

Abstract
In clinical practice, 25–30% of the patients treated with fluoropyrimidines experience severe fluoropyrimidine‐related toxicity. Extensively clinically validated DPYD genotyping tests are available to identify patients at risk of severe toxicity due to decreased activity of dihydropyrimidine dehydrogenase (DPD), the rate limiting enzyme in fluoropyrimidine metabolism. In April 2020, the European Medicines Agency recommended that, as an alternative for DPYD genotype‐based testing for DPD deficiency, also phenotype testing based on pretreatment plasma uracil levels is a suitable method to identify patients with DPD deficiency. Although the evidence for genotype‐directed dosing of fluoropyrimidines is substantial, the level of evidence supporting plasma uracil levels to predict DPD activity in clinical practice is limited. Notwithstanding this, uracil‐based phenotyping is now used in clinical practice in various countries in Europe. We aimed to determine the value of pretreatment uracil levels in predicting DPD deficiency and severe treatment‐related toxicity. To this end, we determined pretreatment uracil levels in 955 patients with cancer, and assessed the correlation with DPD activity in peripheral blood mononuclear cells (PBMCs) and fluoropyrimidine‐related severe toxicity. We identified substantial issues concerning the use of pretreatment uracil in clinical practice, including large between‐center study differences in measured pretreatment uracil levels, most likely as a result of pre‐analytical factors. Importantly, we were not able to correlate pretreatment uracil levels with DPD activity nor were uracil levels predictive of severe treatment‐related toxicity. We urge that robust clinical validation should first be performed before pretreatment plasma uracil levels are used in clinical practice as part of a dosing strategy for fluoropyrimidines.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THIS TOPIC?
☑ Genotyping of DPYD, and adjustment of starting dose in patients with a variant allele, is now widely recommended in clinical practice guidelines. High pretreatment uracil levels (> 16 ng/mL) are associated with higher risk on severe fluoropyrimidine‐related toxicity. Therefore, this is potentially a good alternative for DPYD genotyping. However, this has not been prospectively validated.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ What is the value of measuring pretreatment uracil levels in predicting fluoropyrimidine‐related severe toxicity?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ In this prospective study, the association between pretreatment uracil and both dihydropyrimidine dehydrogenase (DPD) activity in peripheral blood mononuclear cells and fluoropyrimidine‐related toxicity could not be found. Moreover, we observed large between‐center differences in pretreatment uracil levels. We conclude that measuring uracil levels is prone to pre‐analytical errors, and can be affected by circadian rhythm and food intake.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Measurement of pretreatment uracil as a DPD‐phenotyping method is prone to pre‐analytical errors. We therefore urge that robust clinical validation of this phenotyping test is performed, before using this as part of routine clinical practice.
Fluoropyrimidines, including 5‐fluorouracil and its oral prodrug capecitabine, are indispensable drugs in the treatment of different solid tumors. A consistent concern in clinical practice however, is that 25–30% of patients treated with a standard dose experience severe toxicity, which can result in early treatment discontinuation, hospital admission, and even death. 1 , 2 , 3 , 4 , 5 , 6
Deficiency of the main enzyme metabolizing 5‐fluorouracil, dihydropyrimidine dehydrogenase (DPD), strongly increases a patient’s risk of experiencing severe fluoropyrimidine‐related toxicity. 1 , 7 Both genotype‐ and phenotype‐based methods to test for DPD deficiency have been developed, which allow identification of patients at risk of severe toxicity and reduction of their starting dose. 8 , 9 The clinical validity of genotyping‐based tests, which typically test for four DPYD genotypes (DPYD*2A, c.1679T>G, c.2846A>T, and c.1236G>A/HapB) has been established in multiple meta‐analyses and two large prospective studies. 1 , 7 , 10 , 11 , 12 These studies have shown that genotype‐based DPD testing in routine clinical practice leads to improvement of patient safety and is cost‐effective. 13 As a result, DPYD genotyping is now widely recommended in clinical practice guidelines, in predominantly White patient populations where these four DPYD deficient alleles occur at a consistent frequency, and used in different countries in Europe (Table S1 ).
