

Abstract
(Hetero)arylsulfur compounds where the S atom is in the oxidation state VI represent a large percentage of the molecular functionalities present in organic chemistry. More specifically, (hetero)aryl‐SVI fluorides have recently received enormous attention because of their potential as chemical biology probes, as a result of their reactivity in a simple, modular, and efficient manner. Whereas the synthesis and application of the level 1 fluorination at SVI atoms (sulfonyl and sulfonimidoyl fluorides) have been widely studied and reviewed, the synthetic strategies towards higher levels of fluorination (levels 2 to 5) are somewhat more limited. This Minireview evaluates and summarizes the progress in the synthesis of highly fluorinated aryl‐SVI compounds at all levels, discussing synthetic strategies, reactivity, the advantages and disadvantages of the synthetic procedures, the proposed mechanisms, and the potential upcoming opportunities.
Keywords: Fluorine, Pentafluorosulfanyl Arenes, Sulfinyl Trifluorides, Sulfur, Tetrafluorosulfanyl Chlorides
The number of methods that exploit the synthesis of (hetero)arylsulfur(VI) fluorides is steadily growing due to their potential in organic synthesis. This Minireview covers the synthesis of arylsulfur(VI) fluorides from levels 2 to 5 of fluorination, the substrate scope, mechanisms, and the further applicability of these fluorinated compounds.
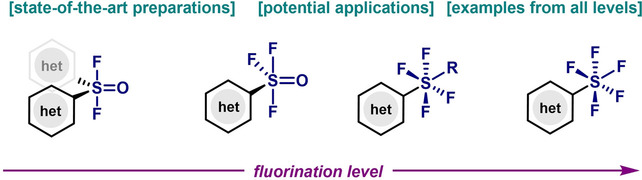
1. Introduction
Aryl‐SVI compounds (including also heteroaryl ones) are ever‐present functionalities in organic synthesis, spanning from the well‐known arylsulfonyl chloride electrophiles [1] to the aryl sulfonamides that are widely present in many pharmaceuticals and agrochemicals. [2] Indeed, the synthesis and applications of these compounds has been widely reviewed and their synthesis is common textbook knowledge. [3] Within the group of aryl‐SVI compounds, however, there is a class of compounds that has received comparatively less attention: these are aryl‐SVI fluorides, where the SVI center is directly attached to F atoms. Indeed, compounds of this class have recently been re‐evaluated, as they have shown interesting properties and applications in various fields of expertise. [4] For example, arylsulfonyl fluorides [5] and arylsulfonimidoyl fluorides [6] have been studied in sulfur–fluoride exchange reactions (SuFEx), and their use as chemical probes for chemical biology has proved highly valuable. [7] However, from a structural and molecular point of view, arylsulfonyl and arylsulfonimidoyl fluorides both represent the lowest level of fluorination at the S atom, with only one fluorine atom attached to the tetrahedral S atom (level 1, Figure 1). Despite the success of level 1, [8] compounds with a S atom at higher levels of fluorination are less well known, but still accessible. For example, diarylsulfur oxide difluorides (level 2) and arylsulfinyl trifluorides (level 3), which present a sulfur atom with a trigonal bypyramidal (TBP) geometry, have been the least studied of the fluorinated aryl‐SVI compounds, with untapped reactivity and applications. Moving up the pyramid, one can find aryltetrafluoro‐λ6‐sulfanyl chlorides and aryltetrafluoro‐λ6‐sulfanes, with various synthetic procedures reported in the literature for their preparation. In particular, aryltetrafluoro‐λ6‐sulfanyl chlorides have been widely studied and applied as precursors for pentafluoro(aryl)‐λ6‐sulfane (Ar−SF5) and deoxyfluorinating agents. [9] In contrast to the lower levels of fluorination, the S atom at level 4 presents an octahedral geometry, which leads to potential structural isomerism. Finally, the highest level (level 5) is occupied by pentafluoro(aryl)‐λ6‐sulfanylarenes (Ar−SF5), which recently gained the attention of the chemistry community because of the unique properties of the SF5 group in material science and medicinal chemistry fields. [10] With an octahedral geometry around the S center and a square‐pyramidal array of fluorine atoms, the symmetrical SF5 group is sterically highly demanding, thus it can be considered as an isostere of the tert‐butyl ( t Bu) and the trifluoromethyl (CF3) groups.[ 11 , 12 ]
Figure 1.
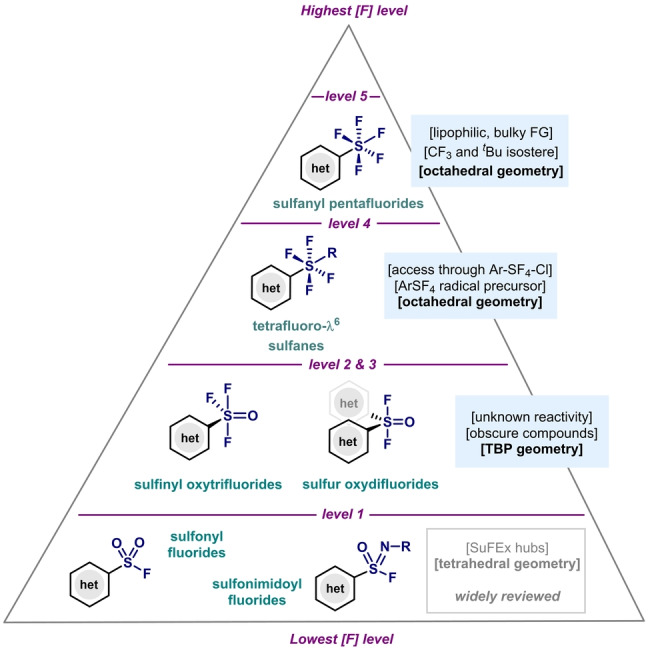
Fluorinated arylsulfur(VI) compounds.
In this Minireview, the synthetic methods, substrate scope, mechanism, and applications of levels 2 to 5 will be evaluated and summarized. Compounds belonging to level 1 (Figure 1) have already been extensively reviewed and will not be included. [8] The organization of this Minireview will follow an increase in the fluorination level, and the different synthetic procedures will be presented and discussed chronologically.
2. Fluorination Level 2
In 1961, Cramer and Coffman reported the reaction of gaseous thionyl tetrafluoride (1, SOF4) with primary amines leading to iminosulfur oxydifluorides 2 in good yields (Scheme 1). [13] This seminal example set a foundation stone for further applications, such as those of Seppelt and Sundermeyer as well as Sharp and co‐workers,[ 14 , 15 ] who capitalized on the strong Si−F bond to react silylated nucleophiles with SOF4. This field, however, remained dormant until Sharpless and co‐workers realized the balanced reactivity and stability of iminosulfur oxydifluorides 2, and their value in organic synthesis and chemical biology (Scheme 1). [16]
Scheme 1.
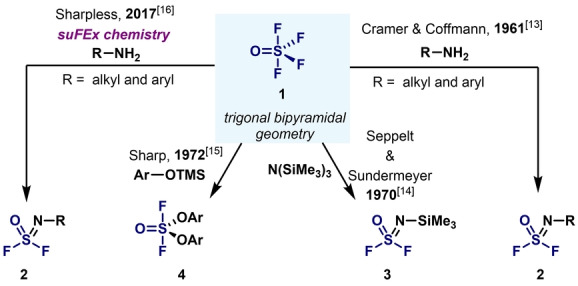
Use of sulfur(VI) fluoride (SOF4) gas as a SuFEx reagent.
In 1978, Clifford et al. reported the synthesis of aryl‐SVI difluorides 7 by reacting thiazyl trifluoride (SNF3; 5) with various aryllithium compounds 6 at −78 °C (Scheme 2). [17] NMR spectroscopy studies revealed that the ‐SNF2 group is more strongly electron‐withdrawing than the ‐NO2 and ‐SF5 groups.
Scheme 2.
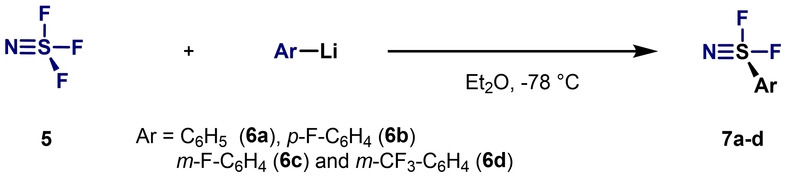
Synthesis of ArSNF2 by Clifford et al. [17]
A year later, Ruppert reported the first examples of diarylsulfur(VI) oxide difluorides. [18] Diaryl sulfoxides 8 could be fluorinated directly by liquid‐phase fluorination using fluorine (F2) at low temperatures (Scheme 3). This synthetic procedure allowed, for the first time, access to diarylsulfur(VI) oxide difluorides 9, which cannot be obtained by the direct arylation of SOF4 (1). The use of other strong fluorinating agents, such as XeF2, failed as alternatives for direct difluorination. The reactivity of the newly synthesized diaryl‐SVI difluorides 9 was also evaluated: they are highly sensitive to moisture and can only be handled under dried protective gas. During these reactivity studies it was found that the Ar2S(O)F2 compounds display high activity as fluorinating agents. Interestingly, in the presence of BF3, oxo‐cationic species 11 were obtained.
Scheme 3.
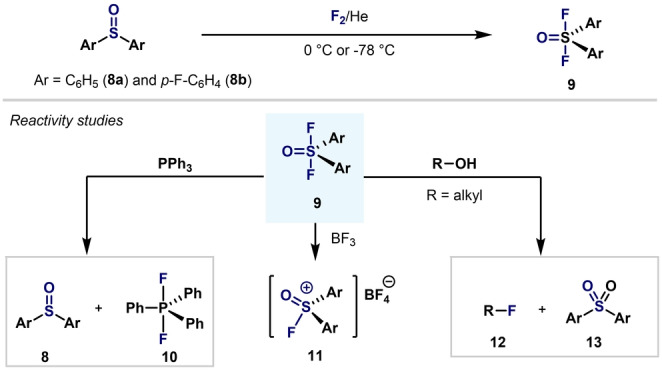
Synthesis and reactivity studies of Ar2SOF2 by Ruppert. [18]
Michalak and Martin reported that sulfurane 14 can be oxidatively fluorinated using an equimolar amount of BrF3 as the difluorinating agent, thereby obtaining all‐trans difluoride 15 (Scheme 4). [19] Surprisingly, when the same difluorination was studied using an excess of BrF3, the cis‐isomer 16 was obtained. Both isomers exhibited the same reactivity towards hydrolysis (highly reactive). Structures 15 and 16 were confirmed by means of NMR spectroscopy and single‐crystal XRD. [20] Whereas heating a solution of 15 did not show isomerization to 16, the presence of catalytic amounts of the Lewis acid SbF5 afforded cis‐isomer 16 quantitatively, presumably via a persulfonium salt intermediate 17. Thus, it was hypothesized that the formation of 16, rather than 15, in the oxidation of the sulfurane 14 with excess of BrF3 arises from the Lewis acidity of BrF3 present in solution leading to the formation of the persulfonium intermediate 17, which leads to isomer 16 after pseudorotation and fluoride capture.
Scheme 4.
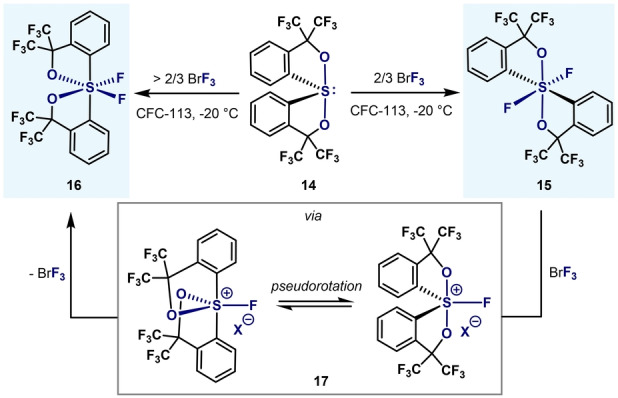
Synthesis of cis‐ and trans‐difluoropersulfuranes by Michalak and Martin.[ 19 , 20 ]
In 1995, Kaneko and co‐workers capitalized on Ruppert′s methodology for the synthesis of α‐fluorosulfones (Scheme 5). Interestingly, when aryl‐alkyl sulfoxides 18 were treated with F2/N2, SVI difluoride species 19 were observed by 19F NMR spectroscopy, and these species readily evolved into the dehydrofluorinated products 20, by virtue of the loss of HF. [21]
Scheme 5.
