

Abstract
Advances made in chimeric antigen receptor (CAR) T cell therapy have revolutionized the treatment and management of certain cancers. Currently, B cell malignancies have been among the few cancers to which CAR T cells have shown persistent and resilient anti‐tumor responses. A growing body of evidence suggests that the persistence of CAR T cells within patients following infusion is linked to the mitochondrial fitness of the CAR T cell, which could affect clinical outcomes. Analysis of CAR T cells from patients undergoing successful treatment has shown an increase in mitochondrial mass and fusion events, and a reduction in aerobic metabolism, highlighting the importance of mitochondria in CAR T cell function. Consequently, there has been recent interest and investment in approaches that focus on mitochondrial programming. In this regard, miRNAs are promising agents in mitochondrial reprogramming for several reasons: (1) natural and artificial miRNAs are non‐immunogenic, (2) one miRNA can simultaneously modulate the expression of multiple genes within a pathway, (3) the small size of a sequence required for producing mature miRNA is ideal for use in viral vectors and (4) different precursor miRNAs (pre‐miRNAs) hairpins can be incorporated into a polycistronic miRNA cluster to create a miRNA cocktail. In this perspective, we describe the latest genetic engineering strategies that can be used to achieve the optimal expression of candidate miRNAs alongside a CAR construct. In addition, we include an in silico analysis of rational candidate miRNAs that could promote the mitochondrial fitness of CAR T cells.
Keywords: CAR T cell, glycolysis, metabolic reprogramming, microRNA, mitochondrial fission
This article discusses the potential use of microRNAs to reprogram chimeric antigen receptor (CAR) T cell metabolism. We provide rationally selected target genes, the miRNAs regulating them, and the criteria for selecting miRNAs. We also describe the genetic constructs to overexpress miRNAs within CAR in T cells.

INTRODUCTION
Advances in cancer therapy have benefited from understanding the complexities of cellular and molecular biological processes, both in terms of the improvements to traditional treatments such as chemo‐ or radiotherapy, as well to more recent treatments such as immunotherapy. Immunotherapies seek to facilitate or engineer the patient’s immune cells to identify and destroy cancerous cells within the body. In this regard, T cells are the primary engineering target of many immunotherapeutic targets owing to their native functions in targeting antigen‐displaying cells for destruction. Chimeric antigen receptor (CAR) T cell therapy utilizes engineering of T cells to express a CAR that recognizes tumor‐associated antigens to enable destruction of the target cell. Upon recognition, activation signals are propagated through ITAMs and costimulatory domains resulting in cytotoxic effects against the target cell, as well the initiation of activation and cell survival mechanisms in CAR T cells. Generally, a CAR consists of an antigen specific single‐chain variable fragment (scFv) from a monoclonal antibody, attached to intracellular signaling domains from the T cell receptor (TCR). 1 , 2
At the time of publication, only selected hematological malignancies, such as acute lymphoblastic leukemia, chronic lymphocytic leukemia, diffuse large B cell lymphoma and multiple myeloma, have been successfully treated with CAR T cell therapy. Failure of CAR T cell therapy may be due to poor long‐term persistence and memory differentiation – factors which are critical in achieving a durable and effective response. 3 The treatment of solid tumors is further hindered by the development of a tumor microenvironment (TME) which imposes metabolic pressures and promotes the formation of dysfunctional CAR T cells as well as regulatory T cells (Treg). 4 , 5 , 6 Recent studies have described a direct link between metabolism and the mitochondrial status of CAR T cells and the effect this has on their persistence. 4 , 7 CAR T cells utilizing oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) as their main metabolic pathways as well as possessing a higher mitochondrial mass as result of mitochondrial fusion, have shown improved patient responses in clinical trials. Consequently, there has been growing interest in the metabolic reprogramming of CAR T cells to improve their efficacy. Approaches shift the metabolic pathway from glycolysis to OXPHOS/FAO or promote mitochondrial fusion by blocking fission factors have shown promising results. 7 Restricting glycolysis to favor OXPHOS and FAO can be achieved through limiting glucose uptake, 8 blocking glycolytic enzymes 9 , 10 or inhibition of the positive regulators of glycolysis. 11 , 12 Such approaches involve the use of loss‐of‐function strategies, small molecule inhibitors, or upregulation of negative regulators. 13 , 14 , 15 , 16 , 17 , 18
MicroRNAs (also known as miRNAs and miRs) are small non‐coding RNAs that regulate gene expression post‐transcriptionally. miRNA biogenesis begins with the transcription of primary miRNA (pri‐miRNA) via RNA polymerase II or RNA polymerase III in some cases. The pri‐miRNA contains 5' cap and 3' polyadenylation and is processed into precursor miRNA (pre‐miRNA) by a microprocessor complex that includes the RNase III enzyme, named Drosha, and the RNA binding protein DiGeorge Syndrome Critical Region 8 (DGCR8) within the nucleus. The pre‐miRNA is a stem‐loop structure, ~85 nt in length with a 5′‐monophosphate and a 3′‐2‐nt overhang. Exportin5 and Ran‐GTPase are responsible for the export of pre‐miRNA from the nucleus to the cytoplasm, where the loop is cleaved by the RNase III Dicer. Next, the double‐strand miRNA duplex (~20‐22 nt) is loaded into the RNA‐induced silencing complex (RISC). One of the strands from the miRNA duplex remains in the RISC complex (guide strand), while the complementary passenger strand one is ejected. Based on the direction of the guide strand in the pre‐miRNA hairpin, the miRNA genes produce −5p or 3‐p mature miRNAs. 19 , 20
MiRNAs predominantly bind mRNA through 3' untranslated regions (UTRs), resulting in mRNA degradation or translational interruption. 21 miRNAs are encoded from distinct miRNA genes or other genomic regions such as introns or exons. 19 , 20 Although most miRNAs regulate gene expression in the cytoplasm, a fraction of miRNA known as mitomiRs are imported into the mitochondria. 22 The mitomiRs have been shown to regulate the expression of genes involved in mitochondrial function and metabolic regulation. 22
T cell metabolism is dynamic and linked to function and differentiation state. Quiescent naïve T cells (TN) have minimal metabolic requirements and use OXPHOS to generate ATP. Following T cell activation, rapidly proliferating effector T cells (TEFF) undergo metabolic reprogramming, switching to glycolytic metabolism to generate both ATP and metabolic precursors needed to meet biosynthetic requirements of activation such as DNA and cell membrane synthesis. Following antigen clearance, T cells undergo contraction leaving only 5% of T cells to differentiate into long‐lived memory cells (TM) responsible for long‐term protection. TM cells utilize OXPHOS metabolism to maintain their cellular processes. 3 , 4
Complete inhibition of metabolic gene expression or function through gene knock‐out or small molecule inhibition can compromise the effector function of T cells. Deletion of glucose uptake receptor, Glut1 23 or AMP‐activated protein kinase (AMPK), 24 have been shown to diminish the in vivo expansion of T cells or to increase the number of suppressive Treg cells, respectively. In a similar fashion, blocking of pyruvate dehydrogenase kinase (PDHK), a positive regulator of glycolysis, with dichloroacetate reduces proinflammatory cytokine production and encourages Treg differentiation. 25 Therefore, due to the relationship of metabolism, cell function and cell fate, immune homeostasis and functionality must be considered when metabolic genes are targeted.
The manipulation of metabolic pathways through miRNA expression is an ideal strategy for manipulation of CAR T cell therapy for several reasons. Firstly, miRNAs fine‐tune rather than completely inhibit gene expression. A single miRNA can modulate the expression of multiple genes simultaneously within a pathway. The small sequence size required to produce mature miRNA is ideal for gene transfer as well as different precursor miRNA (pre‐miRNAs) hairpins can be incorporated into a polycistronic miRNA cluster to create a miRNA “cocktail”. 26 Finally, artificial miRNA can be produced with a lower risk of immunogenicity and unwanted off‐target effects. 26
Few studies have investigated the use of miRNA in CAR T cell therapy; therefore, we will discuss potential miRNAs that can be used to enhance CAR T cell function through metabolic reprogramming. We have focused on genes whose downregulation resulted in a metabolic shift toward OXPHOS/FAO as well as mitochondrial fusion were selected. The functions of target genes are involved in one of four categories: glucose uptake receptors, glycolytic enzymes, positive regulators of glycolysis and mitochondrial fission. miRNAs that have at least one target gene in all categories were identified using bioinformatic analysis alongside published data. Lastly, we discuss genetic engineering approaches to express miRNAs within CAR T cells.
