complementary DNA (cDNA) was amplified and sequenced using Illumina Novaseq. Data were processed with CellRange, version 6.0.1 (10x; Genomics) and the Seurat R package, version 3.2.0. Hallmark gene set in MsigDB, version7.1 (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) was used for gene set enrichment analysis.
Real‐time polymerase chain reaction (PCR)
Total RNA was extracted with an RNeasy Mini kit, followed by cDNA amplification and quantitative PCR (qPCR) analysis with TB Gree Premix Ex Ta II (Takara). Relative messenger RNA expression was normalized to the expression of ACTB and analyzed by the ΔΔCt method.
Antibodies and expression plasmids
Antibodies used and their commercial sources were as follows: β‐actin, STAT2, phosphorylated STAT2, IFN regulatory factor 3 (IRF3), phosphorylated IRF3, STAT1, phosphorylated STAT1, myeloma differentiation–associated protein 5 (MDA5), retinoic acid–inducible gene 1 (RIG‐1), and K48 polyubiquitin (all from Cell Signaling Technology); PSMA7, PSMB4, PSMC5, PSMD1, PSMD2, PSMD3, PSMD12 polyclonal antibody, PSMD13, and PSME2 (all from ABclonal Technology); PSMD12 monoclonal antibody (Santa Cruz Biotechnology); PSMC6 and IFN‐induced protein with tetratricopeptide repeats 3 (Abcam); and Flag (Sigma).
Wild‐type human PSMD12 was cloned from healthy control cDNA and cloned into pXN (Dbf4‐dependent kinase tagged) vector constructed in‐house. Mutant plasmid was constructed by site‐directed mutagenesis.
Western blot analysis
Cells were resuspended for 15 minutes in ice‐cold cell lysis buffer (20 mM Tris HCl [pH 7.4], 150 mM NaCl, 0.5% Nonidet P40, 10% glycerol with protease and phosphatase inhibitors) (ThermoFisher) and centrifuged (13,000g at 4°C for 15 minutes). The supernatants were collected and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (PAGE). Immunoblotting with specific antibodies was performed.
Proteasome activity assay
Proteasome activity was measured with Proteasome‐Glo Chymotrypsin‐Like, Trypsin‐Like, and Caspase‐Like Cell‐Based Assays (Promega). Proteasome inhibition with 5 μM epoxomicin was performed for 2 hours at 37°C. All experiments were conducted on 384‐well plates with 4 technical replicates. Data were collected with a multimode plate reader (BioTek).
Construction of PSMD12 knockdown cell lines
Four different short hairpin RNA (shRNA) sequences (shPSMD12‐1 [CCGGCCTAGCTGTGAAGGATTACATCTCGAGATGTAATCCTTCACAGCTAGGTTTTTG], shPSMD12‐2 [CCGGCCGAATAAGTGGTGACAAGAACTCGAGTTCTTGTCACCACTTATTCGGTTTTTG], shPSMD12‐3 [CCGGCCTTCCTATCAAACTTCGATTCTCGAGAATCGAAGTTTGATAGGAAGGTTTTTG], and shPSMD12‐4 [CCGGGCCAAGTATTATACTCGGATACTCGAGTATCCGAGTATAATACTTGGCTTTTTG]) were constructed into pLKO.1 plasmid and transfected into wild‐type HEK 293T cells, followed by treatment with 1.5 μg/ml puromycin for 5–7 days. Clones were separated and verified by qPCR and Western blot analysis.
Native PAGE
Cells were lysed in TSDG buffer (10 mM Tris HCl, 10 mM NaCl, 1.1 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol, 2 mM ATP, 10% glycerol [pH 7.2]) by freeze‐thawing 15–20 times and suspended in loading buffer (5 mM Tris HCl, 5% glycerol, 0.01% bromphenol blue) followed by electrophoresis on NativePAGE 3–12% Bis‐Tris gels (ThermoFisher). The proteasome subunits were analyzed by immunoblotting with antibodies.
