

Abstract
Optically active cyclopropanes have been widely investigated especially from the views of pharmaceutical and agrochemical industries, and substituting one of the methylenes with the difluoromethylene unit should be promising for developing novel biologically relevant compounds and functional materials. In this paper, the copper‐catalyzed enantioselective hydrosilylation of gem‐difluorocyclopropenes to provide the corresponding chiral gem‐difluorocyclopropanes is presented. The use of copper(I) chloride, chiral ligands including bidentate BINAPs and monodentate phosphoramidites, and silylborane Me2PhSi‐Bpin accompanying sodium tert‐butoxide in methanol was appropriate for the enantioselective hydrosilylation of the strained C=C double bond, and the resultant chiral difluorinated three‐membered ring was unambiguously characterized. Subsequent activation of the silyl groups in enantio‐enriched gem‐difluorocyclopropanes showed substantial reduction of the enantiopurity, indicating cleavage of the distal C−C bond leading to the transient acyclic intermediates.
Keywords: asymmetric synthesis, copper catalysis, cyclopropanes, fluorine, racemization
The copper‐catalyzed enantioselective hydrosilylation of gem‐difluorocyclopropenes providing the chiral gem‐difluorocyclopropanes is presented. The use of copper(I) chloride, chiral ligands including bidentate BINAPs and monodentate phosphoramidites, and silylborane Me2PhSi‐Bpin accompanying NaOtBu in methanol was appropriate for the enantioselective hydrosilylation of the strained C=C double bond. Subsequent activation of the silyl groups in enantio‐enriched gem‐difluorocyclopropanes showed substantial reduction of the enantiopurity, indicating cleavage of the distal C−C bond.

Introduction
Cyclopropanes containing the strained rings show various particular physical properties based on the skeletal bent bonding,[ 1 , 2 ] and have been widely utilized for developing drug candidates based on the structural aspects. [3] In these pharmaceutical and agrochemical studies, the stereochemistry of cyclopropanes is undoubtedly important as indicated by the historical literatures about pyrethrins [4] and 1‐aminocyclopropane carboxylic acid. [5] Additionally, in taking the physicochemical and biological characteristics of fluorine‐containing molecular counterparts promoting bioisosteres via metabolic stability and lipophilicity into account, use of fluorine should be indispensable for further studies on optically active cyclopropanes relating to pharmaceutical and agrochemical research fields.[ 6 , 7 ]
The gem‐difluorocyclopropane skeleton is one of the well‐studied fluorinated cyclopropanes. [8] Whereas a considerable number of gem‐difluorocyclopropane derivatives have been produced so far, examples of the enantio‐controlled synthesis providing optically active gem‐difluorocyclopropanes have been limited. Taguchi et al. reported use of chiral auxiliaries in the [2+1] processes involving Michael addition of lithium enolates. [9] Itoh and co‐workers developed the alternative approach to chiral gem‐difluorocyclopropanes by using kinetic resolution of the racemic mixtures based on lipase‐catalyzed ester hydrolysis. [10] Also, de Meijere utilized enzymatic reactions for synthesis of dispiro compounds containing gem‐difluorocyclopropane. [11] These reported studies employed quantitative amount of the chiral reagent and the racemic mixtures of basically epimerization‐prohibited molecular structures, and thus developing novel and efficient synthetic tools providing optically active gem‐difluorocyclopropanes is highly desirable.