Recently, the European Medicines Agency (EMA) has concluded that product labels needed to be updated. 14 , 15 Since April 2020, based on the EMA’s conclusions, product labels of fluoropyrimidines recommend that:
Patients treated with fluoropyrimidines (fluorouracil, capecitabine, and tegafur) should be tested for DPD deficiency before starting treatment;
Patients with partial DPD deficiency should be treated with an adjusted starting dose;
Genotyping and phenotyping based on plasma uracil levels are currently the most suitable methods to identify patients with DPD deficiency. 14 , 15
The recommendation on DPD phenotyping, specifically on pretreatment uracil levels, is of note considering the absence of both a prospective validation on the uracil threshold as a marker for fluoropyrimidine‐related severe toxicity and evidence that uracil testing improves patient safety when used to individualize dose. Because endogenous plasma uracil is converted into dihydrouracil (DHU) by DPD, the concentration of uracil in plasma is thought to be a proxy for DPD activity, with (exceptionally) elevated levels of endogenous plasma uracil being reflective of a (complete) DPD deficiency and therefore predictive of increased risk for severe toxicity. Consistent with this rationale, it has previously been shown that pretreatment plasma uracil concentrations higher than 15 or 16 ng/mL, depending on the study, were associated with increased risk of severe fluoropyrimidine‐related toxicity. 16 , 17 , 18 However, although the evidence for DPYD genotyping in preventing severe fluoropyrimidine‐related toxicity is extensive and includes data from well‐designed prospective clinical studies data showing that testing leads to improved patient safety, there are no such data to support the use of pretreatment uracil levels. 7 , 9 Moreover, uracil cutoff levels that predict toxicity have not been validated. In addition to this, prior studies have highlighted extensive variability in uracil measurements when different cohorts were compared, which, to date, remains insufficiently explained. 19 Therefore, the evidence available thus far regarding validation of pretreatment uracil and other DPD phenotyping methods appears insufficient to warrant routine use in clinical practice.
In the study reported here, we determined the value of pretreatment uracil levels in predicting fluoropyrimidine‐related severe toxicity and assessed the correlation between pretreatment uracil levels and DPD activity in peripheral blood mononuclear cells (PBMCs)—which is considered the reference assay/gold standard for measuring in vivo DPD activity. 9
PATIENTS AND METHODS
This study was part of the previously reported large prospective multi‐center study in 1103 patients (clinicaltrials.gov identifier NCT02324452). 12 Patient recruitment for this study was open from April 30, 2015, until December 21, 2017. Eligibility criteria have been reported previously 12 ; key criteria were: eligible to start with fluoropyrimidine‐based therapy, age ≥ 18 years, performance status ≤ 2, adequate bone marrow, renal and liver function, and no prior treatment with fluoropyrimidines. Ethical approval was granted by the medical ethical committee of The Netherlands Cancer Institute (NCI), Amsterdam, The Netherlands. All patients provided written informed consent before enrollment.
Pretreatment uracil (U) levels and pretreatment DHU/U ratio were measured in the main study cohort of patients recruited in 17 Dutch hospitals. Protocols for sample collection, handling, and processing for DPD phenotyping were available prior to study start.
The DPD enzyme activity in PBMCs was measured in all DPYD variant allele carriers and in a subgroup of wild type patients. To assess pretreatment DPD enzyme activity and uracil levels, a blood sample was drawn before the start of fluoropyrimidine treatment. The blood samples for pretreatment uracil levels were stored on ice directly and centrifuged within 30 minutes and the plasma stored at −80°C. Uracil levels were measured centrally in the NCI in Amsterdam using a validated bioanalytical method. 20 , 21 Samples for DPD enzyme activity in PBMCs were shipped to the Academic Medical Center (AMC) in Amsterdam for further processing, or processed at the hospital of blood drawn, as described previously. 22 After processing, isolated PBMCs were stored at −80°C before measurement of DPD activity at the AMC in Amsterdam with a validated bioanalytical assay. 21
Patients who received at least one fluoropyrimidine administration were followed for toxicity during the entire treatment period. Association between DPD activity in PBMCs, pretreatment uracil levels, and fluoropyrimidine‐related toxicity was assessed in patients wild type for DPYD variants, as patients who were identified as DPYD variant allele carriers (either DPYD*2A, DPYD*13, c.2846A>T, or c.1236G>A) underwent a per protocol dose adjustment at start of therapy. 12
Toxicity was graded according to the NCI Common Terminology Criteria for Adverse Events (CTC‐AE version 4.03) and severe toxicity was defined as CTC‐AE grade ≥ 3. 23 Toxicities defined by the treating physician as possibly, probably, or definitely related to fluoropyrimidine treatment were taken into account.