Enzyme‐linked immunosorbent assay (ELISA), cytometric bead array system, and IFN response gene score
Interleukin‐6 (IL‐6), tumor necrosis factor (TNF), and IL‐1β in the supernatants of unstimulated PBMCs were quantified by ELISA (R&D Systems). Serum samples were tested with a BDFlex set of human cytokines/chemokines, which includes IL‐8, IL‐1β, IFNγ‐inducible protein 10 (IP‐10), and IFNγ, with buffer kit and normalization beads (BD). The IFN response gene score was calculated as previously described (14).
Statistical analysis
Cell‐based experiments were performed in triplicate. Results were compared by Student's t‐test (unpaired and 2‐tailed). P values less than 0.05 were considered significant.
RESULTS
Patients with the heterozygous PSMD12 nonsense variant
The proband (patient 1) was an 8‐year‐old girl with urticarial skin rashes, severe intellectual disability, and developmental delay. Her father (patient 2) exhibited congenital uveitis and mild intellectual disability. Both subjects displayed mild facial dysmorphic features including frontal bossing, hypertelorism, and nasal bridge depression. Lymphocyte phenotyping revealed increased numbers of CD4+ naive T cells in patient 1 and CD8+ T cells in patient 2. (Supplementary Tables 1 and 2, on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42070). Genomic analysis of patients and their family members by whole‐exome sequencing revealed a novel heterozygous nonsense variant in PSMD12: c.865C>T, p.Arg289*, which was de novo in the father and inherited by his daughter (Figures 1A–C). The probability of loss‐of‐function intolerance of PSMD12 is 1.0, which strongly supports the notion that this loss‐of‐function nonsense variant is pathogenic.
Figure 1.
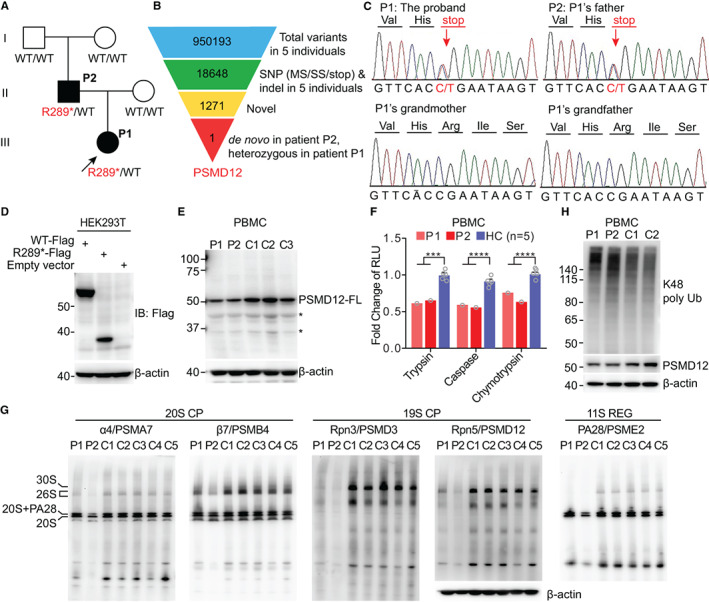
Identification of the PSMD12 variant and proteasome dysfunction in patients with the variant. A, Pedigree of the family carrying the heterozygous novel PSMD12 truncated variant p.R289*. Family members with the variant are patient 1 (P1) (proband [arrow]), an 8‐year‐old girl with urticarial skin rashes, severe intellectual disability, and developmental delay, and her father, patient 2 (P2), with congenital uveitis and mild intellectual disability. B, Schematic representation of the whole‐exome sequencing analysis and variant filtering approach used to identify the pathogenic variant in PSMD12. Single‐nucleotide polymorphism (SNP) includes missense (MS), splice site (SS), and stop codon variants and indel, frameshift, and non‐frameshift insertions and deletions. C, Confirmation of the nonsense variant by Sanger sequencing. D and E, Wild‐type (WT) and truncated PSMD12 expression in transfected HEK 293T cells (D) and in patient and control (C) peripheral blood mononuclear cells (PBMCs) (E). Experiments were performed at least 3 times for each sample. F, Significant differences in 3 types of proteasome proteolytic activity between patient and healthy control (HC) PBMCs. Symbols represent individual samples; for healthy controls, bars show the mean ± SEM. *** = P < 0.001; **** = P < 0.0001. G, Native gel analysis of the proteasome assembly in PBMCs from patients and controls. The assembly of different parts of the proteasome was illustrated using antibodies to the corresponding subunits. The loading amount of each sample is presented relative to the level of β‐actin determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. H, K48 ubiquitin (Ub) levels in PBMCs from the patients and 2 controls. FL = full‐length; RLU = relative luminescence units; CP = core particle; REG = regulatory particle.
