

Abstract
Structural variants (SVs) represent a major source of aberration in tumour genomes. Given the diversity in the size and type of SVs present in tumours, the accurate detection and interpretation of SVs in tumours is challenging. New classes of complex structural events in tumours are discovered frequently, and the definitions of the genomic consequences of complex events are constantly being refined. Detailed analyses of short‐read whole‐genome sequencing (WGS) data from large tumour cohorts facilitate the interrogation of SVs at orders of magnitude greater scale and depth. However, the inherent technical limitations of short‐read WGS prevent us from accurately detecting and investigating the impact of all the SVs present in tumours. The expanded use of long‐read WGS will be critical for improving the accuracy of SV detection, and in fully resolving complex SV events, both of which are crucial for determining the impact of SVs on tumour progression and clinical outcome. Despite the present limitations, we demonstrate that SVs play an important role in tumourigenesis. In particular, SVs contribute significantly to late‐stage tumour development and to intratumoural heterogeneity. The evolutionary trajectories of SVs represent a window into the clonal dynamics in tumours, a comprehensive understanding of which will be vital for influencing patient outcomes in the future. Recent findings have highlighted many clinical applications of SVs in cancer, from early detection to biomarkers for treatment response and prognosis. As the methods to detect and interpret SVs improve, elucidating the full breadth of the complex SV landscape and determining how these events modulate tumour evolution will improve our understanding of cancer biology and our ability to capitalise on the utility of SVs in the clinical management of cancer patients. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: structural variants, whole genome sequencing, tumour evolution, chromothripsis, extrachromosomal DNA, patient stratification
Introduction
During their evolution from normal cells to invasive tumours, cells acquire somatic mutations throughout their entire genome. Structural variants (SVs) represent an underappreciated class of genomic aberration that may amplify, delete, and rearrange genomic regions either focally as a simple change to a single genomic segment, or catastrophically, combining multiple SVs, encompassing large genomic regions and involving multiple chromosomes. Tumours harbour a diverse range of structural events along this spectrum of increasing size and complexity. While copy number alteration (CNA) and aneuploidy have been well studied in tumours [1, 2], the impacts that more complex classes of SV have on tumour development and progression are less well understood. However, the increased availability of whole‐genome sequencing (WGS) data from tumours generated by large‐scale studies such as The Cancer Genome Atlas (TCGA) [3], the Pan‐Cancer Analysis Group of the International Cancer Genome Consortium (PCAWG‐ICGC) [4, 5, 6] and the Hartwig Medical Foundation (HMF) [7], combined with advances in the methods required to interrogate SVs, allow us to examine SVs in tumours at increased depth and scale. Nevertheless, our understanding of the critical role that SVs have in tumourigenesis has been slower to emerge as accurate and comprehensive detection of SVs is challenging due to the diversity in types, sizes, and complexity of SVs in tumours. Some SVs can drive tumourigenesis, while others shed light on the processes of mutation and repair acting during tumour development [8]. Evolutionary histories of cancer genomes reveal the major role of SVs in tumour evolution across many cancer types and the potential of SVs as biomarkers to inform the treatment of cancer patients at various stages of their disease is emerging. In this review, we discuss recent challenges and developments in detecting SVs, present the important roles of SVs in cancer progression and in generating intratumoural heterogeneity, and highlight the emerging clinical applications of SVs as biomarkers in cancer.
The structural complexity of tumour genomes
SVs are frequently found in the normal human genome and contribute greatly to individual human variation [9, 10, 11]. What differentiates tumour genomes from normal genomes are the unusual clusters, types, and sizes of SVs, and in highly rearranged tumours, the increased frequency and complexity of SVs relative to that observed in normal tissues [5]. SVs are formed by the improper repair of double‐stranded DNA breaks [12, 13, 14]. These DNA breaks are triggered by various mechanisms such as exogenous mutagens (DNA damaging chemicals, viral infections, ionising radiation, and ultraviolet light) and cell‐intrinsic mutational processes (oxidative metabolism, telomere dysfunction, and replication stress); in‐depth reviews of the mechanisms contributing to SV formation are detailed elsewhere [13, 14, 15, 16]. SVs can reconfigure DNA sequences across scales from a single genomic locus to multiple entire chromosomes. Simple SVs are single‐segment alterations that include insertions, deletions, duplications, inversions, and translocations (Figure 1A). Complex SVs constitute multiple simple SVs whose co‐occurrence in a genome, within a spatial and/or temporal window, represent the expected genomic consequences of complex events. These consequences, in the form of clusters of SVs, are used to predict the past occurrence of a diverse set of complex events including chromothripsis, chromoplexy, chromoanasynthesis, breakage‐fusion‐bridge (BFB) cycles, extrachromosomal circular DNA (ecDNA), aneuploidy, and whole‐genome duplication (WGD) (Figure 1B). Briefly, chromothripsis is defined by extensive genomic rearrangement localised to a single or sometimes a few chromosomes. The hallmarks of chromothripsis include oscillations between two or three copy number states, random reassembly of fragmented DNA segments, and loss of heterozygosity [17, 18]. Chromoplexy contains interdependent translocations and deletions of multiple chromosomal segments organised in a closed chain [19]. Chromoanasynthesis involves serial microhomology‐based breakage‐induced replication or fork stalling and template switching mechanisms, resulting in localised duplications and triplications and short stretches of microhomologies at breakpoint junctions [20]. BFB cycles occur as a result of telomere loss, dicentric chromosome formation, and mitotic spindle stress [21, 22]. ecDNAs are large, typically more than 1 Mb in length, acentric circular DNAs that are independent from chromosomes and often contain oncogenes and regulatory regions [23, 24]. ecDNAs have previously been described as ‘double minutes’ due to their appearance as paired structures [25]. These ecDNA segments can be reincorporated into the chromosome as large sections of tandem duplications known as homogeneously staining regions (HSRs) [26]. Aneuploidy is the presence of an abnormal number of chromosomes [27], whereas WGD results from the doubling of a complete set of diploid chromosomes [28].
Figure 1.
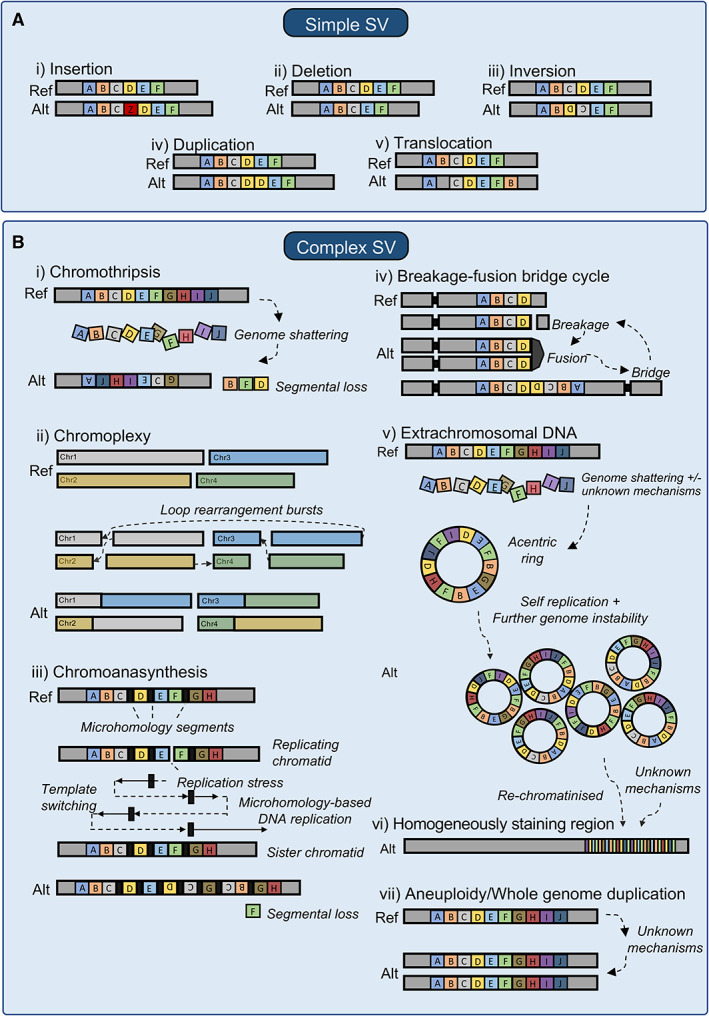
The diverse structural variant (SV) landscape in tumours. (A) Simple SVs include (i) insertions where segments of DNA are added, (ii) deletions where DNA segments are lost, (iii) inversions where DNA segments are in the opposite orientation, (iv) duplications where extra copies of DNA segments are present, and (v) translocations where a section of DNA is joined onto another section at a different genomic location. (B) Complex SVs include: (i) chromothripsis where the genome is shattered and the pieces joined back together at random, (ii) chromoplexy where interdependent translocations and deletions are joined together in a chain, (iii) chromoanasynthesis, which is a pattern of localised amplification and short stretches of breakpoint microhomology, (iv) breakage‐fusion‐bridge cycles, where telomeres are lost, dicentric chromosomes form before breaking due to mitotic stress, (v) extrachromosomal DNAs (ecDNAs), where large segments of DNA form acentric circles that often contain oncogenes and regulatory regions; these ecDNAs may be reincorporated into the chromosome, as (vi) homogeneously staining regions, and (vii) aneuploidy and whole‐genome duplication where some or all chromosomes have additional or double the number of copies, respectively.