Producing chiral gem‐difluorocyclopropanes via enantioselective catalytic processes should contribute to develop the chemistry of fluorinated cyclopropanes. As indicated by the intensive studies on synthesis of chiral cyclopropanes, [12] enantio‐controlled saturation process of the C=C double bond in cyclopropene is a promising and efficient approach to optically active gem‐difluorocyclopropanes. Scheme 1a denotes a recently reported enantioselective synthesis of chiral gem‐difluorocyclopropanes via hydrocupration of homochiral gem‐difluorocyclopropenes followed by treatment with (deuterio)alcohols. [13] Use of polymethylhydrosiloxane (PMHS) as a hydrido source was successful, and the regio‐controlled hydrocupration furnished the corresponding chiral gem‐difluorocyclopropanes as formal reduction products of the cyclopropenes in moderate to good enantiomeric excesses (up to 90 % ee). The regioselectivity of hydrocupration step depends on the steric encumbrance of chiral ligands, and use of sterically encumbered ligands such as DTBM‐SEGPHOS provided two regioisomers in the hydrocupration process. Shortly thereafter, asymmetric catalytic hydrogen transfer process utilizing a Noyori–Ikariya catalyst was developed, and enantiomerically pure gem‐difluorocyclopropanes were produced from gem‐difluorocyclopropenyl esters (Scheme 1b). [14] These catalytic enantioselective reductions of gem‐difluorocyclopropenes are promising to progress the chemistry of fluorinated cyclopropanes based on subsequent conversions of the molecular structures. In fact, the carbonyl functional groups derived from gem‐difluorocyclopropenyl esters can be utilized for chemical conversions such as combining with N‐heterocyclic units. Thus, it should be fruitful to develop synthetic methods for chiral gem‐difluorocyclopropanes enabling additional functionalization processes.
Scheme 1.

Reported catalytic enantioselective reduction of gem‐difluorocyclopropenes affording optically active gem‐difluorocyclopropanes.
In Scheme 1a, exchange of PMHS with other nucleophilic reagents is an attractive strategy for developing synthesis of chiral gem‐difluorocyclopropanes. As mentioned in the recent paper, [13] pinacolborane (HBpin), bis(pinacolato)diboron (pinB‐Bpin), and dimethylzinc (Me2Zn) can be employed. Whereas HBpin gave the hydrocupration product, the diboron and organozinc reagents could generate the corresponding boronated and alkylated gem‐difluorocyclopropane derivatives. Because the boron and zinc reagents required use of sterically encumbered chiral ligands promoting the thermodynamically unfavorable regioselectivity, the synthetic efficiency was not comparable with the case using PMHS. However, these preliminary findings prompted to explore convenient processes for functionalized gem‐difluorocyclopropanes based on the copper‐catalyzed asymmetric process.
In this paper, we examined the copper‐catalyzed asymmetric reactions using a silylborane reagent Me2PhSi‐Bpin as a nucleophile providing silyl‐substituted chiral gem‐difluorocyclopropanes. [15] Hoveyda first confirmed usefulness of silylboranes for enantioselective silylcupration of cyclic conjugated carbonyl compounds. [16] Recently, Oestreich demonstrated enantioselective catalytic synthesis of chiral cyclopropanes via the stereoselective silylcupration process of prochiral cyclopropenes, [17] which encouraged to apply for the enantioselective catalytic hydrosilylation of gem‐difluorocyclopropenes. Our attempt was successful, and the optically active 1,1‐difluoro‐2‐silylcyclopropanes were obtained. The chiral 1,1‐difluorocyclopropane structure was confirmed absolutely, and preliminary conversions of the silyl substituent were attempted. Interestingly, the desilylation process accompanied deviation of the enantiomeric purity, indicating nature of the fluorinated cyclopropane skeleton.[ 8 , 18 , 19 , 20 , 21 , 22 ]
Results and Discussion
Optimization of reaction conditions
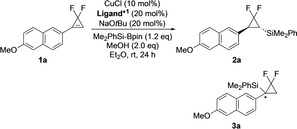
As a starting material, we chose a methoxynaphthyl‐substituted gem‐difluorocyclopropene (1 a) based on the previously reported copper‐mediated process. [13] Taking the enantioselective copper‐catalyzed conversions for cyclopropenes into account, [16] we employed a silylborane Me2PhSi‐Bpin [Bpin=4,4,5,5‐tetramethyl‐1,3,2‐dioxaborolyl (pinacolboryl)] as a nucleophile and utilized combination of copper(I) chloride and a chiral bidentate diphosphine ligand accompanying sodium tert‐butoxide as a chiral catalyst component. In addition, methanol was employed for protonation of the resultant silylcupration intermediate. Table S1 shows screening of solvent with (R)‐SEGPHOS, suggesting that Et2O is the most appropriate solvent for the enantioselective hydrosilylation of 1 a. Also, Table S1 indicates that NHC ligands, promoting the excellent results in the hydrosilylation of conjugated carbonyl compounds, [15] would not give desirable scores. Room temperatures are desirable for the asymmetric hydrosilylation of 1 a (Table S2).