CANDIDATE GENES TO TARGET FOR REPROGRAMMING CAR T CELL METABOLISM
Active metabolic pathways within T cells are linked to both their life cycle and subset differentiation. As such, the metabolic alteration can influence T cell differentiation and function. The promotion of OXPHOS/FAO metabolism or mitochondrial fusion has been shown to increase the number of TM cells and to enhance anti‐tumor activity. 7 Strategies used to induce metabolic reprogramming away from glycolysis and toward OXPHOS/FAO in T cells have included the targeting of glucose uptake receptors, 8 glycolytic enzymes, 9 , 10 metabolic regulators 13 and mitochondrial fission factors to promote mitochondrial fusion. 27 Therefore, 36 genes described here as involved in these four functions could be used as targets to identify candidate miRNAs (Figure 1). It is worth noting that not all receptors or enzymes have a high expression level, or sometimes they are dispensable with their other family members in T cells, and therefore their inhibition may have little or no effect on T cell metabolism. Thus, out of these 36 genes, 20 of them are only high‐value targets that have a predominant function in T cells or express at a higher level (discussed below).
Figure 1.
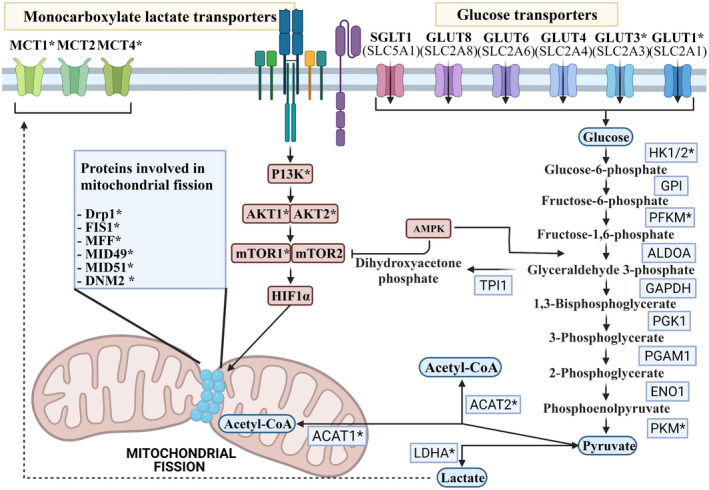
Schematic of T cell metabolic processes and the candidate genes for miRNA targeting. These include glucose transporters, glycolytic enzymes, metabolic regulators and mitochondrial fission factors. * High‐value target genes in T cells. The figure was created with Biorender.com.
Glucose uptake transporters
Glucose uptake in lymphocytes is carried out by five members of the GLUT family of non‐concentrative glucose carriers (Glut 1, 3, 4, 6 and 8) and the Na+‐coupled glucose carrier SGLT1 28 (Figure 1). Glut1 and Glut3 are the main glucose uptake transporters in T cells among these transporters, 29 but others may be expressed following T cell activation or insulin stimulation. 30 , 31 , 32 , 33 Downregulation of Glut1 via overexpression miR143 in Her2‐CAR T cells was shown to increase the number of central memory (TCM) CD8+ T cells, as well as to boost effector function as a result of metabolic reprogramming. 8
Glycolysis enzymes
Glycolysis begins with the phosphorylation of glucose and ends with the conversion of phosphoenolpyruvate to pyruvate through a sequence of enzymatic reactions (Figure 1). The restriction of glycolysis through the targeting of glycolytic enzymes or positive regulators, favors the differentiation of TM subsets and improves the anti‐tumor activity of T cells. 4 , 7 , 9 , 10 Such an effect is seen via inhibition of the first glycolytic enzyme, hexokinase‐1 and 2 (HK) using 2DG, which enhances CD8+ TM cell formation by shifting metabolism toward OXPHOS. 10 In contrast, upregulation of the glycolytic enzyme phosphoglycerate mutase‐1 (PGAM1) diminishes the development of TM cells. 10 It should be noted that only three reactions are rate‐limiting and irreversible among the ten steps of glycolysis. 34 The three key rate‐limiting enzymes include HK, phosphofructokinase 1 (PFKM) and pyruvate kinases (PKM). 34 Changing the level and activity of the reversible steps in glycolysis does not determine the direction of the pathway and is unlikely to have a significant effect on glycolysis. 35
Metabolic regulators
Several metabolic regulators have been recognized in T cells. A metabolic shift toward OXPHOS/FAO metabolism, mitochondrial fusion and TM cell development occurs when these regulators are inhibited. These regulators include mammalian target of rapamycin (mTOR), 13 , 36 , 37 AMP‐activated protein kinase (AMPK), 38 phosphoinositide 3‐kinase (PI3K), 11 , 12 , 17 , 39 , 40 , 41 hypoxia‐inducible factor 1‐alpha (HIF1A), 42 basic leucine zipper ATF‐like transcription factor (BTAF), 43 , 44 lactate dehydrogenase A (LDHA), 45 , 46 monocarboxylate transporters (MCT 1, 2 and 4), 9 AKT serine/threonine kinase (AKT1 and 2) 47 and Acyl‐CoA cholesterol acyltransferase (ACAT1 and 2) 14 , 16 (Figure 1).
Some of the metabolic regulators have a controversial function in T cell development. For instance, deletion of the AMPK gene can cause defective CD8+ TM generation, or an increased level of AMPK promotes the T fitness, expansion and formation of TM cells. 48 , 49 Recently, Mayer et al. showed that AMPK deficiency does not affect T cell fate, clonal diversity, the number of activated T cells and survival in vivo, rather it reduces the magnitude of T cell activation, expansion and protein translational capacity. 50 Conversely, others showed that reducing AMPK signaling via miR17‐92 (indirect effect) or shRNA targeting of AMPK promotes metabolic reprogramming to aerobic glycolysis and restores T cell proliferation in senescent cells. 38 , 51 We omitted AMPK in the high‐value target group due to the lack of research using knockdown approaches, but we cannot rule out the possible positive effects of miRNA targeting of the AMPK based on the scant data available.
Free cholesterols are enriched in microdomains known as lipid rafts in the plasma membrane. In T cells, TCRs and associated signaling molecules cluster at lipid rafts and disruption of lipid rafts impair the TCR signaling and T cell effector functions. 52 ACAT1 and ACAT2 esterify free cholesterol to be stored in the cytoplasmic lipid droplets. Within 6 hours after activation of CD8+ T cell, the ACAT1 mRNA level shows overexpression, whereas the ACAT2 takes 24 hours for an increased level in mRNA. 14 The mRNA level of ACAT1 is nearly 20 times higher in CD8+ T cells and the protein level of ACAT2 is very low. 14 In addition, virus‐specific T cells primarily expressed ACAT1 rather than ACAT2. 53 Deletion of ACAT2 does not affect CD8+ T cell function, suggesting that ACAT1 is the main enzyme in cholesterol esterification in T cells. 14 Inhibition of ACAT1 by small molecules or gene deletion studies showed an increase in effector functions of CD8+ T cells with an increase in TEM cells. 14 , 52 , 53 CAR T cells treated with siRNAs against ACAT1 showed higher cytotoxicity, secretion of proinflammatory cytokines and enhanced tumor regression in vivo. 16 Although we excluded ACAT2 in our high‐value target group, it should be noted that the ACAT2 mRNA level in CD4+ T cells is ≥ 2 fold higher that that in CD8+ T cells. 14 Moreover, the positive impact of ACAT1 deletion is restricted to CD8+ T cells suggesting that ACAT2 may compensate for the ACAT1 lost in CD4+ T cells. 14 , 54 More studies are needed to uncover the function of ACAT2 in CD4+ T cells.
Activation of T cells through TCR engagement, costimulatory molecules or IL‐2 stimulation leads to activation of PI3K. 55 PI3K activates AKT and promotes mTOR signaling (Figure 1). 55 PI3K orchestrates with mTOR and AKT to promote T‐cell glycolytic metabolism and differentiation toward short‐lived TEFF cells, making PI3K an attractive target to enhance the quality of CAR T cell production. So far, several studies have shown that inhibiting PI3K in CAR T cells improves T cell expansion, anti‐tumor activities in vitro and in vivo, reduces the expression of exhaustion markers, TM phenotypes. 11 , 12 , 17 , 39 , 40 , 41
Accumulating evidence suggests the positive effects of the AKT inhibition pathway on the CAR‐T cell performance. 15 , 47 , 56 , 57 Treating the epithelial cell adhesion molecule (EpCAM)‐CAR T cells with an AKT inhibitor, MK2206, promotes CAR T antitumor activity in vivo and increases CAR T cell expansion and the number of TM cells. 15 The treatment was carried out 2 days post‐transduction concurrently with CD3/CD28 stimulation and continued only 3 days after transduction. Interestingly, the authors showed that pre‐treating with AKT inhibitor increases the transduction efficacy of T cells due to upregulation of low‐density lipoprotein receptor that serves as a cellular receptor for the lentivirus with a VSVg envelope. 15 In addition, continuous culture of CAR T cells with AKT inhibitors also showed positive effects in CAR T cells, including lowering the level of glycolysis enzymes and MCT4, while FOXO1‐dependent target genes such as IL7R, KLF4, CD28, ICOS and CD95 showed upregulation. 47 Similar results were obtained where 40% of CAR T cells treated with an AKT inhibitor co‐expressed CD28 and CD62L compared with 10% in untreated CAR T cells. 56
mTOR regulates the T cell function and differentiation, and targeting mTOR has negative and positive impacts on T cells. The immunosuppressive effect of mTOR blockage by small molecule or gene deletion reduces T cell proliferation and increases the generation of non‐functional TM population and CD4+ Treg cells. 58 , 59 Conversely, lowering the level of mTOR by aptamer‐targeted siRNA, IL‐15 treatment or a low level of rapamycin treatment (20 nM vs. 100 nM) enhanced TM phenotypes and antitumor activity of CAR T cells. 13 , 36 , 37 This highlights the potential of knockdown approaches, such as miRNA, for reducing the mTOR level rather than completely abolishing its activity.