Uracil concentrations were compared between hospitals using the Kruskal–Wallis test. The median uracil concentrations in the hospitals were also individually compared with the reference hospital (NCI). The correlation between uracil levels and DPD enzyme activity was assessed by calculating the R 2. Furthermore, the uracil level was compared between patients who developed severe toxicity and patients who did not, using a Wilcoxon signed‐rank test. Last, the uracil levels and DPD enzyme activity were compared between DPYD genotypes using the Kruskal–Wallis test. The threshold for significance was P < 0.05. All statistical analyses were performed using R version 3.6.3.
RESULTS
In total, 1,037 patients participated in this study. Of these, 82 patients were identified as being DPYD variant allele carriers and 955 patients were DPYD wild type. DPD enzyme activity in PBMCs was determined in 138 patients (Figure S1 ).
Pretreatment plasma uracil levels were determined in all patients and were analyzed in relation to DPD activity, DPYD genotype and fluoropyrimidine‐related severe toxicity. Median pretreatment DPD enzyme activity, uracil levels and DHU/U ratios are summarized in Table S2 .
The results from subsequent analyses showed unexpected findings of potential clinical importance. First, there were unexpectedly large between‐center differences in measured pretreatment uracil levels (Kruskal–Wallis test, P < 0.001; Figure 1 ). The median uracil concentration of DPYD wild type patients was 9.63 ng/mL (range: 3.76–188 ng/mL) in the reference hospital (NCI) compared with a range of 7.59–16.30 ng/mL in the other hospitals, with significant differences between hospitals and the reference hospital in eight cases (Figure 1 ). In addition to these between‐center differences there appeared to be an effect of sex on uracil concentrations but this effect was smaller than compared with the effect of the study center (Table S3 ). Age and body surface area were not associated with uracil levels (Table S3 ).
Figure 1.
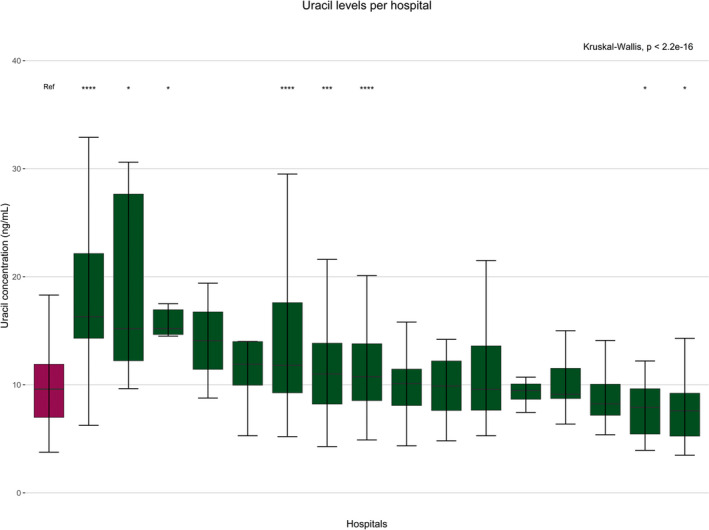
Differences in measured pretreatment uracil levels between hospitals. Differences in uracil concentrations (ng/mL) between the participating hospitals in an explorative substudy of a prospective multicenter study in 955 patients (clinicaltrials.gov identifier NCT02324452). All the samples were measured centrally therefore, the central hospital was chosen to be the reference hospital (indicated in red). Differences between medians were determined using one‐way analysis of variance (Kruskal–Wallis). *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001. [Colour figure can be viewed at wileyonlinelibrary.com]
Second, there was no correlation between pretreatment uracil concentrations and the reference assay (DPD activity in PBMCs; R 2 < 0.01, P = 0.391; Figure 2a ). However, when performing the analyses without the outlier with an uracil concentration of 188.0 ng/mL a significant correlation was found (R 2 < 0.04, P = 0.022). Importantly, there was no association between uracil and severe fluoropyrimidine‐related toxicity, as the median pretreatment uracil level was 10.10 ng/mL in patients without severe toxicity compared with 10.35 ng/mL in the patients with severe toxicity (P = 0.73; Figure 2b ). Multivariable analysis to adjust for other potential risk factors (body surface area, age, sex, treatment regimen, and cancer stage) did not result in a different association between pretreatment uracil levels (both as continuous variable or as dichotomous variable with a cutoff of 16 ng/mL) and severe toxicity (odds ratio: 0.997, 95% confidence interval: 0.97–1.01, P = 0.71). Whereas the epirubicin, cisplatin, and capecitabine/epirubicin, oxaliplatin, and capecitabine treatment regimen, concomitant radiotherapy, and sex are associated with severe toxicity (P = 0.03, P = 0.04, and P = 0.04, respectively). There was no association found between pretreatment DHU/U ratio and severe toxicity (Figure S2 ).