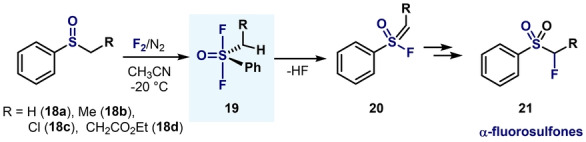
Synthesis of α‐fluorosulfones via aryl‐alkyl‐sulfur(VI) difluoride. [21]
A breakthrough in the synthesis of aryl‐SVI difluorides was made in the same year by Janzen and Ou (Scheme 6). Oxidative fluorination of aryl‐S(IV) compounds such as diphenyl sulfoxide or diphenylsulfur difluoride occurs under mild conditions in the presence of XeF2 and catalytic amounts of chloride anions. [22] In this manner, diarylsulfur(VI) difluorides were obtained in quantitative yields and in short reaction times. Mechanistically, it was postulated that a Cl‐mediated activation of XeF2 for the oxidative fluorination of diarylsulfoxides would initiate the reaction, which would be followed by a radical chain reaction propagated by Ph2S(O⋅)F species. [23]
Scheme 6.
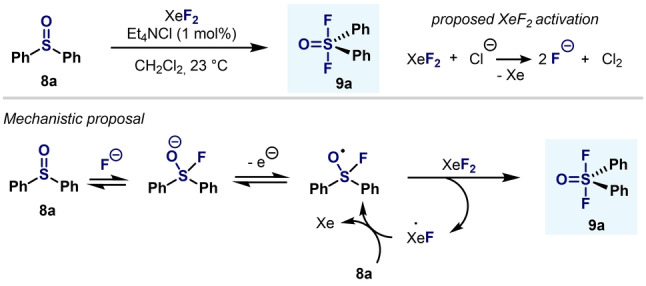
XeF2/Cl− system: synthesis of Ar2SOF2 reported by Janzen and Ou.[ 22 , 23 ]
In 2016, Stephan and co‐workers developed a variety of diaryl fluorosulfoxonium cations (24–27) and applied them to several Lewis acid catalyzed reactions (Scheme 7). [24] Synthetically inspired by the procedure developed by Janzen and Ou, Stephan and co‐workers were able to oxidize diaryl sulfoxides 8 a and 22 to their corresponding diarylsulfur(VI) difluorides in excellent yields and, after fluoride abstraction with either BF3 or [SiEt3][B(C6F5)4], fluorosulfoxonium cations 24–27 could be isolated and characterized by NMR spectroscopy and single‐crystal XRD. Experimental analysis (Gutmann–Beckett method) and theoretical calculations (DFT) confirmed the high Lewis acidity of fluorosulfoxonium cations 24–27. Finally, compound 25 was demonstrated to be highly active in Friedel–Crafts‐type reactions with 1,1‐diphenylethene (Scheme 7, bottom).
Scheme 7.
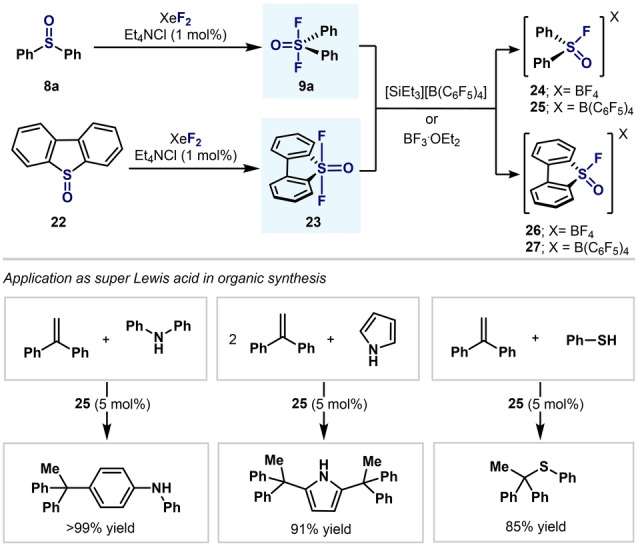
Synthesis and application of diarylfluorosulfoxonium cations by Stephan and co‐workers. [24]
Similarly, in 2021 Panossian and co‐workers applied Lewis acidic sulfoxonium cation 25 in ring‐opening [3+2] and [4+2] annulations (Scheme 8). Excellent yields for a wide substrate scope were obtained, thus validating the excellent Lewis acid character of sulfoxonium cation 25. [25]
Scheme 8.
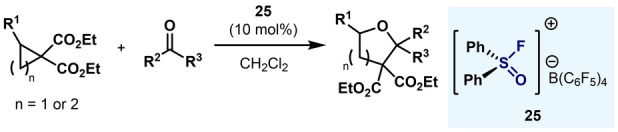
Application of diarylfluorosulfoxonium cation 25 in annulation reactions by Panossian and co‐workers. [25]
3. Fluorination Level 3
Aryl‐SVI trifluorides are the least studied of the fluorination levels known, with few examples described in the literature. To place their development into context, it is important to mention the seminal studies by the groups of Glemser and Sharp in 1971 and 1972: SOF4 (1) can react with trimethylsilylamines [26] and trimethylsilyl aryl ethers [15] to afford the corresponding monosubstituted sulfur(VI) trifluorides 28 and 29 (Scheme 9).
Scheme 9.
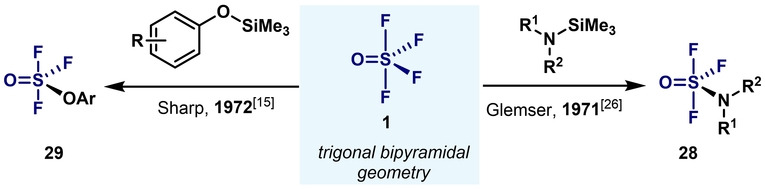
Pioneering syntheses of sulfur(VI) trifluorides.
Similar to aryl‐SVI difluorides, the direct monoarylation of SOF4 (1) is not synthetically viable and, therefore, alternative synthetic routes were required. In 1980, Ruppert showed that aryl‐SVI oxytrifluorides 31 could be obtained by direct fluorine addition to sulfinic fluorides 30 using F2 at very low reaction temperatures. [27] 19F NMR spectroscopy confirmed the finding through the presence of an AX2 pattern, which is in agreement with a trigonal bipyramidal geometry of an S atom with two fluorine atoms occupying the axial positions (Scheme 10). When subjecting the aryl‐SVI oxytrifluoride 31 to BF3, oxocationic species 32 could be obtained, similar to the difluoride analogues (Scheme 3). [18] However, structural evidence was not provided.
Scheme 10.
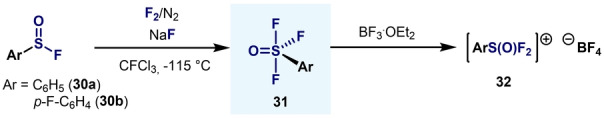
Synthesis of aryl‐SVI oxytrifluorides by Ruppert. [27]
An advance in the area arrived in 2020, when Wang and Cornella developed a new method for the synthesis of ArSOF3 compounds (Scheme 11). [28] This method provided a safer and more general platform to access ArSOF3, with excellent yields obtained for a wide range of compounds. This method capitalizes on the oxidative fluorination of Ar‐S(Phth) (generated in situ from aryl halides) to the corresponding ArSOF3 using easy‐to‐handle reagents and mild reaction conditions. Such compounds exhibit extremely high reactivity and rapidly evolve into their corresponding arylsulfonyl fluoride analogues (ArSO2F) upon exposure to trace amounts of H2O. The high sensitivity of the compounds made all efforts to isolate them unsuccessful. However, the high electrophilicity of ArSOF3 was turned into an advantage, and a wide range of primary aryl‐ and alkylamines were engaged in good yields, thus delivering highly coveted arylsulfonimidoyl fluorides.
Scheme 11.
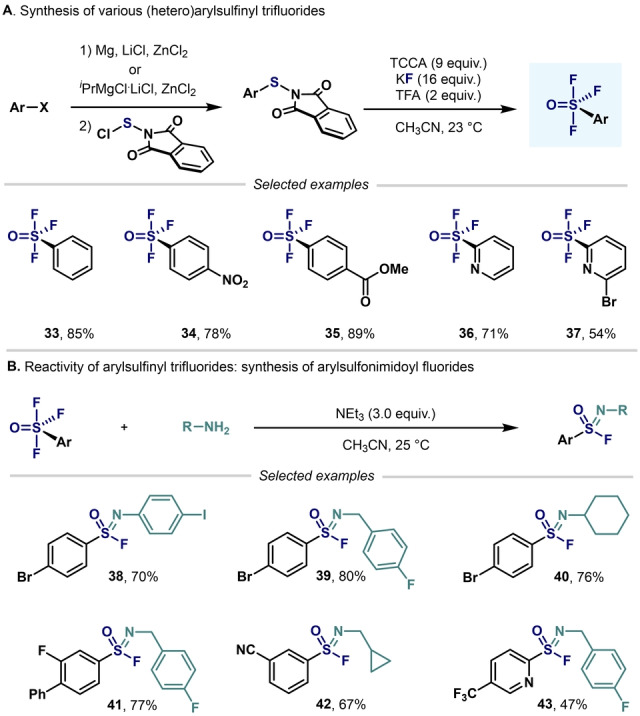
Synthetic procedure for ArSOF3 developed by Wang and Cornella. [28]
4. Fluorination Level 4
The synthesis of aryl‐SVI chlorotetrafluorides (Ar−SF4Cl) has been widely studied due to their potential subsequent applications. Ar−SF4Cl compounds are not only the most commonly utilized precursors to access level 5 (see Section 5), but also widely applied as deoxy‐ and desulfafluorinating agents (Scheme 12). [29] Their high reactivity towards deoxyfluorination makes these reagents a good alternative to deoxyfluorinating agents such as SF4, [30] DAST (diethylaminosulfur trifluoride), [31] PhenoFluor [1,3‐bis(2,6‐diisopropylphenyl)2,2‐difluoro‐2,3‐dihydro‐1H‐imidazole], [32] CpFluor (3,3‐difluoro‐1,2‐diarylcyclopropene), [33] Fluolead (4‐tert‐butyl‐2,6‐dimethylphenylsulfur trifluoride), [34] and PyFluor (2‐pyridinesulfonylfluoride), [35] among others. Tetrafluoro‐λ6‐sulfanyl chlorides 44 do not react with C−O and C−S bonds; however, upon activation with a reductant such as pyridine, ArSF3 45 is generated in situ and acts as a highly effective reagent for the deoxyfluorination of alcohols, aldehydes, ketones, carboxylic acids, and sulfur compounds (Scheme 12).
Scheme 12.
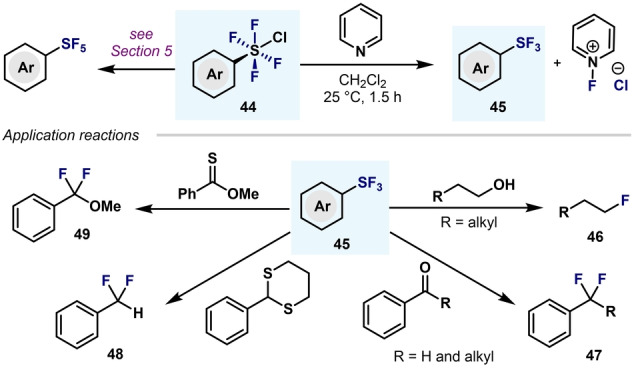
Applications of Ar−SF4Cl in organic synthesis.
Early in the 1950s, aryl‐SVI tetrafluorides were observed in crude mixtures from reactions between organosulfur(II) compounds and anhydrous hydrogen fluoride under electrolytic conditions. [36] The harsh conditions resulted in low‐yielding mixtures of perfluorinated species (Scheme 13A). Years later, the groups of Sharp [37] and Shreeve [38] independently developed strategies to afford perfluoroaryl‐SF4Cl compounds, by using Cl2 or ClF as oxidants (Scheme 13B).