Despite the wide variety in the complex classes of SVs that have been defined, recent large‐scale studies have identified a large fraction of clustered SVs, which are not thought to be the consequence of a particular known complex event as defined in Figure 1B [5, 29]. These unclassified SV clusters represent a gap in our understanding of the highly rearranged tumour genome. It is unclear what proportion of these clusters represent as‐yet uncharacterised classes of complex events or whether they remain unclassified due to significant flexibility in the current genomic definitions of known complex events. For example, arbitrary thresholds are often applied in defining the genomic consequences of complex events such as chromothripsis [18], and complex SVs that do not meet the arbitrary thresholds will not be classified as such. With the increasing availability of long‐read WGS data and improvements in methods for SV detection and analysis, we expect that more classes of complex events will be uncovered and that definitions of the genomic consequences of complex events will be refined. A more recent study [30], using a new algorithm to classify SVs, identified several novel classes of complex SV, the mechanistic origins of which have yet to be explored [31]. These include rigma (large deletions at fragile sites), pyrgo (superenhancer associated duplicated regions) and tyfonas (amplified areas of fold‐back inversions). Further, tracing the sequential formation of complex SVs reveals more complex patterns of rearrangements, such as the recently reported ‘seismic amplifications’ [32]: amplifications which are thought to occur as a result of circular recombination of chromothripsis‐generated ecDNAs. These wave‐like patterns of amplification can be integrated into the chromosome as HSRs or may persist as ecDNAs [33]. Although complex SVs represent a variety of intriguing genomic alterations, this heterogeneous class of mutation is still to be comprehensively characterised, as reflected in the dynamic and often overlapping definitions of these variants. The ability to generate accurate observations of the structural complexity of tumour genomes will benefit the comprehensive analysis of large tumour cohorts and experimental studies of the mechanisms underlying complex events.
New horizons in SV detection
Substantial diversity in the type, size, and complexity of SVs in tumours poses a challenge for accurate detection. Historically, SVs were detected by microscopic karyotyping, fluorescence in situ hybridisation (FISH), and microarray technologies; providing limited SV breakpoint resolution and obscuring complex events. In the last two decades, short‐read WGS has revolutionised SV detection, providing both higher accuracy and resolution; and much of our current knowledge on the diversity of SVs in cancer is derived from short‐read WGS data. In‐depth reviews on short‐read WGS‐based SV detection are discussed elsewhere [8, 34, 35, 36]. Briefly, nucleic acids extracted from tumour tissues and a germline control (usually a blood sample) are PCR (polymerase chain reaction)‐amplified, sequenced as paired‐end reads of 100–300 base pairs in length, and computationally aligned to the reference genome. SV detectionalgorithms (callers) are then used to identify the SVs unique to the tumour sample. Many SV callers have been developed, and their relative merits have been reviewed in detail [8, 37, 38]. SV callers leverage distinct alignment signatures to identify SVs by type and size [39]. Changes in read depth are used together with allelic imbalances to infer CNAs [40, 41, 42]. Clusters of discordantly aligned read pairs (reads that map at abnormal distance or orientation) together with split reads (reads that are partially mapped) can provide precise SV breakpoint locations [43, 44, 45, 46, 47]. SVs can also be detected using a local assembly approach where aberrant reads are reassembled into contigs, before pairwise comparison to the reference genome [46, 47, 48]. Newer SV callers such as GRIDDS‐PURPLE‐LINX [49] and JaBbA [30] incorporate both SV and CNA information to reduce false‐positives and detect the presence of complex SVs. Specialised SV callers have also been developed, tailored to detect specific complex SVs, including ShatterSeek [18] (for chromothripsis) and Amplicon Architect [50] (for ecDNAs).
Irrespective of the strategies used to call SVs, no single short‐read WGS‐based SV caller can identify the complete range of SV types reliably, due to the structural complexity of tumour genomes and the technical limitations of short‐read lengths [51]. Despite our best efforts at optimising SV detection, a large swathe (~15%) of the genome remains inaccessible to short‐read WGS [52]. These genomic regions frequently harbour highly repetitive sequences or extreme GC content [10, 53]. PCR amplification introduces coverage bias across regions with extreme GC content [54]; this bias leads to the artificial reduction of supporting reads that can affect methods used to detect CNAs [40, 41, 42]. In addition, short‐read lengths are not able to span large regions of repetitive sequence in the genome, which are particularly prone to SV formation [14, 52]. Repetitive sequence causes increased alignment ambiguity particularly for short sequencing reads, reducing variant calling accuracy. Similarly, short‐read WGS cannot span most complex SVs in their entirety, leading to the incomplete classification of patterns of single SVs into complex SVs. Due to the read length limitation, short‐read WGS also cannot adequately phase complex SVs into their constituent haplotypes, thereby affecting the ability of short‐read WGS in resolving complex events at allelic resolutions [55]. These challenges to comprehensively detecting SVs, and mapping complex SVs from short‐read WGS suggest that we will have missed many SVs present in tumour genomes and may have underestimated the importance of SVs in tumour progression.
To overcome the limitations of short‐read WGS in fully resolving the structural complexity of tumour genomes, several approaches incorporating longer‐range information have been developed [56, 57, 58]. These include barcoding collections of short reads from the same genomic region to improve assembly accuracy [59, 60, 61, 62, 63, 64, 65, 66, 67, 68], and using fluorescently labelled sequence motifs to image very long contiguous stretches of DNA, termed optical mapping [69]. The former is still subject to the limitations of short‐read lengths and the latter, while useful in identifying complex SVs such as ecDNAs [50, 70, 71], is unable to identify precise breakpoint locations at high resolution. Alternatively, long‐read sequencing approaches, such as those developed by Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) [58] can generate individual reads that are kilobases to megabases in length directly from the native DNA, often spanning SVs in their entirety. In‐depth reviews of the technologies underlying ONT and PacBio long‐read sequencing platforms are detailed elsewhere [56, 57, 58]. Both ONT and PacBio reads can readily traverse the most repetitive regions of the human genome, and lack GC bias, allowing for more comprehensive identification of SVs, including those that span repeat‐rich centromeric and telomeric regions [72, 73, 74, 75, 76, 77, 78]. Most of the SVs discovered using ONT and PacBio reads are novel, lending weight to the notion that a large number of SVs are not detected by short‐read approaches [52, 79, 80, 81]. In addition, longer read lengths allow haplotype phasing and de novo assembly of complex SVs, crucial for the accurate interpretation of the functional impact of these events [82, 83, 84, 85]. Taking advantage of the unique properties of both ONT and PacBio long‐read sequencing, and the development of new whole‐genome assembly methods [78, 86], the Telomere‐to‐Telomere Consortium has recently assembled a complete human genome for the first time [72, 87, 88]. In the future, we expect that this approach will be applied at scale to accurately resolve large complex SV events. With further refinement in long‐read sequencing technologies, including improvements in cost and throughput, and the development of more accurate SV detection algorithms [78, 89, 90], it is likely that long reads will be used more broadly in the future. Large scale initiatives involving the use of long‐read sequencing in cohorts with detailed clinical follow up data, such as Cancer 2.0 by Genomics England [91], represent a valuable opportunity to further our understanding of the full diversity and complexity of SVs and better evaluate their impact on tumour development and progression.
Structural variation as a critical dimension in tumour evolution
Cancers evolve from single cells into genetically distinct and heterogeneous subpopulations of cells. Cells that acquire mutations that confer selective advantages undergo clonal expansion [92, 93]. The dynamics of somatic evolution are thus determined by the rate of mutation and the likelihood that a mutation confers a selective advantage [94]. High rates of mutation provide more opportunity for clonal expansion, while conversely increasing the likelihood of a clone acquiring a deleterious variant. Intratumour heterogeneity, during selective sweeps and in periods of neutral evolution where no cell subpopulation has a fitness advantage over another, offers a reservoir of mutations upon which selection could act in the presence of selective pressures; for example, as a result of therapy [92], driving malignant progression, and therapeutic resistance. Driver mutations can occur early as tumour‐initiating events or present late, contributing to tumour progression and metastasis. In a given tumour sample, these driver mutations might have propagated clonally and be present in all cells or might be subclonal, existing only in a subset.