To determine how the mutation affected protein expression, wild‐type and mutant PSMD12 were transfected into HEK 293T cells, and a truncated PSMD12 containing amino acid residues 1–288 was observed (Figure 1D). Additionally, the expression of PSMD12–full‐length was reduced and the truncated form was not detected in patient PBMCs (Figure 1E), suggesting the possibility of spontaneous degradation of the truncated protein in vivo. Therefore, the novel loss‐of‐function variant resulted in haploinsufficiency of PSMD12.
Proteasome defects associated with PSMD12 haploinsufficiency
Patient PBMCs exhibited significantly reduced proteolytic capacity (Figure 1F), confirming their impaired proteasome functions. Native gel electrophoresis was performed to analyze the assembly of proteasome complex in patient PBMCs. Immunoblotting with antibodies to 19S and 20S proteasome subunits showed markedly decreased levels of 19S and slightly decreased levels of 20S in PBMCs from patients compared to control PBMCs (Figure 1G).
PSMD12‐knockdown HEK 293T cell lines were constructed to evaluate the effect of PSMD12 haploinsufficiency in vitro (Supplementary Figures 1A and B, https://onlinelibrary.wiley.com/doi/10.1002/art.42070). The 3 types of proteasome activities in the knockdown cells were also decreased (Supplementary Figure 1C). Native gel electrophoresis with PSMD12‐knockdown cells demonstrated defective 26S proteasome assembly compared to cells treated with scrambled shRNA (Supplementary Figure 1D). Interactions of the truncated PSMD12 with a set of 19S subunits, including ATPase and non‐ATPase subunits, were all reduced (Supplementary Figure 1E). Impaired proteasome functions led to an accumulation of polyubiquitinated proteins. Both patients showed an accumulation of K48 ubiquitin–modified proteins compared to healthy controls (Figure 1H). Taken together, these data revealed profound disruptions of proteasome dysfunction associated with PSMD12 haploinsufficiency.
Elevated inflammatory signals in patient PBMCs
Proteasome defects are accompanied by ineffective removal of unfolded protein and ubiquitin–protein aggregates, which promotes a type I IFN response. Therefore, disruption of proteasome function can lead to enhanced production of type I IFN (2). To test whether PSMD12 haploinsufficiency is associated with dysregulated IFN production, PBMCs from the 2 patients and 4 control subjects were stimulated with poly(I‐C), and phosphorylation of downstream targets and expression of IFN‐stimulated genes (ISGs) were investigated. Patient 1 displayed enhanced phosphorylation of STAT1, STAT2, and IRF3 and increased expression of MDA5 and RIG‐1, indicating augmented activation of the IFN signaling pathway by poly(I‐C). Patient 2 exhibited enhanced phosphorylation of STAT2 and IRF3 (Figure 2A).
Figure 2.