Table 1 summarizes screening of chiral bidentate ligands. Structures of the ligands Ligand*2 are shown in Figure 1. The initially employed (R)‐SEGPHOS gave the corresponding 1,1‐difluoro‐2‐silylcyclopropane 2 a as a single diastereomer almost quantitatively with a moderate enantioselectivity (Entry 1). Compound 3 a was not observed, indicating that the silylcupration process was almost regioselective under the thermodynamic control promoting the predominant cupration at the arylmethyl position. [13] The 1H NMR spectroscopic data of 2 a characterized the trans configuration. (R)‐DM‐SEGPHOS gave a comparable result with (R)‐SEGPHOS (Entry 2). However, the excess steric encumbrance was not appropriate, and in fact (R)‐DTBM‐SEGPHOS gave a low yield together with a considerable amount of the inseparable kinetic product 3 a (Entry 3). The minor regioisomer 3 a indicated that the excess steric hindrance of the ligand avoided the cupration at the arylmethyl position. [13] Use of (R)‐BINAP slightly improved the enantioselectivity although the yield was low (Entry 4). To our delight, (R)‐Tol‐BINAP was effective to promote a desirable enantioselectivity and a good yield (Entry 5). On the other hand, (R)‐DM‐BINAP was of poor enantioselectivity and regioselectivity (Entry 6), and use of (R)‐Cy‐BINAP resulted in a low yield (Entry 7). (S)‐H8‐BINAP, (R)‐MeO‐BIPHEP, and (S)‐SYNPHOS gave moderate to good yields and enantioselectivity (Entries 8–10). Use of the (S)‐configured ligands gave the enantiomer of 2 a (ent‐2 a). As indicated by Entry 3, (R)‐DTBM‐MeO‐BIPHEP gave a low yield and accompanied a considerable amount of 3 a (Entry 11). Accordingly, we concluded that Tol‐BINAP is the most appropriate bidentate ligand for the copper‐catalyzed enantioselective hydrosilylation of 1 a.
Table 1.
Screening of chiral bidentate ligands.
|
| ||||
|---|---|---|---|---|
|
Entry |
Ligand*2 |
2 a [%][a] |
ee of 2 a [%] |
3 a [%][a] |
|
1 |
(R)‐SEGPHOS |
99 |
73 |
0 |
|
2 |
(R)‐DM‐SEGPHOS |
94 |
75 |
0 |
|
3 |
(R)‐DTBM‐SEGPHOS |
21 |
n.d. |
23 |
|
4 |
(R)‐BINAP |
18[b] |
83 |
0[b] |
|
5 |
(R)‐Tol‐BINAP |
87 |
92 |
0 |
|
6 |
(R)‐DM‐BINAP |
72 |
16 |
17 |
|
7 |
(R)‐Cy‐BINAP |
17[c] |
29 |
trace[c] |
|
8 |
(S)‐H8‐BINAP |
88 |
90[d] |
trace |
|
9 |
(R)‐MeO‐BIPHEP |
99 |
84 |
0 |
|
10 |
(S)‐SYNPHOS |
99 |
65 |
trace |
|
11 |
(R)‐DTBM‐ MeO‐BIPHEP |
23[e] |
n.d. |
23[e] |

[a] Yields were determined by 19F NMR using BTF as an internal standard. [b] 74 % Recovery of 1 a. [c] 60 % Recovery of 1 a. [d] Enantiomer of 2 a (ent‐2 a) was characterized by HPLC analysis. [e] 51 % Recovery of 1 a.
Figure 1.

Structures of bidentate ligands (Ligand*2 ).
The structure of 2 a was confirmed by X‐ray crystallography (Figure 2). [23] Accordingly, the (1R,3S) absolute configuration was confirmed. It is noteworthy that the distal bond (C1−C3) is elongated, and correspondingly the C1−C2−C3 angle is larger than the other skeletal angles. This structural aspect would correlate with the fluorine effects promoting the biradical structure via bond cleavage of the distal bond.[ 8 , 18 , 19 , 20 , 21 , 22 ]
Figure 2.