Figure 2.
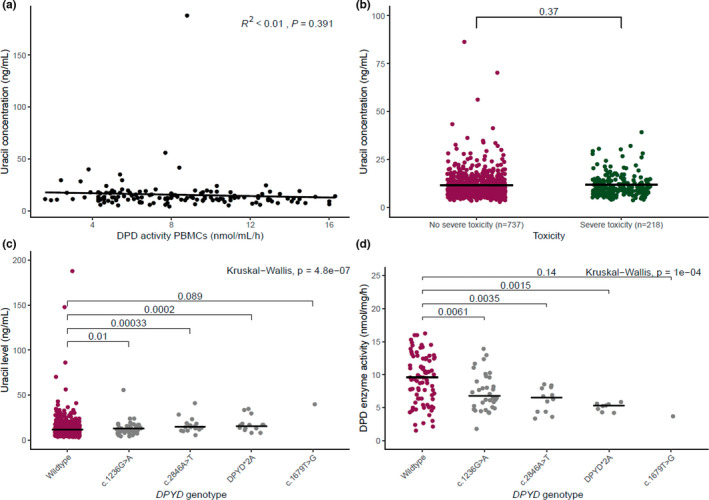
Correlations of endogenous uracil levels, DPD enzyme activity in PBMCs, toxicity, and DPYD genotype. Dots represent individual results. Black lines represent the median of the data. (a) Shows the correlation between endogenous uracil levels and DPD activity. (b) Shows the endogenous uracil concentration in patients with and without severe toxicity. DPYD variants were excluded from the analysis as they received initial dose reductions based on their genotype results. (c) Shows the endogenous uracil levels in patients by DPYD‐genotype. (d) Shows the DPD enzyme activity measured in PBMCs of 138 patients (both DPYD variant carriers and wild type patients). DPD, dihydropyrimidine dehydrogenase; DPYD, gene encoding dihydropyrimidine dehydrogenase; NS, not significant; PBMCs, peripheral blood mononuclear cells; P value; vs, versus. [Colour figure can be viewed at wileyonlinelibrary.com]
Last, and of note, pretreatment uracil levels did differ as expected between DPYD wild types, and DPYD variants c.1236G>A/HapB, c.2846A>T, DPYD*2A, and c.1679T>G with median uracil levels of 10.10, 12.20, 14.60, 16.80, and 40.10 ng/mL, respectively (Figure 2c ). In addition, DPD activity in PBMCs correlated with DPYD genotypes as expected and as previously reported (Figure 2d ). 24
DISCUSSION
In this study, we were not able to confirm that pretreatment uracil levels can predict DPD deficiency and severe fluoropyrimidine‐related toxicity. The results showed no association between pretreatment uracil levels and both DPD activity in PBMCs and occurrence of severe fluoropyrimidine‐related toxicity. More importantly, very large between‐center differences in the uracil measurements were observed. These results are in contrast with the prior single center study that showed a clear correlation between high endogenous uracil levels (> 16 ng/mL) and early severe toxicity 17 and which has been the basis for some of the recommendations in current clinical practice guidelines.