The importance of SVs, in particular for the continued development of the tumour after the last clonal expansion, may be underappreciated due to the substantial technical challenges in identifying subclonal SVs [91]. Recent large‐scale studies suggest that SVs play a significant role in driving tumourigenesis, with more than 50% of cancers harbouring at least one clonal SV driver [91]. Additionally, only 11% of subclones carry a single nucleotide variant (SNV) or small indel driver, suggesting that late tumour development may be disproportionately driven by subclonal large CNAs or SVs [95]. Our ability to detect subclonal SVs is dependent on sequencing depth and quality, tumour cellularity, and background ploidy. The low allelic frequencies of subclonal SVs can present a substantial challenge for both accurate detection and probabilistic assignment of breakpoints to subclones using bulk sequencing methods, where our ability to detect SVs is impaired by sequencing a pooled sample from a heterogeneous population [96]. Single‐cell WGS has greatly helped to accurately determine the clonality of SVs [97, 98, 99, 100]. Regardless, challenges in establishing clonality will inevitably result in a substantial underestimation of the proportion of SVs that are subclonal. Nonetheless, based on current estimates, when SVs occur during tumour evolution shows striking differences across tumour types [101, 102]. For example, pilocytic astrocytomas and non‐Hodgkin lymphomas harbour predominantly clonal SV drivers which are likely to have occurred earlier in tumourigenesis, whereas leiomyosarcomas and ovarian adenocarcinomas contain frequent subclonal SV drivers, which are more likely to contribute to later development [95]. In glioblastomas and medulloblastomas, a substantial fraction of chromosomal gains occur very early in molecular time (within the first 10%), whereas in melanomas, lung cancers, and papillary kidney cancers, chromosomal gains occur late and towards the end of molecular time [101]. Further, driver clonality may depend on the type of SV. For example, there is some suggestion from their predominantly clonal presence in tumour samples that gain‐of‐function SVs, including certain recurrent oncogenic fusions such as TMPRSS2‐ESG and BRAF‐KIAA1549, occur early in tumour development [95].
Genomic instability is a feature of almost all human cancers and allows tumour cells to develop the capabilities required to survive, proliferate, and spread [103, 104, 105, 106]. Conceptually, genomic instability represents an increased tendency for genomic alteration in tumour cells during cell division, occurring as a result of defective surveillance mechanisms governing genomic integrity [107]. Historically, genomic instability in cancer genomes has been thought to follow a stepwise process in which drivers accumulate gradually over time [93, 108, 109]. This is in contrast to the ‘Big Bang’ model of evolution which posits that tumours grow as a single terminal expansion, producing heterogeneous subclones at tumour initiation [110, 111]. In this model, the driver alterations required for tumour initiation are present early and are sufficient to support subsequent expansion [110]. In most tumour types there is a wide temporal window of genomic instability; however, this is not always the case [101]. Recent large‐scale studies suggest that complex SVs such as chromothripsis [17] and chromoplexy [19, 112, 113] typically occur within a single event [17]. Such mutational bursts suggest that cancer cells can alternate long phases of latency with short periods of intense, punctuated events [109, 112, 114], allowing cancer cells to attain greater fitness than would be possible through a gradual or stepwise accumulation of alterations [18]. Within a cancer genome, complex events can occur sequentially, suggesting that they may be mechanistically interlinked, in that the occurrence of one event increases the likelihood of a second. For example, dicentric chromosomes generated by BFB cycles and telomere attrition are precursors to chromothripsis [115, 116]. Similarly, the formation of a complex SV may facilitate the development of another one. For example, chromothripsis frequently occurs after the onset of WGD, with the latter presumably providing cells with sufficient intact genetic material to withstand the onslaught of genomic instability of the former [117]. In support of this, WGD commonly occurs in the intermediate stages of evolution in various cancers [101, 118].
Another form of accelerated evolution is mediated by ecDNAs. Because ecDNA fragments lack centromeres, the molecules are not attached to the mitotic spindle during cell division, leading to uneven segregation into daughter cells [119, 120, 121]. The resultant stochastic non‐Mendelian inheritance leads to rapid and dynamic changes of oncogene content under selection pressure, in response to tumour microenvironment and targeted therapies, leading to treatment resistance [122, 123, 124, 125]. In addition, circular recombination can occur after ecDNA formation, as a result of ongoing mutational processes [32]. In this instance, rapid alterations of the DNA sequence within the ecDNA can occur outside of cell division via complex events such as chromothripsis, providing another dimension to the evolution of structural complexity in the tumour genome [33]. SVs, and particularly complex SVs, play a significant yet underappreciated role in determining how cancer genomes evolve. Single‐cell WGS applied to organoid systems [126, 127] has enabled high‐resolution phylogenetic studies and functional interrogation of subclonal SVs [128]. In addition, longitudinal single‐cell WGS of patient‐derived xenografts from tumour samples has demonstrated that CNAs have an underappreciated impact on clonal fitness [129]. The evolutionary trajectories of structural complexity in tumours significantly impact how tumours progress but vary substantially between tumours and particularly between cancer types. The potential utility of these evolutionary trajectories in informing our understanding of tumour biology and as clinical biomarkers for treatment response and disease progression represents a currently untapped translational opportunity.
The functional impacts of structural complexity
Recent studies indicate that pathogenic SVs occur in at least 30% of all cancers [8, 101, 130]. SVs that are pathogenic disrupt the function of oncogenes or tumour suppressor genes, by increasing or decreasing their expression, respectively. The most well‐studied pathogenic consequences of SVs are their direct impacts on genes, either via gene dosage alteration (Figure 2A) or by the creation of gene fusions (Figure 2B). Gene dosage alterations occur via unbalanced rearrangements from simple SVs such as deletions and duplications, which may encompass entire chromosomes (aneuploidy) or even genomes (WGD). Alternatively, complex SVs such as chromothripsis and ecDNAs generate CNAs [101, 131, 132, 133]. Gene fusions occur via rearrangements that cause juxtaposition of two genes normally at distant loci [112, 134, 135]. A canonical example of a pathogenic oncogenic fusion is the BCR‐ABL1 oncoprotein, highly prevalent in chronic myeloid leukaemia [136, 137]. The functional and clinical impacts of simple CNAs, aneuploidies and fusion genes have been demonstrated across many cancers [138, 139], and form the basis for several highly effective clinical interventions described in the next section. However, WGS has enabled investigation of these events at a much greater depth and scale than was possible before and has highlighted alternative routes to gene dosage alteration that were previously obscured. SVs that occur within noncoding regulatory regions can cause indirect disruption of gene function. For example, in medulloblastoma, different SV classes juxtapose the proto‐oncogenes GFI1 and GFI1B to distal active enhancer elements [140], increasing their expression. This phenomenon has been termed ‘enhancer hijacking’ (Figure 2C). Further, SVs can disrupt the function of oncogenes and tumour suppressor genes via alteration of higher‐order chromatin structure [141, 142, 143] (Figure 2D). A recent study by the PCAWG demonstrated that SVs affect topologically associating domain (TAD) boundaries in a cancer‐specific manner (according to overall SV burden), are able to generate new TAD structures, and can lead to marked changes in chromatin folding [142]. SVs often overlap with binding sites for the transcriptional regulator, CTCF, near proto‐oncogenes of certain cancer types, potentially resulting in disruption of CTCF‐CTCF chromatin folding loops [142]. Both disruption to TAD structures and CTCF‐CTCF chromatin folding loops result in genome restructuring, bringing ectopic enhancers close to proto‐oncogenes, leading to increased expression. Also, different classes of SVs may employ different mechanisms that converge on the same functional outcome, for example, altered gene expression, and these may occur independently or together within the same cancer genome. For example, in glioblastoma, the receptor tyrosine kinase signalling pathway can be altered by disruption of the EGFR oncogene, either via gene fusion (EGFRvIII fusion [144]) or gene dosage increase (EGFR amplification [145, 146]), caused by either simple or complex SVs [147, 148].
Figure 2.

Pathogenic consequences of structural variants (SV). (A) Gene dosage alteration: SVs may (i) increase the expression of an oncogene by having extra copies as a result of amplifications/gains, mediated by various mechanisms such as tandem duplication, breakage‐fusion‐bridge cycle, and extrachromosomal DNA, or (ii) decrease the expression of tumour suppressor genes by reducing their copy number. (B) Gene fusion: translocations may bring two genes together to form a novel gene fusion, which may result in the production of novel fusion proteins beneficial to the cancer. (C) Gene function disruption: SVs may disrupt the function of genes via regulatory hijacking, for example by bringing an enhancer closer to a proto‐oncogene, thereby increasing its expression. (D) Genomic reorganisation: SVs may rearrange the wider chromosomal architecture, for example by rearranging the boundaries of topological associated domains, potentially bringing closer ectopic enhancers that would activate proto‐oncogenes, increasing their expression.
Assigning pathogenicity to a given SV can be challenging, as most SV breakpoints reside within the noncoding region of the genome [149], with no direct impact on the sequences of the coding genome. SVs such as inversions and complex SVs can have unpredictable functional impact, suggesting the presence of complex regulatory effects impacting long range noncoding regulatory elements discussed above rather than simple dosage alteration [150]. In addition, as a single SV commonly spans multiple genes, assigning pathogenicity to disruption of a particular gene can be difficult. Mutations driving tumour progression are usually recurrent, meaning that the same alteration results in the same phenotype across multiple cancer genomes or patients [151, 152]. Given the diversity in the types and sizes of SVs, the discovery of a recurrent distinct SV event is rare, making proving their role in driving tumourigenesis challenging. Furthermore, the combinatorial impacts of multiple SVs can be hard to resolve as phasing SVs, to ensure that they are impacting the same allele, based on short‐read WGS, is almost always impossible. These barriers to determining whether an SV is pathogenic lead to difficulties in determining the clinical impact of such events on patients. Nevertheless, a clinically significant SV does not necessarily have to be a driver of tumourigenesis. SVs can also be the consequence of a particular mutational processes or disrupted DNA repair mechanisms and can reveal insights into the presence or absence of these processes in a given tumour [153, 154]; these genome‐wide patterns of SVs can be clinically useful as biomarkers for treatment response. Lastly, SVs do not always promote tumour development, and in fact can be deleterious to the tumour. For example, deletions of BRCA1/2 leads to DNA damage repair deficiencies in certain tumours [155, 156], and tumours with a large fraction of complex SVs generate vast amounts of neoantigens that may confer susceptibility to immune therapies [157, 158].
