

Abstract
The Genetic Toxicology Technical Committee (GTTC) of the Health and Environmental Sciences Institute (HESI) is developing adverse outcome pathways (AOPs) that describe modes of action leading to potentially heritable genomic damage. The goal was to enhance the use of mechanistic information in genotoxicity assessment by building empirical support for the relationships between relevant molecular initiating events (MIEs) and regulatory endpoints in genetic toxicology. Herein, we present an AOP network that links oxidative DNA damage to two adverse outcomes (AOs): mutations and chromosomal aberrations. We collected empirical evidence from the literature to evaluate the key event relationships between the MIE and the AOs, and assessed the weight of evidence using the modified Bradford‐Hill criteria for causality. Oxidative DNA damage is constantly induced and repaired in cells given the ubiquitous presence of reactive oxygen species and free radicals. However, xenobiotic exposures may increase damage above baseline levels through a variety of mechanisms and overwhelm DNA repair and endogenous antioxidant capacity. Unrepaired oxidative DNA base damage can lead to base substitutions during replication and, along with repair intermediates, can also cause DNA strand breaks that can lead to mutations and chromosomal aberrations if not repaired adequately. This AOP network identifies knowledge gaps that could be filled by targeted studies designed to better define the quantitative relationships between key events, which could be leveraged for quantitative chemical safety assessment. We anticipate that this AOP network will provide the building blocks for additional genotoxicity‐associated AOPs and aid in designing novel integrated testing approaches for genotoxicity.
Abbreviations
- AOP
adverse outcome pathway
- AO
adverse outcome
- AP
apurinic/apyrimidinic
- BER
base excision repair
- BH
Bradford‐Hill
- DSB
double strand break
- GTTC
Genetic Toxicology Technical Committee
- HESI
Health and Environmental Sciences Institute
- HR
homologous recombination
- IATA
integrated approaches to testing and assessment
- KE
key event
- KER
key event relationship
- MIE
molecular initiating event
- MOA
mode of action
- NER
nucleotide excision repair
- NHEJ
non‐homologous end joining
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- SSB
single strand break
- WOE
weight of evidence
1. INTRODUCTION
The Adverse Outcome Pathway (AOP) framework organizes biological knowledge into a linear sequence of events, starting from a molecular initiating event (MIE) and ultimately leading to an adverse outcome (AO: an endpoint of regulatory concern) (Ankley et al., 2010; OECD, 2018). AOPs are purposefully simplified descriptions of toxicological pathways that are chemical‐agnostic (i.e., molecular interaction‐specific, not chemical‐specific) allowing for broad applications (Villeneuve et al., 2014). AOPs provide an effective method for applying new and existing knowledge to demonstrate the relationships between measurable biological events (Key events: KEs) and AOs at all levels of organization: molecular, cellular, tissue, organism, and population. During the process of developing an AOP, all relevant information is considered for assessing biological plausibility and building empirical evidence to support the relationships between the events (key event relationships: KERs). Typically, data are collected from a survey of the literature and vetted against the modified Bradford‐Hill (BH) criteria for causality to evaluate the weight of evidence (WOE) supporting the AOP (OECD, 2018).
The Health and Environmental Sciences Institute (HESI)'s Genetic Toxicology Technical Committee (GTTC) is currently working to develop a series of AOPs ending in potentially heritable genomic damage (e.g., AOs of mutations, chromosomal aberrations, and aneuploidy) to facilitate and promote the application of mechanistic information in genotoxicity assessment (Sasaki et al., 2020). The MIEs investigated by the GTTC thus far include oxidative DNA damage, topoisomerase inhibition, interactions with tubulin, and aurora kinase inhibition.
Herein, we describe an AOP network (Figure 1) developed as part of the HESI‐GTTC project (AOP #296 in the AOPWiki; URL:https://aopwiki.org/aops/296) (OECD, 2018). The AOP network links oxidative DNA damage to two regulatory endpoints in genetic toxicology: the induction of chromosomal aberrations and/or gene mutations. We anticipate that this AOP network will be valuable for designing novel Integrated Approaches to Testing and Assessment (IATA) aimed at identifying oxidative DNA damage as MIE and will be useful for providing key modules when building future AOPs (OECD, 2020a).
FIGURE 1.
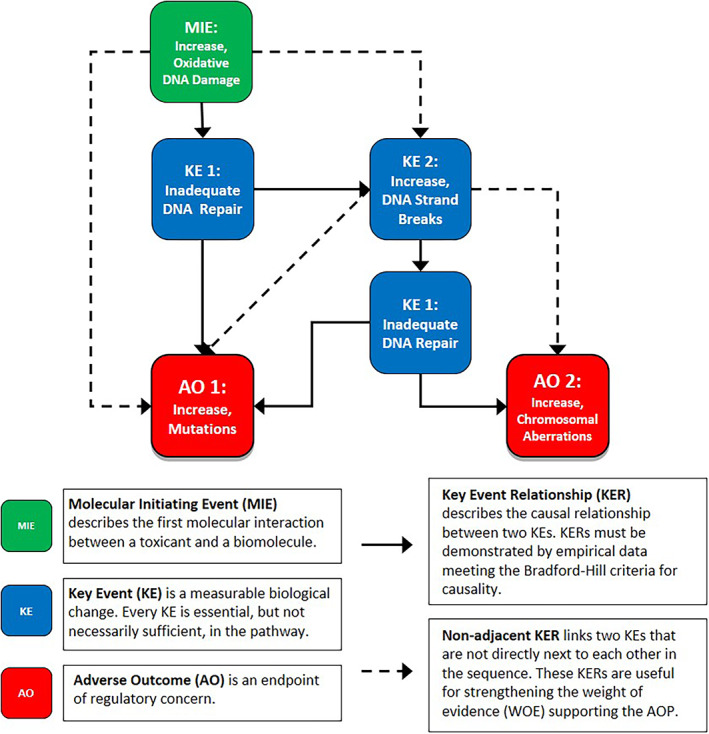
Flow chart of AOP #296: Oxidative DNA damage leading to mutations and chromosomal aberrations. “KE1: Indequate DNA repair” is shown twice in this flow diagram to emphasize that different repair pathways are involved in repairing different type of DNA damage and that “KE2: Increase, DNA strand breaks” is required for the progression to “AO2: Increase, Chromosomal aberrations”
2. ADVERSE OUTCOME PATHWAY (AOP #296)
AOP #296 (Figure 1) links oxidative DNA damage to mutations and chromosomal aberrations. We note that oxidative stress and oxidative DNA damage, as well as DNA strand breaks, mutations, and chromosomal damage, are intermediate processes in diverse pathways. Thus, the modules within this AOP network can be bridged into other AOPs. Indeed, this AOP network already shares KEs and KERs with AOP #15: Alkylation of DNA leading to heritable mutations (URL: https://aopwiki.org/aops/15; [Yauk et al., 2015]) and AOP #272: Direct deposition of ionizing energy onto DNA leading to lung cancer (URL: https://aopwiki.org/aops/272; [Chauhan et al., 2020]).
Strictly speaking, AOP #296 is a network because it contains two separate AOs: mutations and chromosomal aberrations. Table 1 summarizes the KEs that constitute the network and available methods for measuring each KE. Detailed descriptions of each KE are available online in the AOPWiki. The MIE of the network is increases in oxidative DNA damage. This event encompasses the broad range of lesions caused by oxidizing agents including strand breaks caused by direct electrophilic attack on the phosphate backbone and/or oxidatively damaged nitrogenous bases (Cadet et al., 2017; Melis et al., 2013). If the quantity and/or type of oxidative damage exceeds the DNA repair capacity of the cell (both high‐ and low‐fidelity repair pathways), the damage will not be repaired efficiently, completely, and/or accurately, leading to inadequate repair (KE1). Inadequately repaired lesions can lead to an increase in DNA strand breaks (KE2) or insertion of an incorrect base during replication, resulting in elevated numbers of mutations (AO1). Inadequate repair of DNA strand breaks (e.g., joining of two incorrect ends, unincorporated DNA fragment, lack of rejoining) can then lead to structural chromosomal damage and changes in the DNA sequence, resulting in an increase in chromosomal aberrations (AO2) and mutations (AO1), respectively.
TABLE 1.
Summary of key events in the AOP network (AOP #296) and methods of measurement
| Type | Event title | Description | How to measure |
|---|---|---|---|
| Molecular Initiating Event (MIE) | Increases in Oxidative DNA damage | This KE includes:
|
Relative quantification methods
Absolute quantification methods |
| Key Event (KE) | Inadequate DNA repair | This KE includes:
|
The KE can be inferred from retention of DNA lesions or increase in mutations and chromosomal aberrations (AO1 & AO2) which would indicate lack of repair, incomplete repair, or incorrect repair. Thus, methods of measuring this KE are mostly indirect (e.g., time‐course measurement of DNA lesions following exposure, dose–response experiments). Direct methods would include assays that measure a cell's capability to repair DNA damage (e.g., introducing fluorescent reporter construct containing a specific lesion [Mao et al., 2011; Chaim et al., 2017; Nagel et al., 2019]). Models of DNA repair deficiency such as knock‐out cell lines and rodent models (e.g., Agg −/− mice [Kay et al., 2021]) can be used to determine which pathway is critical for repairing the genotoxic lesion (Olivieri et al., 2020). |
| Key Event (KE) | Increases in DNA strand breaks | This KE includes:
|
|
| Adverse Outcome (AO) | Increases in Mutations | This KE includes:
|
|
|
Adverse Outcome (AO) |
Increases in Chromosomal aberrations | This KE includes:
|
|
The GTTC AOP sub‐team developed the AOP network in accordance with the OECD AOP Users' Handbook supplement for guidance in AOP development (OECD, 2018). Below, we provide background relevant to the AOP, summarize the evidence supporting the KERs and KEs, provide an overall AOP assessment, and discuss uncertainties, inconsistencies, and major knowledge gaps.