Molecular structure of 2 a (50 % probability level). Selected bond lengths (Å) and angles (°): C1−C2 1.473(8), C2−C3 1.473(8), C1−C3 1.600(6), C1−F1 1,367(7), C1−F2 1.351(7), C1−Si1 1.897(5), C3−Caryl 1.489(7), C1−C2−C3 65.7(4), C2−C1−C3 57.3(3), C1−C3−C2 57.0(3), F1−C2−F2 106.7(4). CCDC‐2145088.
Next, we examined screenings of chiral monodentate ligands. All the screenings of 25 monodentate ligands are tabulated in Tables S3–S6. Tables S3–S5 summarize the conditions using 12 mol % of ligands. Table 2 shows the selected results using 20 mol % of monodentate phosphorus ligands. In addition, Table S6 shows all results using 20 mol % monodentate ligands. Structures of monodentate ligands Ligand*1 are listed in Figure 3. The binaphthyl‐based phosphoramidite ligands L‐1/2/3/4 showed moderate to good yields but the lower enantioselectivity compared with the bidentate ligands (Entries 1–4). Installing substituents at the 3,3’‐positions was not effective (Table S4). Ligands L‐5 and L‐6 bearing the octahydrobinaphthyl skeleton did not improve the scores (Entry 5,6). On the other hand, the yields and enantioselectivity were excellent when using L‐7/8/9 (Entries 7–9), and especially L‐8 could be decided as the most effective monodentate ligand for the enantioselective hydrosilylation of 1 a. It should be noted that the absolute configuration of 2 a produced in the presence of L‐1 to L‐9 was identical to the product of the R‐configured bidentate ligands such as (R)‐Tol‐BINAP.
Table 2.
Screening of chiral monodentate ligands.
|
| ||||
|---|---|---|---|---|
|
Entry |
Ligand*1 |
2 a [%][a] |
ee of 2 a [%] |
3 a [%][a] |
|
1 |
L‐1 |
92 |
44 |
trace |
|
2 |
L‐2 |
74[b] |
43 |
trace[b] |
|
3 |
L‐3 |
76[c] |
46 |
5[c] |
|
4 |
L‐4 |
93 |
33 |
7 |
|
5 |
L‐5 |
37[d] |
28 |
1[d] |
|
6 |
L‐6 |
82 |
17 |
4 |
|
7 |
L‐7 |
78 |
88 |
trace |
|
8 |
L‐8 |
90 |
96 |
1 |
|
9 |
L‐9 |
89 |
88 |
trace |
[a] Yields were determined by 19F NMR using BTF as an internal standard. [b] 10 % recovery of 1 a. [c] 11 % recovery of 1 a. [d] 55 % recovery of 1 a.
Figure 3.

Structures of monodentate ligands (Ligand*1 ). Cy=cyclohexyl.
Substrate scope
As described above, we succeeded in optimizing both bidentate and monodentate chiral ligands for the copper‐mediated hydrosilylation of 1 a. Next, we worked about scope and limitation of the optimized conditions for synthesis of chiral gem‐difluorocyclopropanes (Scheme 2). The naphthyl‐substituted substates 1 a–c gave the corresponding product 2 a‐c in good yields and enantioselectivity. Whereas both Condition A with (R)‐Tol‐BINAP and Condition B with L‐8 could be employed for the p‐chlorinated and p‐methoxycarbonylated substrates 1 e and 1 f, 1 d and 1 g permitted only use of Condition B or Condition A, respectively. Both Tol‐BINAP and L‐8 gave poor results in the reactions using nitro‐substituted substrate 1 h and p‐methoxyphenyl derivative 1 i. In the synthesis of 2 h, inseparable syn isomer was accompanied, indicating that the electron‐withdrawing group might stabilize the cupration intermediate extremely and/or promote the complicated electron‐transfer process. Whereas 1 i could not provide the corresponding product, the m‐ and o‐methoxy substrates 1 j and 1 k were tolerant to give 2 j and 2 k, respectively. The p‐methoxy substrate 1 i was reluctant to interact with the nucleophilic silylcopper species, which was in sharp contrast with the achiral synthesis using Xantphos. [15] Although the reason has not been clarified yet, the electron‐rich nature of the C=C unit and/or the steric properties might suppress the silylation step from the silylcopper species bearing the chiral phosphorus ligands. It should be noted that 1 i showed low yield even in the reaction with chiral copper hydride reagent. [13] The quinoline‐substituted derivative 2 l could be synthesized by employing Condition A, indicating that the electronic character of L‐8 might not be suitable to facilitate the interaction between the silylcopper species and the cyclopropane 1 l. The findings about synthesis of 2 d, 2 g, and 2 l suggest that the chiral bidentate and monodentate ligands can be utilized complementary according to the gem‐difluorocyclopropene substrates.