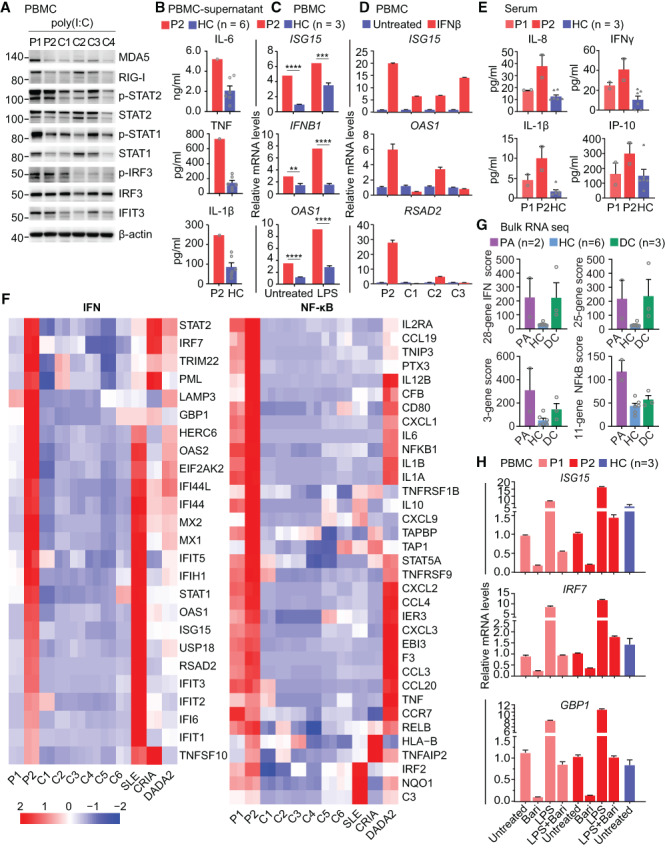
Elevated inflammatory signals in PBMCs from patients with the PSMD12 variant. A, Western blots of interferon (IFN) signaling pathway–involved proteins in poly(I‐C)–stimulated PBMCs from patients 1 and 2 and control subjects. Results shown are representative of 2 independent experiments. B, Levels of the cytokines interleukin‐6 (IL‐6), tumor necrosis factor (TNF), and IL‐1β in cultured supernatants of PBMCs from patient 2 and 6 control subjects, measured by enzyme‐linked immunosorbent assay. C and D, Expression levels of IFN‐stimulated genes in PBMCs from patient 2 and 3 control subjects. PBMCs were treated with lipopolysaccharide (LPS) (C) or IFNβ (D) or were left untreated. ** = P < 0.01; *** = P < 0.001; **** = P < 0.0001. E, Concentrations of the proinflammatory cytokines IL‐8, IFNγ, and IL‐1β and the chemokine IFNγ‐inducible protein 10 (IP‐10) in serum from patients 1 and 2 and from control subjects, determined by cytometric bead array analysis. Two samples from each subject, obtained at different times, were assessed. F, Expression patterns of genes involved in the IFN and NF‐κB signaling pathways in PBMCs from patients 1 and 2, healthy control subjects, and disease controls, determined by RNA sequencing. G, IFN response gene scores (28‐gene, 25‐gene, and 3‐gene) and NF‐κB score (11‐gene) in the 2 patients (PA), healthy controls, and disease controls (DC), determined using bulk RNA sequencing data. H, Expression levels of IFN pathway genes in LPS‐stimulated PBMCs from patients 1 and 2 and healthy controls, with and without baricitinib (Bari) treatment. In B–E, G, and H, symbols represent individual samples; for healthy controls (and for studies in which >1 sample from each of the 2 patients was assessed), bars show the mean ± SEM. MDA5 = myeloma differentiation–associated protein 5; RIG‐I = retinoic acid–inducible gene I; IRF3 = IFN regulatory factor 3; IFIT3 = IFN‐induced protein with tetratricopeptide repeats 3; SLE = systemic lupus erythematosus; CRIA = cleavage‐resistant receptor‐interacting protein kinase 1–induced autoinflammatory; DADA2 = deficiency of adenosine deaminase 2; (see Figure 1 for other definitions).
We next examined the production of proinflammatory cytokines in the supernatants of PBMCs from patient 2, by ELISA. Basal production of IL‐6, TNF, and IL‐1β was significantly higher in this patient compared to PBMCs from controls (Figure 2B). Correspondingly, transcription levels of ISGs, including ISG15, IFNB1, and OAS1, were elevated in PBMCs from patient 2, with or without LPS stimulation (Figure 2C). In addition, PBMCs from patient 2 were hyperresponsive to IFNβ stimulation, with increased levels of ISG expression (Figure 2D). Cytometric bead array analysis in serum from patients and controls showed that levels of proinflammatory cytokines such as IL‐8, IFNγ, and IL‐1β, as well as the chemokine IP‐10, were consistently increased in the 2 patients, especially in patient 2 (Figure 2E). The heterogeneity of inflammatory signatures between patient 1 and patient 2 was probably due to different disease activity and inflammatory conditions during sample collection.