Scheme 13.
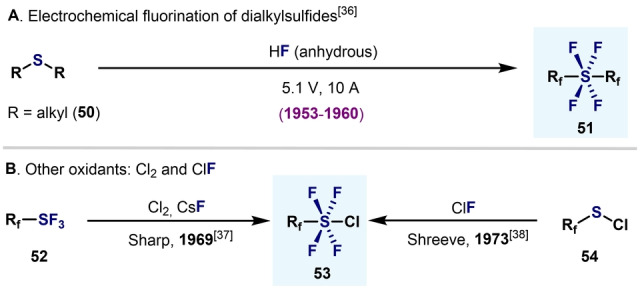
Early examples of perfluoroalkyltetrafluorosulfanes.
It was in 1973 when Denney et al. developed a method for the synthesis of diaryl‐SVI tetrafluorides using CF3OF as the oxidant (Scheme 14). [39] Whereas dialkyl sulfides 50 reacted quickly at low temperatures, diphenylsulfide (55) required the presence of a large excess of CF3OF. Based on 19F NMR spectroscopy results, it was proposed that after an initial oxidation to the corresponding diaryl‐S(IV) difluoride 57, cationic intermediate 58 forms, which eventually leads to 56 upon warming.
Scheme 14.
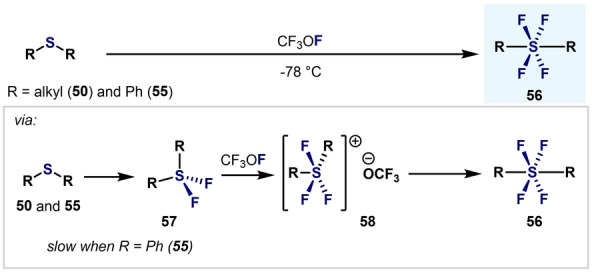
Early examples of alkyl‐ and aryltetrafluorosulfanes by Denney et al. [39]
A breakthrough in the synthesis of Ar−SF4Cl arrived in 1997 when Janzen and co‐workers reported the oxidation of aryldisulfides with XeF2 in the presence of [Et4N][Cl] (Scheme 15). [40] A large excess of XeF2 favored the formation of the trans isomer (method A), whereas lower amounts of oxidant favored the cis isomer (method B). Regardless of the method employed, analytically pure stereoisomers could not be obtained.
Scheme 15.
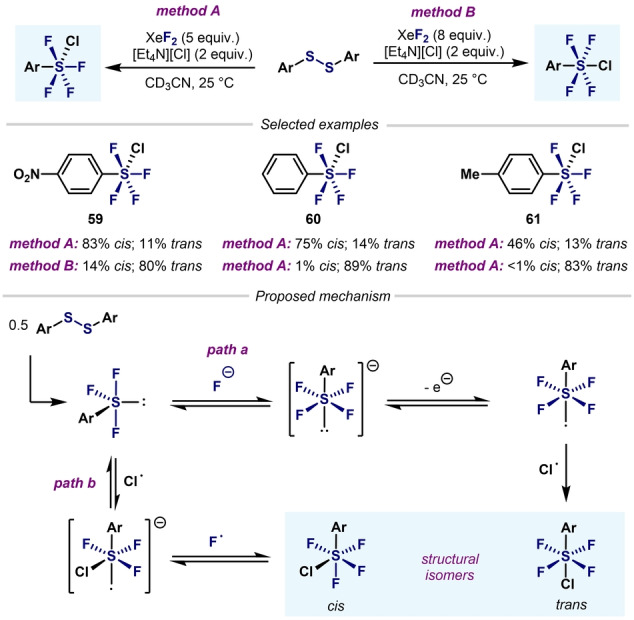
Synthesis of Ar−SF4Cl by Janzen and co‐workers. [40]
It was proposed that after oxidation of the disulfide, Ar−SF3 species are formed. At this point, two pathways can be envisaged. As already reported by Janzen and Ou,[ 22 , 23 ] chlorine radicals (or Cl2) and fluoride anions are formed when XeF2 is mixed with chloride anions. Therefore, in path a (Scheme 15), fluoride anions react with Lewis acidic Ar−SF3, and after oxidation and radical coupling with a chlorine radical, the trans isomer is formed. On the other hand, Ar−SF3 can initially react with a chlorine radical (Scheme 15, path b) followed by radical coupling with a fluorine radical, thereby leading to the cis isomer. It was observed that over time, the trans isomer isomerizes to the cis isomer under the reaction conditions. Unfortunately, the authors did not provide an explanation for the effect of the Cl anions and excess XeF2 in the slow trans‐to‐cis isomerization.
Kirsch et al. reported the direct fluorination of diarylsulfides using F2/N2, which produced a mixture of cis and trans Ar2SF4 isomers (Scheme 16). [41] Importantly, single‐crystal XRD structures were obtained for both isomers. Ab initio and DFT calculations suggested that a cis‐to‐trans isomerization can occur from a BF3‐based catalytic process via a sulfuranonium cation intermediate 65, thus precluding thermal isomerization.
Scheme 16.
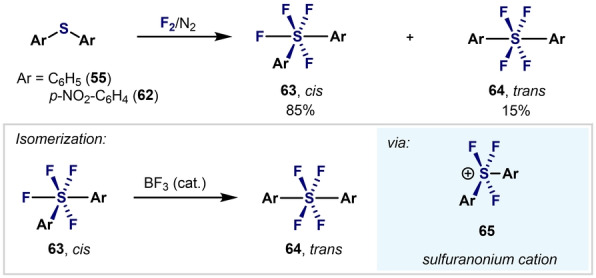
Synthesis of cis‐ and trans‐diaryltetrafluorosulfane mixture reported by Kirsch et al. [41]
An advance in this area was reported in 2012 by Umemoto et al. (Scheme 17), [42] who used mild reaction conditions and a Cl2/KF system to convert a wide range of aryldisulfides or arylthiols into the corresponding aryltetrafluoro‐λ6‐sulfanyl chlorides in excellent yields and trans selectivity. Interestingly, when polyfluoroaryl‐SF4Cl was synthesized, mixtures of trans and cis isomers were obtained. Since thermal isomerization did not occur over time, the authors concluded that each isomer was formed through each isomeric salt (trans and cis form).
Scheme 17.
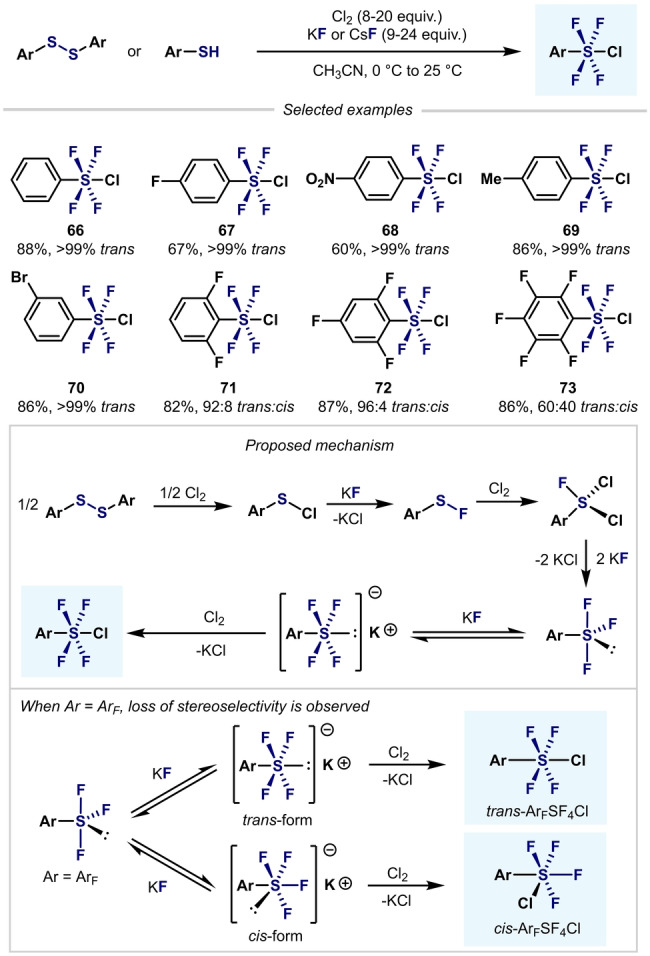
Synthesis of trans‐Ar−SF4Cl by Umemoto et al. [42]
Inspired by the procedure developed by Umemoto et al., Kanishchev and Dolbier and co‐workers synthesized a wide range of 2‐pyridyl‐SVI chlorotetrafluorides with excellent trans stereoselectivity (Scheme 18). [43] However, the presence of ortho substituents (F, Me, and Cl) to the S atom decreased the yield considerably (79–81), with the corresponding heteroaryl‐SF3 compounds afforded as side products in almost all cases.
Scheme 18.
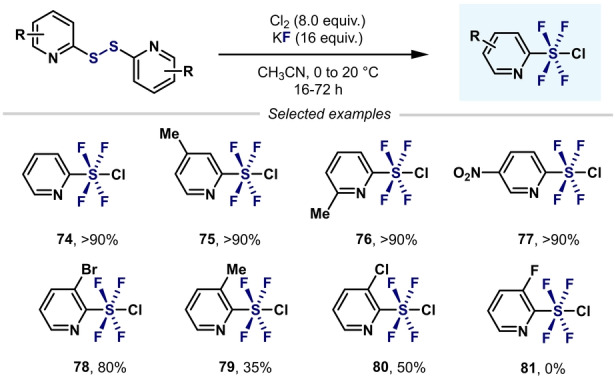
Synthesis of 2‐pyridyl‐SF4Cl by Kanishchev and Dolbier. [43]
Shibata and co‐workers expanded this reactivity to 3‐ and 4‐pyridyl‐SF4Cl (Scheme 19). [44] The presence of fluorine atoms on the pyridine ring is essential for the successful conversion of pyridyldisulfides into the corresponding meta‐ and para‐aryl‐SF4Cl compounds. The presence of another substituent (H, Me, or Cl) on the pyridine moiety (87–89) led to the undesired heteroaryl‐SF3 being obtained. The authors hypothesized that the presence of an electron‐withdrawing group (EWG) such as fluorine would decrease the nucleophilicity of the N atom in the pyridine ring and, hence, stabilize the SF4Cl group.
Scheme 19.
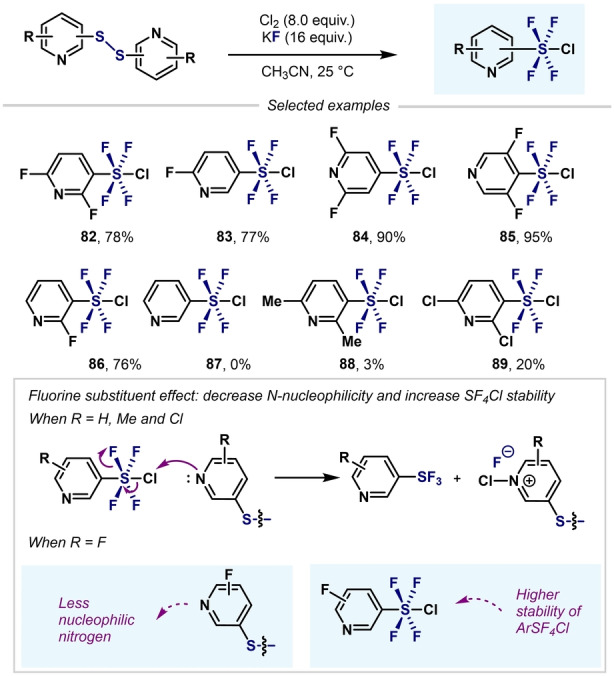
Effect of a fluorine substituent on the synthesis of pyridyl‐SF4Cl. [44]
A remarkable breakthrough in this topic came in 2019 when Togni, Santschi, Pitts, and co‐workers presented the first approach to aryl‐SF4Cl that avoided the use of hazardous, gaseous oxidizing agents (e.g. F2 and Cl2). The method featured the easy‐to‐handle solid trichloroisocyanuric acid (TCCA), potassium fluoride (KF), and catalytic amounts of trifluoroacetic acid (TFA). This simple synthetic method permitted the synthesis of a wide range of aryl‐ and heteroaryl‐SF4Cl compounds in good yields (Scheme 20). [45]
Scheme 20.