Targeting lactate transporters (MCTs) to restrict glycolysis has also been investigated in the context of immunotherapy. T cells express three monocarboxylate transporters, MCT1, 2 and 4. 60 However, studies showed that MCT1 and MCT4 are the primary lactate transporter in T cells. 61 Upon T cell activation, MCT1 expression peaks at 12 hours while the MCT4 level induces with a delay, sometimes between 48 and 72 hours post‐stimulation. 61 Blocking MCT1 and MCT4 seems a safe approach for reducing glycolysis and improving immunotherapy without compromising the anti‐tumor activities of T cells. 9 , 62 , 63 , 64
HIF‐1, a member of the HIF transcription factor family, binds hypoxia response element (HRE) in the genome. Activation of HIF1 triggers a transcriptional program resulting in the adaptation of cells to the low oxygen level in hypoxia condition by minimizing oxygen consumption via promoting glycolytic program. 65 HIF‐1 directly upregulates the expression of several glycolytic enzymes, LDHA, pyruvate dehydrogenase kinase 1 (PDK1), shifting the metabolic program away from TCA to glycolysis to generate ATP. 65 In combination with the PI3K‐AKT‐mTOR pathway following T cell activation, HIF‐1 is a key modulator in the transition to glycolysis in T cells. 42 HIF‐1 is critical for T cell's effector functions and promotes differentiation of T cells toward terminally differentiated TEFF cells. 42 Genetic deletion of HIF‐1 in T cells impaired acquisition of effector function. 66 , 67 Meanwhile, persistently elevated levels of HIF‐1 cause lethal immunopathology due to the augmented effector capacity of CTLs. Plus, constitutive overexpression of HIF‐1 results in the upregulation of the exhaustion markers (e.g. PD‐1, CTLA‐4, LAG‐3 and TIM3) while T‐bet, Emoes and TCF‐1 are downregulated. Therefore, due to the variety of HIF‐1 target genes, it seems wise to use knockdown approaches to reduce the HIF‐1 in T cells for the purpose of metabolic reprogramming.
Mitochondria have several functions within T cells which are vital for the elimination of cancer. These functions include energy generation, T cell activation, biosynthesis, cell fate, cellular survival and cellular migration. 4 Mitochondrial morphology refers to the fused or fragmented state of the mitochondria within a cell. The morphological state of the mitochondria is coupled with its function in T cells and as such different T cell subsets possess different mitochondrial morphologies which best suit their function. Mitochondrial fission factors include dynamin‐related protein 1 (Drp1), fission mitochondrial 1 (FIS1), mitochondrial fission factor (MFF), mitochondrial dynamics protein of 49 Kda (MID49), mitochondrial dynamics protein of 51 Kda (MID51) and dynamin 2 (DNM2). 68
Potential miRNA candidates
We will discuss miRNAs identified using DIANA‐microTCDS and TarBase v.8, as well as published miRNAs experimentally verified through qRT‐PCR, western blot and reporter assay (Supporting Information 1). There are 455 miRNAs with a 7 ‐ 8‐mer match in their seed sequence to the 36 target genes involved within the categories mentioned above. Of the 36 target genes, only five genes, Glut6, SGLT1, BTAF, FIS1 and MID49, had fewer than ten potential miRNAs. As mentioned before, 20 target genes are either highly expressed in T cells, or have a crucial function in T cell metabolism, making them more suitable as miRNA targets (Supporting Information 1; Figure 1).
In general miRNAs often target more than one gene. The implication of this is critical when determining which miRNA to overexpress within CAR T cells to prevent unintended gene regulation. In our analysis, approximately two‐thirds of identified miRNAs modulate more than two target genes (Figure 2). Potentially, 22 miRNAs have ≥ 10 target genes and 59 of them had at least one target within all gene categories (Supporting Information 1; Figure 2).
Figure 2.
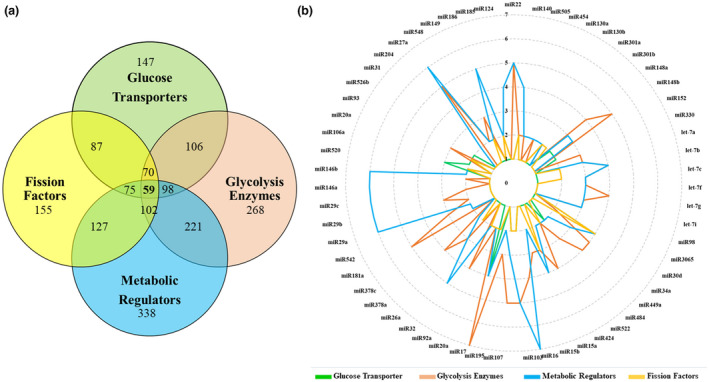
Most of the identified miRNAs have shared targets. (a) The Venn diagram shows the number of miRNAs identified for each category of genes and the number of shared miRNAs between groups. (b) The radar chart illustrates the 59 miRNAs with at least one target gene in each group of genes. The circular lines represent the number of target genes that a miRNA is predicted to target in each category. TEFF cells have fragmented mitochondria that utilize glycolytic metabolism, while TN and TM cells have fused mitochondria which use OXPHOS to generate ATP. Therefore, miRNAs that target glycolytic enzymes might be under‐expressed in TEFF cells. To determine this, we looked at the miRNA profile expressed during TN → TEFF → TM stages of development based on published data. 75 , 79 , 115 A comparison of our identified miRNAs with the miRNA expression profile of each T cell subset found 12 miRNAs that are expressed at a low level in TEFF cells, whilst being upregulated in TM cells. These miRNAs have several targets among genes involved in glucose uptake, glycolysis and mitochondrial fission (Table 1).
Table 1.
Changes in miRNA expression of T cells during TN → TEFF → TM development. These miRNAs potentially target several genes involved in glycolytic pathway and mitochondrial fission.
| miRNA | Targets |
|---|---|
| miR15a | Glut3, HK1, ALDOA, PKM, PI3K, AKT1, AKT2, Drp1, FIS1 & DNM2 |
| miR15b | Glut3, HK1, ALDOA, PKM, PI3K, MCT1 & Drp1 |
| miR26a | Glut3, TIP1, GAPDH, PGK1, ACAT2, AMPK, MCT1, MFF & MID51 |
| miR26b | Glut3, TIP1, PGK1, LDHA & AMPK |
| miR146a, b | Glut3, ALDOA, PGK1, AMPK, AKT1, MCT4, HIF1A & DNM2 |
| miR101 | TIP1, mTOR, MCT, MFF & DNM2 |
| Let‐7f | Glut3, ALDOA, HK2, PGK1, AMPK, AKT2, MCT4 & MID51 |
| miR142 | Glut3, GPI, PFKM, ALDOA, PI3K, AKT2, MCT2 & HIF1A |
| miR150 | Glut3, GAPDH, PKM, LDHA & MCT1 |
| miR16 | Glut4, HK1, ALDOA, PGK1, PKM, LDHA, AMPK, PI3K, MCT1, MCT2, MCT4, HIF1A & Drp1 |
| miR29a | Glut3, GPI, ALDOA, PI3K, AKT2, MCT1, 2, 4, HIF1A & MID51 |
It is not within the purview of this paper to elucidate the functions and implications of all miRNAs identified, but previous research has investigated the function of miR146a and miR29 both of which have target genes within all gene groups. 69 The expression of miR146a is upregulated following TCR stimulation to support the TM establishment. miR146a has minimal expression in TN cells but increased expression in effector memory (TEM) and TCM cells. 69 The role of miR146a in TM development is unknown, though targeting IL‐2 production has been proposed as a mechanism. 69 In addition, ectopic expression of miR146 protects T cells from activation‐induced cell death (AICD) by directly targeting the Fas‐associated death domain (FADD). 69 In our analysis, miR146a has seven target genes (Glut3, PGK1, AMPK, AKT2, MCT4, HIF1A and DNM2) which make it among the few miRNAs that have at least one target in all gene groups (Supporting Information 2; Figure 2b).