The emerging clinical applications of SVs in cancer
Despite the many challenges in identifying clinically relevant SVs, recent findings uncover the translational potential of SVs as both causes (direct functional impact) and consequences (reflection of mutational processes) of cancer. While SV products such as gene fusions and amplifications are well known for their translational impacts (for example, as treatment targets [137, 159]), the potential clinical application of other SVs, particularly complex SVs, remain relatively unexplored. In the last decade, many studies have demonstrated proof‐of‐concept data highlighting the potential roles that SVs can play in every stage of cancer, from screening to treatment to prognostication (Figure 3). Premalignancy, driver SVs can exist decades before the onset of symptoms, generating opportunities for screening and early detection [160, 164, 165, 166, 167, 168]. The TRACERx group [160] showed that chromosome 3p loss is often the initiating driver in clear cell renal cell carcinoma and is predicted to arise 30–50 years before diagnosis (Figure 3A). In lung adenocarcinomas, driver fusion oncogenes, often derived from complex SVs, could occur decades before disease onset [164]. These findings suggest that in some cancer types a long latent window exists in which an effective screening strategy could be employed to allow earlier detection of cancer. However, it is important to consider when employing such a strategy that a single initiating driver may not be sufficient for cancer to develop and that for some individuals a necessary secondary event may never occur. SVs can also be used to predict progression to tumour from a premalignant state. In multiple myeloma, Oben et al [165] showed that the presence of myeloma‐defining genomic events, including chromothripsis in the rarely progressing precursor state, monoclonal gammopathy of undetermined significance (MGUS), can define a more progressive subset. In oesophageal cancer, complex SVs and CNAs are the most accurate predictors of malignant transformation from the Barrett's oesophagus state [166, 167, 168].
Figure 3.
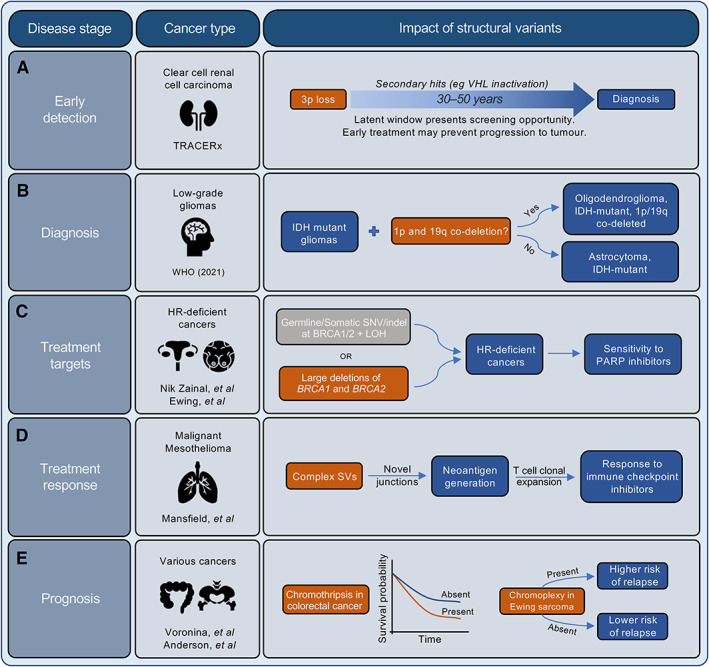
The clinical importance of structural variants (SVs) across all stages of the patient journey. SVs have significant potential in influencing the management of cancer patients throughout the disease trajectory. Some examples of the potential impact of SVs include: (A) Early detection: the TRACERx study on clear cell renal cell carcinoma [160] identified that 3p loss was frequently generated via chromothripsis, and that the loss could occur decades before diagnosis, suggesting the opportunity for early detection and intervention. (B) Diagnosis: the stratification of low‐grade gliomas (isocitrate dehydrogenase [IDH] mutant gliomas) has recently been updated by the WHO to include those with or without concurrent 1p and 19q co‐deletion [161]. (C) Treatment targets: tumours with large deletions of both BRCA1 and BRCA2 loci form a subset of HR‐deficient tumours that are exquisitely sensitive to PARP inhibitors [154, 155, 162]. (D) Treatment response: Mansfield et al [157, 158] demonstrated that malignant mesothelioma harbours neoantigens that are generated by complex SVs, and that their generation was associated with the clonal expansion of T cells, indicating that complex SV burden can be used to predict response to immune checkpoint inhibitors. (E) Prognosis: the presence of complex SVs are associated with poor prognosis. For example, patients with chromothripsis‐harbouring colorectal tumours have poorer survival rates compared to those without chromothripsis [163], and patients with Ewing sarcoma that contain chromoplexy have a higher risk of relapse following first‐line therapy [112].
Following malignant transformation, the presence of specific SVs is pathognomonic of certain cancer types and can act as a diagnostic tool. For example, the prototypical oncoprotein BCR‐ABL1 is diagnostic of chronic myeloid leukaemia [169]. Other diagnostic fusion genes include PML‐RARA in acute promyelocytic leukaemia [170], TMPRSS2‐ERG in prostate adenocarcinoma [171, 172, 173], and EWSR1‐ETS in Ewing sarcoma [112, 135, 174]. Other classes of SVs have been proposed as part of the established diagnostic criteria of certain cancers, including focal amplification or deletion (EGFR amplification in glioblastoma and CDKN2A/B deletion in IDH mutant astrocytoma [161]) and chromosomal gains and losses (chromosome 7 gain or 10 loss in glioblastoma, 1p and 19q losses in oligodendroglioma (Figure 3B) [175, 176]). Diagnosis based on these types of events is often indicative of their critical role in driving the development of the tumour, and as such many of the features used for diagnosis are effective targets for treatment.
Several oncogenic gene fusions are currently being used or explored as therapeutic targets in various cancer types [139]. The prototypical example of this is the use of imatinib mesylate to treat haematological cancers generating BCR‐ABL fusion oncoprotein [137, 169, 177]. More recently, targeting fusion transcripts has expanded into other cancer types. These include the application of inhibitors of ALK‐containing fusions in lung cancers [178] and anaplastic large‐cell lymphomas [169], NTRK‐containing fusions in various solid tumours [179, 180, 181], and FCGR‐TACC fusions in glioblastoma [182, 183] and other malignancies [184]. However, the utility of SVs as therapeutic targets is not limited to gene fusions. Significant progress has been made in some settings by targeting treatment based on oncogene amplification, including ERBB2 in breast and ovarian cancers [159, 185], EGFR in breast, colorectal, and lung cancers [186, 187, 188], and MYC in neuroblastoma [189]. As evidence accumulates on the role of complex SVs, in particular of ecDNAs, on oncogene amplification, we would expect our grasp of the clinical utility of these events to strengthen. On the flip side, tumour suppressor loss is also an effective target for treatment. While restoring tumour suppressor function is challenging [190], the greatest clinical impact has come from exploiting vulnerabilities in cancer cells that lack functional tumour suppressor genes. This is the case when BRCA1/2 function is lost in homology‐directed repair (HR) deficient breast, ovarian, prostate, and pancreatic cancers, conferring the tumours' sensitivity to PARP inhibition via synthetic lethality. BRCA1/2 loss consists of a pathogenic SNV/indel in BRCA1/2 in either the germline or somatic DNA followed by loss of the other allele via copy number neutral loss of heterozygosity or deletion [154, 162]. However, recent evidence has emerged from WGS that indicates that tumours with large heterozygous deletions spanning the length of both BRCA1 and BRCA2, in the absence of SNVs/indels, are also more likely to be HR‐deficient and may also benefit from PARP inhibitors [155] (Figure 3C). This is an exciting exemplar, where the clinical impact of a known therapeutic target may be extended to a greater number of patients when we comprehensively account for the full structural complexity of tumour genomes.