3. BACKGROUND
Oxidative stress generally refers to an imbalance between oxidants and antioxidants within a cell, where oxidants overwhelm the antioxidants. Elevated levels of cellular reactive oxygen species (ROS) and reactive nitrogen species (RNS) have been associated with genomic instability (Hanahan & Weinberg, 2011; Panieri & Santoro, 2016; Roszkowski et al., 2011). Indeed, the ability to induce excessive ROS and other free radicals is a common characteristic of carcinogens (Krewski et al., 2019; Smith et al., 2016). Therefore, the potential for chemicals to induce oxidative DNA damage can be an important consideration in chemical genotoxicity and carcinogenicity safety assessment.
Oxidants can arise from both endogenous and exogenous sources. Many endogenous biochemical processes involve redox reactions; at steady state, cellular respiration and various metabolic activities (e.g., oxidative phosphorylation in the mitochondria, NADPH oxidase, monoamine oxidase, and CYP450 monooxygenase activities) continuously generate ROS and other free radicals (Liou & Storz, 2010). Furthermore, the role that ROS and RNS play as secondary signaling molecules in various pathways indicates that certain amounts of these reactive species (e.g., H2O2) are essential for proper cellular functions (D'Autreaux & Toledano, 2007; Finkel, 2011; Weidinger & Kozlov, 2015). Healthy cells are capable of tightly regulating oxidant levels at steady state using endogenous antioxidants (e.g., N‐acetylcysteine, glutathione) and enzymes (e.g., glutathione‐S‐transferases) and have antioxidant responses for countering elevations in ROS and other oxidants to a certain extent (e.g., via catalase), to maintain non‐cytotoxic levels of oxidants.
All biomolecules, including nuclear and mitochondrial DNA, are susceptible to oxidative damage (Cadet et al., 2017; Markkanen, 2017). Oxidative stress occurs when free radicals overwhelm the cellular antioxidant capacity, which can occur under certain physiological conditions, such as inflammation, and via exogenous stressors that are direct oxidants and/or induce oxidative species. Perturbation of cellular respiration (e.g., electron transport chain disruptors [Wang et al., 2015]), depletion of endogenous antioxidants (e.g., glutathione [Beddowes et al., 2003]), generation and/or amplification of reactive species (e.g., redox cycling of quinones (Penning, 2017), Fenton reaction of metals (Cu2+, Fe2+, Ni2+) with molecular oxygen (Lloyd & Phillips, 1999)), and direct oxidation of cellular components (e.g., ionizing and non‐ionizing radiation, oxidants such as potassium bromate [Murata et al., 2001]) are ways in which toxicants cause oxidative stress and damage in the cell.
ROS and other free radicals cause a broad range of oxidative damage to DNA. Direct electrophilic attack on the phosphate backbone can induce DNA strand breaks. Additionally, all nitrogenous bases are vulnerable to oxidative damage (Cadet & Wagner, 2013; Cooke et al., 2003). Oxidative adducts form mainly on C5 and less frequently on C6 of thymine and cytosine (e.g., thymine glycol, 5‐hydroxy‐2′‐deoxycytidine [5‐OHdC]), and on C8 of guanine and adenine (e.g., 8‐oxo‐deoxyguanine (8‐oxo‐dG), 8‐oxo‐deoxyadenine,2,6‐diamino‐4‐hydroxy‐5‐formamidopyrimidine (FapyG), 8‐nitroguanine, 4,6‐diamino‐5‐formamidopyrimidine (FapyA)) (Berquist & Wilson III, 2012; Hiraku, 2010; Whitaker et al., 2017). Guanine is most prone to oxidation due to its low oxidation potential (i.e., most easily loses electrons) (Jovanovic & Simic, 1986). In fact, 8‐oxo‐dG is the most mutagenic and abundant oxidative DNA lesion; between 2400 and 10,000 8‐oxo‐dG are estimated to be present in the genome at steady state due to endogenous oxidants (Kalam et al., 2006; Ohno et al., 2006; Swenberg et al., 2011). In addition, abasic (AP; apurinic or apyrimidinic) sites are abundant in dsDNA at steady state as they can arise from spontaneous hydrolytic reactions and the removal of nitrogenous bases by glycosylases during base damage repair (Swenberg et al., 2011). Oxidative degradation of certain cellular components can also give rise to toxic products and exacerbate the damage in the cell during oxidative stress; for example, lipid peroxidation generates DNA‐reactive aldehydes, such as malondialdehyde and acrolein, that form mutagenic malondialdehyde‐2′‐deoxyguanosine (M1G) and γ‐hydroxy‐1,N2‐propano‐2′‐deoxyguanosine adducts, respectively (Gentile et al., 2017; Zhou et al., 2005).
For practical reasons, AOP #296 equates 8‐oxo‐dG formation with ‘oxidative DNA damage’, and the MIE. The fate of guanine lesions has been most extensively researched and is well understood (Cadet et al., 2017; Markkanen, 2017; Roszkowski et al., 2011; Whitaker et al., 2017). In human biomonitoring studies, 8‐oxo‐dG is an accepted biomarker of oxidative stress and oxidative DNA damage (Cooke et al., 2008; Li et al., 2014). We note that 8‐oxo‐dG is not a terminal product of oxidative damage; 8‐oxo‐dG can be further oxidized to additional mutagenic intermediates such as spiroiminodihydantoin and guanidinohydantoin (Jena & Mishra, 2012). However, as with many other oxidative lesions on pyrimidines and adenine, these lesions are estimated to be small fractions compared to 8‐oxo‐dG (Cooke et al., 2008; Yu et al., 2005).
When DNA damage is caused by uncertain mechanisms, oxidative stress is often suspected because it is observed under diverse chemical insults such as tert‐butylhydroquinone (Eskandani et al., 2014; Ramadan & Suzuki, 2012), heavy metals (Patlolla et al., 2009a; Patlolla et al., 2009b; Skipper et al., 2016), and carbon tetrachloride (Beddowes et al., 2003). Oxidative DNA base damage has been implicated in genomic instability, which is one of the hallmarks of cancer (Hanahan & Weinberg, 2011). Furthermore, oxidative DNA damage is observed in many non‐neoplastic diseases including neurodegenerative (Coppede & Migliore, 2015), cardiovascular (Ishida et al., 2014; Shah et al., 2018), and inflammatory airway diseases (Tzortzki et al., 2012). Thus, measurement of 8‐oxo‐G is an effective strategy to assess a chemical's potential to induce oxidative DNA damage (through direct and indirect processes) and can provide insight into the causal genotoxic mechanism.
4. KEY EVENT RELATIONSHIPS (KERs)
KERs describe the causal relationships between upstream and downstream KEs. The WOE analysis of KERs involves an assessment of biological plausibility, empirical evidence including evidence supporting the essentiality of each KE in the AOP, and consideration of uncertainties and inconsistencies. In AOP #296, two sets of KERs connect the KEs in the network – adjacent and non‐adjacent (Figure 1). Adjacent KERs link two KEs that are directly upstream and downstream of each other in the pathway. Non‐adjacent KERs link two KEs that are separated by one or more intermediate KEs; these KERs are particularly useful for strengthening the evidence for the overall pathway when there is a limited number of studies supporting the adjacent KERs (e.g., when one KE is not routinely measured). Tables 2 and 3 summarize the adjacent and non‐adjacent KERs within the AOP network, respectively. These tables describe each of the KERs and provide examples of empirical evidence supporting the relationships. Extended sets of empirical evidence for each KER are available online in the AOPWiki, and collectively, the empirical evidence for both types of KERs provides WOE for the overall AOP (OECD, 2018).
TABLE 2.
Summary of adjacent key event relationships (KER)
| Key event relationship (KER) title | Description | Examples of empirical evidence |
|---|---|---|
| Increases in Oxidative DNA damage leads to Inadequate DNA repair | At steady state, oxidative lesions generated by endogenous free radicals are readily repaired by basal repair mechanisms, mainly base excision repair (BER), to maintain baseline levels. However, if the level of oxidative DNA lesions (i.e., oxidized bases, abasic sites, strand breaks) increases above a cell's ability to detoxify, an exceeded repair capacity can lead to lack of, or faulty repair (i.e., increase in unrepaired lesions, repair intermediates, mispaired bases) – all indicators of inadequate DNA repair. |
Concentration/Dose Concordance
Temporal Concordance
Other types of evidence |
| Inadequate DNA repair leads to Increases in DNA strand breaks | Exceeded BER capacity due to an increase in oxidative lesions can lead to an accumulation of repair intermediates, including AP sites and SSBs. Increase in the number of SSBs elevates the risk of two SSBs occurring in close proximity to each other; if two SSBs occur on opposite strands, it may be converted into a DSB, exacerbating the damage. Increase in unrepaired lesions and repair intermediates due to inadequate repair can further impede the repair of other damaged sites nearby. BER intermediates are known to be replication blocks that can cause a replication fork to stall and collapse. Collapsing of replication forks can cause DSBs, the most toxic type of DNA lesion. |
Concentration/Dose and Incidence Concordance
Dose and Temporal Concordance
|
| Increases in DNA strand breaks leads to Inadequate DNA repair |
Increase in the number of strand breaks can exceed the repair capacity (DSB: NHEJ or HR; SSB: SSBR), resulting in prolonged presence of strand breaks (lack of repair). Increase in the occurrence of NHEJ may also increase the incidence of two incorrect ends being joined, altering the DNA sequence. DSBs may also lead to mutagenic salvage DNA repair pathways such as break‐induced replication (BIR) and microhomology‐mediated break‐induced replication (MMBIR). |
Concentration and Incidence Concordance
Temporal Concordance
|
| Inadequate DNA repair leads to Increases in Mutations |
Higher incidences of NHEJ and mutagenic salvage repair can increase the chance of incorrect joining of two broken ends, altering the DNA sequence. Unrepaired base lesions can lead to point mutations, especially if they are able to form stable base pairs with incoming nucleotides during replication (e.g., 8‐oxo‐dG base pairing with adenine). |
Incidence and Concentration Concordance
Temporal Concordance
|
| Inadequate DNA repair leads to Increases in Chromosomal Aberrations | If DSBs are not repaired in a timely manner, the ends can shift away from the original position, reducing the likelihood of error‐free repair. Unrepaired strand breaks and mis‐joined ends by incorrect repair can result in translocation, inversion, deletion of sections (unincorporated fragments), and other structural aberrations of the chromosome (e.g., ring and loop formation). |
Concentration/Dose and Incidence Concordance
Temporal Concordance
|
TABLE 3.