The miR29 family is amongst the most highly expressed miRNAs in TN and TM populations. 70 The miR29 family includes three members: miR29a and miR29b located at chromosome 7 and expressed as a polycistronic primary transcript, whereas miR29c is located in chromosome 1. The expression of all miR29 members is upregulated in response to IL‐21, a cytokine used to support CAR T cell expansion and to increase the TCM and TSCM development. 71 Overexpression of miR29 in CD8+ T cells reduces the TEFF cell number while boosting the frequency of TM cells. 72 Three members of the miR29 family were among the miRNAs with the highest target number in our analysis (≥ 10 targets, Supporting Information 1). In addition, miR29 has at least one target in all gene groups (Supporting Information 2). However, the role of miR29 in T cell subsets is controversial due to the potential targeting of TBET, Eomes and IFN‐γ. 72
It is always possible that selected miRNA may have undesirable off‐targets, particularly in regulating the expression of genes involved in effector functions and master regulators of TM development. Due to the vast number of potential genes, transcription factors and regulators involved in such processes, it is time‐consuming to screen via bioinformatic tools. However, if researchers choose to perform screening prior to the functional assays, software such as DIANA‐microT‐CDS and TargetScan are helpful in providing extensive lists of potential target genes. A single complementary sequence with a weak binding (< 7‐mer) in a distal 3'UTR site may not be considered off‐target. In contrast, multiple complementary sequences throughout the 3'UTR, or a strong binding site in the proximal region of 3'UTR (> 7‐mer, with full seed complementary) will likely be considered off‐target.
We instead propose that it is more relevant to screen for deleterious off‐targets by monitoring CAR T cell function following miRNA overexpression. It is arguably less relevant to be concerned about off‐target effects of miRNA that still yield the desired outcomes. Endpoints must be carefully selected to predict maximal clinical effects. Such desirable endpoints should include enhanced anti‐tumor effects against a range of blood and solid cancers, as well as optimal T cell longevity and memory cell formation. To this end, an approach of using cluster pooled miRNA is particularly helpful. If the high number of potential miRNAs will be tested (such as all the proposed 59 miRNAs), we suggest using them in the backbone of miRNA clusters, for example based on a miR17‐92 backbone (see Figure 5). Hence, only < 10 final constructs will be tested rather than ~60 constructs. Observing any negative effects within individual clusters makes it easier to narrow an analysis down to find the culpable miRNA.
Figure 5.
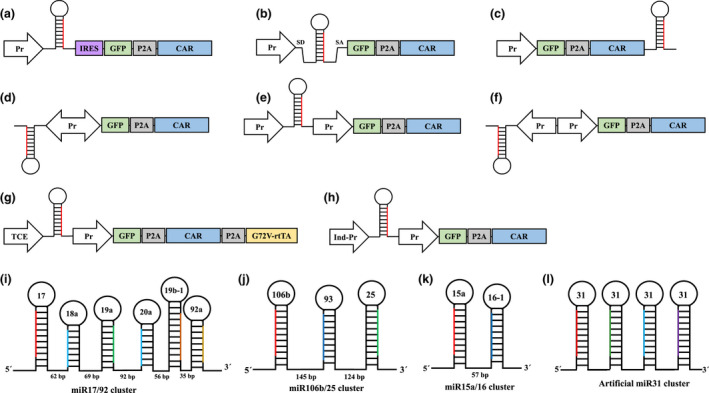
Strategies to express miRNA in CAR T cells. One single promoter drives both miRNA and CAR genes in a, b, c & d strategies. (a) A promoter derives miRNA and green fluorescent protein (GFP)‐P2A‐CAR coding sequence and internal ribosome entry site (IRES) sequence enhance the translation, (b) Intronic miRNAs use alternative splicing for maturation result in 5'UTR of mRNA vital for preserved translation, (c) placing miRNA downstream of CAR sequence, (d) bidirectional promoters to express miRNA and GFP‐P2A‐CAR with a single promoter. Alternatively, miRNAs and GFP‐P2A‐CAR can be expressed separately using two promoters in (e) uni‐directional or (f) reverse‐orientation fashion. Controlled miRNA expression using (g) Tet‐On system with G72V‐rtTA or (h) an auto‐inducible promoter. Clusters of natural miRNAs to express (i) six miRNAs, (j) three miRNAs, or (k) two miRNAs. (l) Structure of artificial polycistronic miR31 by joining several repeats of pri‐miR31 sequences.
miRNAs that translocate to the mitochondria are referred to as mitochondrial miRNAs or mitomiRs. The mitomiRs have been shown to influence various mitochondrial functions such as OXPHOS, TCA, lipid and amino acid metabolism and Ca2+ homeostasis by targeting mitochondrial transcripts or nuclear‐encoded genes inside the mitochondria. 22 , 73 The mitomiRs can cause metabolic reprogramming by regulating gene expression at the pre‐translational level. 74 So far, several human mitomiRs and their targets have been recognized. 73 There are around 60 potential mitomiRs that are predicted to alter gene expression and mitochondrial activity (Supporting Information 1). Most of these mitomiRs have several potential target genes, with at least one target in each of our target categories (Supporting Information 2). Such miRNAs may have a more direct role in metabolic and mitochondrial regulations.
CONSIDERATIONS WHEN SELECTING miRNAS FOR EXPRESSION IN CAR T CELLS
In T cells, miRNAs are precisely regulated during the lifespan and during subset differentiation, therefore selection of miRNA for overexpression within T cells should take into account not only the potential of multiple target genes, but also the timing and magnitude of miRNA expression. A large number of target genes are targeted by miRNAs belonging to families such as miR17‐92 and miR15‐16 (Supporting Information 1). The use of these families is appealing as they not only target genes of interest but they also express polycistronic clusters that make it easier to express multiple miRNAs with a single DNA cassette. However, previous studies have shown that continuous expression of these families might impair TM development. 75 , 76 For example, exogenous upregulation of miR15‐16 family members restricts the TM development by downregulation Eomes and CD127. 77 Moreover, only a transient expression of miR19‐72 during expansion time is required for normal memory formation. 75
Another example of the temporal regulation of miRNAs during TM development is the let‐7 family. It has been shown that the let‐7 family is expressed in TN cells, downregulated in TEFF cells and re‐expressed in TM cells. 78 , 79 Downregulation of the let‐7 family during the expansion phase is necessary for T cell proliferation and expansion. 80 Loss of let‐7 increases T cell proliferation and effector function while it also promotes differentiation of terminal effector cells, mitochondria fission and AICD in T cells. 78 , 80
Many studies investigating the role of miRNAs in T cell function utilize murine models for infection settings. Whilst conserved miRNAs tend to have comparable targets and functions both in humans and mice, some miRNAs are divergent in both their function and targets. For example, in the murine T cell model, the downregulation of miR17‐92 after the initial expansion phase is necessary for TM development. 75 In contrast, data from human T cells showed that there is continued expression of several members of the miR17‐92 in human TM phenotypes, including, miR20a, miR19b and miR92 which are preferentially expressed in human CD8+ TCM cells. 81 In addition, miR17‐92 clusters were experimentally verified to increase T cell survival and persistence by downregulating the proapoptotic protein Bim. 82
The expansion of CAR T cells is not an equivalent process to the expansion seen in physiological T cells. 3 Under physiological conditions, following antigen triggered TCR activation, T cells differentiate to TEFF cells which then expand to a higher number. After antigen clearance, TEFF cells undergo a contraction phase as a result of AICD leaving only ~5% of cells as a potential pool to differentiate to TM cells. The environmental conditions contributing to each physiological T cell expansion event is influenced by surrounding immune cells that produce a unique combination and concentration of cytokines and other activating molecules to influence T cell response and cell fate. In contrast, CAR T cells undergo expansion both in vitro and in vivo. The initial expansion involves isolated patient T cells which are activated in the absence of antigen via CD3 and CD28 antibodies for a period of 2 to 3 days. During this time, T cells are also cultured with one or more gamma‐chain cytokines such as IL‐2, IL‐7, IL‐15 and IL‐21 to support the homeostatic proliferation of T cells. Currently, only IL‐2 has the FDA approval for the CAR T cell therapy, although IL‐7/IL‐15 and IL‐21 have been studied in some clinical trials and pre‐clinical studies. 83 , 84 After transduction, commonly using retro‐ or lentiviral vectors, T cells are expanded with gamma‐chain cytokines for another ~10 days to provide sufficient numbers for infusion. 1 , 3 In other words, in the absence of antigen, the expansion phase is continued in vitro by controlling the media, cell number and cytokine treatment (Figure 3).
Figure 3.