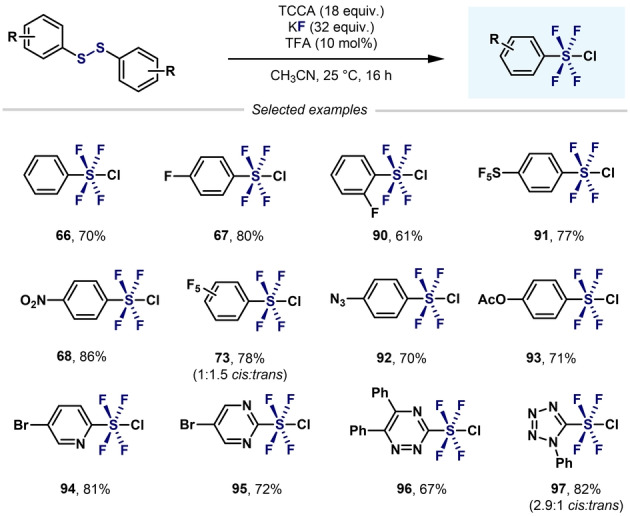
Synthesis of aryl‐ and heteroaryl‐SF4Cl developed by Pitts, Togni, Santschi et al.. [45]
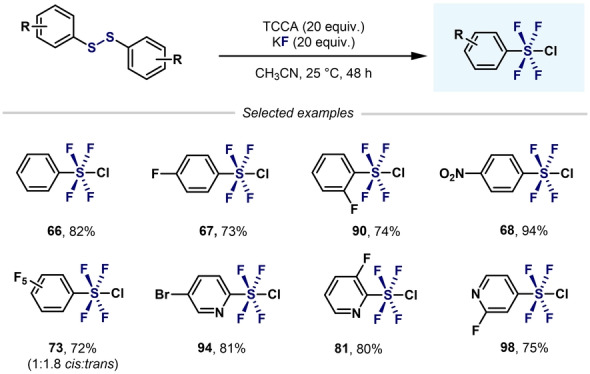
Shibata and co‐workers used the method developed by Togni and co‐workers—without TFA—to obtain various (hetero)aryl‐SF4Cl (Scheme 21). [46] However, longer reactions times were required.
Scheme 21.
Synthesis of aryl‐ and heteroaryl‐SF4Cl from aryl halides developed by Wang and Cornella. [28]
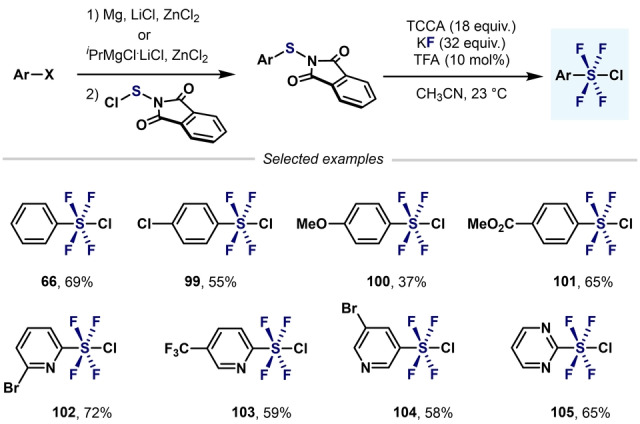
Another improvement in the synthesis or aryl‐ and heteroaryl‐SF4Cl has recently been reported by Wang and Cornella (Scheme 22). [28] Inspired by the synthetic method developed by Togni, Santschi, Pitts, and co‐workers, the authors were able to convert (hetero)aryl halides into the corresponding tetrafluoro‐λ6‐sulfanyl chlorides in excellent yields. It is important to highlight that, by slightly modifying the oxidation step, both level 3 (Scheme 11) and level 4 aryl‐SVI fluorides could be obtained in excellent yields from the same Ar‐S(Phth) starting materials.
Scheme 22.
Synthesis of aryl‐ and (hetero)aryl‐SF4Cl developed by Shibata and co‐workers. [46]
The same group, also reported the use of arylphosphorothiolates as convergent substrates for the synthesis of Ar−SF4Cl (Scheme 23). [47] In this regard, similar yields were obtained as with the previous procedure using Ar‐S(Phth) (Scheme 22) as starting materials. [28]
Scheme 23.
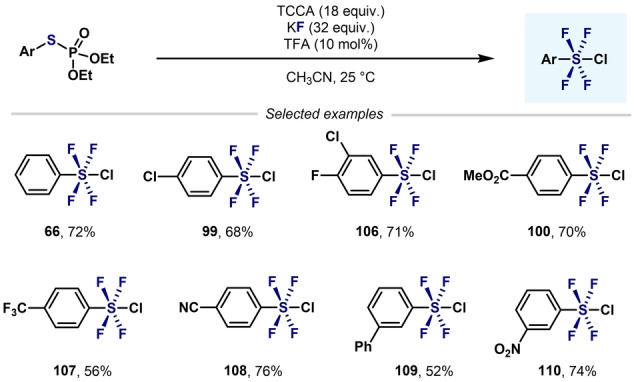
Synthesis of aryl‐SF4Cl from arylphosphorothiolates developed by Wang and Cornella. [47]
Recently, Pascali and co‐workers reported a strategy to obtain aryltetrafluoro‐λ6‐sulfanyl chlorides by flow microfluidic technology. [48] Unfortunately, this preliminary study only provided the Ar−SF4Cl compounds in low yields (5–10 %) together with undesired compounds such as ArSO2F and ArSOF.
As mentioned before (Scheme 12), (hetero)aryltetrafluoro‐λ6‐sulfanyl chlorides have attracted great attention as precursors of (hetero)aryl‐SF5 and as deoxyfluorinating agents. [29] However, Ar−SF4Cl compounds have also been utilized in the synthesis of alkynyl‐ and alkenyl‐aryltetrafluoro‐λ6‐sulfanes, by capitalizing on the labile S−Cl bond (Scheme 24).
Scheme 24.
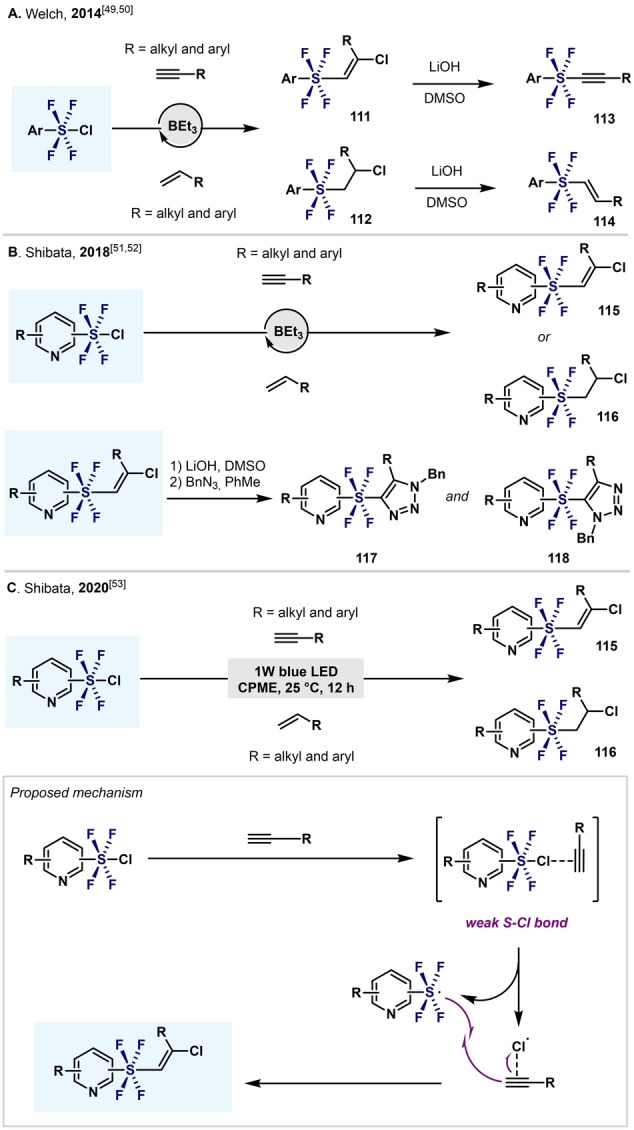
Application of Ar−SF4Cl in the synthesis of alkyl‐ and alkenyltetrafluoro‐λ6‐sulfanes and their derivatization.
In 2014, Welch and co‐workers developed a BEt3‐catalyzed direct addition of Ar−SF4Cl to primary alkynes and alkenes through a S−Cl homolytic cleavage (Scheme 24A).[ 49 , 50 ] Single‐crystal XRD revealed an octahedral geometry at the S atom, with all the fluorine atoms in the axial positions. [49] Moreover, dehydrochlorination of the addition products (111, 112) with lithium hydroxide formed the alkynyl 113 and (E)‐alkenyl‐aryltetrafluoro‐λ6‐sulfanes 114 in excellent yields, with no decomposition of the SF4 group. In 2018, Shibata and co‐workers synthesized pyridyltetrafluoro‐λ6‐sulfanes with alkenyl 115 or alkyl 116 substituents through a radical addition of pyridine‐SF4Cl to terminal alkynes and alkenes (Scheme 24B). [51] By means of single‐crystal XRD and DFT calculations, the authors disclosed an octahedral geometry with a trans configuration of the hypervalent SVI center. Furthermore, the trans‐tetrafluoro‐λ6‐sulfanes bearing an alkenyl group 115 were further derivatized through a thermal Huisgen 1,3‐dipolar cycloaddition to provide a wide range of three‐dimensional building blocks with two independent N‐heterocycles (117, 118). [52]
In 2020 Shibata and co‐workers also reported the addition of Py‐SF4Cl to terminal alkynes and alkenes under irradiation with light (1 W blue LED; Scheme 24C). This procedure is an excellent alternative to BEt3‐catalyzed processes, as the borane is often the source of undesired side reactivity or substrate decomposition. In agreement with previous reports, the authors proposed a radical process to explain the reactivity observed. [53]
5. Fluorination Level 5
In this level, only one type of compound reigns sovereign: namely, Ar−SF5. Although known for many years, it was only recently that the pentafluorosulfanyl group (SF5) became an interesting fluorine‐containing building block because of its thermal and chemical stability [54] as well as inertness under physiological conditions. [55] The electrostatic surface presented by the SF5 moiety is comparable to that of CF3 and its electron‐withdrawing effect suggests that the effects of SF5 and CF3 groups are similar in magnitude. [56] The electronegativity of the SF5 group has been measured to be as high as 3.65, compared to a value of 3.36 for the CF3 group. [57] The SF5 group is the newest member of a short list of functional groups that possess both high electronegativity and high lipophilicity, two properties that are generally juxtaposed. As a result of such unique properties, aryl‐SF5 compounds have attracted attention, and synthetic efforts towards their preparation have been the focus of intensive research. [11] Investigations on Ar−SF5 span from applications as t Bu and CF3 isosteres in medicinal chemistry,[ 10 , 58 ] optoelectronic materials, [59] or agrochemicals, [60] to their ability to function as push‐pull fluoro‐ [61] and choromophores. [62] Before discussing Ar−SF5 compounds, it is important to mention that the first syntheses of C−SF5 compounds were reported in the 1950s by Cady and co‐workers (Scheme 25).[ 63 , 64 ] The authors reported the conversion of either methylmercaptan (119) or carbon disulfide (120) into CF3‐SF5 (121) using CoF3 and F2 [63] or HF in an electrochemical setup. [64] Although the yields and purity of the mixtures were low, these synthetic procedures truly opened the door to a new era of SVI pentafluoride chemistry. [65] Equally important is the synthesis of SF5Cl (123) reported by Nyman and Roberts by the direct oxidation of SF4 (122) with ClF. [66] Nowadays, SF5Cl (123) gas has become a benchmark reagent for the synthesis of SF5 compounds, and efforts toward its practical usage are of great interest. [67]
Scheme 25.