Bulk RNA sequencing of PBMCs demonstrated that both patients had elevated basal expression of genes in the IFN signaling pathway (Figure 2F) compared to 6 healthy controls, and the top 20 genes that were differentially expressed between patient 2 and healthy controls included some ISGs (Supplementary Table 3, on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42070). These findings were further confirmed by the higher IFN response gene score in the 2 patients (Figure 2G). The IFN signature in patient 2 was as strong as that in a patient with active systemic lupus erythematosus and higher than that in patients with other autoinflammatory diseases (disease controls) such as CRIA (cleavage‐resistant receptor‐interacting protein kinase 1–induced autoinflammatory) (15) and DADA2 (deficiency of adenosine deaminase 2) (16) (Figure 2F). NF‐κB signaling was also elevated in both patients compared to healthy controls, and the signaling was higher than that observed in a CRIA patient during a febrile episode (Figure 2F). These results demonstrated systemic inflammation with activation of IFN and NF‐κB signaling in the setting of PSMD12 haploinsufficiency.
We further found that type I IFN signaling in PBMCs from the 2 patients could be suppressed by treatment with the JAK inhibitor baricitinib in vitro (Figure 2H). These data suggest that JAK inhibition may be a potential strategy to control the inflammatory response in patients with PSMD12 deficiency, in accordance with the utility of this class of medication for the treatment of PRAAS and other interferonopathies.
Single‐cell RNA sequencing was performed in both patients and in 2 age‐ and sex‐matched controls, and identified a total of 13 different cell clusters (Figure 3A). Patient 1 expressed increased levels of ISGs, such as STAT1, ISG15, and OAS1, in different cell types, especially in CD14+ classic monocytes and CD16+ nonclassic monocytes (Figure 3B). Genes that mediate proinflammatory responses were prominently expressed in patient monocytes, with markedly greater expression of genes in the type I IFN pathway compared to that in controls (Figure 3C and Supplementary Figure 1F [https://onlinelibrary.wiley.com/doi/10.1002/art.42070]).
Figure 3.
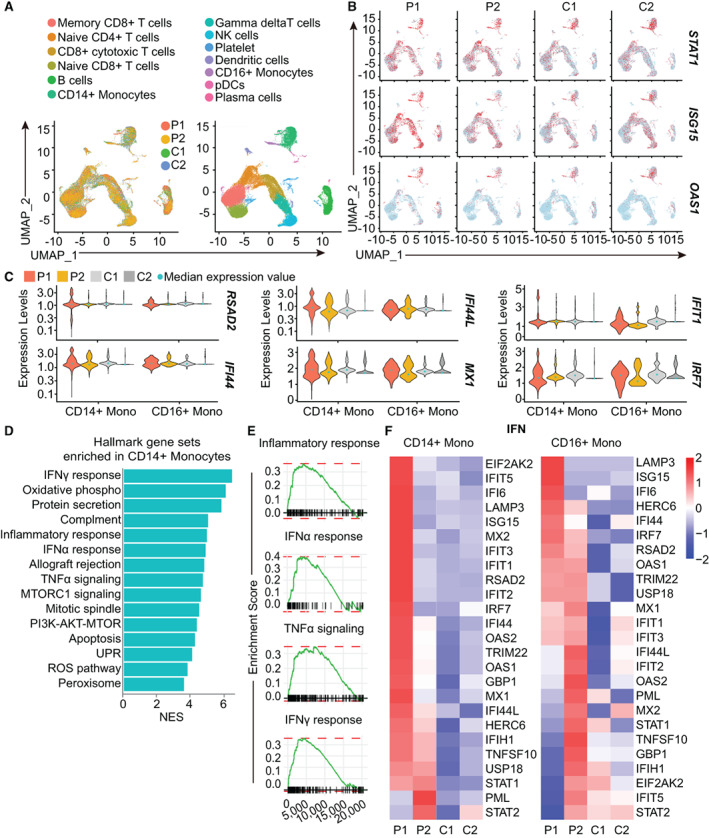
Enrichment of inflammatory pathways in monocytes from patients with the PSMD12 variant. A, Single‐cell RNA sequencing analysis of cell type distributions in PBMCs from the 2 patients and 2 healthy controls (left), and different cell clusters in the patients and controls (right). Control 1 was age‐ and sex‐matched to patient 1, and control 2 was age‐ and sex‐matched to patient 2. B, FeaturePlots of interferon (IFN)–related genes, created using single‐cell RNA sequencing data. Colored dots indicate single cells in the UMAP plot; red color indicates a higher level of gene expression. C, Violin plots showing the expression of IFN‐stimulated genes in CD14+ and CD16+ monocytes (Mono) from the 2 patients and 2 healthy control subjects. Light blue dots show the median expression level of all cells. D, Enriched hallmark gene set pathways of pathways in CD14+ monocytes analyzed by gene set enrichment analysis (GSEA). E, GSEA plot of inflammatory response, as well as IFNα, tumor necrosis factor (TNF), and IFNγ responses in CD14+ monocytes from the patients and controls. F, Comparison of type I IFN signaling pathways in CD14+ and CD16+ monocyte subsets from patients and controls, determined by single‐cell RNA sequencing. NK = natural killer; pDCs = plasmacytoid dendritic cells; mTORC1 = mechanistic target of rapamycin complex 1; PI3K = phosphatidylinositol 3‐kinase; UPR = unfolded protein response; ROS = reactive oxygen species; NES = normalized enrichment score (see Figure 1 for other definitions).