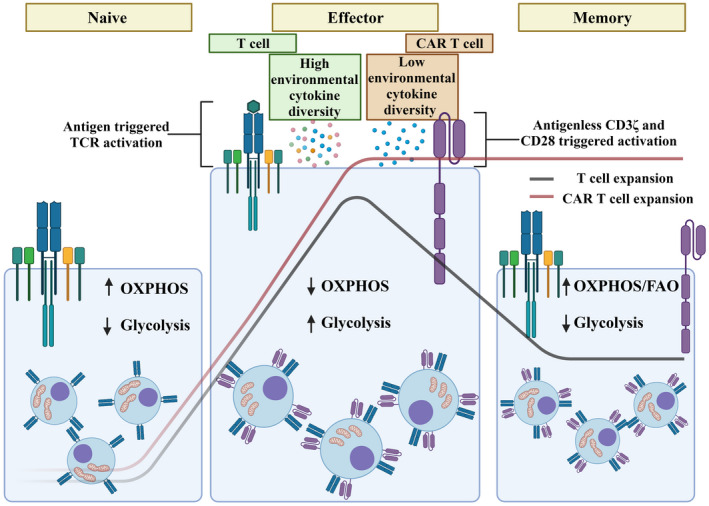
Physiological T cell activation and expansion is distinct from CAR T cell culture. The mitochondria within naïve T cells utilizes a OXPHOS and FAO which is reprogrammed when T cells are activated through antigen presentation and a diverse range of cytokines. Following activation effector T cells possess fragmented mitochondria with a glycolytic metabolism to facilitate effector function. Contraction (the green line) of these effector T cells into a memory population once again reprograms the mitochondria toward OXPHOS and FAO. In contrast CAR T cells are activated through CD3 and CD28 antibody stimulation in the presence of one or more cytokines. Furthermore, CAR T cell expansion is maintained, without allowing the natural contraction of the population (the red line). The figure was created with Biorender.com.
Further variations between CAR T cell expansion and physiological T cell expansion include the composition of the CAR T cell being implemented. There are several CAR T cell generations, predominantly categorized based on the number of costimulatory domains. Second‐generation CAR T cells have CD3ζ with only one costimulatory domain, while third‐generation CAR T cells are composed of CD3ζ with two costimulatory domains. 1 Each costimulatory domain confers different functions to the CAR T cell and promotes different metabolic programs. 1 CD28, ICOS and OX40 costimulation results in a more pronounced glycolytic phenotype. 1 whilst CD137 (4‐1BB) promotes less efficient glycolysis while more efficiently enhancing mitochondrial respiration and fusion. 85 Furthermore, cytokines used during ex vivo expansion impact metabolism and mitochondrial functions. For example, IL‐2 drives T cells toward effector‐like phenotypes and a metabolic program characterized by enhanced glycolysis. 86 Whilst CAR T cells expanded under IL‐15 have a higher mitochondria mass, spare respiratory capacity (SRC) and FAO metabolism. 13 The implications of the expansion conditions on the efficacy of miRNA action in CAR T cells has not been thoroughly investigated; however, the effect of these conditions on the metabolic state of the CAR T cell should be considered when choosing miRNA for overexpression.
It should also be noted that T cell stimulation is sustained in CAR T cells due to CAR tonic signaling in an antigen‐independent manner. 87 Continuous tonic signaling from different costimulatory domains will impact CAR T cell metabolism and the anti‐tumor function of CAR T cells. 88 Therefore, the overexpression of a miRNA may exhibit differing effects based on the CAR design and have a different effect on murine or human T cell models.
The abundance of target gene transcripts, as well as the number of miRNA binding sites influence the effectiveness of miRNA. It has been suggested that miRNAs set a threshold in which, at a low level of target transcript, miRNAs act as a “switch” to repress the gene expression (Figure 4a). When the target gene transcript levels are high, miRNAs act instead as a fine‐tuner. 89 , 90 Interestingly previous research has shown that when the abundance of target gene transcripts is low, the presence of two and seven miRNA binding sites in the 3'UTR resulted in 2‐fold and 10‐fold reporter inhibition, respectively. 89 However, when target gene transcripts levels are high, regardless of the number of complementary binding sites, the gene repression stayed still at 2‐fold. 89 Therefore, it would be expected that glycolytic inhibiting miRNAs would act as a fine‐tuner in TEFF cells where the glycolysis is at its highest. This might be an advantage of using miRNAs since abolishing glycolysis negatively affects the effector function and tumor killing ability of T cells. 66 , 67
Figure 4.
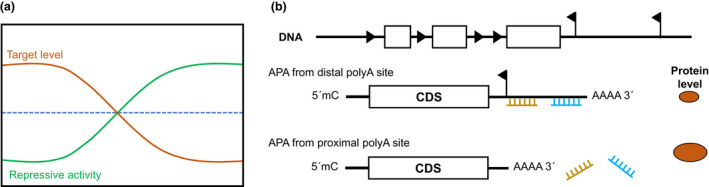
(a) miRNAs are more potent in repressing genes when the target gene is expressed below the miRNA threshold. (b) miRNA binding sites may be lost due to alternative polyadenylation (APA) mechanisms. This attenuation of miRNA sites can affect the protein expression of the target gene. Potential alternative poly‐A sites of 36 candidate genes using 3' end sequencing data 114 may be predicted by APAatlas 93 (Supporting Information 3). Therefore, the selection of miRNAs with several binding sites for expression within CAR T cells, is crucial to circumvent the possible remove of target regions by alternative poly adenylation sites.
Lastly, the location of the miRNA complementary binding sequence within the 3'UTR also influences the inhibition of the target. It is well known that many genes have different mRNA isoforms that vary in their 3'UTR length. In addition, genes in cells with higher proliferation (such as T cells during expansion) tend to have a shorter 3'UTR due to proximal polyA site usage caused by alternative polyadenylation (APA). 91 The 3'UTR harbors regulatory sequences including miRNA binding sites and AU‐rich elements that negatively regulate gene expression. 92 Hence, a longer 3'UTR is more likely to possess an abundance of miRNA interacting elements (Figure 4b). The length of 3'UTRs vary among tissues, genes in tissue such as the brain tend to have longer 3'UTR, while genes in blood cells prefer isoforms with shorter 3'UTR. 93 It is therefore possible that miRNA used to inhibit a gene within hepatocytes may not be able to inhibit the same gene within T cells.
STRATEGIES TO OVEREXPRESS miRNA IN CAR T CELLS
The primary miRNA (pri‐miRNA) sequence along with flanking can be expressed under RNA polymerase II promoters to produce mature miRNAs. 19 , 20 Following transcription, pri‐miRNA is cleaved by Drosha to produce a ~85 bp stem‐loop structure known as the pre‐miRNA. A subset of intronic miRNAs produces pre‐miRNA during RNA splicing which is independent from Drosha. 94 The DNA sequence necessary for optimal Drosha cleavage contains pri‐miRNA and a flanking region, which vary in size depending on the miRNA. 95
The overexpression of miRNA within CAR T cells can be achieved through a variety of strategies. The use of dual transduction to express a miRNA and a CAR within a T cell involves the use of two separate viral vectors, one containing the CAR and a reporter gene such as green fluorescent protein (GFP), whilst the other contains the miRNA and a second reporter gene such as red fluorescent protein. 96 This approach is costly, time consuming and, due to the use of multiple reporters to identify successful dual‐transduced T cells, limits the available fluorescent channels for phenotypic analysis by flow cytometry. 97 Due to these disadvantages, we will focus on alternative approaches which involve the selected miRNA and CAR being encoded within a single DNA cassette.
As detailed in Figure 5, these strategies involve the use of either a single or dual promoter construct within a lenti/retroviral or transposon gene transfer system. The main advantage of single promoter constructs is the reduced size of the overall cassette, which increases the efficiency of gene transfer. 98 Additional promoters within viral vectors decreases the viral titration and therefore efficacy. 97
miRNA can be positioned upstream or downstream of the CAR coding sequence. However, placing a miRNA at 5' of the mRNA diminishes the expression of coding gene, as pri‐miRNA cleavages by Drosha in the nucleus removes the 5'mC from mRNA necessary from mRNA exportation to cytoplasm. 99 It has been shown that placing the internal ribosome entry site (IRES) sequence upstream of the coding region (Figure 5a) leads to optimal production of miRNA and translation of the coding gene. 100 However, some reports still showed sporadic reporter gene expression in this design. 99 Alternatively, intronic miRNA can be used to preserve the 5'UTR after miRNA excision (Figure 5b). Several known single or cluster intronic miRNAs are included in our identified miRNAs. Using intronic miRNAs yields a high level of mature miRNA and coding protein. 101 However, miR26b and miR208a may induce lower exon ligation leading to a low protein level of coding genes. 101 Lastly, positioning miRNA at the 3' end of mRNA (Figure 5c) also produces a sufficient level of both coding gene and miRNA simultaneously. 99
Bidirectional promoters may be used to express miRNAs and a CAR with a single, compact promoter (Figure 5d). Recently, we and others have shown that several commonly used human and viral promoters have bidirectional activities in human cell lines. 102 , 103 Synthetic bidirectional promoters also can be used by fusing two minimal promoters back‐to‐back, or by duplicating TATA and other core elements in the reverse direction. 104 This might be useful when one direction is more robust and therefore allows a control over the expression level of miRNA or CAR.
Dual‐promoter constructs have been used widely to express two GOI where both promoters are constitutive (Figure 5e, f) or inducible (Figure 5g, h). In a constitutive manner, we have compared the function of EF1 (driving CAR and GFP) and hPGK (driving miR429) in both uni‐ or reverse directions in the lentiviral system. Although both orientation CAR and GFP expression were similar, the level of mature miR429 was slightly higher in the reverse orientation (Unpublished data Rad SMAH and McLellan AD 2020).