Summary of non‐adjacent key event relationships (KER)
| Key event relationship (KER) title | Description | Empirical evidence |
|---|---|---|
| Increases in Oxidative DNA damage leads to Increases in DNA strand breaks | Biologically plausible mechanisms linking increase in oxidative DNA lesions to increase in strand breaks include: 1) Incomplete BER due to an imbalance in the glycosylase and AP site endonuclease (APE) activities resulting in AP site and SSB accumulation; 2) increase in oxidative lesions impeding the repair of neighboring lesions (possibly SSBs); 3) occurrence of SSBs in close proximity to each other during BER; 4) collision of replication fork with BER proteins and intermediates. |
Concentration/Dose Concordance
Temporal Concordance
|
| Increases in Oxidative DNA damage leads to Increases in Mutations | It has been extensively demonstrated that 8‐oxodG, the most abundant oxidative DNA lesion, preferentially forms base pairs with incoming dA during replication causing G to T transversions, which are characteristic of oxidative DNA damage. |
Temporal and Incidence Concordance
Concentration Concordance
|
| Increases in DNA strand breaks leads to Increases in Mutations | Increases in SSBs and DSBs can lead to a higher incidence of erroneous repair by NHEJ and mutagenic salvage repair pathways. | Concentration/Dose and Incidence Concordance
|
| Increases in DNA strand breaks leads to Increases in Chromosomal Aberrations | Mechanistically, DSBs must occur for chromosomal aberrations to occur. If DSBs are not rejoined in a timely manner, the ends may shift away from their original position, resulting in loss of segments or rearrangement of sections. | Temporal Concordance
|
To develop this AOP, the team members were assigned modules (i.e., KEs or KERs) and reviewed the literature to identify key methodologies for measuring the KEs within their modules. For each KER, a literature search was conducted independently of other KERs to maintain modularity. The modified BH criteria for causality guided the selection of studies that were used as empirical evidence. Initially, the BH criteria were created to facilitate the evaluation of causal associations in epidemiological studies. The BH criteria used herein have been modified for applications in WOE assessment of AOPs (Becker et al., 2015).
The three main criteria considered when evaluating empirical support for the KERs include: concordance in concentration/dose, temporality, and incidence of the KEs (Becker et al., 2015; OECD, 2018). Concentration or dose concordance indicates that the upstream KE is observed at the same or lower stressor concentrations or doses than the downstream KE within the KER. Similarly, incidence concordance indicates that the upstream KE occurs more frequently (or at the same frequency), or at a higher (or the same) incidence, than the downstream KE. Temporal concordance is demonstrated by the detection of the upstream KE at an earlier (or identical) time point to the downstream KE. Both in vivo and in vitro data were considered and diverse experimental methods were used within these studies (Tables 2 and 3). Thus, to evaluate empirical support for the AOP, studies that measured more than one KE from the AOP were considered.
5. BIOLOGICAL PLAUSIBILITY
Proper repair of oxidative DNA lesions is critical for maintaining genomic integrity. It is well‐established knowledge that DNA repair capacity in a cell is limited and that an increase in oxidative DNA lesions above a cell's threshold for effective repair can lead to damage of the genome (Brenerman et al., 2014; Cadet & Wagner, 2013; Yang et al., 2006). Overall, based on the current mechanistic understanding of mutation and chromosomal aberration induction by ROS and other oxidizing agents, biological plausibility of this AOP network is strong.
The mutagenicity of certain oxidative base lesions has been extensively studied (Riva et al., 2020). Many of the oxidative base lesions can form stable base pairs with incorrect dNTPs during replication via translesion synthesis (TLS), giving rise to base substitutions (Cadet et al., 2017; Maddukuri et al., 2014; Shimizu et al., 2007). For example, FapyA and 8‐oxo‐dA can base pair with C, giving rise to A to C transversions (Kalam et al., 2006). Similarly, dA can be inserted opposite of 8‐oxo‐dG or FapyG by replicative polymerases and TLS polymerases, η and κ, leading to G to T transversions (Markkanen et al., 2012; Shimizu et al., 2007; Taggart et al., 2014). Since 8‐oxo‐G is the predominant oxidative DNA lesion, G to T transversions are the most predominant base substitutions observed in cells exposed to various oxidative stress inducers (Arai et al., 2002; Klungland et al., 1999; Unfried et al., 2002).
Oxidative damage to nitrogenous bases is primarily repaired by base excision repair (BER) and, to a lesser extent, by nucleotide excision repair (NER) (Scott et al., 2014; Shafirovich et al., 2016; Whitaker et al., 2017). Several different DNA glycosylases initiate the repair of oxidative base lesions: OGG1, MYH (MUTYH), NTHL1, NEIL1, NEIL2, and NEIL3 (Markkanen, 2017; Shafirovich et al., 2016). BER glycosylases are either mono‐functional (glycosylase only) or bifunctional (glycosylase and lyase). DNA glycosylases remove oxidized nitrogenous bases by hydrolyzing the N‐glycosidic bond, giving rise to an abasic site (AP). The lyase function of bifunctional glycosylases creates a SSB 3′ to the AP site after the removal of the oxidized nitrogenous base (Jacobs & Schar, 2012; Minko et al., 2016).
Following BER initiation by glycosylases, the AP endonuclease (APE1) then creates a SSB 5′ to the AP site. A single nucleotide gap will form if a bifunctional glycosylase preceded APE1, as there will be a nick 3′ to the AP site created earlier by the lyase function. If BER is initiated by a mono‐functional glycosylase, the AP site will remain after the APE1 reaction. Flap endonuclease (FEN1) is then required to excise the AP site by creating a nick 3′ to the site, giving rise to a single nucleotide gap. DNA polymerase β then synthesizes over the gap and DNA ligase forms a new phosphodiester bond that seals the SSB (Whitaker et al., 2017). Overwhelmed BER capacity may cause an imbalance in the initiating steps of BER, resulting in incomplete repair of oxidative lesions (Cabelof et al., 2004; Parrish et al., 2018; Wang, Li, et al., 2018; Yang et al., 2006). If BER is disrupted before completion, intermediates such as AP sites and SSBs can persist in the genome (Cabelof et al., 2004; Parrish et al., 2018; Sheng et al., 2012).
Both strand breaks and AP sites can arise directly due to reactive species and as BER intermediates (Cadet et al., 2017). SSBs and AP sites are known to block DNA replication; if a replication fork encounters such lesions, it can stall and collapse, resulting in a DSB (Ensminger et al., 2014; Kidane et al., 2014; Kuzminov, 2001). Furthermore, unrepaired oxidative base lesions, SSBs, and AP sites are more toxic and difficult to repair when in closer proximity to each other (Pearson et al., 2004; Sedletska et al., 2013; Yang et al., 2004). Oxidative base lesions, especially 8‐oxo‐dG, can impede the repair of neighboring SSBs, increasing the risk of DSB formation (Calsou et al., 1996; Lomax et al., 2004). Thus, if such base lesions persist in the genome due to inadequate repair, the repair of existing and new strand breaks can be impaired, leading to a net increase in strand breaks (Caglayan & Wilson, 2015). In addition, if two SSBs occur on opposite strands within a sufficiently small distance during BER, the SSBs may be converted to a DSB. Some studies suggest that multiple DNA lesions within one or two helical turns can increase the rate of DSB formation (Cannan & Pederson, 2016).
While BER is essential for maintaining genomic integrity, it is known to have both protective and damaging effects on the genome during oxidative stress. For example, MUTYH, a glycosylase that removes dA opposite 8‐oxo‐dG, may undergo futile cycles of dA removal and reinsertion by polymerases, potentially increasing AP sites in the genome (Hashimoto et al., 2004). Moreover, knock‐out of OGG1, a bifunctional glycosylase that primarily targets 8‐oxo‐dG lesions opposite dC, prevented AP site accumulation in human cells infected with H. pylori monitored over a 72‐h period (Kidane et al., 2014). AP sites are electrophilic and chemically unstable and, thus, are prone to strand breakage (Greenberg, 2014). In addition, an increase in the number of damaged sites where BER is in progress may increase the risk of DSB generation due to collision with the replication fork (Ensminger et al., 2014). Other studies have shown exacerbated strand breaks in BER‐proficient cells following acute chemical exposures (Banda et al., 2017; Sheng et al., 2012; Wang, Li, et al., 2018). However, a compromised BER capability (due to glycosylase variants or knock‐out) leads to an increase in spontaneous mutation frequency and structural damage (DSBs and MN formation) under chronic chemical exposures (Arai et al., 2002; Galick et al., 2013; Hu et al., 2005; Minowa et al., 2000; Ondovcik et al., 2012). Collectively, these studies suggest that elevated BER glycosylase activity in response to increases in oxidative lesions can give rise to higher incidences of incomplete repair but a timely clearance of oxidative base lesions is ultimately critical for maintaining genomic stability.