Scheme 2.

Substrate scope. The yields were determined by 19F NMR using BTF as an internal standard.
Possible reaction mechanisms
Based on the published literatures,[ 17 , 24 , 25 ] it would be possible to propose a catalytic procedure shown in Scheme 3. The silylcopper species [L*2Cu‐SiPhMe2] generated from copper(I) alkoxide [L*2Cu‐OMe, initially L*2Cu‐OtBu], could coordinate on the C=C double bond in 1 enantioselectively, and the production via Re approach was predominant in using the (R)‐configured chiral bidentate ligands as well as monodentate ligands L1‐9. Subsequent stereoselective syn addition of the Si−Cu bond provided Int‐2 followed by protonation furnishing 2 and spontaneous regeneration of the catalytic species.
Scheme 3.

A proposed reaction mechanism.
It is quite hard to clarify the difference between the bidentate and monodentate ligands in the catalytic process, which induces the remarkable substrate dependence. It is plausible that the catalytic process includes the complicated equilibrium including the reaction intermediates. Attempts to simulate the complicated equilibrium experimentally and theoretically are in progress.
Desilylation accompanying reduction of enantiopurity
The silyl‐substituted chiral gem‐difluorocyclopropanes are attractive because of possible subsequent transformations. In this study, we examined desilylation of 2 as a preliminary attempt toward synthesis of functional chiral gem‐difluorocyclopropanes from 2. Scheme 4 displays the desilylation of 2 a, 2 b, and 2 l by TBAF. As one of the comparable reports suggested, [26] desilylation with fluoride ion proceeds via retention of configuration. However, all the examined substrates provided the corresponding gem‐difluorocyclopropane 4 a, 4 b, and 4 l in moderate yields but accompanied decrease of enantiomeric purity. Silyl‐substituted gem‐difluorocyclopropanes 2 a and 2 b showed reduction of ee of 5–12 %. On the other hand, 2 l bearing the electron‐deficient quinoline substituent provided the corresponding desilylation product 4 l with the considerable decrease of ee from 2 l (Δee=43 %).
Scheme 4.

Desilylation of the selected silyl‐substituted gem‐difluorocyclopropanes with decrease of enantiopurity.
The substantial drops of ee should relate with the physical properties of the gem‐difluorocyclopropane skeleton bearing the aryl substituents. Attempted DFT calculations for the difluorocyclopropyl anions [25] generated from 2 a, 2 b, and 2 l indicated that the distal bond of difluorocyclopropyl anion leading to 4 l is elongated (Table S7), which might correlate with the decrease of ee in Scheme 4. Although intensive study should be requisite, it could be predicted that the electron‐withdrawing substituents in gem‐difluorocyclopropane might promote elongation of the distal bond probably because of partial delocalization of the negative charge up to the alfa position facilitating the transient biradical state. As indicated by the X‐ray structure of 2 a in Figure 2, the distal bond can be homolytically cleaved leading to the corresponding biradical species,[ 8 , 18 , 19 , 20 , 21 , 22 ] which would relate with the reduction of enantiopurity. Effects of the methoxy group are complicated, but further investigations will clarify the particular substituent effect.