We next sought to identify gene expression patterns in monocytes and their relationship to the elevated inflammatory response in the patients. Of note, we observed a significant enrichment of inflammatory response and IFN response gene sets in CD14+ monocytes identified by single‐cell RNA sequencing using gene set enrichment analysis (Figures 3D and E). Additionally, CD14+ classic monocytes and CD16+ nonclassic monocytes from the patients exhibited elevated expression of genes in the type I IFN pathway (Figure 3F). These results suggest that monocytes may have a prominent role in propagating the inflammatory response in PSMD12 haploinsufficiency.
DISCUSSION
The proteasome is a protease complex and an important cellular machinery that regulates protein turnover; the integrity of each of its subunits is of great importance to the overall function of this sophisticated complex. Biallelic pathogenic mutations in genes encoding proteasome subunits can cause PRAAS due to excess type I IFN signaling. However, the mechanism by which disrupted proteasome function translates into type I IFN production remains unclear. The potential contributions of oxidative stress and endoplasmic reticulum stress to IFN production have been considered as possible explanations (3).
Unlike most other forms of PRAAS that require biallelic mutations, haploinsufficiency of PSMD12 is sufficient to cause type I IFN up‐regulation and autoinflammation. Previous studies have focused on the association of PSMD12 variants, including microdeletions and point mutations, with neurodevelopmental disorders, and autoinflammation has frequently not been addressed in such neurology‐based investigations. This is the first study to identify strong IFN and NF‐κB signaling, as found in typical autoinflammatory diseases and systemic lupus erythematosus, in patients with PSMD12, establishing a correlation between PSMD12 haploinsufficiency and PRAAS. Importantly, our results demonstrate that defective proteasome subunits, in addition to PSMB4, PSMB8, PSMB9, PSMB10, PSMA3, PSMG2, and POMP (17), are linked to interferonopathy. With the identification of additional patients with PSMD12 loss‐of‐function mutations in the future, more systemic autoinflammation phenotypes may be reported. Our results also implicate the likelihood of latent or subclinical autoinflammation in patients with Stankiewicz‐Isidor syndrome and those with predominantly neurodevelopmental disorders who have proteasome defects due to deficiency of PSMB1 and PSMC3. It is possible that such inflammation may be a pathogenic factor in these neurodevelopmental disorders.
In addition, reduced penetrance in patients with the PSMD12 mutation needs to be taken into consideration, and may explain the variable disease expressivity and severity among individuals with mutations in the same gene. Reduced penetrance has been reported in some type I interferonopathies, including Aicardi Goutières syndrome (due to dominantly inherited mutations in IFIH1) and STING‐associated vasculitis with onset in infancy (caused by gain‐of‐function mutations in TMEM173) (18, 19, 20).