Failed repair of oxidative lesions (e.g., replication fork stalling and collapsing due to BER intermediates or unrepaired SSBs during the S phase) can lead to the formation of DSBs. The active repair pathway and the efficiency of repair can vary depending on the cell cycle stage, and these pathways can be error‐prone. Non‐homologous end joining (NHEJ), the fastest DSB repair pathway, is an error‐prone process that ligates two broken ends generally based on microhomologies (≤4 nt) of single‐strand overhangs and is active across all cell cycle stages (Chang et al., 2017; Lieber et al., 2010; Rodgers & McVey, 2016). The level of NHEJ activity has been observed to increase with the progression in cell cycle, reaching the highest level in the G2 phase; concordantly, the efficiency of DSB repair has been reported to be the highest in the G2/M phase (Mao et al., 2008). Comparatively, NHEJ is often a longer process than homologous recombination (HR), which requires homologous sequences in the sister chromatid or homologous chromosome as a template to ensure fidelity of the reconstructed strands; HR is mostly restricted to the S and G2 phases of the cell cycle and is most active in the S phase (Brandsma & van Gent, 2012; Fugger & West, 2016; Gandhi et al., 2012).
As with oxidative base lesions, the repair capacity for DSBs is limited and repair kinetics are compromised with increasing DSBs; thus, the risk of faulty or lack of repair and, consequently, structural chromosomal damage (AO2) increases with the number of DSBs (Calsou et al., 1996; Cannan & Pederson, 2016). The presence of DNA strand breaks over a prolonged period of time increases the chance of the two broken ends diffusing away from the original position, hampering restoration of the original structure (Schipler & Iliakis, 2013). In a cell under chemical insult, DSBs may also lead to the induction of salvage DNA repair pathways with high error rates such as microhomology‐mediated end joining (MMEJ), break‐induced replication (BIR), and microhomology‐mediated break‐induced replication (MMBIR) (Kramara et al., 2018; Sakofsky et al., 2015; Seol et al., 2018). While salvage repair pathways and NHEJ promote survival of the cell, the inadequate repair of DSBs can ultimately result in structural aberrations (AO2) (i.e., large deletions and insertions, translocations, ring formation) and mutations (AO1) (Ferguson & Alt, 2001; Iliakis et al., 2004). In addition, chromosomal aberrations can also arise from HR, which is typically a highly accurate repair pathway; for example, a crossover event between the damaged site and a region of sequence homology on another chromosome can result in exchange‐type structural aberrations causing translocations, radial formation, and/or dicentric chromosomes with accompanying acentric fragments (Obe et al., 2002). Thus, DSBs in areas of repetitive sequences have a higher risk of misalignment with an incorrect chromosome region during homologous recombination (Brandsma & van Gent, 2012).
6. ESSENTIALITY OF KEY EVENTS
The essentiality of each KE in the pathway can be demonstrated by modulating one KE (e.g., via overexpression or inhibition of an enzyme) and observing concordance in the downstream KEs. The essentiality of the MIE, “Increase, Oxidative DNA Damage”, has been demonstrated by studies inhibiting or exacerbating ROS by treating cells with an antioxidant (e.g., N‐acetylcysteine) or depleting endogenous glutathione (using buthionine sulphoximine). For example, pre‐treatment with an antioxidant reduced the quantity of oxidative base lesions (MIE) that are formed following exposure to a ROS inducer, with concordant decreases in DNA strand breaks (KE2: Increase, DNA Strand Breaks) and MN (AO2: Increase, Chromosomal Aberrations) observed at later time points (Dong et al., 2014; Federici et al., 2015; Kopp et al., 2020; Reliene et al., 2004). Similarly, depletion of glutathione resulted in elevated levels of both 8‐oxo‐dG and DNA strand breaks (Beddowes et al., 2003).
The essentiality of KE1, “Inadequate DNA Repair”, has been demonstrated by studies that modulated DNA repair capacity by knocking out various BER glycosylases (Hu et al., 2005; Nallanthighal et al., 2017; Suzuki et al., 2010; Wang, Li, et al., 2018). For example, steady‐state levels of oxidative lesions (e.g., 8‐oxo‐dG, FapyG, FapyA) and spontaneous G to T transversions (AO1: Increase, Mutations) increased in mammalian cells deficient in various BER glycosylases (Hu et al., 2005; Nallanthighal et al., 2017; Suzuki et al., 2010). OGG1 deficiency had different effects on cells exposed for different durations. After a 30 min‐exposure to hydrogen peroxide, significantly fewer strand breaks (KE2) were observed in OGG1‐deficient cells compared to wild type cells, suggesting an accumulation of SSBs due to incomplete BER in wild type cells (Wang, Li, et al., 2018). A seven‐day continuous exposure to silver nanoparticles in mice induced significant concordant increases in 8‐oxo‐dG, DSBs (KE2), and MN (AO2) in Ogg1‐deficient mice compared to wild type mice (Nallanthighal et al., 2017). An increase in OGG1 activity may lead to incomplete BER and an acute exacerbation of strand breaks; however, persistent oxidative DNA lesions resulting from aberrant glycosylase activity may induce MN in daughter cells.
The essentiality of KE2, “Increase, DNA Strand Breaks”, has been shown by studies that examined both mutations (AO1) arising from incorrect rejoining of DSBs and chromosomal damage indicated by MN induction (AO2) (Kurashige et al., 2017; Tatsumi‐Miyajima et al., 1993). Tatsumi‐Miyajima et al. (1993) introduced plasmids with or without a DSB in the supF gene to five different human fibroblast cell lines and monitored supF gene mutation frequency in a colony formation assay. The plasmids that contained a DSB produced a higher frequency of supF mutants than the undamaged plasmids; up to 50% of the analyzed colonies were supF gene mutants indicating mutagenic repair of the DSB by human fibroblasts. To demonstrate the essentiality of KE2 in inducing structural damage (AO2), studies examining strand breaks and MN were considered. In a study by Kurashige et al. (2017), rat thyroid cells were irradiated with X‐rays with or without NAC pre‐treatment; a reduced number of strand breaks and a concordant reduction in MN frequency were observed in NAC‐treated cells. Together, these two studies demonstrate the effect of modulating DNA strand breaks on the two AOs of the AOP network.
7. UNCERTAINTIES AND KNOWLEDGE GAPS
During the process of developing this AOP network, we identified areas requiring further understanding and empirical evidence. Most importantly, the quantitative relationships between the KEs within the AOP network require development. In order to facilitate predictive toxicology, the level to which oxidative DNA damage occurs in order to result in mutations and chromosomal aberrations must be determined. Currently, empirical evidence to establish quantitative relationships is limited; there is a lack of studies specifically measuring DNA strand breaks, chromosomal aberrations, and mutations in relation to the quantities of oxidative DNA lesions detected at early time points. Fully evaluating the modified BH criteria was a challenge for certain KERs because the methods used to measure the MIE, KEs, and AOs differ vastly in sensitivity and analytical sampling times (e.g., comet assay for strand breaks vs. gene mutation and MN assays). Thus, a better understanding of the dose–response curves (thresholds and shapes) of different commonly used assays and the quantitative relationships between the responses measured by these assays are critical for establishing stronger causal relationships between the KEs.
Current knowledge of the consequences of oxidative base lesions is largely based on studies focusing on 8‐oxo‐dG. Indeed, the literature on the mutagenicity and repair of oxidative base lesions is overrepresented by studies on 8‐oxo‐dG, while the mutagenic and clastogenic potential of other oxidative lesions, such as 8‐oxo‐dA and thymine glycol, are not as well defined (Bellon et al., 2009; Kalam et al., 2006). Furthermore, the mechanisms through which oxidative base lesions cause clastogenicity and mutagenicity involve several different repair pathways. While BER is the primary repair pathway for oxidative lesions, MMR and NER may also play important roles in repairing oxidative damage (Brierley & Martin, 2013; Melis et al., 2012). For example, MMR removes oxidized bases that are inserted into DNA during replication, predominantly 8‐oxodGMP paired to dC, as well as non‐oxidized bases inserted opposite oxidatively damaged bases, such as dAMP inserted opposite 8‐oxodG (Bridge et al., 2014; Colussi et al., 2002). NER is responsible for repairing bulkier, helix‐distorting lesions induced by free radicals, such as 8,5′‐cyclopurine‐2′‐deoxynucleosides and intra‐ and inter‐strand crosslinks (Melis et al., 2012). In cells with defective BER glycosylases, NER has been observed to provide additional support in repairing oxidatively altered bases that do not disturb the helical structure (Melis et al., 2012). Furthermore, there are genetic factors that may modulate a cell's susceptibility to oxidative stress and associated DNA damage. For example, cells that are deficient in BRCA1 or BRCA2, proteins involved in genome maintenance, have been observed to have elevated levels of intracellular ROS and to be more sensitive to oxidative DNA damage in the S phase due to compromised HR (Fridlich et al., 2015; Renaudin et al., 2021). Overall, the interplay of different repair pathways in modulating the KEs downstream of the MIE requires further research to better define both the mechanistic and quantitative understanding of the AOP.