Conclusion
We have presented the copper‐mediated enantioselective and regioselective hydrosilylation of gem‐difluorocyclopropenes affording the corresponding optically active silyl‐substituted difluorocyclopropanes. Both bidentate (Tol‐BINAP) and monodentate (TADDOL‐type) ligands can be utilized complementary. Structural elucidate of the silyl‐substituted difluorocyclopropanes was completed, and the reaction mechanisms were discussed accordingly. As a characteristic finding on gem‐difluorocyclopropanes, we observed notable decrease of enantiomeric purity in the fluoride‐mediated desilylation procedure, indicating notable characters of the negatively charged gem‐difluorocyclopropane unit. Although intensive studies should be requisite for understanding the unexpected physical properties of the silylated gem‐difluorocyclopropanes, the findings will be fruitful to synthesize functional chiral gem‐difluorocyclopropanes for developing such as pharmaceutical and agrochemical sciences.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported in part by Grants‐in‐Aid for Scientific Research (No. 19H02685) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Financial supports were from Nissan Chemical Corporation.
K. Sekine, D. Akaishi, K. Konagaya, S. Ito, Chem. Eur. J. 2022, 28, e202200657.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Wiberg K. B., Angew. Chem. Int. Ed. 1986, 25, 312–322; [Google Scholar]; Angew. Chem. 1986, 98, 312–322. [Google Scholar]
- 2.O. G. Kulinkovich, Cyclopropanes in Organic Synthesis, Wiley, Hoboken, 2015.
- 3. Talele T. T., J. Med. Chem. 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Staudinger H., Ruzicka L., Helv. Chim. Acta 1924, 7, 177–201; [Google Scholar]
- 4b. Aratani T., Pure Appl. Chem. 1985, 57, 1839–1844. [Google Scholar]
- 5. Yang S. F., Hoffman N. E., Annu. Rev. Plant Physiol. 1984, 35, 155–189. [Google Scholar]
- 6.
- 6a.I. Ojima Eds., Fluorine in Medicinal Chemistry and Chemical Biology Wiley, Chichester, 2009;
- 6b.P. Kirsch, Modern Fluoroorganic Chemistry Wiley, Weinheim, 2013;
- 6c.V. P. Reddy, Organofluorine Compounds in Biology and Medicine Elsevier, Amsterdam, 2015.
- 7.
- 7a. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]
- 7b. Hagmann W. K., J. Med. Chem. 2008, 51, 4359–4369; [DOI] [PubMed] [Google Scholar]
- 7c. Xing L., Blakemore D. C., Narayanan A., Unwalla R., Lovering F., Denny R. A., Zhou H., Bunnage M. E., ChemMedChem 2015, 10, 715–726. [DOI] [PubMed] [Google Scholar]
- 8. Adekenova K. S., Wyatt P. B., Adekenov S. M., Beilstein J. Org. Chem. 2021, 17, 245–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Taguchi T., Sasaki H., Shibuya A., Morikawa T., Tetrahedron Lett. 1994, 35, 913–916; [Google Scholar]
- 9b. Taguchi T., Shibuya A., Sasaki H., Endo J.-I., Morikawa T., Shiro M., Tetrahedron: Asymmetry 1994, 5, 1423–1426; [Google Scholar]
- 9c. Shibuya A., Kurishita M., Ago C., Taguchi T., Tetrahedron 1996, 52, 271–278. [Google Scholar]
- 10.
- 10a. Mitsukura K., Korekiyo S., Itoh T., Tetrahedron Lett. 1999, 40, 5739–5742; [Google Scholar]
- 10b. Itoh T., Ishida N., Mitsukura K., Hayase S., Ohashi K., J. Fluorine Chem. 2004, 125, 775–783; [Google Scholar]
- 10c. Itoh T., Kanbara M., Nakajima S., Sakuta Y., Hayase S., Kawatsura M., Kato T., Miyazawa K., Uno H., J. Fluorine Chem. 2009, 130, 1157–1163. [Google Scholar]
- 11. Miyazawa K., Yufit D. S., Howard J. A. K., de Meijere A., Eur. J. Org. Chem. 2000, 4109–4117. [Google Scholar]
- 12.