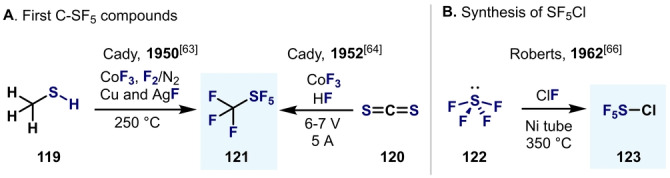
Pioneering syntheses of C−SF5 and SF5Cl.
The synthesis of arylsulfanyl pentafluorides (Ar−SF5) can be classified on the basis of the synthetic approach. Therefore, we have organized this section in three subsections.
5.1. Direct Oxidation of Diaryldisulfides or Arylthiols
The direct oxidation of diaryl disulfides or aryl thiols with strong fluorinating agents was the first approach toward the synthesis of arylsulfanyl pentafluorides (Ar−SF5). [68] In all cases, low yields and narrow substrate scope were common features of those pioneering methods (Scheme 26). The first synthetic procedure for aryl‐SF5 compounds was reported by Sheppard et al. in 1960. [68a] When phenylsulfur trifluoride (124, PhSF3) is heated gradually to 130 °C with AgF2 in a reactor made of copper or Teflon, phenylsulfur pentafluoride (128, Ph‐SF5) is obtained, albeit in low yields (10–13 %, Scheme 26).
Scheme 26.
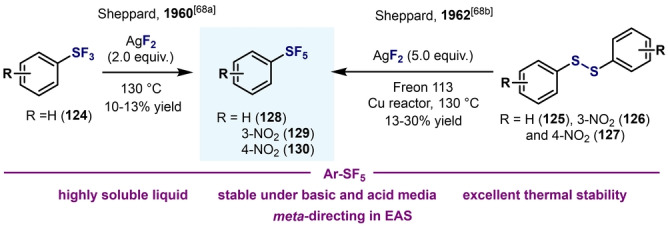
Early syntheses of Ar−SF5: synthesis and study of the physical and chemical properties by Sheppard.[ 68 , 69 ]
Two years later, the same author slightly modified the previous procedure by adding aryl disulfides 125–127 to five molar equivalents of AgF2 suspended in 1,1,2‐trichloro‐1,2,2‐trifluoroethane (“Freon” 113) in a copper reactor. [68b] This modified procedure was found to be particularly effective when NO2‐substituted Ar−SF5 126 and 127 were utilized (Scheme 26). Several important conclusions arose from this work: 1) Ph‐SF5 is a colorless liquid that is soluble in common organic solvents, even hydrocarbons; 2) Ph‐SF5 shows excellent stability under basic conditions (NaOH) and acidic conditions (H2SO4), being hydrolyzed under the latter conditions only above 100 °C; 3) Ph‐SF5 also shows excellent thermal stability, even at 400 °C; 4) Ph‐SF5 directs nitration at the meta‐position; 5) the SF5 group is stable under catalytic hydrogenation conditions. Although low yields were obtained in all cases, this pioneering study from Sheppard et al. opened a new pathway in the synthesis or arylsulfur(VI) fluorides [69] and the further application to several fields such as biology and medicinal chemistry. [11]
Another direct oxidation procedure was developed by Karstev and co‐workers (Scheme 27), [70] who synthesized polychloropyridine‐SF5 132 by the direct oxidation of the corresponding thiols 131 with IF5. However, this procedure also suffered from low yields and narrow scope.
Scheme 27.
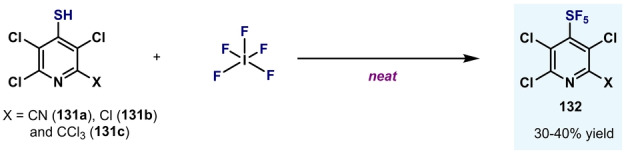
Other preliminary synthetic methods for the synthesis of Ar−SF5. [70]
A few years later, the groups of Spink and Philp independently reported the use of F2 for the conversion of diaryldisulfides 126 and 127 into the corresponding Ar−SF5 compounds 129 and 130 (Scheme 28). Whereas Chambers and Spink developed the oxidation in flow conditions, [71] Philp and co‐workers performed the oxidation in batch. [72] Both methods led to improved yields and milder reaction conditions compared to Sheppard's AgF2‐based procedure. [68]
Scheme 28.
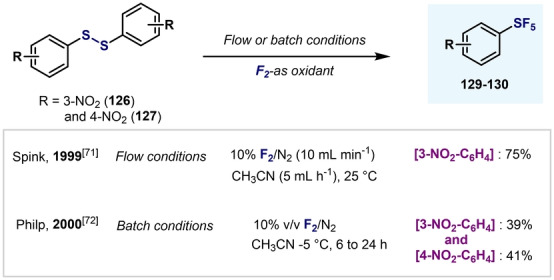
First examples of the synthesis of Ar−SF5 by direct oxidation using F2.
In 2000, Ou and Janzen applied the XeF2‐Et4NCl system to the conversion of a limited number of diaryldisulfides (125 and 133) into Ar−SF5 compounds (Scheme 29). In this case, low‐to‐moderate yields were obtained along with trans‐Ar−SF4Cl as the main by‐product. [23b]
Scheme 29.
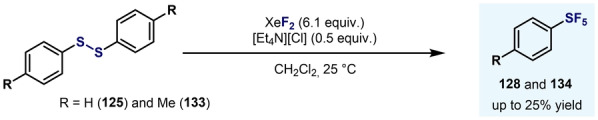
Synthesis of Ar−SF5 by direct oxidation using XeF2‐Et4NCl. [23b]
In 2016 and 2019, Beier and co‐workers reported the direct fluorination of ortho‐, meta‐, and para‐substituted aromatic thiols and disulfides using F2 (Scheme 30). [73] By comparing the synthetic performance under batch and flow conditions, it was found that a hybrid batch‐flow process (synthesis of Ar−SF3 in batch, then Ar−SF5 in flow) provided good yields. [73b] By benchmarking experimental data with DFT calculations, the authors ruled out three nonradical pathways for the conversion of Ar−SF3 into Ar−SF5. It was finally proposed that the reaction proceeds through a radical mechanism after homolytic cleavage of the F−F bond. Propagation and termination steps are almost barrierless and the reaction depends on the stability of the Ar−SF4 radical species. However, further mechanistic insights are required to elucidate the mechanism for the direct fluorination of diaryldisulfides using F2.
Scheme 30.
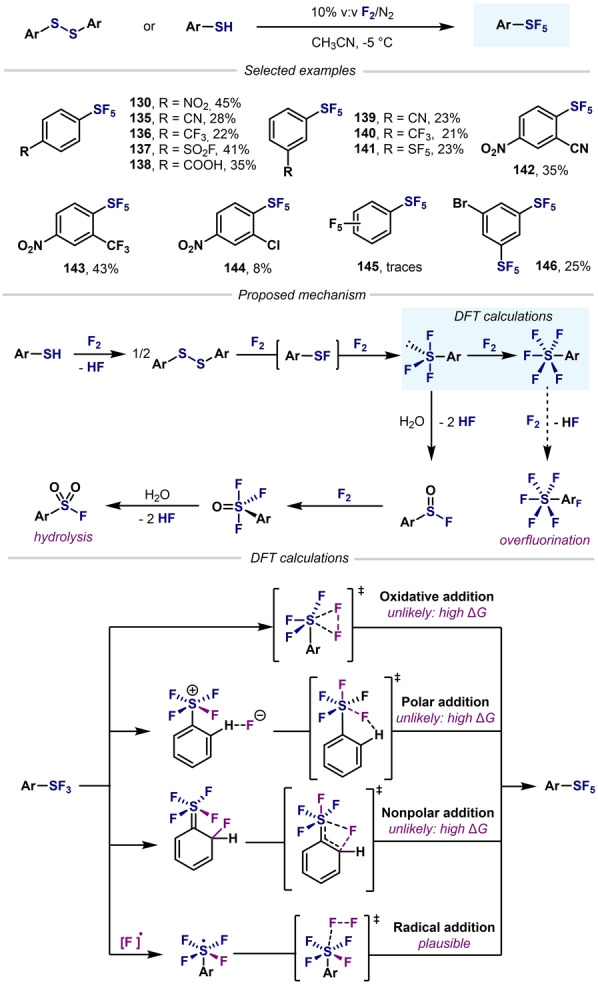
Synthesis of Ar−SF5 by direct oxidation using F2, as developed by Beier and co‐workers. [73]
5.2. Synthesis of Ar−SF5 from Ar−SF4Cl
As mentioned in Section 4, Ar−SF4Cl species have been used as synthetic precursors of Ar−SF5 through Cl−F exchange. Compared to the direct fluorination of thiols or diaryldisulfides, the use of Ar−SF4Cl as precursors leads to higher yields of the desired Ar−SF5 under milder and safer conditions. In 2012, Umemoto et al. converted a wide range of Ar−SF4Cl into their corresponding Ar−SF5 compounds in good to excellent yields under mild reaction conditions, through a Cl–F exchange using either ZnF2‐HF (KHF2) or SbIII/V fluorides (Scheme 31). [42] Since then, several Cl–F exchange methods have been reported to expand the scope of possibilities to access Ar−SF5, and accommodate more functional groups (Scheme 32).
Scheme 31.
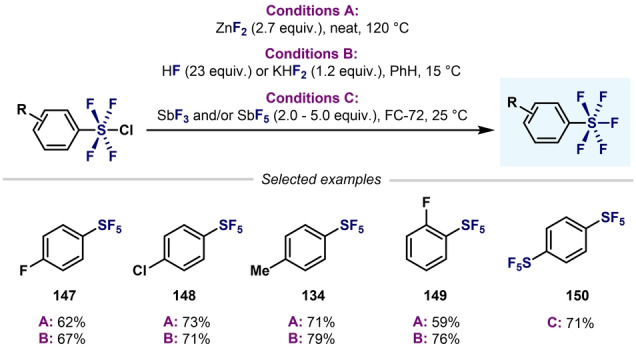
First synthesis of Ar−SF5 from Ar−SF4Cl by Umemoto et al. [42]
Scheme 32.

Conversion of Ar−SF4Cl into Ar−SF5: state‐of‐the‐art.
Beier and co‐workers reported that a combination of KHF2 and TFA at room temperature was optimal to convert various Ar−SF4Cl into their corresponding Ar−SF5 products (Scheme 32A). [74] In this regard, comparable yields were obtained in almost all cases to those obtained with Umemoto's methodology. Kanishchev and Dolbier reported the conversion of 2‐pyridyl‐SF4Cl into the corresponding 2‐pyridyl−SF5 by using AgF as the fluoride source (Scheme 32B). These were pioneering examples of the efficient synthesis of highly sought after N‐heterocyclic−SF5 compounds. [43] In 2016, Shibata and co‐workers reported a similar method for the conversion of a wide range of fluoro‐containing 3‐ and 4‐pyridyl−SF4Cl compounds, with moderate to good yields obtained (Scheme 32C). [44] When treating 2‐fluoropyrdine‐SF5 compounds with different N‐ and O‐nucleophiles, nucleophilic aromatic substitution occurred, thus illustrating the great EWG ability of SF5. IF5 was also demonstrated to be a good fluoride source for the same purpose (Scheme 32D). [75] Several years later, the same group reported a novel strategy for the synthesis of aryl‐ and heteroaryl‐SVI pentafluorides by a Ag2CO3‐induced Cl–F exchange (Scheme 32E). [76] Remarkably, this fluorination does not require any external fluoride sources; rather, the reaction proceeds through a self‐immolative mechanism of Ar−SF4Cl. In 2019, Togni and co‐workers also converted a wide variety of Ar−SF4Cl compounds into the corresponding Ar−SF5 derivatives by using AgF as the classic external fluoride source (Scheme 32F). [45] Recently, Guzyr et al. reported the synthesis of aryl‐ and heteroaryl‐SVI pentafluorides using HgO and HF (Scheme 32G). [77] Very recently, the Cornella and Shibata groups independently showed that AgBF4 is also a valid source of F for the synthesis or Ar−SF5 compounds (Scheme 32H).[ 47 , 78 ] The authors proposed activation of the Cl atom of (hetero)aryl‐SF4Cl by Ag+, and subsequent attack of the fluoride atom of the BF4 anion to the S center by either a concerted or stepwise mechanism.