Studies on oxidative damage to nuclear DNA (nDNA) were examined exclusively when developing this AOP network. However, we note that oxidative damage occurs not only in nDNA but also in the deoxynucleotide (dNTP) and ribonucleotide (rNTP) triphosphate pools and in mitochondrial DNA (mtDNA). mtDNA is more susceptible to oxidative damage than nDNA due to its location and crosstalk exists between the nucleus and mitochondria during oxidative stress; DNA glycosylases that initiate BER such as OGG1 are encoded by nDNA and the enzymes translocate to the mitochondria to repair oxidative mtDNA damage (Cha et al., 2015; Saki & Prakash, 2017). BER is essential for maintaining mitochondrial genome stability as it is for the nuclear genome (Cha et al., 2015). Oxidized dNTPs and rNTPs, especially 8‐oxodGTP and r8oxoG, can be inserted into both genomes during replication and excision repair, giving rise to mismatches and impeding the repair of existing damage, respectively; both scenarios can directly lead to inadequate DNA repair, promoting the progression of this AOP network (Caglayan et al., 2017; Colussi et al., 2002; Malfatti et al., 2017; Russo et al., 2004). Moving forward, KEs addressing oxidative damage to the dNTP pool and mtDNA are necessary to build a more complete map of oxidative stress‐related genotoxicity and to expand the AOP network to other related AOs.
8. OVERALL WEIGHT OF EVIDENCE ASSESSMENT (SUMMARY) OF THE AOP NETWORK
8.1. Biological plausibility: Strong
Rationale: Considering the current literature and mechanistic understanding of oxidative DNA lesions, the repair pathways, and the induction of the two AOs, the biological plausibility of the AOP network is strong. Extensive research has been performed to investigate the toxicity of 8‐oxo‐dG; this guanine lesion is known to be abundant and is an accepted biomarker of oxidative stress and DNA damage. Many studies have reported a strong positive correlation between the levels of 8‐oxo‐dG and G to T transversions. Mechanistically, 8‐oxo‐dG is known to base pair with both dC and dA, but with a stronger preference for dA. Indeed, G to T transversions are the most prevalent base substitutions observed under oxidative stress‐inducing chemical exposures and in BER glycosylase knock‐out cells and rodents. Moreover, BER, the primary repair pathway for oxidized base lesions, and strand break repair pathways (HR and NHEJ) have been extensively studied and the existence of a limit in repair capacity of DNA repair is well known. It was established that inadequate BER and strand break repair can both lead to increased strand breaks downstream. Finally, inadequate repair of strand breaks leading to structural genomic damage has been observed in numerous studies and is a mechanistically plausible event.
8.2. Essentiality of the KEs: Strong
Rationale: The essentiality of the MIE and the two KEs has been demonstrated by studies that modulated these events in both in vitro and in vivo models. An increase or decrease in oxidative DNA lesions, DNA repair capacity (via gene knock‐out), or DNA strand breaks led to concordant changes in the downstream KEs and the AOs in a variety of model systems.
8.3. Empirical evidence supporting the KERs: Moderate
Rationale: Empirical evidence supporting individual KERs varies between weak and strong. The two KERs with strong WOE, Increases in Oxidative DNA Damage leading to Mutations and Inadequate DNA repair leading to Mutations, are supported by data from both in vivo and in vitro studies (e.g., OGG1 deficiency or overexpression in human cells and rodents) that demonstrate concentration/dose, incidence, and temporal concordances in the two events. However, the extent of evidence available to evaluate the BH criteria for certain KERs (especially strand breaks leading to the two AOs) was low because most studies are not designed to take this into consideration and do not measure all the necessary endpoints to support a KER within a single study. Moreover, concentration and temporal concordance analysis was not feasible in many cases because of the use of methods to measure strand breaks that differed in sensitivity, dynamic range, and measurement time points.
8.4. Quantitative understanding: Weak
Rationale: There is very limited quantitative understanding of the amount of oxidative DNA damage that exceeds repair capacity and leads to mutations and chromosomal aberrations; few studies have specifically investigated these relationships using a quantitative approach. In order to address the current gaps in quantitative understanding, future studies should be designed to concurrently measure the KEs of this AOP over increasing stressor concentrations and time in different model systems. Such experiments would allow quantitative analysis of the changes in the KEs in relation to one another.
9. CONCLUDING REMARKS
Oxidative stress is a cellular hazard that contributes to DNA damage during chemical insult; indeed, the ability to induce oxidative stress and genotoxicity are two prevalent characteristics of carcinogens. The AOP network described here provides WOE that links early oxidative DNA damage to regulatory endpoints in genetic toxicology (Heflich et al., 2020). As the genotoxicity testing paradigm moves toward integrating more quantitative and predictive in vitro methods, incorporation of mechanistic information will become necessary in chemical safety assessment. The AOP framework provides an effective method for organizing mechanistic knowledge and identifying knowledge gaps. As demonstrated in the development of the AOP network, the framework can be used to coordinate information from various mechanism‐based assays to assess the probability that a chemical will cause oxidative DNA damage leading to mutations and/or chromosomal aberrations. As such, AOPs that describe genotoxic pathways can provide the foundation for designing IATA for genotoxicity. With new mechanistic assays measuring non‐traditional genotoxicity endpoints emerging, AOPs will help contextualize their utility in applied genetic toxicology. Incorporating AOPs in genotoxicity assessment will facilitate the transition toward predictive, mechanism‐based testing paradigms for more streamlined and more efficient assessment of chemical safety.
10. STATEMENT OF AUTHOR CONTRIBUTIONS
All authors contributed to determining the KEs in the AOP and the structure of the flow diagram. EC provided first drafts of KE(R) and the overall assessment of the AOP, received and consolidated input from co‐authors, revised accordingly, and managed all information in the AOPWiki. All authors provided content during KE(R) development, reviewed and agreed to the final AOP, and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
ACKNOWLEDGMENTS
This project was supported by funds from Health Canada's Genomics Research and Development Initiative awarded to C.Y. CY thanks the Canada Research Chairs program. Stipend support for E.C. was also provided through the Research in Environmental and Analytical Chemistry and Toxicology (REACT) program funded under the Natural Science and Engineering Council of Canada (NSERC)’s Collaborative Research and Training (CREATE) Program. The authors would like to acknowledge the Health and Environmental Sciences Institute (HESI) Genetic Toxicology Technical Committee (GTTC) for the contributions and support, Jan van Benthem as a co‐leader of the Mode of Action Working Group for his support, and the internal reviewers who provided excellent suggestions to improve this manuscript.
Cho, E. , Allemang, A. , Audebert, M. , Chauhan, V. , Dertinger, S. , Hendriks, G. et al. (2022) AOP report: Development of an adverse outcome pathway for oxidative DNA damage leading to mutations and chromosomal aberrations. Environmental and Molecular Mutagenesis, 63(3), 118–134. Available from: 10.1002/em.22479
Accepted by: J. OBrien
Funding information Natural Science and Engineering Council of Canada (NSERC)’s Collaborative Research and Training (CREATE) Program; Health Canada's Genomics Research and Development Initiative
REFERENCES
- Amente, S. , Di Palo, G. , Scala, G. , Castrignano, T. , Gorini, F. , Cocozza, S. et al. (2019) Genome‐wide mapping of 8‐oxo‐7,8‐dihydro‐2 ‐deoxyguanosine reveals accumulation of oxidatively‐generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Research, 47, 221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankley, G. , Bennett, R. , Erickson, R. , Hoff, D. , Hornung, M. , Johnson, R. et al. (2010) Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environmental Toxicology and Chemistry, 29, 730–741. [DOI] [PubMed] [Google Scholar]
- Arai, T. , Kelly, V.P. , Minowa, O. , Noda, T. & Nishimura, S. (2002) High accumulation of oxidative DNA damage, 8‐hydroxyguanine, in Mmh/Ogg1 deficient mice by chronic oxidative stress. Carcinogenesis, 23, 2005–2010. [DOI] [PubMed] [Google Scholar]