- 12a. Dian L., Marek I., Chem. Rev. 2018, 118, 8415–8434; [DOI] [PubMed] [Google Scholar]
- 12b. Müller D. S., Marek I., Chem. Soc. Rev. 2016, 45, 4552–4566; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Vicente R., Synthesis 2016, 48, 2343–2360; [Google Scholar]
- 12d. Edwards A., Rubina M., Rubin M., Curr. Org. Chem. 2016, 20, 1862–1877; [Google Scholar]
- 12e. Raiguru B. P., Nayak S., Mishra D. R., Das T., Mohapatra S., Mishra N. P., Asian J. Org. Chem. 2020, 9, 1088–1132; [Google Scholar]
- 12f. Zhu Z.-B., Wei Y., Shi M., Chem. Soc. Rev. 2011, 40, 5534–5563; [DOI] [PubMed] [Google Scholar]
- 12g. Marek I., Simaan S., Masarwa A., Angew. Chem. Int. Ed. 2007, 46, 7364–7376; 2008, 47, 1982; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7508–7520; 2008, 120, 2008; [Google Scholar]
- 12h. Rubin M., Rubina M., Gevorgyan V., Chem. Rev. 2007, 107, 3117–3179; [DOI] [PubMed] [Google Scholar]
- 12i. Rubin M., Rubina M., Gevorgyan V., Synthesis 2006, 1221–1245; [Google Scholar]
- 12j. Wasa M., Engle K. M., Lin D. W., Yoo E. J., Yu J.-Q., J. Am. Chem. Soc. 2011, 133, 19598–19601; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12k. Kubota A., Sanford M. S., Synthesis 2011, 2579–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sekine K., Ushiyama A., Endo Y., Mikami K., J. Org. Chem. 2020, 85, 7916–7924. [DOI] [PubMed] [Google Scholar]
- 14. Yamani K., Pierre H., Archambeau A., Meyer C., Cossy J., Angew. Chem. Int. Ed. 2020, 59, 18505–18509; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18663–18667. [Google Scholar]
- 15.Achiral synthesis using Xantphos: Zhao X., Xu S., He J., Zhou Y., Cao S., Org. Chem. Front. 2019, 6, 2539–2543. [Google Scholar]
- 16.
- 16a. Lee K.-s., Hoveyda A. H., J. Am. Chem. Soc. 2010, 132, 2898–2900; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Lee K.-s., Wu H., Haeffner F., Hoveyda A. H., Organometallics 2012, 31, 7823–7826; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c. Pace V., Rae J. P., Harb H. Y., Procter D. J., Chem. Commun. 2013, 49, 5150–5152; [DOI] [PubMed] [Google Scholar]
- 16d. Pace V., Rae J. P., Procter D. J., Org. Lett. 2014, 16, 476–479. [DOI] [PubMed] [Google Scholar]
- 17. Zhang L., Oestreich M., Chem. Eur. J. 2019, 25, 14304–14307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferreno J. C., De Staricco E. A. R., Staricco E. H., J. Phys. Chem. 1975, 79, 1242–1246. [Google Scholar]
- 19. Jefford C. W., Mareda J., Gehret J.-C. E., Kabengele T., Graham W. D., Burger U., J. Am. Chem. Soc. 1976, 98, 2585–2593. [Google Scholar]
- 20. W. R. Dolbier Jr. , Acc. Chem. Res. 1981, 14, 195–200. [Google Scholar]
- 21. Getty S. J., Hrovat D. A., Xu J. D., Barker S. A., Borden W. T., J. Chem. Soc. Faraday Trans. 1994, 90, 1689–1701. [Google Scholar]
- 22. Tian F., Lewis S. B., Bartberger M. D., W. D. Dolbier Jr. , Borden W. T., J. Am. Chem. Soc. 1998, 120, 6187–6188. [Google Scholar]
- 23.Deposition Number 2145088 (for 2 a) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 24. Oestreich M., Hartmann E., Mewald M., Chem. Rev. 2013, 113, 402–441. [DOI] [PubMed] [Google Scholar]
- 25. Sawamura M., Ito H., Carbon-Boron and Carbon-Silicon Bond Formation, in Copper-Catalyzed Asymmetric Synthesis (Eds.: Alexakis A., Krause N., Woodward S.), Wiley, Weinheim, 2014, pp. 157–177. [Google Scholar]
- 26. Chan T. H., Lau P. W. K., Li M., Tetrahedron Lett. 1976, 2667–2670. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.