This study had some limitations. Skin biopsy specimens or histologic images from the patients were unavailable as the skin features appeared very early in the clinical course and were not assessed histologically. We were also unable to acquire sufficient blood samples from the patients during disease flares, for more comprehensive exploration of disease mechanisms. With the small number of patients, it was also difficult to explore the heterogeneity of inflammatory features among patients. Therefore, further studies are needed to investigate the relationship of proteasome dysfunction and elevated type I IFN signatures.
In conclusion, we have identified a novel PSMD12 truncating variant that impairs proteasome subunit assembly and reduces proteasome catalytic activity. We have described autoinflammatory features of PSMD12 haploinsufficiency and defined, for the first time, the inflammatory signals at the single‐cell level. Taken together, our findings provide further insights into the biology of proteasomes and the pathologic basis of PRAAS.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Zhou and Dong had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Yan, Zhang, Tao, Zhou.
Acquisition of data
Yan, Zhang, Zhou, Dong.
Analysis and interpretation of data
Yan, Zhang, Lee, Tao, J. Wang, S. Wang, Zhou.
Supporting information
Disclosure Form
Figure S1 Defect of proteasome assembly and function in PSMD12 haploinsufficiency.
A, Expression levels of endogenous PSMD12 in the NC and two PSMD12 knock‐down HEK293T cell lines. NC, scrambling shRNA treated wild‐type HEK293T cells.
B, qPCR analysis on PSMD12 expression in the NC and two PSMD12 knock down HEK293T cell lines.
C, Significant difference of three types of proteasome proteolytic activity between NC and two PSMD12 knock down HEK293T cell lines. **P < 0.01, ****P < 0.0001.
D, Native gel analysis of the proteasome assembly in the NC and two PSMD12 knock down HEK293T cells. The assembly of different parts of the proteasome was illustrated using antibodies of the subunits correspondingly. CP, core particle; RP, regulatory particle.
E, The interactions between PSMD12 (wild‐type or truncated) and 19S proteasome subunits in wild‐type HEK293T cells. Cells were transfected with flag‐tagged wild‐type or Arg289* PSMD12 plasmid before lysed and immunoprecipitated with flag antibody.
F, Violin plots showing the expression of ISGs in patients and two controls in different cell types.
Supplementary Table 1 Clinical manifestations of patients.
Supplementary Table 2. Immunophenotypes of patients.
Supplementary Table 3. Top 20 differentially expressed genes in bulk RNAseq between patient P2 and healthy controls.
Appendix S1: Supporting information
ACKNOWLEDGMENTS
We thank the patients and control subjects for their support during the study.
Mr. Yan's work was supported by the Zhejiang Provincial Natural Science Foundation of China (grant LQ19H040016). Dr. Zhou's work was supported by the National Key Research and Development Program of China (grant 2018YFC1004903), the National Natural Science Foundation of China (grants 31771548 and 81971528), and the Zhejiang Provincial Natural Science Foundation of China (grant LR19H100001). Dr. Dong's work was supported by the Key Research and Development Program of Zhejiang Province (grant 2019C03025).
Mr. Yan and Ms. Zhang contributed equally to this work. Drs. Zhou and Dong contributed equally to this work.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42070&file=art42070‐sup‐0001‐Disclosureform.pdf.
Contributor Information
Qing Zhou, Email: zhouq2@zju.edu.cn.
Minyue Dong, Email: dongmy@zju.edu.cn.