In cases where SVs are not directly impacting tumour development or where the functional consequences of the SVs have not yet been determined, such as those SVs with breakpoints within a noncoding region, SVs can be used as biomarkers for treatment response. Tumours harbouring complex SVs such as chromothripsis and chromoplexy can generate various novel fusion junctions that result in the generation of neoantigens when expressed [112, 113, 135, 164, 191, 192]. For example, in malignant mesothelioma widespread complex SVs are predicted to generate neoantigens and correlate with clonal expansion of tumour infiltrating T lymphocytes [157, 193], suggesting that the presence of these complex SV events may be a useful biomarker for response to immune checkpoint inhibitors (Figure 3D) [194]. Further, tumours with ecDNAs may be more resistant to targeted oncogenic therapies due to their inherent plasticity and the significant contribution of ecDNAs to intratumoural heterogeneity [124]. In addition, overall genomic instability itself, rather than the presence of a particular SV, may be exploited as a treatment target [195]. A recent study suggested that drug inhibition against KIF18A, a key protein in the maintenance of spindle dynamics, resulted in antiproliferative effects specifically in tumours with increased chromosomal instability [196]. A further area of biomarker discovery lies in the application of mutational signatures [154, 162, 197]. These genome‐wide patterns of mutation have been extensively studied in the context of SNVs and are particularly useful for predicting exposure to mutational processes and DNA repair deficiencies. Although SV signatures are currently less well explored due to challenges in classifying SVs as a result of their diversity, patterns of tandem duplication and deletion are strongly associated with HR deficiency and can be used, together with the rest of the mutational landscape, as a proxy for treatment response [153, 154, 162]. We expect the clinical utility of SV signatures as biomarkers to expand as our understanding of the SV landscape develops and strategies for informative classification emerge.
Increased genomic instability as a result of ongoing chromosomal rearrangements has long been associated with poor prognosis in many cancer types [92, 198]. This association is likely driven by a greater propensity for treatment resistance as a result of the significant contribution of genomic instability to intratumoural heterogeneity [105]. Further, the association may also reflect analogous patterns of cell tolerance to extensive genomic rearrangements as to treatment onslaught. On the other hand, extreme levels of genomic instability, where most of the tumour genome is subject to chromosomal rearrangements and CNAs, are associated with better prognosis [199, 200]. In this instance, tumour cells with an overwhelming burden of genomic instability are on the brink of death, as so much of their genome is adversely altered, making successful replication unlikely. In addition, high levels of genomic instability render the tumour cells more immunogenic, resulting in a more efficient immune response and tumour clearance [201]. These seemingly conflicting findings suggest that a threshold of genomic instability may exist, such that tumour growth and adaptation are enhanced up to a certain level of instability, but that beyond this level, growth and survival may be compromised. This phenomenon suggests that tumour cells rely on an exquisite balance of genomic instability in ensuring their survival. More specifically, the impact of individual structural events on patient prognosis, other than where they are a target for treatment, is less clear. Complex SVs such as chromothripsis are associated with poor prognosis in multiple cancer subtypes (Figure 3E) [132, 163], including multiple myeloma [202], malignant melanoma [203], medulloblastoma [204], neuroblastoma [205], osteosarcoma [206], metastatic colorectal cancer [163, 207], and acute myeloid leukaemia [208, 209]. Similarly, ecDNAs are associated with poor outcome in aggressive cancers such as glioblastoma [210], neuroblastoma [211], medulloblastoma [70], and breast cancer [159]. Chromoplexy events in Ewing sarcoma portend the high risk of relapse [112] (Figure 3E). SVs are also a marker of late‐stage disease in malignant melanoma [212] and prostate cancer [213]. In general, those tumours that acquire a burst of mutations, perhaps through the generation of a complex SV, in a short period of time tend to proliferate rapidly and metastasize early to many differing sites, resulting in poorer clinical outcomes [92]. Overall, despite the significant challenges in identifying and interpreting the impact of SVs, their potential for substantial clinical impact, whether as diagnostic tools, therapeutic targets, or biomarkers for treatment response or patient prognosis, is clear.
Conclusions and future perspectives
The SV landscape of a tumour genome in all its complexity holds tremendous potential for clinical impact at all stages of the patient's treatment journey, from early detection to predicting survival outcomes. However, if we are to capitalise on this opportunity to maximise the resultant benefit for patients, barriers with respect to the accurate detection and interpretation of the entire diverse SV landscape must be overcome. Our current understanding of this facet of tumour biology is based on insight gleaned from short‐read WGS from large tumour cohorts, which, due to technical limitations will necessarily underestimate the scale and complexity of these harder to study mutational events. Expanded use of long‐read sequencing technologies combined with improvements in analytical methods designed to encapsulate the full complexity of these events will address some of the current limitations. Nevertheless, challenges to interpreting the impact of these events will remain, to say nothing of the significant challenges in translating resultant insights to a clinical setting, which are the subject of many dedicated reviews [214, 215, 216].
Despite current limitations, existing studies provide an exciting glimpse into the emerging aspects of the genomic landscape of tumours, which represent opportunities to expand our understanding of tumour biology and provide novel avenues for translational exploitation in the future. Our rapidly developing appreciation for the impact that complex SVs have on tumour evolution and patient outcome is still in its infancy, but as the field develops we expect that these events along with the rest of the SV landscape will be crucial to unravelling the evolutionary trajectories that cells take on their routes to malignancy and metastases.
Author contributions statement
Both authors contributed to all processes involved in writing this review and approved the final article.
Acknowledgements
AH is supported by Cancer Research UK and the Edinburgh Clinical Academic Training (ECAT) programme; AE is supported by a University of Edinburgh Chancellor's Fellowship and core funding from the MRC Human Genetics Unit.
No conflicts of interest were declared.
References
- 1. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy‐number alteration across human cancers. Nature 2010; 463: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shlien A, Malkin D. Copy number variations and cancer. Genome Med 2009; 1: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cancer Genome Atlas Research Network , Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan‐Cancer analysis project. Nat Genet 2013; 45: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. ICGC/TCGA Pan‐Cancer Analysis of Whole Genomes Consortium . Pan‐cancer analysis of whole genomes. Nature 2020; 578: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Y, Roberts ND, Wala JA, et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020; 578: 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodriguez‐Martin B, Alvarez EG, Baez‐Ortega A, et al. Pan‐cancer analysis of whole genomes identifies driver rearrangements promoted by LINE‐1 retrotransposition. Nat Genet 2020; 52: 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Priestley P, Baber J, Lolkema MP, et al. Pan‐cancer whole‐genome analyses of metastatic solid tumours. Nature 2019; 575: 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Belzen IAEM, Schönhuth A, Kemmeren P, et al. Structural variant detection in cancer genomes: computational challenges and perspectives for precision oncology. NPJ Precis Oncol 2021; 5: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. 1000 Genomes Project Consortium , Abecasis GR, Altshuler D, et al. A map of human genome variation from population‐scale sequencing. Nature 2010; 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sudmant PH, Rausch T, Gardner EJ, et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015; 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collins RL, Brand H, Karczewski KJ, et al. A structural variation reference for medical and population genetics. Nature 2020; 581: 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Currall BB, Chiang C, Talkowski ME, et al. Mechanisms for structural variation in the human genome. Curr Genet Med Rep 2013; 1: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang W‐J, Li L‐Y, Cui J‐W. Chromosome structural variation in tumorigenesis: mechanisms of formation and carcinogenesis. Epigenetics Chromatin 2020; 13: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carvalho CMB, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 2016; 17: 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang L, Luquette LJ, Gehlenborg N, et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 2013; 153: 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yi K, Ju YS. Patterns and mechanisms of structural variations in human cancer. Exp Mol Med 2018; 50: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144: 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell 2013; 152: 1226–1236. [DOI] [PubMed] [Google Scholar]
- 19. Shen MM. Chromoplexy: a new category of complex rearrangements in the cancer genome. Cancer Cell 2013; 23: 567–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pellestor F, Gatinois V. Chromoanasynthesis: another way for the formation of complex chromosomal abnormalities in human reproduction. Hum Reprod 2018; 33: 1381–1387. [DOI] [PubMed] [Google Scholar]
- 21. Gisselsson D, Pettersson L, Höglund M, et al. Chromosomal breakage‐fusion‐bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci U S A 2000; 97: 5357–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McClintock B. The stability of broken ends of chromosomes in Zea Mays. Genetics 1941; 26: 234–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Verhaak RGW, Bafna V, Mischel PS. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer 2019; 19: 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu S, Turner KM, Nguyen N, et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 2019; 575: 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cox D, Yuncken C, Spriggs A. Minute chromatin bodies in malignant tumours of childhood. Lancet 1965; 286: 55–58. [DOI] [PubMed] [Google Scholar]