- Arlt, M.F. , Wilson, T.E. & Glover, T.W. (2012) Replication stress and mechanisms of CNV formation. Current Opinion in Genetics & Development, 22, 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balajee, A.S. (2014) Multicolour FISH analysis of ionising radiation induced micronucleus formation in human lymphocytes. Mutagenesis, 29, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda, D. , Nunez, N. , Burnside, M. , Bradshaw, K. & David, S. (2017) Repair of 8‐oxoG:a mismatches by the MUTYH glycosylase: mechanism, Metals & Medicine. Free Radical Biology & Medicine, 107, 202–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda, M. , Recio, L. & Parsons, B.L. (2013) ACB‐PCR measurement of spontaneous and furan‐induced H‐ras codon 61 CAA to CTA and CAA to AAA mutation in B6C3F1 mouse liver. Environmental and Molecular Mutagenesis, 54, 659–667. [DOI] [PubMed] [Google Scholar]
- Becker, R. , Ankley, G. , Edwards, S. , Kennedy, S. , Linkov, I. , Meek, B. et al. (2015) Increasing scientific confidence in adverse outcome pathways:application of tailored Bradford‐Hill considerations for EvaluatingWeight of evidence. Regulatory Toxicology and Pharmacology, 72, 514–537. [DOI] [PubMed] [Google Scholar]
- Beddowes, E. , Faux, S. & Chipman, J.K. (2003) Chloroform, carbon tetrachloride and glutathione depletion induce secondary genotoxicity in liver cells via oxidative stress. Toxicology, 187, 101–115. [DOI] [PubMed] [Google Scholar]
- Bellon, S. , Shikazono, N. , Cunniffe, S. , Lomax, M. & O'Neill, P. (2009) Processing of thymine glycol in a clustered DNA damage site: mutagenic or cytotoxic. Nucleic Acids Research, 37, 4430–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berquist, B. & Wilson, D., III . (2012) Pathways for repairing and tolerating the Spectrum of oxidative DNA lesions. Cancer Letters, 327, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsma, I. & van Gent, D.C. (2012) Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integrity, 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenerman, B. , Illuzzi, J. & Wilson, D., III . (2014) Base excision repair capacity in informing healthspan. Carcinogenesis, 35, 2643–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton, J. , Sichel, F. , Bainchini, F. & Prevost, V. (2003) Measurement of 8‐Hydroxy‐2′‐Deoxyguanosine by a commercially available ELISA test: comparison with HPLC/electrochemical detection in calf thymus DNA and determination in human serum. Analytical Letters, 36, 123–134. [Google Scholar]
- Bridge, G. , Rashid, S. & Martin, S.A. (2014) DNA mismatch repair and oxidative DNA damage: implications for cancer biology and treatment. Cancers, 6, 1597–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley, D.J. & Martin, S.A. (2013) Oxidative stress and the DNA mismatch repair pathway. Antioxidants & Redox Signaling, 18, 2420–2428. [DOI] [PubMed] [Google Scholar]
- Bryce, S. , Bemis, J. , Mereness, J. , Spellman, R. , Moss, J. , Dickinson, D. et al. (2014) Interpreting in vitro micronucleus positive results: simple biomarker matrix discriminates clastogens, aneugens, and misleading positive agents. Environmental and Molecular Mutagenesis, 55, 542–555. [DOI] [PubMed] [Google Scholar]
- Bryce, S. , Bernacki, D. , Bemis, J. & Dertinger, S. (2016) Genotoxic mode of action predictions from a multiplexed flow cytometric assay and a machine learning approach. Environmental and Molecular Mutagenesis, 57, 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelof, D. , Raffoul, J. , Nakamura, J. , Kapoor, D. , Abdalla, H. & Heydari, A. (2004) Imbalanced Base excision repair in response to folate deficiency is accelerated by polymerase β Haploinsufficiency. The Journal of Biological Chemistry, 279, 36504–36513. [DOI] [PubMed] [Google Scholar]
- Cadet, J. , Davies, K. , Medeiros, M. , Di Mascio, P. & Wagner, J.R. (2017) Formation and repair of oxidatively generated damage in cellular DNA. Free Radical Biology & Medicine, 107, 13–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet, J. & Wagner, J.R. (2013) DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harbor Perspectives in Biology, 5, a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caglayan, M. , Horton, J.K. , Dai, D.P. , Stefanick, D.F. & Wilson, S.H. (2017) Oxidized nucleotide insertion by pol β confounds ligation during base excision repair. Nature Communications, 8, 14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caglayan, M. & Wilson, S. (2015) Oxidant and environmental toxicant‐induced effects compromise DNA ligation during base excision DNA repair. DNA Repair, 35, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calsou, P. , Frit, P. & Salles, B. (1996) Double strand breaks in DNA inhibit nucleotide excision repair in vitro. The Journal of Biological Chemistry, 271, 27601–27607. [DOI] [PubMed] [Google Scholar]
- Cannan, W.J. & Pederson, D.S. (2016) Mechanisms and consequences of double‐Strand DNA break formation in chromatin. Journal of Cellular Physiology, 231, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, M. , Kim, D.K. & Mook‐Jung, I. (2015) The role of mitochondrial DNA mutation on neurodegenerative diseases. Experimental & Molecular Medicine, 47, e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaim, I.A. , Nagel, Z.D. , Jordan, J.J. , Mazzucato, P. , Ngo, L.P. & Samson, L.D. (2017) In vivo measurements of interindividual differences in DNA glycosylases and APE1 activities. Proceedings of the National Academy of Sciences of the United States of America, 114, E10379–E10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, H. , Pannunzio, N. , Adachi, N. & Lieber, M. (2017) Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nature Reviews Molecular Cell Biology, 18, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan, V. , Sherman, S. , Said, Z. , Yauk, C.L. & Stainforth, R. (2020) A case example of a radiation‐relevant adverse outcome pathway to lung cancer. International Journal of Radiation Biology, 97, 68–84. [DOI] [PubMed] [Google Scholar]
- Chepelev, N. , Kennedy, D. , Gagne, R. , White, T. , Long, A. , Yauk, C. et al. (2015) HPLC measurement of the DNA oxidation biomarker, 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine, in cultured cells and animal tissues. Journal of Visualized Experiments, 102, e52697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernikova, S.B. , Wells, R.L. & Elkind, M. (1999) Wortmannin sensitizes mammalian cells to radiation by inhibiting the DNA‐dependent protein kinase‐mediated rejoining of double‐Strand breaks. Radiation Research, 151, 159–166. [PubMed] [Google Scholar]
- Collins, A.R. (2000) Comparison of different methods of measuring 8‐oxoguanine as a marker of oxidative DNA damage. Free Radical Research, 32, 333–341. [DOI] [PubMed] [Google Scholar]
- Collins, A.R. (2004) The comet assay for DNA damage and repair. Molecular Biotechnology, 26, 249–262. [DOI] [PubMed] [Google Scholar]
- Colussi, C. , Parlanti, E. , Degan, P. , Crescenzi, M. , Dogliotti, E. & Bignami, M. (2002) The mammalian mismatch repair pathway removes DNA 8‐oxodGMP incorporated from the oxidized dNTP Pool. Current Biology, 12, 912–918. [DOI] [PubMed] [Google Scholar]
- Cooke, M. , Evans, M. , Dizdaroglu, M. & Lunec, J. (2003) Oxidative DNA damage: mechanisms, mutation, and disease. The FASEB Journal, 17, 1195–1214. [DOI] [PubMed] [Google Scholar]
- Cooke, M. , Olinski, R. & Loft, S. (2008) Measurement and meaning of oxidatively modified DNA lesions in urine. Cancer Epidemiology, Biomarkers & Prevention, 17, 3–14. [DOI] [PubMed] [Google Scholar]
- Coppede, F. & Migliore, L. (2015) DNA damage in neurodegenerative diseases. Mutation Research ‐ Fundamental and Molecular Mechanisms of Mutagenesis, 776, 84–97. [DOI] [PubMed] [Google Scholar]
- Dahle, J. , Brunborg, G. , Svendsrud, D. , Stokke, T. & Kvam, E. (2008) Overexpression of human OGG1 in mammalian cells decreases ultraviolet a induced mutagenesis. Cancer Letters, 267, 18–25. [DOI] [PubMed] [Google Scholar]
- D'Autreaux, B. & Toledano, M. (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature Reviews Molecular Cell Biology, 8, 813–824. [DOI] [PubMed] [Google Scholar]
- Deferme, L. , Briede, J.J. , Claessen, S.M. , Jennen, D.G. , Cavill, R. & Kleinjans, J.C. (2013) Time series analysis of oxidative stress response patterns in HepG2: a toxicogenomics approach. Toxicology, 306, 24–34. [DOI] [PubMed] [Google Scholar]
- Dertinger, S.D. , Kraynak, A.R. , Wheeldon, R.P. , Bernacki, D.T. , Bryce, S.M. , Hall, N. et al. (2019) Predictions of genotoxic potential, mode of action, molecular targets, and potency via a tiered multiflow® assay data analysis strategy. Environmental and Molecular Mutagenesis, 60, 513–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikomey, E. & Brammer, I. (2000) Relationship between cellular radiosensitivity and non‐repaired double‐strand breaks studied for different growth states, dose rates and plating conditions in a normal human fibroblast line. International Journal of Radiation Biology, 76, 773–781. [DOI] [PubMed] [Google Scholar]
- Dobrovolsky, V.N. , Shaddock, J.G. & Heflich, R.H. (2014) Analysis of in vivo mutation in the Hprt and Tk genes of mouse lymphocytes. Methods in Molecular Biology, 1105, 255–270. [DOI] [PubMed] [Google Scholar]
- Dong, H. , Xu, D. , Hu, L. , Li, L. , Song, E. & Song, Y. (2014) Evaluation of N‐acetyl‐cysteine against tetrachlorobenzoquinoneinduced genotoxicity and oxidative stress in HepG2 cells. Food and Chemical Toxicology, 64, 291–297. [DOI] [PubMed] [Google Scholar]
- Ensminger, M. , Iloff, L. , Ebel, C. , Nikolova, T. , Kaina, B. & Lobrich, M. (2014) DNA breaks and chromosomal aberrations arise when replication meets base excision repair. The Journal of Cell Biology, 206, 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandani, M. , Hamishehkar, H. & Dolatabadi, J.E.N. (2014) Cytotoxicity and DNA damage properties of tert‐butylhydroquinone (TBHQ) food additive. Food Chemistry, 153, 315–320. [DOI] [PubMed] [Google Scholar]