For a controlled expression, inducible promoters can be utilized either through drug or auto‐inducible promoters. The tetracycline inducible system is one of the tightest rheostats for controlling gene expression in mammals (Figure 5g). This system is composed of two elements, the TCE (tet‐responsive) promoter and the rtTA (reverse tetracycline‐controlled trans‐activator). In the presence of tetracycline or doxycycline, conformational changes in rtTA make it able to bind and drive the transcription from the TCE promoter. Recently, our group developed a Tet‐On system for CAR T cell applications. We showed that introducing a G72V mutation in rtTA (G72V‐rtTA) described previously for yeast, 105 significantly enhanced the Tet‐On system function in large gene cassettes containing a CAR. 106 Such a system might be beneficial when the expression of a miRNA needs to be regulated during T cell differentiation. For instance, let‐7 has an increased expression within TN and TM cells whilst is downregulated in TEFF cells. The downregulation of let‐7 during expansion is necessary for TM development. 75 , 76 , 80
There have been several auto‐inducible promoters investigated in T cells with potential use in CAR T cell therapy. Nuclear factor of activated T‐cells (NFAT), nuclear receptor subfamily 4 group A member 1 (NR4A1) and CD69 promoters are the leading examples of auto‐inducible promoters. 107 The activity of these promoters depends on the activation status of T cells; ON when T cells are engaged with antigen and activated, OFF when T cells are in resting condition. These promoters are ideal when the expression of GOI or miRNA is needed only during activated T cells (Figure 5h).
Finally, a cocktail of miRNAs can be used to inhibit the expression of several genes by using natural or artificial polycistronic miRNAs (Figure 5i–l). Because miRNA families tend to have shared seed sequences and common targets, mature miRNA sequences could replace the natural miRNAs. It should be noted that these replacements should not change the nucleotide compositions critical for the miRNA maturation process. Further sequence optimization may be applied to achieve a desirable level of mature miRNAs. 101
Interestingly, the degree of processing individual miRNAs within the cluster might be distinct, which gives the advantage of a less inhibitory effect on sensitive targets. For instance, in the miR19‐72 cluster (Figure 5i), miR17, miR19a and miR20a have a higher level of mature miRNA than miR18a, miR19b and miR92a. 108 Examples of natural miRNA clusters with potential application in CAR T cells are shown in Figure 5i–k. Multimeric miRNA systems have been used to inhibit HIV‐1 and HCV replication by replacing the miRNA sequence with small interfering RNAs (siRNA) sequences in the miR17‐92 backbone. 109 , 110 A more significant intrinsic inhibitory activity with multimeric miRNA was achieved compared with conventional short hairpin (shRNA) design. 109 Artificial polycistronic miRNAs by joining several repeats of a single pri‐miRNA sequence and is another way to get a ubiquitous inhibition effect on all targets (Figure 5l). 110
CONCLUSION
Metabolism impacts on T cell function and differentiation. Promoting T cell metabolism toward OXPHOS/FAO and mitochondrial fusion has been shown to improve the TM differentiation and anti‐cancer effects of CAR T cell therapy. However, complete inhibition of gene expression or activity can compromise the effector function of T cells. In this regard, miRNA‐mediated gene downregulation offers an alternative strategy to boost metabolically reprogramming of CAR T cells toward a fitter mitochondria and metabolism. However, our understanding of the function of miRNAs in T cell metabolism remains minimal and therefore, there is need for identification and characterization of miRNA functions in both T cells and CAR T cells. In this study, we identified potential miRNAs that target the genes involved in glycolytic metabolism and mitochondrial fission. We focused our analysis on miRNAs that are conserved and experimentally validated. However, miRNA acts in a cell and tissue specific manner, therefore the tissue or cells used in their validation must be considered. In addition, the differences between CAR T cells and native T cells adds to the complexity of the miRNA functions. The location of the miRNA complementary sequence in 3'UTR is another consideration and target sequences in proximal regions are less likely to be affected by APA. Moreover, multiple miRNAs expressed in a clustered manner against either a common target or differing targets may be beneficial to enhance the effect of miRNA‐mediated gene modulation. Costimulatory molecules and culture condition also impact the CAR T cell metabolism; hence it is necessary to validate candidate miRNA function in the conditions relevant to their final application. Therefore, in the search for candidate miRNA or combination of miRNA candidates, we suggest that several miRNA candidates should be investigated in the context of CAR T cells.
METHODS
Potential miRNAs were identified using three bioinformatics software and verified published data. TargetScan7.2 111 was used to predict miRNAs with a high binding probability to target genes, filtering conserved miRNA with a seed match 7mer‐m8 and context++ score percentile 80%. Verified miRNAs were identified using high‐throughput techniques by DIANA‐microT‐CDS at threshold 0.7 112 and TarBase v.8 filtered for Homo sapiens, negative regulation, validated as positive and, direct validation. 113 Experimentally published miRNAs with references are provided in Supporting Information 1.
The polyA sites for each gene were collected from 3' end sequencing data available in PolyASite2.0 114 containing the location of the polyA site (chromosome, position and strand). Sites supported by more than one 3' end sequencing protocol are shown in bold.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
Seyed Mohammad Rad: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Software; Supervision; Writing – original draft; Writing – review & editing. Joshua Colin Hosseini Halpin: Data curation; Formal analysis; Methodology; Writing – original draft; Writing – review & editing. Supannikar Tawinwung: Conceptualization; Formal analysis; Project administration; Supervision; Writing – review & editing. Koramit Suppipat: Conceptualization; Investigation; Resources; Supervision; Writing – review & editing. Nattiya Hirankarn: Conceptualization; Funding acquisition; Methodology; Project administration; Resources; Supervision; Writing – review & editing. Alexander Donald McLellan: Conceptualization; Formal analysis; Funding acquisition; Investigation; Project administration; Resources; Supervision; Writing – original draft; Writing – review & editing.
FUNDING
This research was funded by the Royal Society of New Zealand Marsden Fund, grant number UOO1806. This research project also was supported by the Second Century Fund (C2F), Chulalongkorn University.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Contributor Information
Seyed Mohammad Ali Hosseini Rad, Email: a.hoseini.rad@gmail.com.
Nattiya Hirankarn, Email: Nattiya.H@chula.ac.th.
Alexander D McLellan, Email: alex.mclellan@otago.ac.nz.