- 26. Storlazzi CT, Lonoce A, Guastadisegni MC, et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res 2010; 20: 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 2009; 10: 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bielski CM, Zehir A, Penson AV, et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet 2018; 50: 1189–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rustad EH, Yellapantula VD, Glodzik D, et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov 2020; 1: 258–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hadi K, Yao X, Behr JM, et al. Distinct classes of complex structural variation uncovered across thousands of cancer genome graphs. Cell 2020; 183: 197–210.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dahiya R, Hu Q, Ly P. Mechanistic origins of diverse genome rearrangements in cancer. Semin Cell Dev Biol 2022; 123: 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosswog C, Bartenhagen C, Welte A, et al. Chromothripsis followed by circular recombination drives oncogene amplification in human cancer. Nat Genet 2021; 53: 1673–1685. [DOI] [PubMed] [Google Scholar]
- 33. Pellman D, Zhang C‐Z. Decoding complex patterns of oncogene amplification. Nat Genet 2021; 53: 1626–1627. [DOI] [PubMed] [Google Scholar]
- 34. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next‐generation sequencing technologies. Nat Rev Genet 2016; 17: 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gong T, Hayes VM, Chan EKF. Detection of somatic structural variants from short‐read next‐generation sequencing data. Brief Bioinform 2021; 22: bbaa056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ho SS, Urban AE, Mills RE. Structural variation in the sequencing era. Nat Rev Genet 2020; 21: 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mahmoud M, Gobet N, Cruz‐Dávalos DI, et al. Structural variant calling: the long and the short of it. Genome Biol 2019; 20: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kosugi S, Momozawa Y, Liu X, et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol 2019; 20: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet 2011; 12: 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boeva V, Zinovyev A, Bleakley K, et al. Control‐free calling of copy number alterations in deep‐sequencing data using GC‐content normalization. Bioinformatics 2011; 27: 268–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Talevich E, Shain AH, Botton T, et al. CNVkit: genome‐wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 2016; 12: e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raine KM, Van Loo P, Wedge DC, et al. ascatNgs: identifying somatically acquired copy‐number alterations from whole‐genome sequencing data. Curr Protoc Bioinformatics 2016; 56: 15.9.1–15.9.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rausch T, Zichner T, Schlattl A, et al. DELLY: structural variant discovery by integrated paired‐end and split‐read analysis. Bioinformatics 2012; 28: i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Layer RM, Chiang C, Quinlan AR, et al. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 2014; 15: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen X, Schulz‐Trieglaff O, Shaw R, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016; 32: 1220–1222. [DOI] [PubMed] [Google Scholar]
- 46. Cameron DL, Schröder J, Penington JS, et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res 2017; 27: 2050–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cameron DL, Baber J, Shale C, et al. GRIDSS2: comprehensive characterisation of somatic structural variation using single breakend variants and structural variant phasing. Genome Biol 2021; 22: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wala JA, Bandopadhayay P, Greenwald NF, et al. SvABA: genome‐wide detection of structural variants and indels by local assembly. Genome Res 2018; 28: 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shale C, Baber J, Cameron DL, et al. Unscrambling cancer genomes via integrated analysis of structural variation and copy number. bioRxiv 2020: 2020.12.03.410860 [Not peer reviewed]. [DOI] [PMC free article] [PubMed]
- 50. Deshpande V, Luebeck J, Nguyen NPD, et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun 2019; 10: 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Menzel P, Frellsen J, Plass M, et al. On the accuracy of short read mapping. Methods Mol Biol 2013; 1038: 39–59. [DOI] [PubMed] [Google Scholar]
- 52. Zhao X, Collins RL, Lee W‐P, et al. Expectations and blind spots for structural variation detection from long‐read assemblies and short‐read genome sequencing technologies. Am J Hum Genet 2021; 108: 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ebbert MTW, Jensen TD, Jansen‐West K, et al. Systematic analysis of dark and camouflaged genes reveals disease‐relevant genes hiding in plain sight. Genome Biol 2019; 20: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Benjamini Y, Speed TP. Summarizing and correcting the GC content bias in high‐throughput sequencing. Nucleic Acids Res 2012; 40: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sedlazeck FJ, Lee H, Darby CA, et al. Piercing the dark matter: bioinformatics of long‐range sequencing and mapping. Nat Rev Genet 2018; 19: 329–346. [DOI] [PubMed] [Google Scholar]
- 56. Amarasinghe SL, Su S, Dong X, et al. Opportunities and challenges in long‐read sequencing data analysis. Genome Biol 2020; 21: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. De Coster W, Weissensteiner MH, Sedlazeck FJ. Towards population‐scale long‐read sequencing. Nat Rev Genet 2021; 22: 572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Logsdon GA, Vollger MR, Eichler EE. Long‐read human genome sequencing and its applications. Nat Rev Genet 2020; 21: 597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zheng GXY, Lau BT, Schnall‐Levin M, et al. Haplotyping germline and cancer genomes with high‐throughput linked‐read sequencing. Nat Biotechnol 2016; 34: 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang F, Christiansen L, Thomas J, et al. Haplotype phasing of whole human genomes using bead‐based barcode partitioning in a single tube. Nat Biotechnol 2017; 35: 852–857. [DOI] [PubMed] [Google Scholar]
- 61. Wang O, Chin R, Cheng X, et al. Efficient and unique cobarcoding of second‐generation sequencing reads from long DNA molecules enabling cost‐effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res 2019; 29: 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sanders AD, Falconer E, Hills M, et al. Single‐cell template strand sequencing by Strand‐seq enables the characterization of individual homologs. Nat Protoc 2017; 12: 1151–1176. [DOI] [PubMed] [Google Scholar]
- 63. Falconer E, Hills M, Naumann U, et al. DNA template strand sequencing of single‐cells maps genomic rearrangements at high resolution. Nat Methods 2012; 9: 1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kuleshov V, Jiang C, Zhou W, et al. Synthetic long‐read sequencing reveals intraspecies diversity in the human microbiome. Nat Biotechnol 2016; 34: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Peters BA, Kermani BG, Sparks AB, et al. Accurate whole‐genome sequencing and haplotyping from 10 to 20 human cells. Nature 2012; 487: 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lieberman‐Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science 2009; 326: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Viswanathan SR, Ha G, Hoff AM, et al. Structural alterations driving castration‐resistant prostate cancer revealed by linked‐read genome sequencing. Cell 2018; 174: 433–447.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nordlund J, Marincevic‐Zuniga Y, Cavelier L, et al. Refined detection and phasing of structural aberrations in pediatric acute lymphoblastic leukemia by linked‐read whole‐genome sequencing. Sci Rep 2020; 10: 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yuan Y, Chung CY‐L, Chan T‐F. Advances in optical mapping for genomic research. Comput Struct Biotechnol J 2020; 18: 2051–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chapman OS, Luebeck J, Wani S, et al. The landscape of extrachromosomal circular DNA in medulloblastoma. bioRxiv 2021: 2021. doi.org/10.18.464907 [Not peer reviewed].
- 71. Luebeck J, Coruh C, Dehkordi SR, et al. AmpliconReconstructor integrates NGS and optical mapping to resolve the complex structures of focal amplifications. Nat Commun 2020; 11: 4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Miga KH, Koren S, Rhie A, et al. Telomere‐to‐telomere assembly of a complete human X chromosome. Nature 2020; 585: 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thibodeau ML, O'Neill K, Dixon K, et al. Improved structural variant interpretation for hereditary cancer susceptibility using long‐read sequencing. Genet Med 2020; 22: 1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Audano PA, Sulovari A, Graves‐Lindsay TA, et al. Characterizing the major structural variant alleles of the human genome. Cell 2019; 176: 663–675.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. De Coster W, De Rijk P, De Roeck A, et al. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res 2019; 29: 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jain M, Koren S, Miga KH, et al. Nanopore sequencing and assembly of a human genome with ultralong reads. Nat Biotechnol 2018; 36: 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Norris AL, Workman RE, Fan Y, et al. Nanopore sequencing detects structural variants in cancer. Cancer Biol Ther 2016; 17: 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nurk S, Walenz BP, Rhie A, et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high‐fidelity long reads. Genome Res 2020; 30: 1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chaisson MJP, Huddleston J, Dennis MY, et al. Resolving the complexity of the human genome using single‐molecule sequencing. Nature 2015; 517: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. De Coster W, Van Broeckhoven C. Newest methods for detecting structural variations. Trends Biotechnol 2019; 37: 973–982. [DOI] [PubMed] [Google Scholar]
- 81. Sedlazeck FJ, Rescheneder P, Smolka M, et al. Accurate detection of complex structural variations using single‐molecule sequencing. Nat Methods 2018; 15: 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Beyter D, Ingimundardottir H, Oddsson A, et al. Long‐read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet 2021; 53: 779–786. [DOI] [PubMed] [Google Scholar]
- 83. Stancu MC, Van Roosmalen MJ, Renkens I, et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun 2017; 8: 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Roberts HE, Lopopolo M, Pagnamenta AT, et al. Short and long‐read genome sequencing methodologies for somatic variant detection; genomic analysis of a patient with diffuse large B‐cell lymphoma. Sci Rep 2021; 11: 6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sakamoto Y, Zaha S, Suzuki Y, et al. Application of long‐read sequencing to the detection of structural variants in human cancer genomes. Comput Struct Biotechnol J 2021; 19: 4207–4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rautiainen M, Marschall T. MBG: minimizer‐based sparse de Bruijn graph construction. Bioinformatics 2021; 37: 2476–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Logsdon GA, Vollger MR, Hsieh P, et al. The structure, function and evolution of a complete human chromosome 8. Nature 2021; 593: 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. bioRxiv 2021. doi.org/10.1101/2021.05.26.445798 [Not peer reviewed].