We identified potential pitfalls in the clinical use of pretreatment uracil levels to test for DPD deficiency. As uracil concentration in whole blood samples is stable for at least 4 hours when stored at 2–8°C, and in heparin plasma for at least 5 days when stored at 2–8°C, 21 , 25 the observed large variability between study centers could therefore probably be explained by differences in the duration of pre‐analytical sample handling at room temperature and processing among the 17 hospitals that participated in the prospective study. 21 , 25 The current study and the prior retrospective study 17 that was performed at one of the participating centers used the same validated bioanalytical assay, which was performed centrally, and it is therefore unlikely that the results are explained by the bioanalytical method. 21 Our hypothesis therefore is that between‐center differences in pre‐analytical sample processing are the main cause for the observed unexpected results. Second, the influence of circadian rhythm and food intake cannot be excluded. 25 , 26 In this study, both the time of sampling and the time of last meal before blood drawl was not standardized in all patients, which has been shown to affect DPD enzyme activity. 26 Hence, we feel that differences in pre‐analytical sample handling and processing are the main causes of the variability seen in pretreatment uracil concentration between hospitals. Previous data have also raised potential concerns regarding between‐center variability in observed measurements for pretreatment uracil/dihydrouracil ratio. 19 This should therefore be regarded as a note of caution for institutes that are currently using these DPD phenotyping tests.
The measurement of uracil levels prior to fluoropyrimidine‐based treatment is now advised by health authorities, reimbursed, and used in at least two countries in Europe (Supplementary Table S1 ). 27 , 28 The EMA’s recommendations will possibly further increase the uptake of pretreatment uracil tests. The concerns raised in this study and the fact that previous studies also raised concerns about between‐center variability in observed measurements add to the uncertainty around the test. 19 Considering the above, prospective validation of DPD phenotyping tests, including implementation of robust sample handling procedures and a personalized dosing advice in patients with high uracil concentration, is urgently needed. In addition, bioanalytical cross validation of the uracil test should be conducted. A large prospective study investigating the effect of phenotype‐guided dosing based on pretreatment uracil levels is currently being conducted (clinicaltrials.gov identifier NCT04194957). In this study, the time among sampling, processing, storage, and transportation is standardized to avoid pre‐analytical errors as much as possible. In addition, blood samples are taken between 8 and 10 in the morning and patients are required to be fasted to minimize the influence of circadian rhythm on the DPD enzyme and food intake on the uracil levels, respectively. This study may provide further insight into the validity of pretreatment uracil levels as a way to prevent severe fluoropyrimidine‐related toxicity. Besides pretreatment uracil, various other DPD phenotyping tests have been explored to identify patients with DPD deficiency, which include measurement of DPD enzyme activity in PBMCs, a 2‐13C uracil‐based breath test, and a uracil test dose. 29 The level of clinical validation in the predicting of severe toxicity of these tests varies, but is currently considerably lower compared with pretreatment uracil levels. Therefore, further research will be needed to understand the clinical utility of these tests.
CONCLUSION
In conclusion, the most important learning from this study is that measurement of pretreatment uracil concentration as a DPD‐phenotyping method to predict severe toxicity, is prone to pre‐analytical error. This is in contrast to genotyping methods which have shown to yield consistent results across centers and regions using available standardized protocols. Misclassification of patients in terms of DPD deficiency will have potentially relevant impact on patients’ safety and treatment outcome. We therefore urge that before using pretreatment uracil levels as part of routine clinical practice to adjust starting doses of fluoropyrimidines, robust clinical validation is performed, standardized protocols for sample handling and processing are developed, and bioanalytical cross validation is conducted.
FUNDING
C.A.T.C.L. was supported by an unrestricted grant from Roche Pharmaceuticals. L.M.H., C.A.T.C.L., and this study were sponsored by the Dutch Cancer Society (Alpe‐d’HuZes/KWF‐fund NKI2013‐6249). There was no involvement from any of the funding sources in the study design, data collection, analysis, or interpretation of the data.