- Federici, C. , Drake, K. , Rigelsky, C. , McNelly, L. , Meade, S. , Comhair, S. et al. (2015) Increased mutagen sensitivity and DNA damage in pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine, 192, 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, D. & Alt, F.W. (2001) DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene, 20, 5572–5579. [DOI] [PubMed] [Google Scholar]
- Finkel, T. (2011) Signal transduction by reactive oxygen species. The Journal of Cell Biology, 194, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlich, R. , Annamalai, D. , Roy, R. , Bernhein, G. & Powell, S.N. (2015) BRCA1 and BRCA2 protect against oxidative DNA damage converted into double‐strand breaks during DNA replication. DNA Repair, 30, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugger, K. & West, S.C. (2016) Keeping homologous recombination in check. Cell Research, 26, 397–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galick, H. , Kathe, S. , Liu, M. , Robey‐Bond, S. , Kidane, D. , Wallace, S. et al. (2013) Germ‐line variant of human NTH1 DNA glycosylase induces genomic instability and cellular transformation. Proceedings of the National Academy of Sciences of the United States of America, 110, 14314–14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, M. , Evdokimova, V.N. , Cuenco, K. , Nikiforova, M. , Kelly, L. , Stringer, J. et al. (2012) Homologous chromosomes make contact at the sites of double‐strand breaks in genes in somatic G0/G1‐phase human cells. Proceedings of the National Academy of Sciences of the United States of America, 109, 9454–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Canton, C. , Anadon, A. & Meredith, C. (2013) Assessment of the in vitro p‐H2AX assay by high content screening asa novel genotoxicity test. Mutation Research, 757, 158–166. [DOI] [PubMed] [Google Scholar]
- Ge, J. , Ngo, L. , Kaushal, S. , Tay, I.J. , Thadhani, E. , Kay, J.E. et al. (2021) CometChip enables parallel analysis of multiple DNA repair activities. DNA Repair, 106, 103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile, F. , Arcaro, A. , Pizzimenti, S. , Daga, M. , Cetrangolo, C. , Dianzani, C. et al. (2017) DNA damage by lipid peroxidation products: implications in cancer, inflammation and autoimmunity. AIMS Genetics, 4, 103–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, K.A. , Hada, M. , Jackson, L.J. , Elliott, T. , Kawata, T. , Pluth, J.M. , & Cucinotta, F.A. (2009). Dose response of γ rays and iron nuclei for induction of chromosomal aberrations in normal and repair‐deficient cell lines. Radiation Research, 171(6), 752–763. Available from: 10.1667/rr1680.1 [DOI] [PubMed] [Google Scholar]
- Gollapudi, B.B. , Lynch, A.M. , Heflich, R.H. , Dertinger, S.D. , Dobrovolsky, V.N. , Froetschl, R. et al. (2015) The in vivo pig‐a assay: a report of the international workshop on Genotoxicity testing (IWGT) workgroup. Mutation Research, 783, 23–35. [DOI] [PubMed] [Google Scholar]
- Greenberg, M.M. (2014) Abasic and oxidized abasic site reactivity in DNA: enzyme inhibition, cross‐linking, and nucleosome catalyzed reactions. Accounts of Chemical Research, 47, 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. & Weinberg, R. (2011) Hallmarks of cancer: the next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hashimoto, K. , Tominaga, Y. , Nakabeppu, Y. & Moriya, M. (2004) Futile short‐patch DNA base excision repair of adenine:8‐oxoguanine mispair. Nucleic Acids Research, 32, 5928–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heflich, R.H. , Johnson, G.E. , Zeller, A. , Marchetti, F. , Douglas, G.R. , Witt, K.L. et al. (2020) Mutation as a toxicological endpoint for regulatory decision‐making. Environmental and Molecular Mutagenesis, 61, 34–41. [DOI] [PubMed] [Google Scholar]
- Hendriks, G., Atallah, M., Morolli B., Calléja, F., Ras‐Verloop, N. , Raamsman, M. , Huijskens, I. , van de Water, B. , & Vrieling, H. (2012). The toxtracker assay: novel GFP reporter systems that provide mechanistic insight into the genotoxic properties of chemicals. Toxicological Sciences, 125(1), 285–298. Available from: 10.1093/toxsci/kfr281 [DOI] [PubMed] [Google Scholar]
- Hiraku, Y. (2010) Formation of 8‐nitroguanine, a nitrative DNA lesion, in inflammation‐related carcinogenesis and its significance. Environmental Health and Preventive Medicine, 15, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , de Souza‐Pinto, N.C. , Haraguchi, K. , Hogue, B. , Jaruga, P. , Greenberg, M.M. et al. (2005) Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. The Journal of Biological Chemistry, 280, 40544–40551. [DOI] [PubMed] [Google Scholar]
- Iliakis, G. , Wang, H. , Perrault, A.R. , Boecker, W. , Rosidi, B. , Windhofer, F. et al. (2004) Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenetic and Genome Research, 104, 14–20. [DOI] [PubMed] [Google Scholar]
- Ishida, T. , Ishida, M. , Tashiro, S. , Yoshizumi, M. & Kihara, Y. (2014) Role of DNA damage in cardiovascular disease. Circulation Journal, 78, 42–50. [DOI] [PubMed] [Google Scholar]
- Jacobs, A. & Schar, P. (2012) DNA glycosylases: in DNA repair and beyond. Chromosoma, 121, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena, N.R. & Mishra, P.C. (2012) Formation of ring‐opened and rearranged products of guanine: mechanisms and biological significance. Free Radical Biology and Medicine, 53, 81–94. [DOI] [PubMed] [Google Scholar]
- Jovanovic, S. & Simic, M. (1986) One‐electron redox potential of purines and pyrimidines. The Journal of Physical Chemistry, 90, 974–978. [Google Scholar]
- Kalam, M.A. , Haraguchi, K. , Chandani, S. , Loechler, E. , Moriya, M. , Greenberg, M.M. et al. (2006) Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring‐opened formamidopyrimidines (Fapy lesions) and 8‐oxo‐purines in simian kidney cells. Nucleic Acids Research, 34, 2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima, Y. , Yamaguchi, N. , Teshima, R. , Narahara, H. , Yamaoka, Y. , Anai, H. et al. (2017) Detection of DNA double‐strand breaks by pulsed‐field gel electrophoresis. Genes to Cells, 22, 84–93. [DOI] [PubMed] [Google Scholar]
- Kay, J. E. , Corrigan, J. J. , Armijo, A. L. , Nazari, I. S. , Kohale, I. N. , Torous, D. K. , Avlasevich, S. L. , Croy, R. G. , Wadduwage, D. N. , Carrasco, S. E. , Dertinger, S. D. , White, F. M. , Essigmann, J. M. , Samson, L. D. , & Engelward, B. P. (2021). Excision of mutagenic replication‐blocking lesions suppresses cancer but promotes cytotoxicity and lethality in nitrosamine‐exposed mice. Cell Reports, 34(11), 108864. 10.1016/j.celrep.2021.108864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury, L. , Zalko, D. , & Audebert, M. (2013) Validation of high‐throughput genotoxicity assay screening using γH2AX in‐cell western assay on HepG2 cells. Environmental and Molecular Mutagenesis, 54(9), 737–746. Available from: 10.1002/em.21817 [DOI] [PubMed] [Google Scholar]
- Kidane, D. , Murphy, D. & Sweasy, J. (2014) Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during helicobacter pylori infection. Oncogene, 3, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungland, A. , Rosewell, I. , Hollenbach, S. , Larsen, E. , Daly, G. , Epe, B. et al. (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA, 96, 13300–13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp, B. , Sanders, P. , Alassane‐Kpembi, I. , Fessard, V. , Zalko, D. , Le Hegarat, L. et al. (2020) Synergic toxic effects of food contaminant mixtures in human cells. Mutagenesis, 35, 415–424. [DOI] [PubMed] [Google Scholar]
- Kramara, J. , Osia, B. & Malkova, A. (2018) Break‐induced replication: the where, the why, and the how. Trends in Genetics, 34, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krewski, D. , Bird, M. , Al‐Zoughool, M. , Birkett, N. , Billard, M. , Milton, B. et al. (2019) Key characteristics of 86 agents known to cause cancer in humans. Journal of Toxicology and Environmental Health, 22, 244–263. [DOI] [PubMed] [Google Scholar]
- Kuhne, M. , Rothkamm, K. & Lobrich, M. (2000) No dose‐dependence of DNA double‐strand break misrejoining following a‐particle irradiation. International Journal of Radiation Biology, 76, 891–900. [DOI] [PubMed] [Google Scholar]
- Kuhne, M. , Urban, G. , Frankenberg, D. & Lobrich, M. (2005) DNA double‐strand break misrejoining after exposure of primary human fibroblasts to CK characteristic X rays, 29 kVp X rays and 60Co gamma rays. Radiation Research, 164, 669–676. [DOI] [PubMed] [Google Scholar]
- Kurashige, T. , Shimamura, M. & Nagayama, Y. (2017) N‐acetyl‐l‐cysteine protects thyroid cells against DNA damage induced by external and internal irradiation. Radiation and Environmental Biophysics, 56, 405–412. [DOI] [PubMed] [Google Scholar]
- Kuzminov, A. (2001) Single‐strand interruptions in replicating chromosomes cause double‐strand breaks. Proc Natl Acad Sci USA, 95, 8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, I.B. , Singer, T.M. , Boucher, S.E. & Douglas, G.R. (2005) Detailed review of transgenic rodent mutation assays. Mutation Research, 590, 1–280. [DOI] [PubMed] [Google Scholar]