REFERENCES
- 1. Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol 2019; 8: e1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang C, Zhuang Q, Liu J, Liu X. Synthetic biology in chimeric antigen receptor T (CAR T), cell engineering. ACS Synth Biol 2022; 11: 1–15. [DOI] [PubMed] [Google Scholar]
- 3. McLellan AD, Hosseini RA. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol 2019; 97: 664–674. [DOI] [PubMed] [Google Scholar]
- 4. Hosseini Rad A, Halpin JC, Mollaei M, Smith Bell SW, Hirankarn N, McLellan AD. Metabolic and mitochondrial functioning in chimeric antigen receptor (CAR)–T cells. Cancers (Basel) 2021; 13: 1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. White LG, Goy HE, Rose AJ, McLellan AD. Controlling cell trafficking: addressing failures in CAR T and NK cell therapy of solid tumours. Cancers (Basel) 2022; 14: 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hou AJ, Chen LC, Chen YY. Navigating CAR‐T cells through the solid‐tumour microenvironment. Nat Rev Drug Discov 2021; 20: 531–550. [DOI] [PubMed] [Google Scholar]
- 7. Xu X, Gnanaprakasam J, Sherman J, Wang R. A metabolism toolbox for CAR T therapy. Front Oncol 2019; 9: 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang T, Zhang Z, Li F, et al. miR‐143 regulates memory T cell differentiation by reprogramming T cell metabolism. J Immunol 2018; 201: 2165–2175. [DOI] [PubMed] [Google Scholar]
- 9. Renner K, Bruss C, Schnell A, et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep 2019; 29(135–150): e139. [DOI] [PubMed] [Google Scholar]
- 10. Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Investig 2013; 123: 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perkins MR, Grande S, Hamel A, et al. Manufacturing an enhanced CAR T cell product by inhibition of the PI3K/Akt pathway during T cell expansion results in improved in vivo efficacy of anti‐BCMA CAR T cells. Blood 2015; 126: 1893.26232170 [Google Scholar]
- 12. Petersen CT, Hassan M, Morris AB, et al. Improving T‐cell expansion and function for adoptive T‐cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists. Blood Adv 2018; 2: 210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alizadeh D, Wong RA, Yang X, et al. IL15 enhances CAR‐T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res 2019; 7: 759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang W, Bai Y, Xiong Y, et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016; 531: 651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Q, Ding J, Sun S, et al. Akt inhibition at the initial stage of CAR‐T preparation enhances the CAR‐positive expression rate, memory phenotype and in vivo efficacy. Am J Cancer Res 2019; 9: 2379. [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao L, Liu Y, Zhao F, et al. Inhibition of cholesterol esterification enzyme enhances the potency of human chimeric antigen receptor T cells against pancreatic carcinoma. Mol Ther Oncolytics 2020; 16: 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng W, Carol E, Alli R, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor‐modified T cells. Leukemia 2018; 32: 1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng W, Jones LL, Geiger TL. Modulation of PI3K signaling to improve CAR T cell function. Oncotarget 2018; 9: 35807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Annese T, Tamma R, De Giorgis M, Ribatti D. microRNAs biogenesis, functions and role in tumor angiogenesis. Front Oncol 2020; 10: 2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng Y, Croce CM. The role of microRNAs in human cancer. Signal Transduct Target Ther 2016; 1: 15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hosseini Rad A, Bavarsad MS, Arefian E, Jaseb K, Shahjahani M, Saki N. The role of microRNAs in stemness of cancer stem cells. Oncol Rev 2013; 7: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Srinivasan H, Das S. Mitochondrial miRNA (MitomiR): a new player in cardiovascular health. Can J Physiol Pharmacol 2015; 93: 855–861. [DOI] [PubMed] [Google Scholar]
- 23. Macintyre AN, Gerriets VA, Nichols AG, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 2014; 20: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beezhold K, Moore N, Chiaranunt P, Brown R, Byersdorfer CA. Deletion of AMP‐activated protein kinase (AMPK) in donor T cells protects against graft‐versus‐host disease through control of regulatory T cell expansion and target organ infiltration. Blood 2016; 128: 806. [Google Scholar]
- 25. Gerriets VA, Kishton RJ, Nichols AG, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Investig 2015; 125: 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bofill‐De Ros X, Gu S. Guidelines for the optimal design of miRNA‐based shRNAs. Methods 2016; 103: 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simula L, Campanella M, Campello S. Targeting Drp1 and mitochondrial fission for therapeutic immune modulation. Pharmacol Res 2019; 146: 104317. [DOI] [PubMed] [Google Scholar]
- 28. Lang F, Singh Y, Salker MS, et al. Glucose transport in lymphocytes. Pflug Arch Eur J Physiol 2020; 472: 1401–1406. [DOI] [PubMed] [Google Scholar]
- 29. Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol 2004; 172: 4661–4665. [DOI] [PubMed] [Google Scholar]
- 30. Fischer HJ, Sie C, Schumann E, et al. The insulin receptor plays a critical role in T cell function and adaptive immunity. J Immunol 2017; 198: 1910–1920. [DOI] [PubMed] [Google Scholar]
- 31. Maratou E, Dimitriadis G, Kollias A, et al. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur J Clin Invest 2007; 37: 282–290. [DOI] [PubMed] [Google Scholar]
- 32. McCarthy SA, Mufson RA, Pearce EJ, Rathmell JC, Howcroft TK. Metabolic reprogramming of the immune response in the tumor microenvironment. Cancer Biol Ther 2013; 14: 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kavanagh Williamson M, Coombes N, Juszczak F, et al. Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV‐1. Viruses 2018; 10: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun X, Peng Y, Zhao J, Xie Z, Lei X, Tang G. Discovery and development of tumor glycolysis rate‐limiting enzyme inhibitors. Bioorg Chem 2021; 112: 104891. [DOI] [PubMed] [Google Scholar]
- 35. Zuo J, Tang J, Lu M, et al. Glycolysis rate‐limiting enzymes: novel potential regulators of rheumatoid arthritis pathogenesis. Front Immunol 2021; 12: 779787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer‐targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Investig 2014; 124: 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nian Z, Zheng X, Dou Y, et al. Rapamycin pretreatment rescues the bone marrow AML cell elimination capacity of CAR‐T cells. Clin Cancer Res 2021; 27: 6026–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lanna A, Henson SM, Escors D, Akbar AN. AMPK‐TAB1 activated p38 drives human T cell senescence. Nat Immunol 2014; 15: 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dwyer CJ, Arhontoulis DC, Rangel Rivera GO, et al. Ex vivo blockade of PI3K gamma or delta signaling enhances the antitumor potency of adoptively transferred CD8+ T cells. Eur J Immunol 2020; 50: 1386–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stock S, Kluever A‐K, Endres S, Kobold S. Enhanced chimeric antigen receptor T cell therapy through co‐application of synergistic combination partners. Biomedicines 2022; 10: 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang S, Funk CR, Ravindranathan S, Chen K, Waller EK. PI3K δ/γ inhibition enhances the expansion and anti‐tumor cytotoxicity of CART cells for CLL patients. Blood 2021; 138: 4795. [Google Scholar]
- 42. Phan AT, Goldrath AW. Hypoxia‐inducible factors regulate T cell metabolism and function. Mol Immunol 2015; 68: 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kagoya Y, Nakatsugawa M, Yamashita Y, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Investig 2016; 126: 3479–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Man K, Gabriel SS, Liao Y, et al. Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory‐like T cells during chronic infection. Immunity 2017; 47(1129–1141): e1125. [DOI] [PubMed] [Google Scholar]
- 45. Hermans D, Gautam S, García‐Cañaveras JC, et al. Lactate dehydrogenase inhibition synergizes with IL‐21 to promote CD8+ T cell stemness and antitumor immunity. Proc Natl Acad Sci USA 2020; 117: 6047–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tu VY, Ayari A, O'Connor RS. Beyond the lactate paradox: how lactate and acidity impact T cell therapies against cancer. Antibodies 2021; 10: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klebanoff CA, Crompton JG, Leonardi AJ, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor‐engineered adoptive immunotherapy. JCI Insight 2017; 2: e95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Braverman EL, Dobbs A, Monlish DA, Byersdorfer C. Increasing AMPK activity in human T cells enhances memory subset formation without sacrificing in vitro expansion. Blood 2020; 136: 38–39. [Google Scholar]
- 49. Lepez A, Pirnay T, Denanglaire S, et al. Long‐term T cell fitness and proliferation is driven by AMPK‐dependent regulation of reactive oxygen species. Sci Rep 2020; 10: 21673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mayer KA, Smole U, Zhu C, et al. The energy sensor AMPK orchestrates metabolic and translational adaptation in expanding T helper cells. FASEB J 2021; 35: e21217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Izreig S, Samborska B, Johnson RM, et al. The miR‐17∼92 microRNA cluster is a global regulator of tumor metabolism. Cell Rep 2016; 16: 1915–1928. [DOI] [PubMed] [Google Scholar]
- 52. Kidani Y, Bensinger SJ. Modulating cholesterol homeostasis to build a better T cell. Cell Metab 2016; 23: 963–964. [DOI] [PubMed] [Google Scholar]
- 53. Schmidt NM, Wing PA, Diniz MO, et al. Targeting human Acyl‐CoA: cholesterol acyltransferase as a dual viral and T cell metabolic checkpoint. Nat Commun 2021; 12: 2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stone EL, D'Cruz LM. Tumor‐infiltrating T cells are invigorated by modulating cholesterol metabolism. Transl Cancer Res 2016; S303–S305. [Google Scholar]
- 55. Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kδ and primary immunodeficiencies. Nat Rev Immunol 2016; 16: 702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Urak R, Walter M, Lim L, et al. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J ImmunoTher Cancer 2017; 5: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mousset CM, Hobo W, Ji Y, et al. Ex vivo AKT‐inhibition facilitates generation of polyfunctional stem cell memory‐like CD8+ T cells for adoptive immunotherapy. Oncoimmunology 2018; 7: e1488565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. El Hage A, Dormond O. Combining mTOR inhibitors and T cell‐based immunotherapies in cancer treatment. Cancers (Basel) 2021; 13: 1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Valmori D, Tosello V, Souleimanian NE, et al. Rapamycin‐mediated enrichment of T cells with regulatory activity in stimulated CD4+ T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4+ T cells. J Immunol 2006; 177: 944–949. [DOI] [PubMed] [Google Scholar]