CONFLICT OF INTEREST
J.H.M.S. and J.H.B. are (part time) employees, stock‐, and patent holders of Modra Pharmaceuticals, a spin out company developing oral taxane formulations; J.H.M.S. is also part time employee of Byondis bv and received consultancy fees from Debiopharm all not related to the contents of the manuscript. D.M. is a current full‐time employee, and shareholder of AstraZeneca, not related to the contents of the manuscript. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
M.W., J.K., J.J.S., and D.M. wrote the manuscript. C.A.T.C.L., L.M.H., J.H.M.S., H.G., A.C., H.J.G., R.H.J.M., J.J.S., and D.M. designed the research. F.M.M., C.A.T.C.L., L.M.H., A.L.T.I., J.H.M.S., H.G., A.C., H.J.G., and R.H.J.M. performed the research. M.W., J.K., F.M.M., C.A.T.C.L., L.M.H., A.B.P.K., J.G.M., M.C.S., N.V., H.R., J.H.B., D.P., and R.H.N.S. analyzed the data. A.B.P.K., H.R., J.H.B., and A.M. contributed new reagents/analytical tools.
Supporting information
Fig S1
Fig S2
Table S1
Table S2
Table S3
- 1. Meulendijks, D. et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine‐associated toxicity: a systematic review and meta‐analysis of individual patient data. Lancet Oncol. 16, 1639–1650 (2015). [DOI] [PubMed] [Google Scholar]
- 2. Hoff, P.M. et al. Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first‐line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. J. Clin. Oncol. 19, 2282–2292 (2001). [DOI] [PubMed] [Google Scholar]
- 3. Koopman, M. et al. Sequential versus combination chemotherapy with capecitabine, irinotecan, and oxaliplatin in advanced colorectal cancer (CAIRO): a phase III randomised controlled trial. Lancet 370, 135–142 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Van Cutsem, E. et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: results of a large phase III study. J. Clin. Oncol. 19, 4097–4106 (2001). [DOI] [PubMed] [Google Scholar]
- 5. Sharma, B.B. , Rai, K. , Blunt, H. , Zhao, W. , Tosteson, T.D. & Brooks, G.A. Pathogenic DPYD variants and treatment‐related mortality in patients receiving fluoropyrimidine chemotherapy: a systematic review and meta‐analysis. Oncologist 26, 1008–1016 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barin‐Le Guellec, C. et al. Toxicities associated with chemotherapy regimens containing a fluoropyrimidine: a real‐life evaluation in France. Eur. J. Cancer 124, 37–46 (2020). [DOI] [PubMed] [Google Scholar]
- 7. Deenen, M.J. et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J. Clin. Oncol. 34, 227–234 (2016). [DOI] [PubMed] [Google Scholar]
- 8. van Staveren, M.C. , Guchelaar, H.J. , van Kuilenburg, A.B. , Gelderblom, H. & Maring, J.G. Evaluation of predictive tests for screening for dihydropyrimidine dehydrogenase deficiency. Pharmacogenomics J. 13, 389–395 (2013). [DOI] [PubMed] [Google Scholar]
- 9. Knikman, J.E. , Gelderblom, H. , Beijnen, J.H. , Cats, A. , Guchelaar, H.J. & Henricks, L.M. Individualized dosing of fluoropyrimidine‐based chemotherapy to prevent severe fluoropyrimidine‐related toxicity: what are the options? Clin. Pharmacol. Ther. 109, 591–604 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosmarin, D. et al. Genetic markers of toxicity from capecitabine and other fluorouracil‐based regimens: investigation in the QUASAR2 study, systematic review, and meta‐analysis. J. Clin. Oncol. 32, 1031–1039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Terrazzino, S. , Cargnin, S. , Del Re, M. , Danesi, R. , Canonico, P.L. & Genazzani, A.A. DPYD IVS14+1G>A and 2846A>T genotyping for the prediction of severe fluoropyrimidine‐related toxicity: a meta‐analysis. Pharmacogenomics 14, 1255–1272 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Henricks, L.M. et al. DPYD genotype‐guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 19, 1459–1467 (2018). [DOI] [PubMed] [Google Scholar]
- 13. Henricks, L.M. et al. A cost analysis of upfront DPYD genotype‐guided dose individualisation in fluoropyrimidine‐based anticancer therapy. Eur. J. Cancer 107, 60–67 (2019). [DOI] [PubMed] [Google Scholar]
- 14. Pharmacovigilance Risk Assessment Committee (PRAC) of the European Medicines Agency (EMA) . Assessment report: Fluorouracil and fluorouracil related substances (capecitabine, tegafur and flucytosine) containing medicinal products <https://www.ema.europa.eu/en/documents/referral/fluorouracil‐fluorouracil‐related‐substances‐article‐31‐referral‐assessment‐report_en.pdf> (2020). Accessed August 11, 2021.