5.3. Alternative Syntheses Using SF5Cl
In Sections 5.1 and 5.2 it was shown that direct oxidative fluorination and F–Cl exchange from Ar−SF4Cl represents the standard approach for the synthesis of Ar−SF5 compounds. Nevertheless, other alternative synthetic procedures involving the de novo synthesis of the aromatic ring have appeared in the literature. For example, Ar−SF5 can be assembled through a Diels–Alder reaction of ethynylsulfur pentafluoride (182) with different dienes, as demonstrated by Hoover and Coffmann (Scheme 33). [79]
Scheme 33.
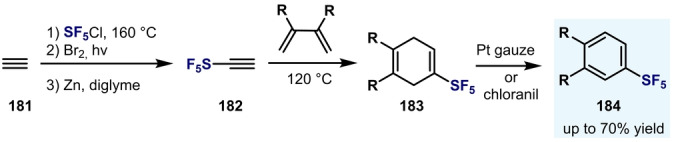
Synthesis of Ar−SF5 developed by Hoover and Coffmann. [79]
Sergeeva and Dolbier developed a convenient three‐step synthesis of Ph‐SF5 from 1,4‐cyclohexadiene (165) with an overall yield of 70 %. The key step in this synthesis is the radical addition of SF5Cl to an alkene, thereby forging a C−SF5 bond in almost quantitative yield (Scheme 34A). [80]
Scheme 34.
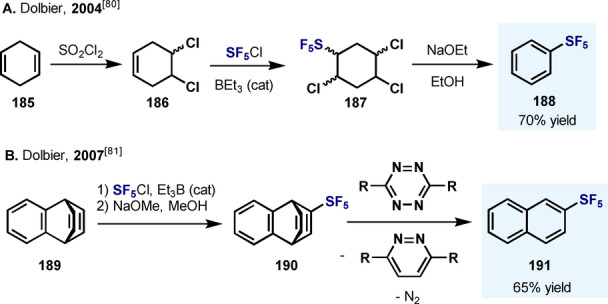
Synthesis of Ar−SF5 developed by Sergeeva and Dolbier. [80]
The same group also developed a three‐step synthesis of 2‐pentafluorosulfanylnaphthalene (191) by the initial addition of SF5Cl to benzobarralene 189. Elimination of the ethylene bridge by a cycloaddition/retro‐cycloaddition sequence with 3,6‐bis‐(2‐pyridyl)‐1,2,4,5‐tetrazine afforded the Ar−SF5 191 in good yield (Scheme 34B). [81]
Ponomarenko et al. reported the Et3B‐catalyzed SF5Cl radical addition reactions of substituted aryl‐ and naphthyl‐SF5 from 7‐oxanorbornene derivatives 192 and 195 (Scheme 35A). [82] The high regioselectivity observed for the formation of 2‐SF5‐1‐naphthol (194) is consistent with ab initio computational studies, which revealed a SF5⋅⋅⋅HO hydrogen bond that renders additional stabilization to the product. Duda and Lentz prepared pentafluoro(3,3,3‐trifluoroprop‐1‐yn‐1‐yl)‐λ6‐sulfane (201) in high yields in two steps from 3,3,3‐trifluoropropyne (199) by the addition of SF5Br followed by a dehydrobromination reaction (Scheme 35B). [83] This compound was demonstrated to be a good dienophile in Diels–Alder reactions, as shown by its reaction with pyranone. Importantly, this procedure permits the introduction of the SF5 group at the ortho position of arenes (202). Carreira and co‐workers reported a synthetic strategy for preparing SF5‐containing N‐heterocyclic building blocks, such as quinolinones, quinolines, and pyridines (Scheme 35C). [84] Benzyl SF5‐acetate (204) proved to be an excellent candidate to participate in aldol reactions, providing a wide range of 3‐SF5‐quinolinones in good to excellent yields. These compounds can be rapidly converted into the corresponding quinolines 205 using either POCl3 or POBr3.
Scheme 35.

Synthesis of different aryl‐SF5 and heteroaryl‐SF5 compounds.
Since the SF5 group is considered a bioisostere of the CF3 and t Bu groups, the authors compared the physicochemical properties of 3‐SF5‐quinolinone with its CF3 and t Bu analogues. Preliminary collected data showed that SF5 exhibits higher lipophilicity than CF3, but lower than the t Bu group. The membrane permeability increases in the order SF5< t Bu<CF3. In terms of pK a values, the SF5‐quinolinone was the most acidic. Concerning the solubility, both the CF3 and SF5 compounds are considerably more soluble than the t Bu counterpart.
Kanishchev and Dolbier generated ortho‐SF5‐benzyne (208) by a lithiation/elimination sequence starting from 2‐fluoro‐SF5‐benzene 207 (Scheme 35D). [85] The highly reactive SF5‐benzyne intermediate underwent Diels–Alder cyclization with furan in situ, and the product 209 was subjected to a series of further chemical transformations en route to 210 and 211.
All these synthetic alternatives allow access to ortho‐substituted aryl‐SF5 compounds in good yields, in contrast to direct oxidative fluorination (Section 5.1), where the ortho‐substituted diaryldisulfides are still the main limitation to expand the substrate scope.
6. Conclusion and Outlook
This Minireview highlights different approaches toward the synthesis aryl‐SVI fluorides, where the central S atom is substituted with 2, 3, 4, or 5 F atoms, thus defining the fluorination level. Level 2 fluorinated compounds have been successfully prepared by using XeF2/Et3NCl as an oxidative fluorinating system, a method that still remains in use nowadays. Remarkably, these compounds have gained much attention recently, as the corresponding sulfoxonium cations have been shown to be super‐Lewis acids for organic transformations.
Level 3 fluorination is still an underdeveloped platform, with untapped potential for synthesis. It is clear that ArSOF3 compounds are good linchpin reagents for the synthesis of sulfonimidoyl fluorides.
Whereas levels 2 and 3 are still underdeveloped, their higher fluorinated analogues (levels 4 and 5) have been widely studied and their synthesis widely explored. In this regard, recent advances in the use of Ar−SF4Cl compounds have shown that strong oxidants such as F2 and Cl2 can be replaced by the safer and easy‐to‐handle TCCA/KF system.
Finally, the synthesis of arylsulfanyl pentafluorides have been the most studied and improved, as a result of the recent interest of Ar−SF5 compounds in medicinal chemistry. The current synthetic approaches rely on the Cl–F exchange from the corresponding Ar−SF4Cl compounds. Alternatively, other synthetic procedures that employ SF5Cl gas as a precursor to the SF5 group have also been successful, although the synthesis of the aryl ring is required.
Despite the excellent advances in the synthesis of (hetero)arylsulfur(VI) fluorides and their successful applications, the reported approaches still suffer from several disadvantages, such as narrow substrate scope and harsh reaction conditions. Thus, we envision that innovative work in this area is likely to arise from new greener and milder synthetic methods. Moreover, as a result of the high interest in the SF5 group in medicinal chemistry and biology, this area will also evolve to avoid the use of toxic SF5Cl as a SF5 radical precursor, and the use of the greener, but less reactive, SF6 gas. The very few examples of this latter approach already show the promising synthetic utility of SF6 gas as a building block for sulfur(VI) fluoride synthesis. [86]
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Marc Magre earned his PhD under the supervision of Prof. Montserrat Diéguez and Prof. Oscar Pàmies at Universitat Rovira i Virgili (Spain) in 2016, working on tailor‐made chiral Pd‐ and Ir‐based catalysts for enantioselective transformations. In 2017, he joined the group of Prof. Magnus Rueping (RWTH Aachen; Germany) as a postdoctoral researcher, where he developed novel magnesium‐catalyzed hydrofunctionalizations of unsaturated systems. In 2020, he joined the Max Planck Institut für Kohlenforschung (Mülheim an der Ruhr, Germany) as a postdoctoral researcher in the group of Dr. Josep Cornella, where he focuses on bismuth catalysis.

Biographical Information
Shengyang Ni was born in Yancheng (China), in 1992. He earned his PhD under the supervision of Prof. Yi Pan at Nanjing University (China) in 2019, working on Ni‐catalyzed reductive cross‐coupling reactions. During this time, he was a visiting PhD student (2017–2019) in the group of Prof. Phil Baran at Scripps Research Institute (US). After his PhD, he stayed in the same group to continue his postdoctoral research. In 2021, he joined the Max Planck Institut für Kohlenforschung (Mülheim an der Ruhr, Germany) as a postdoctoral researcher in the group of Dr. Josep Cornella, where he focuses on Ni‐catalyzed N2O activation.

Biographical Information
Josep Cornella (Pep) studied chemistry at the University of Barcelona (2008) and completed his PhD in 2012 at Queen Mary University (UK) under the supervision of Prof. Igor Larrosa. He then pursued postdoctoral studies in the groups of Prof. Ruben Martin (ICIQ, Spain) as a Marie Curie Postdoctoral Fellow and Prof. Phil S. Baran (The Scripps Research Institute, California, USA) as a Beatriu de Pinos Fellow. In 2017, he was appointed as a Max Planck Research Group Leader in the Department of Organometallic Chemistry at the Max‐Planck‐Institut für Kohlenforschung in Mülheim an der Ruhr, Germany, where he leads the Sustainable Catalysis Laboratory.

Acknowledgements
Financial support for this work was provided by the Max‐Planck‐Gesellschaft, Max‐Planck‐Institut für Kohlenforschung and Fonds der Chemischen Industrie (FCI‐VCI),Boehringer Ingelheim Foundation (Exploration Grant, BIS) and the European Union's Horizon 2020 research and innovation programme under Agreement No. 850496 (ERC Starting Grant, J.C.). We thank Prof. Dr. A. Fürstner for insightful discussions and generous support. Open Access funding enabled and organized by Projekt DEAL.