- 60. Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev 2012; 249: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rundqvist H, Veliça P, Barbieri L, et al. Lactate potentiates differentiation and expansion of cytotoxic T cells. SSRN Journal 2019. 10.2139/ssrn.3411249 [DOI] [Google Scholar]
- 62. Rostamian H, Khakpoor‐Koosheh M, Jafarzadeh L, et al. Restricting tumor lactic acid metabolism using dichloroacetate improves T cell functions. BMC Cancer 2022; 22: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Murray CM, Hutchinson R, Bantick JR, et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat Chem Biol 2005; 1: 371–376. [DOI] [PubMed] [Google Scholar]
- 64. Kumagai S, Koyama S, Itahashi K, et al. Lactic acid promotes PD‐1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 2022; 40: 201–218.e9. [DOI] [PubMed] [Google Scholar]
- 65. Tao J‐H, Barbi J, Pan F. Hypoxia‐inducible factors in T lymphocyte differentiation and function. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol 2015; 309: C580–C589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Palazon A, Tyrakis PA, Macias D, et al. An HIF‐1α/VEGF‐A axis in cytotoxic T cells regulates tumor progression. Cancer Cell 2017; 32(669–683): e665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Doedens AL, Phan AT, Stradner MH, et al. Hypoxia‐inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat Immunol 2013; 14: 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee H, Yoon Y. Mitochondrial fission and fusion. Biochem Soc Trans 2016; 44: 1725–1735. [DOI] [PubMed] [Google Scholar]
- 69. Curtale G, Citarella F, Carissimi C, et al. An emerging player in the adaptive immune response: microRNA‐146a is a modulator of IL‐2 expression and activation‐induced cell death in T lymphocytes. Blood 2010; 115: 265–273. [DOI] [PubMed] [Google Scholar]
- 70. Adoro S, Cubillos‐Ruiz JR, Chen X, et al. IL‐21 induces antiviral microRNA‐29 in CD4 T cells to limit HIV‐1 infection. Nat Commun 2015; 6: 7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Štach M, Musil J, Cetkovsky P, Otahal P. Interleukin 21 enhances survival and expansion of CAR T cells via inhibition of their terminal differentiation during interaction with tumor target cells. Blood 2018; 132: 4545. [Google Scholar]
- 72. Gagnon JD, Ansel KM. MicroRNA regulation of CD8+ T cell responses. Noncoding RNA Investig 2019; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Borralho PM, Rodrigues CM, Steer CJ. microRNAs in mitochondria: an unexplored niche. microRNA: Basic Science . Springer: 2015; 31–51. [DOI] [PubMed] [Google Scholar]
- 74. Fan S, Tian T, Chen W, et al. Mitochondrial miRNA determines chemoresistance by reprogramming metabolism and regulating mitochondrial transcription. Cancer Res 2019; 79: 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu T, Wieland A, Araki K, et al. Temporal expression of microRNA cluster miR‐17‐92 regulates effector and memory CD8+ T‐cell differentiation. Proc Natl Acad Sci USA 2012; 109: 9965–9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Khan AA, Penny LA, Yuzefpolskiy Y, Sarkar S, Kalia V. MicroRNA‐17∼92 regulates effector and memory CD8 T‐cell fates by modulating proliferation in response to infections. Blood 2013; 121: 4473–4483. [DOI] [PubMed] [Google Scholar]
- 77. Zhang Z, Li F, Tian Y, et al. Metformin enhances the antitumor activity of CD8+ T lymphocytes via the AMPK–miR‐107–Eomes–PD‐1 pathway. J Immunol 2020; 204: 2575–2588. [DOI] [PubMed] [Google Scholar]
- 78. Pobezinskaya EL, Wells AC, Angelou CC, et al. Survival of naïve T cells requires the expression of let‐7 miRNAs. Front Immunol 2019; 10: 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wu H, Neilson JR, Kumar P, et al. miRNA profiling of naive, effector and memory CD8 T cells. PLoS One 2007; 2: e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wells AC, Daniels KA, Angelou CC, et al. Modulation of let‐7 miRNAs controls the differentiation of effector CD8 T cells. Elife 2017; 6: e26398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Salaun B, Yamamoto T, Badran B, et al. Differentiation associated regulation of microRNA expression in vivo in human CD8+ T cell subsets. J Transl Med 2011; 9: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR‐17‐92 expression in lymphocytes. Nat Immunol 2008; 9: 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Xu Y, Zhang M, Ramos CA, et al. Closely related T‐memory stem cells correlate with in vivo expansion of CAR. CD19‐T cells and are preserved by IL‐7 and IL‐15. Blood 2014; 123: 3750–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang Z, Miao L, Ren Z, Tang F, Li Y. Gene‐edited interleukin CAR‐T cells therapy in the treatment of malignancies: present and future. Front Immunol 2021; 12: 718686. [DOI] [PMC free article] [PubMed]
- 85. Teijeira A, Labiano S, Garasa S, et al. Mitochondrial morphological and functional reprogramming following CD137 (4–1BB) costimulation. Cancer Immunol Res 2018; 6: 798–811. [DOI] [PubMed] [Google Scholar]
- 86. van der Windt GJ, Everts B, Chang C‐H, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012; 36: 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gavriil A, Barisa M, Halliwell E, Anderson J. Engineering solutions for mitigation of chimeric antigen receptor T‐cell dysfunction. Cancers (Basel) 2020; 12: 2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Long AH, Haso WM, Shern JF, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat Genet 2011; 43: 854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jeker LT, Bluestone JA. Micro RNA regulation of T‐cell differentiation and function. Immunol Rev 2013; 253: 65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jia X, Yuan S, Wang Y, et al. The role of alternative polyadenylation in the antiviral innate immune response. Nat Commun 2017; 8: 14605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tamaddon M, Shokri G, Rad SMAH, Rad I, Razavi ÀE, Kouhkan F. Involved microRNAs in alternative polyadenylation intervene in breast cancer via regulation of cleavage factor "CFIm25". Sci Rep 2020; 10: 11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hong W, Ruan H, Zhang Z, et al. APAatlas: decoding alternative polyadenylation across human tissues. Nucleic Acids Res 2020; 48: D34–D39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J 2007; 26: 775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Saini HK, Enright AJ, Griffiths‐Jones S. Annotation of mammalian primary microRNAs. BMC Genom 2008; 9: 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Frimpong K, Spector S. Cotransduction of nondividing cells using lentiviral vectors. Gene Ther 2000; 7: 1562–1569. [DOI] [PubMed] [Google Scholar]
- 97. Amendola M, Venneri MA, Biffi A, Vigna E, Naldini L. Coordinate dual‐gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat Biotechnol 2005; 23: 108–116. [DOI] [PubMed] [Google Scholar]
- 98. Hosseini Rad A, Poudel A, Tan GMY, McLellan AD. Promoter choice: Who should drive the CAR in T cells? PLoS One 2020; 15: e0232915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hu T, Chen P, Fu Q, et al. Comparative studies of various artificial microRNA expression vectors for RNAi in mammalian cells. Mol Biotechnol 2010; 46: 34–40. [DOI] [PubMed] [Google Scholar]
- 100. Trujillo RD, Yue SB, Tang Y, O'Gorman WE, Chen C‐Z. The potential functions of primary microRNAs in target recognition and repression. EMBO J 2010; 29: 3272–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Seyhan AA. A multiplexed miRNA and transgene expression platform for simultaneous repression and expression of protein coding sequences. Mol Biosyst 2016; 12: 295–312. [DOI] [PubMed] [Google Scholar]
- 102. He K, Rad S, Poudel A, McLellan AD. Compact bidirectional promoters for dual‐gene expression in a Sleeping Beauty transposon. Int J Mol Sci 2020; 21: 9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Anno Y‐N, Myslinski E, Ngondo‐Mbongo RP, et al. Genome‐wide evidence for an essential role of the human Staf/ZNF143 transcription factor in bidirectional transcription. Nucleic Acids Res 2011; 39: 3116–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Vogl T, Kickenweiz T, Pitzer J, et al. Engineered bidirectional promoters enable rapid multi‐gene co‐expression optimization. Nat Commun 2018; 9: 3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Roney IJ, Rudner AD, Couture J‐F, Kærn M. Improvement of the reverse tetracycline transactivator by single amino acid substitutions that reduce leaky target gene expression to undetectable levels. Sci Rep 2016; 6: 27697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hosseini Rad A, Poudel A, Tan GMY, McLellan AD. Optimisation of Tet‐On inducible systems for Sleeping Beauty‐based chimeric antigen receptor (CAR) applications. Sci Rep 2020; 10: 13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kulemzin SV, Matvienko DA, Sabirov AH, et al. Design and analysis of stably integrated reporters for inducible transgene expression in human T cells and CAR NK‐cell lines. BMC Med Genomics 2019; 12: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chaulk SG, Xu Z, Glover MJ, Fahlman RP. MicroRNA miR‐92a‐1 biogenesis and mRNA targeting is modulated by a tertiary contact within the miR‐17∼ 92 microRNA cluster. Nucleic Acids Res 2014; 42: 5234–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu YP, Haasnoot J, Ter Brake O, Berkhout B, Konstantinova P. Inhibition of HIV‐1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic Acids Res 2008; 36: 2811–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bourhill T, Arbuthnot P, Ely A. Successful disabling of the 5′ UTR of HCV using adeno‐associated viral vectors to deliver modular multimeric primary microRNA mimics. J Virol Methods 2016; 235: 26–33. [DOI] [PubMed] [Google Scholar]
- 111. Agarwal V, Bell GW, Nam J‐W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015; 4: e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Paraskevopoulou MD, Georgakilas G, Kostoulas N, et al. DIANA‐microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res 2013; 41: W169–W173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, et al. DIANA‐TarBase v8: a decade‐long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res 2018; 46: D239–D245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Herrmann CJ, Schmidt R, Kanitz A, Artimo P, Gruber AJ, Zavolan M. PolyASite 2.0: a consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res 2020; 48: D174–D179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhang Z, Zhang C, Li F, Zhang B, Zhang Y. Regulation of memory CD8+ T cell differentiation by microRNAs. Cell Physiol Biochem 2018; 47: 2187–2198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material