REFERENCES
- 1. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 2017;18:832–42. [DOI] [PubMed] [Google Scholar]
- 2. Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss‐of‐function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 2015;125:4196–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poli MC, Ebstein F, Nicholas SK, de Guzman MM, Forbes LR, Chinn IK, et al. Heterozygous truncating variants in POMP escape nonsense‐mediated decay and cause a unique immune dysregulatory syndrome. Am J Hum Genet 2018;102:1126–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Jesus AA, Brehm A, vanTries R , Pillet P, Parentelli AS, Sanchez GA, et al. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J Allergy Clin Immunol 2019;143:1939–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarrabay G, Mechin D, Salhi A, Boursier G, Rittore C, Crow Y, et al. PSMB10, the last immunoproteasome gene missing for PRAAS. J Allergy Clin Immunol 2020;145:1015–7. [DOI] [PubMed] [Google Scholar]
- 6. Çetin G, Klafack S, Studencka‐Turski M, Kruger E, Ebstein F. The ubiquitin‐proteasome system in immune cells. Biomolecules 2021;11:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kanazawa N, Hemmi H, Kinjo N, Ohnishi H, Hamazaki J, Mishima H, et al. Heterozygous missense variant of the proteasome subunit β‐type 9 causes neonatal‐onset autoinflammation and immunodeficiency. Nat Commun 2021;12:6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kroll‐Hermi A, Ebstein F, Stoetzel C, Geoffroy V, Schaefer E, Scheidecker S, et al. Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol Med 2020;12:e11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ansar M, Ebstein F, Ozkoc H, Paracha SA, Iwaszkiewicz J, Gesemann M, et al. Biallelic variants in PSMB1 encoding the proteasome subunit β6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum Mol Genet 2020;29:1132–43. [DOI] [PubMed] [Google Scholar]
- 10. Kury S, Besnard T, Ebstein F, Khan TN, Gambin T, Douglas J, et al. De novo disruption of the proteasome regulatory subunit PSMD12 causes a syndromic neurodevelopmental disorder. Am J Hum Genet 2017;100:689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Naud ME, Tosca L, Martinovic J, Saada J, Metay C, Drevillon L, et al. Prenatal diagnosis of a 2.5 Mb de novo 17q24.1q24.2 deletion encompassing KPNA2 and PSMD12 genes in a fetus with craniofacial dysmorphism, equinovarus feet, and syndactyly. Case Rep Genet 2017;2017:7803136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khalil R, Kenny C, Hill RS, Mochida GH, Nasir R, Partlow JN, et al. PSMD12 haploinsufficiency in a neurodevelopmental disorder with autistic features. Am J Med Genet B Neuropsychiatr Genet 2018;177:736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palumbo P, Palumbo O, Di Muro E, Leone MP, Castellana S, Biagini T, et al. Expanding the clinical and molecular spectrum of PSMD12‐related neurodevelopmental syndrome: an additional patient and review. Arch Clin Med Case Rep 2019;03. [Google Scholar]
- 14. De Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest 2020;130:1669–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, et al. A dominant autoinflammatory disease caused by non‐cleavable variants of RIPK1. Nature 2020;577:109–14. [DOI] [PubMed] [Google Scholar]
- 16. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early‐onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 2014;370:911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rice GI, Duany YD, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, et al. Gain‐of‐function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 2014;46:503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng S, Lee PY, Wang J, Wang S, Huang Q, Huang Y, et al. Interstitial lung disease and psoriasis in a child with Aicardi‐Goutieres syndrome. Front Immunol 2020;11:985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING‐activating mutation underlies a familial inflammatory syndrome with lupus‐like manifestations. J Clin Invest 2014;124:5516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Figure S1 Defect of proteasome assembly and function in PSMD12 haploinsufficiency.
A, Expression levels of endogenous PSMD12 in the NC and two PSMD12 knock‐down HEK293T cell lines. NC, scrambling shRNA treated wild‐type HEK293T cells.
B, qPCR analysis on PSMD12 expression in the NC and two PSMD12 knock down HEK293T cell lines.
C, Significant difference of three types of proteasome proteolytic activity between NC and two PSMD12 knock down HEK293T cell lines. **P < 0.01, ****P < 0.0001.
D, Native gel analysis of the proteasome assembly in the NC and two PSMD12 knock down HEK293T cells. The assembly of different parts of the proteasome was illustrated using antibodies of the subunits correspondingly. CP, core particle; RP, regulatory particle.
E, The interactions between PSMD12 (wild‐type or truncated) and 19S proteasome subunits in wild‐type HEK293T cells. Cells were transfected with flag‐tagged wild‐type or Arg289* PSMD12 plasmid before lysed and immunoprecipitated with flag antibody.
F, Violin plots showing the expression of ISGs in patients and two controls in different cell types.
Supplementary Table 1 Clinical manifestations of patients.
Supplementary Table 2. Immunophenotypes of patients.
Supplementary Table 3. Top 20 differentially expressed genes in bulk RNAseq between patient P2 and healthy controls.
Appendix S1: Supporting information