M. Magre, S. Ni, J. Cornella, Angew. Chem. Int. Ed. 2022, 61, e202200904; Angew. Chem. 2022, 134, e202200904.
References
- 1.For recent examples of aryl sulfonyl chloride reactivity, see
- 1a. Dubbaka S. R., Vogel P., Angew. Chem. Int. Ed. 2005, 44, 7674–7684; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7848–7859; [Google Scholar]
- 1b. Gómez-Palomino A., Cornella J., Angew. Chem. Int. Ed. 2019, 58, 18235–18239; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18403–18407; [Google Scholar]
- 1c. Hell S. M., Meyer C. F., Misale A., Sap J. B. I., Christensen K. E., Willis M. C., Trabanco A. A., Gouverneur V., Angew. Chem. Int. Ed. 2020, 59, 11620–11626; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 11717–11723; [Google Scholar]
- 1d. Lu X., Yi Q., Pan X., Wang P., Vessally E. J., J. Sulfur Chem. 2020, 41, 210–228. [Google Scholar]
- 2.For recent examples concerning the bioactivity of sulphonamides, see
- 2a. Drug Discovery: Practices, Processes, and Perspectives (Eds.: J. J. Li, E. J. Corey), Wiley, Hoboken, 2013;
- 2b. Scott K. A., Njardarson J. T. in Sulfur Chemistry (Ed.: Jiang X.), Springer International Publishing, Cham, 2018, pp. 1–34; [Google Scholar]
- 2c. Zhao F., Li P., Liu X., Jia X., Wang J., Liu H., Molecules 2019, 24, 164–186; [Google Scholar]
- 2d. Shah F., Bell I. M., J. Med. Chem. 2020, 63, 7447–7457. [DOI] [PubMed] [Google Scholar]
- 3.For recent developments in the synthesis of arylsulfur(VI) compounds, see
- 3a. Wiezorek S., Lamers P., Bolm C., Chem. Soc. Rev. 2019, 48, 5408–5423; [DOI] [PubMed] [Google Scholar]
- 3b. Liang C., Collet F., Robert-Peillard F., Mueller P., Dodd R. H., Dauban P., J. Am. Chem. Soc. 2008, 130, 343–350; [DOI] [PubMed] [Google Scholar]
- 3c. Li Z., Yu H., Bolm C., Angew. Chem. Int. Ed. 2017, 56, 9532–9535; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9660–9663; [Google Scholar]
- 3d. Yu H., Li Z., Bolm C., Angew. Chem. Int. Ed. 2018, 57, 12053–12056; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12229–12232; [Google Scholar]
- 3e. Chen Y., Murray P. R., Davies A. T., Willis M. C., J. Am. Chem. Soc. 2018, 140, 8781–8787; [DOI] [PubMed] [Google Scholar]
- 3f. Bremerich M., Conrads C. M., Langletz T., Bolm C., Angew. Chem. Int. Ed. 2019, 58, 19014–19020; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 19190–19196; [Google Scholar]
- 3g. Zhang Z. X., Davies T. Q., Willis M. C., J. Am. Chem. Soc. 2019, 141, 13022–13027; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3h. Li Y., Chen S., Wang M., Jiang X., Angew. Chem. Int. Ed. 2020, 59, 8907–8911; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 8992–8996; [Google Scholar]
- 3i. Davies T. Q., Tilby M. J., Ren J., Parker N. A., Skolc D., Hall A., Duarte F., Willis M. C., J. Am. Chem. Soc. 2020, 142, 15445–15453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For selected applications of arylsulfur(VI) fluorides, see
- 4a. Dong J., Sharpless K. B., Kwisnek L., Oakdale J. S., Fokin V. V., Angew. Chem. Int. Ed. 2014, 53, 9466–9470; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9620–9624; [Google Scholar]
- 4b. Chen W., Dong J., Plate L., Mortenson D. E., Brighty G. J., Li S., Liu Y., Galmozzi A., Lee P. S., Hulce J. J., J. Am. Chem. Soc. 2016, 138, 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For recent syntheses of arylsulfonyl fluorides, see
- 5a. Davies W., Henry Dick J. H., J. Chem. Soc. 1931, 2104–2109; [Google Scholar]
- 5b. Davies A. T., Curto J. M., Bagley S. W., Willis M. C., Chem. Sci. 2017, 8, 1233–1237; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Chen Y., Willis M. C., Chem. Sci. 2017, 8, 3249–3253; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Tribby A. L., Rodríguez I., Shariffudin S., Ball N. D., J. Org. Chem. 2017, 82, 2294–2299; [DOI] [PubMed] [Google Scholar]
- 5e. Lee C., Ball N. D., Sammis G. M., Chem. Commun. 2019, 55, 14753–14756; [DOI] [PubMed] [Google Scholar]
- 5f. Laudadio G., Bartolomeu A. A., Verwijlen L. M. H. M., Cao Y., De Oliveira K. T., Noël T., J. Am. Chem. Soc. 2019, 141, 11832–11836; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5g. Lou T. S.-B., Bagley S. W., Willis M. C., Angew. Chem. Int. Ed. 2019, 58, 18859–18863; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 19035–19039; [Google Scholar]
- 5h. Tony Lo P. K., Chen Y., Willis M. C., ACS Catal. 2019, 9, 10668–10673; [Google Scholar]
- 5i. Liu Y., Yu D., Guo Y., Xiao J.-C., Chen Q.-Y., Liu C., Org. Lett. 2020, 22, 2281–2286; [DOI] [PubMed] [Google Scholar]
- 5j. Pérez-Palau M., Cornella J., Eur. J. Org. Chem. 2020, 2497–2500; [Google Scholar]
- 5k. Magre M., Cornella J., J. Am. Chem. Soc. 2021, 143, 21497–21502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For examples of the synthesis of arylsulfonimidoyl fluorides, see
- 6a. Johnson C. R., Bis K. G., Cantillo J. H., Meanwell N. A., Reinhard M. F. D., Zeller J. R., Vonk G. P., J. Org. Chem. 1983, 48, 1–3; [Google Scholar]
- 6b. Kowalczyk R., Edmunds A. J. F., Hall R. G., Bolm C., Org. Lett. 2011, 13, 768–771; [DOI] [PubMed] [Google Scholar]
- 6c. Guo J., Kuang C., Rong J., Li L., Ni C., Hu J., Chem. Eur. J. 2019, 25, 7259–7264; [DOI] [PubMed] [Google Scholar]
- 6d. Greed S., Briggs E. L., Idiris F. I. M., White A. J. P., Lücking U., Bull J. A., Chem. Eur. J. 2020, 26, 12533–1538; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Liu Y., Pan Q., Hu X., Guo Y., Chen Q.-Y., Liu C., Org. Lett. 2021, 23, 3975–3980; [DOI] [PubMed] [Google Scholar]
- 6f. Tony Lo P. K., Willis M. C., J. Am. Chem. Soc. 2021, 143, 15576–15581. [DOI] [PubMed] [Google Scholar]
- 7.For selected examples of suFEx chemistry, see
- 7a. Dong J., Krasnova L., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2014, 53, 9430–9448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9584–9603; [Google Scholar]
- 7b.Ref. [4a];
- 7c. Qin H.-L., Zheng Q., Bare G. A. L., Wu P., Sharpless K. B., Angew. Chem. Int. Ed. 2016, 55, 14155–14158; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14361–14364; [Google Scholar]
- 7d. Guo T., Meng G., Zhan X., Yang Q., Ma T., Xu L., Sharpless K. B., Dong J., Angew. Chem. Int. Ed. 2018, 57, 2605–2610; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2635–2640; [Google Scholar]
- 7e. Xu R., Xu T., Yang M., Cao T., Liao S., Nat. Commun. 2019, 10, 3752–3758; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7f. Liang D.-D., Streefkerk D. E., Jordaan D., Wagemakers J., Baggerman J., Zuilhof H., Angew. Chem. Int. Ed. 2020, 59, 7494–7500; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7564–7570. [Google Scholar]
- 8.For recent reviews, see
- 8a. Barrow A. S., Smedley C. J., Zheng Q., Li S., Dong J., Moses J. E., Chem. Soc. Rev. 2019, 48, 4731–4758; [DOI] [PubMed] [Google Scholar]
- 8b. Lee C., Cook A. J., Elisabeth J. E., Friede N. C., Sammis G. M., Ball N. D., ACS Catal. 2021, 11, 6578–6589; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Chelagha A., Louvel D., Taponard A., Berthelon R., Tlili A., Catalysts 2021, 11, 830; for a recent review on fluorinated sulfoximines, see [Google Scholar]
- 8d. Bizet V., Kowalczyk R., Bolm C., Chem. Soc. Rev. 2014, 43, 2426–2438. [DOI] [PubMed] [Google Scholar]
- 9. Cui B., Shibata N. in Fluorination (Eds.: Hu J., Umemoto T.), Springer Singapore, Singapore, 2020, pp. 20–29. [Google Scholar]
- 10.For the biological application of ArSF5, see Sowaileh M. F., Hazlitt R. A., Colby D. A., ChemMedChem 2017, 12, 1481–1490. [DOI] [PubMed] [Google Scholar]
- 11.For recent reviews on ArSF5, see
- 11a. Savoie P. R., Welch J. T., Chem. Rev. 2015, 115, 1130–1190; [DOI] [PubMed] [Google Scholar]
- 11b. Das P., Tokunaga E., Shibata N., Tetrahedron Lett. 2017, 58, 4803–4815; [Google Scholar]
- 11c. Kanishchev O. S., Dolbier W. R. in Advances in Heterocyclic Chemistry, Vol. 120 (Eds.: Scriven E. F. V., Ramsden C. A.), Academic Press, New York, 2016, pp. 1–42; [Google Scholar]
- 11d. Beier P. in Emerging Fluorinated Motifs (Eds.: Cahard D., Ma J.-A.), Wiley-VCH, Weinheim, 2020, pp. 551–570. [Google Scholar]
- 12. Westphal M. V., Wolfstädter B. T., Plancher J.-M., Gatfield J., Carreira E. M., ChemMedChem 2015, 10, 461–469. [DOI] [PubMed] [Google Scholar]
- 13. Cramer R., Coffman D. D., J. Org. Chem. 1961, 26, 4010–4014. [Google Scholar]
- 14. Seppelt K., Sundermeyer W., Angew. Chem. Int. Ed. Engl. 1970, 9, 905–905; [Google Scholar]; Angew. Chem. 1970, 82, 931–931. [Google Scholar]
- 15. Ross D. S., Sharp D. W. A., J. Chem. Soc. Dalton Trans. 1972, 34–37. [Google Scholar]
- 16.For selected examples of SuFEx chemistry using SOF4, see
- 16a. Li S., Wu P., Moses J. E., Sharpless K. B., Angew. Chem. Int. Ed. 2017, 56, 2903–2908; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2949–2954; [Google Scholar]
- 16b. Gao B., Li S., Wu P., Moses J. E., Sharpless K. B., Angew. Chem. Int. Ed. 2018, 57, 1939–1943; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1957–1961; [Google Scholar]
- 16c. Kitamura S., Zheng Q., Woehl J. L., Solania A., Chen E., Dillon N., Hull M. V., Kotaniguchi M., Cappiello J. R., Kitamura S., Nizet V., Sharpless K. B., Wolan D. W., J. Am. Chem. Soc. 2020, 142, 10899–10904; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Li S., Li G., Gao B., Pujari S. P., Chen X., Kim H., Zhou F., Klivansky L. M., Liu Y., Driss H., Liang D.-D., Lu J., Wu P., Zuilhof H., Moses J., Sharpless K. B., Nat. Chem. 2021, 13, 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clifford A. F., Howell J. L., Wooton D. L., J. Fluorine Chem. 1978, 11, 433–439. [Google Scholar]
- 18. Ruppert I., Angew. Chem. Int. Ed.. 1979, 18, 880–881; [Google Scholar]; Angew. Chem. 1979, 91, 941–943. [Google Scholar]
- 19. Michalak R. S., Martin J. C., J. Am. Chem. Soc. 1981, 103, 214–215. [Google Scholar]
- 20. Michalak R. S., Martin J. C., J. Am. Chem. Soc. 1982, 104, 1683–1692. [Google Scholar]
- 21. Chiba J., Sugihara T., Kaneko C., Chem. Lett. 1995, 24, 581–582. [Google Scholar]
- 22. Janzen A. F., Ou X., J. Fluorine Chem. 1995, 71, 207–207. [Google Scholar]
- 23.
- 23a. Ou X., Janzen A. F., Can. J. Chem. 1996, 74, 2002–2007; [Google Scholar]
- 23b. Ou X., Janzen A. F., J. Fluorine Chem. 2000, 101, 279–283. [Google Scholar]
- 24. Tsao F. A., Waked A. E., Cao L., Hofmann J., Liu L., Grimme S., Stephan D. W., Chem. Commun. 2016, 52, 12418–12421. [DOI] [PubMed] [Google Scholar]
- 25. Manel A., Berreur J., Leroux F. R., Panossian A., Org. Chem. Front. 2021, 8, 5289–5295. [Google Scholar]
- 26. von Halasz S. P., Glemser O., Chem. Ber. 1971, 104, 1256–1263. [Google Scholar]
- 27. Ruppert I., Chem. Ber. 1980, 113, 1047–1052. [Google Scholar]
- 28. Wang L., Cornella J., Angew. Chem. Int. Ed. 2020, 59, 23510–23515; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 23716–23721. [Google Scholar]
- 29. Umemoto T., Singh R. P., J. Fluorine Chem. 2012, 140, 17–27. [Google Scholar]
- 30. Hasek W. R., Smith W. C., Engelhardt V. A., J. Am. Chem. Soc. 1960, 82, 543–551. [Google Scholar]
- 31. Lal G. S., Pez G. P., Pesaresi R. J., Prozonic F. M., Cheng H., J. Org. Chem. 1999, 64, 7048–7054. [Google Scholar]
- 32. Sladojevich F., Arlow S. I., Tang P., Ritter T., J. Am. Chem. Soc. 2013, 135, 2470–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li L., Ni C., Wang F., Hu J., Nat. Commun. 2016, 7, 13320–13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Umemoto T., Singh R. P., Xu Y., Saito N., J. Am. Chem. Soc. 2010, 132, 18199–18205. [DOI] [PubMed] [Google Scholar]
- 35. Nielsen M. K., Ugaz C. R., Li W., Doyle A. G., J. Am. Chem. Soc. 2015, 137, 9571–9574. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Clifford A. F., El-Shamy H. K., Emeléus H. J., Haszeldine R. N., J. Chem. Soc. Abstr. 1953, 2372–2375; [Google Scholar]
- 36b. Hoffmann F. W., Simmons T. C., Beck R. B., Holler H. V., Katz T., Koshar R. J., Larsen E. R., Mulvaney J. E., Rogers F. E., Singleton B., Sparks R. S., J. Am. Chem. Soc. 1957, 79, 3424–3429; [Google Scholar]
- 36c. Dresdner R. D., Young J. A., J. Am. Chem. Soc. 1959, 81, 574–577; [Google Scholar]
- 36d. Dresdner R., T. Reed III , Taylor T., Young J. A., J. Org. Chem. 1960, 25, 1464–1466. [Google Scholar]
- 37. Darragh J. I., Sharp D. W. A., J. Chem. Soc. Chem. Commun. 1969, 864–865. [Google Scholar]
- 38. Abe T., Shreeve J. M., J. Fluorine Chem. 1973, 3, 187–196. [Google Scholar]
- 39. Denney D. B., Denney D. Z., Hsu Y. F., J. Am. Chem. Soc. 1973, 95, 8191–8192. [Google Scholar]
- 40. Ou X., Bernard G. M., Janzen A. F., Can. J. Chem. 1997, 75, 1878–1884. [Google Scholar]
- 41.