- 89. Tham CY, Tirado‐Magallanes R, Goh Y, et al. NanoVar: accurate characterization of patients' genomic structural variants using low‐depth nanopore sequencing. Genome Biol 2020; 21: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jiang T, Liu Y, Jiang Y, et al. Long‐read‐based human genomic structural variation detection with cuteSV. Genome Biol 2020; 21: 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cancer 2.0 Initiative Genomics England 2022. [Accessed 21 April 2022]. Available at https://www.genomicsengland.co.uk/initiatives/cancer
- 92. Turajlic S, Sottoriva A, Graham T, et al. Resolving genetic heterogeneity in cancer. Nat Rev Genet 2019; 20: 404–416. [DOI] [PubMed] [Google Scholar]
- 93. Greaves M. Evolutionary determinants of cancer. Cancer Discov 2015; 5: 806–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nowell PC. The clonal evolution of tumor cell populations. Science 1976; 194: 23–28. [DOI] [PubMed] [Google Scholar]
- 95. Dentro SC, Leshchiner I, Haase K, et al. Characterizing genetic intratumor heterogeneity across 2,658 human cancer genomes. Cell 2021; 184: 2239–2254.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cmero M, Yuan K, Ong CS, et al. Inferring structural variant cancer cell fraction. Nat Commun 2020; 11: 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Fan X, Yang C, Li W, et al. SMOOTH‐seq: single‐cell genome sequencing of human cells on a third‐generation sequencing platform. Genome Biol 2021; 22: 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Funnell T, O'Flanagan CH, Williams MJ, et al. The impact of mutational processes on structural genomic plasticity in cancer cells. bioRxiv 2021: 2021.06.03.446999 [Not peer reviewed].
- 99. Salehi S, Dorri F, Chern K, et al. Cancer phylogenetic tree inference at scale from 1000s of single cell genomes. bioRxiv 2020. doi.org/10.1101/2020.05.06.058180 [Not peer reviewed].
- 100. Williams MJ, Funnell T, O'Flanagan CH, et al. Evolutionary tracking of cancer haplotypes at single‐cell resolution. bioRxiv 2021: 2021.06.04.447031 [Not peer reviewed].
- 101. Gerstung M, Jolly C, Leshchiner I, et al. The evolutionary history of 2,658 cancers. Nature 2020; 578: 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Notta F, Chan‐Seng‐Yue M, Lemire M, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016; 538: 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov 2022; 12: 31–46. [DOI] [PubMed] [Google Scholar]
- 104. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 105. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 106. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability‐‐an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11: 220–228. [DOI] [PubMed] [Google Scholar]
- 107. Yao Y, Dai W. Genomic instability and cancer. J Carcinog Mutagen 2014; 5: 1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hansford S, Huntsman DG. Boveri at 100: Theodor Boveri and genetic predisposition to cancer. J Pathol 2014; 234: 142–145. [DOI] [PubMed] [Google Scholar]
- 109. Vendramin R, Litchfield K, Swanton C. Cancer evolution: Darwin and beyond. EMBO J 2021; 40: e108389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sottoriva A, Kang H, Ma Z, et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015; 47: 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sun R, Hu Z, Curtis C. Big bang tumor growth and clonal evolution. Cold Spring Harb Perspect Med 2018; 8: a028381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Anderson ND, de Borja R, Young MD, et al. Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors. Science 2018; 361: eaam8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Baca SC, Prandi D, Lawrence MS, et al. Punctuated evolution of prostate cancer genomes. Cell 2013; 153: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Umbreit NT, Zhang CZ, Lynch LD, et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020; 368: eaba0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Li Y, Schwab C, Ryan S, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 2014; 508: 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Maciejowski J, Li Y, Bosco N, et al. Chromothripsis and kataegis induced by telomere crisis. Cell 2015; 163: 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. López S, Lim EL, Horswell S, et al. Interplay between whole‐genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat Genet 2020; 52: 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Connor AA, Gallinger S. Pancreatic cancer evolution and heterogeneity: integrating omics and clinical data. Nat Rev Cancer 2022; 22: 131–142. [DOI] [PubMed] [Google Scholar]
- 119. de Carvalho AC, Kim H, Poisson LM, et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet 2018; 50: 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lange JT, Chen CY, Pichugin Y, et al. Principles of ecDNA random inheritance drive rapid genome change and therapy resistance in human cancers. bioRxiv 2021: 2021.06.11.447968 [Not peer reviewed].
- 121. Yi E, Gujar AD, Guthrie M, et al. Live‐cell imaging shows uneven segregation of extrachromosomal DNA elements and transcriptionally active extrachromosomal DNA hubs in cancer. Cancer Discov 2022; 12: 468–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Alt FW, Kellems RE, Bertino JR, et al. Selective multiplication of dihydrofolate reductase genes in methotrexate‐resistant variants of cultured murine cells. J Biol Chem 1978; 253: 1357–1370. [PubMed] [Google Scholar]
- 123. Haber DA, Schimke RT. Unstable amplification of an altered dihydrofolate reductase gene associated with double‐minute chromosomes. Cell 1981; 26: 355–362. [DOI] [PubMed] [Google Scholar]
- 124. Nathanson DA, Gini B, Mottahedeh J, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014; 343: 72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Turner KM, Deshpande V, Beyter D, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017; 543: 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Youk J, Kwon HW, Kim R, et al. Dissecting single‐cell genomes through the clonal organoid technique. Exp Mol Med 2021; 53: 1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jager M, Blokzijl F, Sasselli V, et al. Measuring mutation accumulation in single human adult stem cells by whole‐genome sequencing of organoid cultures. Nat Protoc 2018; 13: 59–78. [DOI] [PubMed] [Google Scholar]
- 128. Roerink SF, Sasaki N, Lee‐Six H, et al. Intratumour diversification in colorectal cancer at the single‐cell level. Nature 2018; 556: 457–462. [DOI] [PubMed] [Google Scholar]
- 129. Salehi S, Kabeer F, Ceglia N, et al. Clonal fitness inferred from time‐series modelling of single‐cell cancer genomes. Nature 2021; 595: 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dixon JR, Xu J, Dileep V, et al. Integrative detection and analysis of structural variation in cancer genomes. Nat Genet 2018; 50: 1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Weischenfeldt J, Dubash T, Drainas AP, et al. Pan‐cancer analysis of somatic copy‐number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet 2017; 49: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cortés‐Ciriano I, Lee JJ‐K, Xi R, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole‐genome sequencing. Nat Genet 2020; 52: 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Morton AR, Dogan‐Artun N, Faber ZJ, et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell 2019; 179: 1330–1341.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med 1999; 341: 164–172. [DOI] [PubMed] [Google Scholar]
- 135. Gorthi A, Romero JC, Loranc E, et al. EWS–FLI1 increases transcription to cause R‐loops and block BRCA1 repair in Ewing sarcoma. Nature 2018; 555: 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR‐ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001; 344: 1038–1042. [DOI] [PubMed] [Google Scholar]
- 137. Hochhaus A, Larson RA, Guilhot F, et al. Long‐term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med 2017; 376: 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012; 13: 189–203. [DOI] [PubMed] [Google Scholar]
- 139. Mertens F, Johansson B, Fioretos T, et al. The emerging complexity of gene fusions in cancer. Nat Rev Cancer 2015; 15: 371–381. [DOI] [PubMed] [Google Scholar]
- 140. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole‐genome landscape of medulloblastoma subtypes. Nature 2017; 547: 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Sadowski M, Kraft A, Szalaj P, et al. Spatial chromatin architecture alteration by structural variations in human genomes at the population scale. Genome Biol 2019; 20: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Akdemir KC, Le VT, Chandran S, et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat Genet 2020; 52: 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Hawley JR, Zhou S, Arlidge C, et al. Reorganization of the 3D genome pinpoints noncoding drivers of primary prostate tumors. Cancer Res 2021; 81: 5833–5848. [DOI] [PubMed] [Google Scholar]
- 144. Koga T, Li B, Figueroa JM, et al. Mapping of genomic EGFRvIII deletions in glioblastoma: insight into rearrangement mechanisms and biomarker development. Neuro Oncol 2018; 20: 1310–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Furgason JM, Koncar RF, Michelhaugh SK, et al. Whole genome sequence analysis links chromothripsis to EGFR, MDM2, MDM4, and CDK4 amplification in glioblastoma. Oncoscience 2015; 2: 618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Szerlip NJ, Pedraza A, Chakravarty D, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A 2012; 109: 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Brennan CW, Verhaak RGW, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Liu F, Hon GC, Villa GR, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol Cell 2015; 60: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhang X, Meyerson M. Illuminating the noncoding genome in cancer. Nat Cancer 2020; 1: 864–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chiang C, Scott AJ, Davis JR, et al. The impact of structural variation on human gene expression. Nat Genet 2017; 49: 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Bailey MH, Tokheim C, Porta‐Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018; 174: 1034–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Martínez‐Jiménez F, Muiños F, Sentís I, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer 2020; 20: 555–572. [DOI] [PubMed] [Google Scholar]
- 153. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature 2020; 578: 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 2017; 23: 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ewing A, Meynert A, Churchman M, et al. Structural variants at the BRCA1/2 loci are a common source of homologous repair deficiency in high‐grade serous ovarian carcinoma. Clin Cancer Res 2021; 27: 3201–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Stok C, Kok YP, van den Tempel N, et al. Shaping the BRCAness mutational landscape by alternative double‐strand break repair, replication stress and mitotic aberrancies. Nucleic Acids Res 2021; 49: 4239–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Mansfield AS, Peikert T, Smadbeck JB, et al. Neoantigenic potential of complex chromosomal rearrangements in mesothelioma. J Thorac Oncol 2019; 14: 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Mansfield AS, Peikert T, Vasmatzis G. Chromosomal rearrangements and their neoantigenic potential in mesothelioma. Transl Lung Cancer Res 2020; 9: S92–S99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Vicario R, Peg V, Morancho B, et al. Patterns of HER2 gene amplification and response to anti‐HER2 therapies. PLoS One 2015; 10: e0129876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Mitchell TJ, Turajlic S, Rowan A, et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell 2018; 173: 611–623.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 2021; 23: 1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Nik‐Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature 2016; 534: 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Voronina N, Wong JKL, Hübschmann D, et al. The landscape of chromothripsis across adult cancer types. Nat Commun 2020; 11: 2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Lee JJ‐K, Park S, Park H, et al. Tracing oncogene rearrangements in the mutational history of lung adenocarcinoma. Cell 2019; 177: 1842–1857.e21. [DOI] [PubMed] [Google Scholar]
- 165. Oben B, Froyen G, Maclachlan KH, et al. Whole‐genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat Commun 2021; 12: 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Newell F, Patel K, Gartside M, et al. Complex structural rearrangements are present in high‐grade dysplastic Barrett's oesophagus samples. BMC Med Genomics 2019; 12: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Killcoyne S, Fitzgerald RC. Evolution and progression of Barrett's oesophagus to oesophageal cancer. Nat Rev Cancer 2021; 21: 731–741. [DOI] [PubMed] [Google Scholar]
- 168. Paulson T, Galipeau P, Oman K, et al. Somatic whole genome dynamics of precancer in Barrett's Esophagus. Research Square 2021; doi.org/10.21203/rs.3.rs‐257949/v1 [Not peer reviewed].