- 15. European Medicines Agency (EMA) . EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine <https://www.ema.europa.eu/en/news/ema‐recommendations‐dpd‐testing‐prior‐treatment‐fluorouracil‐capecitabine‐tegafur‐flucytosine> (2020). Accessed September 13,2021.
- 16. Boisdron‐Celle, M. et al. 5‐Fluorouracil‐related severe toxicity: a comparison of different methods for the pretherapeutic detection of dihydropyrimidine dehydrogenase deficiency. Cancer Lett. 249, 271–282 (2007). [DOI] [PubMed] [Google Scholar]
- 17. Meulendijks, D. et al. Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine‐associated toxicity. Br. J. Cancer 116, 1415–1424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Etienne‐Grimaldi, M.C. et al. New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS One 12, e0175998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sistonen, J. et al. Predicting 5‐fluorouracil toxicity: DPD genotype and 5,6‐dihydrouracil:uracil ratio. Pharmacogenomics 15, 1653–1666 (2014). [DOI] [PubMed] [Google Scholar]
- 20. Coudoré, F. et al. Validation of an ultra‐high performance liquid chromatography tandem mass spectrometric method for quantifying uracil and 5,6‐dihydrouracil in human plasma. J. Chromatogr. Sci. 50, 877–884 (2012). [DOI] [PubMed] [Google Scholar]
- 21. Jacobs, B.A. et al. Development and validation of a rapid and sensitive UPLC‐MS/MS method for determination of uracil and dihydrouracil in human plasma. J. Pharm. Biomed. Anal. 126, 75–82 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Van Kuilenburg, A.B.P. , Van Lenthe, H. , Tromp, A. , Veltman, P.C.J. & Van Gennip, A.H. Pitfalls in the diagnosis of patients with a partial dihydropyrimidine dehydrogenase deficiency. Clin. Chem. 46, 9–17 (2000). [PubMed] [Google Scholar]
- 23. NCI . National Cancer Institute: Common Terminology Criteria for Adverse Events v4.03 <https://evs.NCI.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010‐06‐14_QuickReference_8.5x11.pdf> (2010). Accessed August 2021.
- 24. van Kuilenburg, A.B. et al. Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5‐fluorouracil‐associated toxicity: identification of new mutations in the DPD gene. Clin. Cancer Res. 6, 4705–4712 (2000). [PubMed] [Google Scholar]
- 25. Capiau, S. , Van Landschoot, A. , Reyns, T. & Stepman, H. Pre‐analytical considerations for the analysis of uracil and 5,6‐dihydrouracil in heparin plasma. Clin. Chem. Lab. Med. 60, e112–e115 (2022). [DOI] [PubMed] [Google Scholar]
- 26. Jacobs, B.A. et al. Pronounced between‐subject and circadian variability in thymidylate synthase and dihydropyrimidine dehydrogenase enzyme activity in human volunteers. Br. J. Clin. Pharmacol. 82, 706–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Casneuf, V. et al. Joint Belgian recommendation on screening for DPD‐deficiency in patients treated with 5‐FU, capecitabine (and tegafur). Acta Clin. Belg. 77, 346–352 (2022). [DOI] [PubMed] [Google Scholar]
- 28. Haute Autorité de Santé . Des recommandations pour prévenir certaines toxicités sévères des chimiothérapies par fluoropyrimidines <https://www.has‐sante.fr/jcms/c_2892234/fr/des‐recommandations‐pour‐prevenir‐certaines‐toxicites‐severes‐des‐chimiotherapies‐par‐fluoropyrimidines> (2018). Accessed March 12, 2021.
- 29. Meulendijks, D. , Cats, A. , Beijnen, J.H. & Schellens, J.H. Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity ‐ Ready for clinical practice? Cancer Treat. Rev. 50, 23–34 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Table S2
Table S3