- 41a. Kirsch P., Bremer M., Kirsch A., Osterodt J., J. Am. Chem. Soc. 1999, 121, 11277–11280; [Google Scholar]
- 41b. Kirsch P., Hahn A., Eur. J. Org. Chem. 2006, 1125–1131. [Google Scholar]
- 42. Umemoto T., Garrick L. M., Saito N., Beilstein J. Org. Chem. 2012, 8, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kanishchev O. S., W. R. Dolbier Jr. , Angew. Chem. Int. Ed. 2015, 54, 280–284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 282–286. [Google Scholar]
- 44. Kosobokov M., Cui B., Balia A., Matsuzaki K., Tokunaga E., Saito N., Shibata N., Angew. Chem. Int. Ed. 2016, 55, 10781–10785; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10939–10943. [Google Scholar]
- 45. Pitts C. R., Bornemann D., Liebing P., Santschi N., Togni A., Angew. Chem. Int. Ed. 2019, 58, 1950–1954; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1970–1974. [Google Scholar]
- 46. Saidalimu I., Liang Y., Niina K., Tanagawa K., Saito N., Shibata N., Org. Chem. Front. 2019, 6, 1157–1161. [Google Scholar]
- 47. Wang L., Ni S., Cornella J., Synthesis 2021, 53, 4308–4312. [Google Scholar]
- 48. Surjadinata G., Hunter L., Matesic L., Pascali G., J. Flow Chem. 2021, 11, 107–115. [Google Scholar]
- 49. Zhong L., Savoie P. R., Filatov A. S., Welch J. T., Angew. Chem. Int. Ed. 2014, 53, 526–529; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 536–539. [Google Scholar]
- 50. Zhong L., Filatov A. S., Welch J. T., J. Fluorine Chem. 2014, 167, 192–197. [Google Scholar]
- 51. Das P., Takada M., Tokunaga E., Saito N., Shibata N., Org. Chem. Front. 2018, 5, 719–724. [Google Scholar]
- 52. Das P., Niina K., Hiromura T., Tokunaga E., Saito N., Shibata N., Chem. Sci. 2018, 9, 4931–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Niina K., Tanagawa K., Sumii Y., Saito N., Shibata N., Org. Chem. Front. 2020, 7, 1276–1282. [Google Scholar]
- 54.
- 54a. Sheppard W. A., J. Am. Chem. Soc. 1962, 84, 3072–3076; [Google Scholar]
- 54b. Kirsch P., Bremer M., Heckmeier M., Tarumi K., Angew. Chem. Int. Ed. 1999, 38, 1989–1992; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 2174–2178; [Google Scholar]
- 54c. Kirsch P., Bremer M., Angew. Chem. Int. Ed. 2000, 39, 4216–4235; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 4384–4405; [Google Scholar]
- 54d. Kirsch P., Hahn A., Eur. J. Org. Chem. 2005, 3095–3100. [Google Scholar]
- 55.
- 55a. Wipf P., Mo T., Geib S. J., Caridha D., Dow G. S., Gerena L., Roncal N., Milner E. E., Org. Biomol. Chem. 2009, 7, 4163–4165; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55b. Altomonte S., Baillie G. L., Ross R. A., Riley J., Zanda M., RSC Adv. 2014, 4, 20164–20176 . [Google Scholar]
- 56. True J. E., Thomas T. D., Winter R. W., Gard G. L., Inorg. Chem. 2003, 42, 4437–4441. [DOI] [PubMed] [Google Scholar]
- 57. Saethre L. J., Berrah N., Bozek J. D., Boerve K. J., Carroll T. X., Kukk E., Gard G. L., Winter R., Thomas T. D., J. Am. Chem. Soc. 2001, 123, 10729–10737. [DOI] [PubMed] [Google Scholar]
- 58. Sumii Y., Sasaki K., Tsuzuki S., Shibata N., Molecules 2019, 24, 3610–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chan J. M. W., J. Mater. Chem. C 2019, 7, 12822–12834. [Google Scholar]
- 60. Kim J. G., Kang O.-Y., Kim S. M., Issabayeva G., Oh I. S., Lee Y., Lee W. H., Lim H. J., Park S. J., Molecules 2020, 25, 5536–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gautam P., Yu C. P., Zhang G., Hillier V. E., Chan J. M. W., J. Org. Chem. 2017, 82, 11008–11020. [DOI] [PubMed] [Google Scholar]
- 62. Gautam P., Wang Y., Zhang G., Sun H., Chan J. M. W., Chem. Mater. 2018, 30, 7055–7066. [Google Scholar]
- 63. Silvey G. A., Cady G. H., J. Am. Chem. Soc. 1950, 72, 3624–3626. [Google Scholar]
- 64. Silvey G. A., Cady G. H., J. Am. Chem. Soc. 1952, 74, 5792–5793. [Google Scholar]
- 65. Huang H. N., Lagow R. J., Roesky H., Inorg. Chem. 1991, 30, 789–794. [Google Scholar]
- 66. Nyman F., Roberts H. L., J. Chem. Soc. 1962, 3180–3183. [Google Scholar]
- 67.For examples on the use of SF5Cl for organic synthesis, see
- 67a. Henkel T., Krügerke T., Seppelt K., Angew. Chem. Int. Ed. Engl. 1990, 29, 1128–1129; [Google Scholar]; Angew. Chem. 1990, 102, 1171–1172; [Google Scholar]
- 67b. Aït-Mohand S., Dolbier W. R., Org. Lett. 2002, 4, 3013–3015; [DOI] [PubMed] [Google Scholar]
- 67c. Dolbier W. R., Aït-Mohand S., Schertz T. D., Sergeeva T. A., Cradlebaugh J. A., Mitani A., Gard G. L., Winter R. W., Thrasher J. S., J. Fluorine Chem. 2006, 127, 1302–1310; [Google Scholar]
- 67d. Welch J., Ngo S., Lim D., WO 2009026191A1, 2009;
- 67e. Pitts C. R., Santschi N., Togni A., WO2019229103A1, 2019;
- 67f. Shou J. Y., Xu X. H., Qing F. L., Angew. Chem. Int. Ed. 2021, 60, 15271–15275; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 15399–15403; [Google Scholar]
- 67g. Debrauwer V., Leito I., Lõkov M., Tshepelevitsh S., Parmentier M., Blanchard N., Bizet V., ACS Org. Inorg. Au 2021, 1, 43–50; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67h. Feng F.-F., Ma J.-A., Cahard D., J. Org. Chem. 2021, 86, 13808–13816. [DOI] [PubMed] [Google Scholar]
- 68.
- 68a. Sheppard W. A., J. Am. Chem. Soc. 1960, 82, 4751–4752; [Google Scholar]
- 68b. Sheppard W. A., J. Am. Chem. Soc. 1962, 84, 3064–3072. [Google Scholar]
- 69.For similar studies on direct oxidation using AgF2, see
- 69a. Williams A. G., Foster N. R., WO9422817, 1994;
- 69b. Sipyagin A. M., Bateman C. P., Tan Y.-T., Thrasher J. S., J. Fluorine Chem. 2001, 112, 287–295; [Google Scholar]
- 69c. Sipyagin A. M., Enshov V. S., Kashtanov S. A., Bateman C. P., Mullen B. D., Tana Y.-T., Thrasher J. S., J. Fluorine Chem. 2004, 125, 1305–1316; [Google Scholar]
- 69d. Sipyagin A. M., Bateman C. P., Matsev A. V., Waterfeld A., Jilek R. E., Key C. D., Szulczewski G. J., Thrasher J. S., J. Fluorine Chem. 2014, 167, 203–210. [Google Scholar]
- 70. Sipyagin A. M., Pomytkin I. A., Paltsun S. V., Aleinikov N. N., Kartsev V. G., J. Fluorine Chem. 1991, 54, 115–115. [Google Scholar]
- 71. Chambers R. D., Spink R. C. H., Chem. Commun. 1999, 883–884. [Google Scholar]
- 72. Bowden R. D., Comina P. J., Greenhall M. P., Kariuki B. M., Loveday A., Philp D., Tetrahedron 2000, 56, 3399–3408. [Google Scholar]
- 73.
- 73a. Ajenjo J., Greenhall M., Zarantonello C., Beier P., Beilstein J. Org. Chem. 2016, 12, 192–197; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73b. Ajenjo J., Klepetářová B., Greenhall M., Bím D., Culka M., Rulíšek L., Beier P., Chem. Eur. J. 2019, 25, 11375–11382. [DOI] [PubMed] [Google Scholar]
- 74. Lummer K., Ponomarenko M. V., Röschenthaler G.-V., Bremer M., Beier P., J. Fluorine Chem. 2014, 157, 79–83. [Google Scholar]
- 75. Cui B., Kosobokov M., Matsuzaki K., Tokunaga E., Shibata N., Chem. Commun. 2017, 53, 5997–6000. [DOI] [PubMed] [Google Scholar]
- 76. Cui B., Jia S., Tokunaga E., Saito N., Shibata N., Chem. Commun. 2017, 53, 12738–12741. [DOI] [PubMed] [Google Scholar]
- 77. Guzyr O. I., Kozel V. N., Rusanov E. B., Rozhenko A. B., Fetyukhin V. N., Shermolovich Y. G., J. Fluorine Chem. 2020, 239, 109635. [Google Scholar]
- 78. Tanagawa K., Zhao Z., Saito N., Shibata N., Bull. Chem. Soc. Jpn. 2021, 94, 1682–1684. [Google Scholar]
- 79. Hoover F. W., Coffman D. D., J. Org. Chem. 1964, 29, 3567–3570. [Google Scholar]
- 80. Sergeeva T. A., Dolbier W. R., Org. Lett. 2004, 6, 2417–2419. [DOI] [PubMed] [Google Scholar]
- 81. Dolbier W. R., Mitani A., Warren R. D., Tetrahedron Lett. 2007, 48, 1325–1326. [Google Scholar]
- 82. Ponomarenko M. V., Lummer K., Fokin A. A., Serguchev Y. A., Bassil B. S., Röschenthaler G.-V., Org. Biomol. Chem. 2013, 11, 8103–8112. [DOI] [PubMed] [Google Scholar]
- 83. Duda B., Lentz D., Org. Biomol. Chem. 2015, 13, 5625–5628. [DOI] [PubMed] [Google Scholar]
- 84. Joliton A., Plancher J.-M., Carreira E. M., Angew. Chem. Int. Ed. 2016, 55, 2113–2117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2153–2157. [Google Scholar]
- 85. Kanishchev O. S., Dolbier W. R., J. Org. Chem. 2016, 81, 11305–11311. [DOI] [PubMed] [Google Scholar]
- 86.
- 86a. Rombach D., Birenheide B., Wagenknecht H.-A., Chem. Eur. J. 2021, 27, 8088–8093; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86b. Rombach D., Wagenknecht H.-A., ChemCatChem 2018, 10, 2955–2961; [Google Scholar]
- 86c. Rombach D., Wagenknecht H.-A., Angew. Chem. Int. Ed. 2020, 59, 300–303; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 306–310. [Google Scholar]