- 169. Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020; 34: 966–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Sanz MA, Fenaux P, Tallman MS, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019; 133: 1630–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Carver BS, Tran J, Gopalan A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet 2009; 41: 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Yao Y, Wang H, Li B, et al. Evaluation of the TMPRSS2:ERG fusion for the detection of prostate cancer: a systematic review and meta‐analysis. Tumour Biol 2014; 35: 2157–2166. [DOI] [PubMed] [Google Scholar]
- 173. Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005; 310: 644–648. [DOI] [PubMed] [Google Scholar]
- 174. Blay J‐Y, De Pinieux G, Gouin F. Ewing's sarcoma. N Engl J Med 2021; 384: 1477. [DOI] [PubMed] [Google Scholar]
- 175. Rushing EJ. WHO classification of tumors of the nervous system: preview of the upcoming 5th edition. Memo 2021; 14: 188–191. [Google Scholar]
- 176. Gonzalez Castro LN, Wesseling P. The cIMPACT‐NOW updates and their significance to current neuro‐oncology practice. Neurooncol Pract 2021; 8: 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI‐571 inhibition of abelson tyrosine kinase. Science 2000; 289: 1938–1942. [DOI] [PubMed] [Google Scholar]
- 178. Sasaki T, Rodig SJ, Chirieac LR, et al. The biology and treatment of EML4‐ALK non‐small cell lung cancer. Eur J Cancer 2010; 46: 1773–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Cocco E, Scaltriti M, Drilon A. NTRK fusion‐positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018; 15: 731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Harada G, Santini FC, Wilhelm C, et al. NTRK fusions in lung cancer: from biology to therapy. Lung Cancer 2021; 161: 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Vaishnavi A, Capelletti M, Le AT, et al. Oncogenic and drug‐sensitive NTRK1 rearrangements in lung cancer. Nat Med 2013; 19: 1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012; 337: 1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Mata DA, Benhamida JK, Lin AL, et al. Genetic and epigenetic landscape of IDH‐wildtype glioblastomas with FGFR3‐TACC3 fusions. Acta Neuropathol Commun 2020; 8: 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Krook MA, Reeser JW, Ernst G, et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br J Cancer 2021; 124: 880–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kaushik Tiwari M, Colon‐Rios DA, Tumu HCR, et al. Direct targeting of amplified gene loci for proapoptotic anticancer therapy. Nat Biotechnol 2022; 40: 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Sordella R, Bell DW, Haber DA, et al. Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Science 2004; 305: 1163–1167. [DOI] [PubMed] [Google Scholar]
- 187. Kato S, Okamura R, Mareboina M, et al. Revisiting epidermal growth factor receptor (EGFR) amplification as a target for anti‐EGFR therapy: analysis of cell‐free circulating tumor DNA in patients with advanced malignancies. JCO Precis Oncol 2019; 3: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Li Q‐H, Wang Y‐Z, Tu J, et al. Anti‐EGFR therapy in metastatic colorectal cancer: mechanisms and potential regimens of drug resistance. Gastroenterol Rep (Oxf) 2020; 8: 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Madden SK, de Araujo AD, Gerhardt M, et al. Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c‐Myc. Mol Cancer 2021; 20: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Morris LGT, Chan TA. Therapeutic targeting of tumor suppressor genes. Cancer 2015; 121: 1357–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Yang W, Lee K‐W, Srivastava RM, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med 2019; 25: 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Yarchoan M, Johnson BA 3rd, Lutz ER, et al. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017; 17: 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Yoshikawa Y, Emi M, Hashimoto‐Tamaoki T, et al. High‐density array‐CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc Natl Acad Sci U S A 2016; 113: 13432–13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Carbone M, Yang H, Gaudino G. Does chromothripsis make mesothelioma an immunogenic cancer? J Thorac Oncol 2019; 14: 157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Bielski CM, Taylor BS. Homing in on genomic instability as a therapeutic target in cancer. Nat Commun 2021; 12: 3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Marquis C, Fonseca CL, Queen KA, et al. Chromosomally unstable tumor cells specifically require KIF18A for proliferation. Nat Commun 2021; 12: 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Koh G, Degasperi A, Zou X, et al. Mutational signatures: emerging concepts, caveats and clinical applications. Nat Rev Cancer 2021; 21: 619–637. [DOI] [PubMed] [Google Scholar]
- 198. McGranahan N, Burrell RA, Endesfelder D, et al. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep 2012; 13: 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Andor N, Maley CC, Ji HP. Genomic instability in cancer: teetering on the limit of tolerance. Cancer Res 2017; 77: 2179–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Jamal‐Hanjani M, A'Hern R, Birkbak NJ, et al. Extreme chromosomal instability forecasts improved outcome in ER‐negative breast cancer: a prospective validation cohort study from the TACT trial. Ann Oncol 2015; 26: 1340–1346. [DOI] [PubMed] [Google Scholar]
- 201. Aguadé‐Gorgorió G, Solé R. Genetic instability as a driver for immune surveillance. J Immunother Cancer 2019; 7: 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Magrangeas F, Avet‐Loiseau H, Munshi NC, et al. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 2011; 118: 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Hirsch D, Kemmerling R, Davis S, et al. Chromothripsis and focal copy number alterations determine poor outcome in malignant melanoma. Cancer Res 2013; 73: 1454–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Rausch T, Jones DTW, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012; 148: 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Molenaar JJ, Koster J, Zwijnenburg DA, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012; 483: 589–593. [DOI] [PubMed] [Google Scholar]
- 206. Smida J, Xu H, Zhang Y, et al. Genome‐wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int J Cancer 2017; 141: 816–828. [DOI] [PubMed] [Google Scholar]
- 207. Skuja E, Kalniete D, Nakazawa‐Miklasevica M, et al. Chromothripsis and progression‐free survival in metastatic colorectal cancer. Mol Clin Oncol 2017; 6: 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Fontana MC, Marconi G, Feenstra JDM, et al. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia 2018; 32: 1609–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Rücker FG, Dolnik A, Blätte TJ, et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica 2018; 103: e17–e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Kim H, Nguyen N‐P, Turner K, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet 2020; 52: 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Koche RP, Rodriguez‐Fos E, Helmsauer K, et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet 2020; 52: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Vergara IA, Mintoff CP, Sandhu S, et al. Evolution of late‐stage metastatic melanoma is dominated by aneuploidy and whole genome doubling. Nat Commun 2021; 12: 1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018; 175: 889. [DOI] [PubMed] [Google Scholar]
- 214. Damodaran S, Berger MF, Roychowdhury S. Clinical tumor sequencing: opportunities and challenges for precision cancer medicine. Am Soc Clin Oncol Educ Book 2015; 35: e175–e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Rosenquist R, Cuppen E, Buettner R, et al. Clinical utility of whole‐genome sequencing in precision oncology. Semin Cancer Biol 2021. doi.org/10.1016/j.semcancer.2021.06.018 [DOI] [PubMed] [Google Scholar]
- 216. Macintyre G, Ylstra B, Brenton JD. Sequencing structural variants in cancer for precision therapeutics. Trends Genet 2016; 32: 530–542. [DOI] [PubMed] [Google Scholar]