- Lankoff, A. , Wojcik, A. , Fessard, V. & Meriluoto, J. (2006) Nodularin‐induced genotoxicity following oxidative DNA damage and aneuploidy in HepG2 cells. Toxicology Letters, 164, 239–248. [DOI] [PubMed] [Google Scholar]
- Li, H.H. , Chen, R. , Hyduke, D.R. , Williams, A. , Frötschl, R. , Ellinger‐Ziegelbauer, H. , O'Lone, R. , Yauk, C.L. , Aubrecht, J. , & Fornace, A.J. (2017). Development and validation of a high‐throughput transcriptomic biomarker to address 21st century genetic toxicology needs. Proceedings of the National Academy of Sciences, 114(51), E10881–E10889. Available from: 10.1073/pnas.1714109114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P. , Gu, Y. , Yu, S. , Li, Y. , Yang, J. & Jia, G. (2014) Assessing the suitability of 8‐OHdG and micronuclei as genotoxic biomarkers in chromate‐exposed workers: a cross‐sectional study. BMJ Open, 4, e005979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Song, M. , Kasai, H. & Kawai, K. (2013) Generation and threshold level of 8‐OHdG as oxidative DNA damage elicited by low dose ionizing radiation. Genes Environ, 35, 88–92. [Google Scholar]
- Lieber, M. , Gu, J. , Lu, H. , Shimazaki, N. & Tsai, A.G. (2010) Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Sub‐Cellular Biochemistry, 50, 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou, G.Y. & Storz, P. (2010) Reactive oxygen species in cancer. Free Radical Research, 44, 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, D.R. & Phillips, D.H. (1999) Oxidative DNA damage mediated by copper(II), iron(II) and nickel(II) Fenton reactions: evidence for site‐specific mechanisms in the formation of double‐strand breaks, 8‐hydroxydeoxyguanosine and putative intrastrand cross‐links. Mutation Research, Fundamental and Molecular Mechanisms of Mutagenesis, 424, 23–36. [DOI] [PubMed] [Google Scholar]
- Lobrich, M. , Kuhne, M. , Wetzel, J. & Rothkamm, K. (2000) Joining of correct and incorrect DNA double‐Strand break ends in Normal human and ataxia telangiectasia fibroblasts. Genes, Chromosomes & Cancer, 27, 59–68. [PubMed] [Google Scholar]
- Lomax, M. , Cunniffe, S. & O'Neill, P. (2004) 8‐OxoG retards the activity of the ligase III/XRCC1 complex during the repair of a single‐strand break, whne present within a clustered DNA damage site. DNA Repair, 3, 289–299. [DOI] [PubMed] [Google Scholar]
- Maddukuri, L. , Ketkar, A. , Eddy, S. , Zafar, M. & Eoff, R. (2014) The Werner syndrome protein limits the error‐prone 8‐oxo‐dG lesion bypass activity of human DNA polymerase kappa. Nucleic Acids Research, 42, 12027–12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfatti, M.C. , Balachander, S. , Antoniali, G. , Koh, K.D. , Saint‐Pierre, C. , Gasparutto, D. et al. (2017) Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2. Nucleic Acids Research, 45, 11193–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, Z. , Bozzella, M. , Seluanov, A. & Gorbunova, V. (2008) DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle, 7, 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, Z. , Hine, C. , Tian, X. , Van Meter, M. , Au, M. , Vaidya, A. et al. (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science, 332, 1443–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markkanen, E. (2017) Not breathing is not an option: how to deal with oxidative DNA damage. DNA Repair, 59, 82–105. [DOI] [PubMed] [Google Scholar]
- Markkanen, E. , Castrec, B. , Vilani, G. & Hubscher, U. (2012) A switch between DNA polymerases δ and λ promotes error‐free bypass of 8‐oxo‐G lesions. Proceedings of the National Academy of Sciences of the United States of America, 27, 931–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon, S. , Schuemann, J. , Paganetti, H. & Prise, K. (2016) Mechanistic modelling of DNA repair and cellular survival following radiation‐induced DNA damage. Scientific Reports, 6, 33290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis, J.P. , van Steeg, H. & Luijten, M. (2012) Role of nucleotide excision repair proteins in oxidative DNA damage repair: an updating. Antioxidants & Redox Signaling, 18, 2409–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis, J.P. , van Steeg, H. & Luijten, M. (2013) Oxidative DNA damage and nucleotide excision repair. Antioxidants & Redox Signaling, 18, 2409–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko, I. , Jacobs, A. , de Leon, A. , Gruppi, F. , Donley, N. , Harris, T. et al. (2016) Catalysts of DNA Strand cleavage at Apurinic/Apyrimidinic sites. Scientific Reports, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa, O. , Arai, T. , Hirano, M. , Monden, Y. , Nakai, S. , Fukuda, M. et al. (2000) Mmh/Ogg1 gene inactivation results in accumulation of 8‐hydroxyguanine in mice. Proceedings of the National Academy of Sciences of the United States of America, 97, 4156–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller P., Jantzen K., Løhr M., Andersen M. H., Jensen D. M., Roursgaard M. , Danielsen, P. H. , Jensen, A. , & Loft, S. (2018). Searching for assay controls for the Fpg‐ and hOGG1‐modified comet assay. Mutagenesis, 33(1), 9–19. Available from: 10.1093/mutage/gex015 [DOI] [PubMed] [Google Scholar]
- Murata, M. , Bansho, Y. , Inoue, S. , Ito, K. , Ohnishi, S. , Midorikawa, K. et al. (2001) Requirement of glutathione and cysteine in guanine‐specific oxidation of DNA by carcinogenic potassium bromate. Chemical Research in Toxicology, 14, 678–685. [DOI] [PubMed] [Google Scholar]
- Murata, M. , Thanan, R. , Ma, N. & Kawanishi, S. (2012) Role of Nitrative and oxidative DNA damage in inflammation‐related carcinogenesis. Journal of Biomedicine & Biotechnology, 2012, 623019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel, Z.D. , Beharry, A.A. , Mazzucato, P. , Kitange, G.J. , Sarkaria, J.N. , Kool, E.T. et al. (2019) Fluorescent reporter assays provide direct, accurate, quantitative measurements of MGMT status in human cells. PLoS ONE, 14, e0208341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallanthighal, S. , Chan, C. , Murray, T. , Mosier, A. , Cady, N. & Reliene, R. (2017) Differential effects of silver nanoparticles on DNA damage and DNA repair gene expression in Ogg1‐deficient and wild type mice. Nanotoxicology, 11, 996–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo, L.P. , Kaushal, S. , Chaim, I.A. , Mazzucato, P. , Ricciardi, C. , Samson, L.D. et al. (2021) CometChip analysis of human primary lymphocytes enables quantification of inter‐individual differences in the kinetics of repair of oxidative DNA damage. Free Radical Biology & Medicine, 174, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obe, G. , Pfeiffer, P. , Savage, J.R. , Johannes, C. , Goedecke, W. , Jeppesen, P. et al. (2002) Chromosomal aberrations: formation, identification and distribution. Mutation Research, Fundamental and Molecular Mechanisms of Mutagenesis, 504, 17–36. [DOI] [PubMed] [Google Scholar]
- OECD . (2014a) Test no. 487: in vitro mammalian cell micronucleus Test OECD guideline for the testing of chemicals, section 4. Paris, France: OECD Publishing. [Google Scholar]
- OECD . (2014b) Test no. 489: in vivo mammalian alkaline comet assay OECD guideline for the testing of chemicals, section 4. Paris, France: OECD Publishing. [Google Scholar]
- OECD . (2015) Test no. 483: mammalian Spermatogonial chromosomal aberration Test OECD guideline for the testing of chemicals, section 4. Paris, France: OECD Publishing. [Google Scholar]
- OECD . (2016a) Test no. 473: in vitro mammalian chromosomal aberration Test OECD guideline for the testing of chemicals, section 4. Paris, France: OECD Publishing. [Google Scholar]
- OECD . (2016b) Test no. 474: mammalian erythrocyte micronucleus test OECD guideline for the testing of chemicals, section 4. Paris, France: OECD Publishing. [Google Scholar]
- OECD . (2016c) Test no. 475: mammalian bone marrow chromosomal aberration Test OECD guideline for the testing of chemicals, section 4. Paris, France: OECD Publishing. [Google Scholar]
- OECD . (2018) Users' handbook supplement to the guidance document for developing and assessing AOPs,OECD series on adverse outcome pathways, no. 1. Paris, France: OECD Publishing. [Google Scholar]
- OECD . (2020a) Overview of concepts and available guidance related to integrated approaches to testing and assessment (IATA),OECD series on testing and assessment, no. 329. Paris, France: OECD Publishing. [Google Scholar]
- OECD . (2020b) Test no. 488: transgenic rodent somatic and germ cell mutation assays OECD guideline for the testing of chemicals, section 4. Paris, France: OECD Publishing. [Google Scholar]
- Ohno, M. , Miura, T. , Furuichi, M. , Tominaga, Y. , Tsuchimoto, D. , Sakumi, K. et al. (2006) A genome‐wide distribution of 8‐oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Research, 16, 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri, M. , Cho, T. , Álvarez‐Quilón, A. , Li, K. , Schellenberg, M.J. , Zimmermann, M. , Hustedt, N. , Rossi, S.E. , Adam, S. , Melo, H. , Heijink, A.M. , Sastre‐Moreno, G. , Moatti, N. , Szilard, R.K. , McEwan, A. , Ling, A.K. , Serrano‐Benitez, A. , Ubhi, T. , Feng, S. , Pawling, J. , Delgado‐Sainz, I. , Ferguson, M.W. , Dennis, J.W. , Brown, G.W. , Cortés‐Ledesma, F. , Williams, R.S. , Martin, A. , Xu, D. , & Durocher, D. (2020). A genetic map of the response to DNA damage in human cells. Cell, 182(2), 481–496.e21. Available from: 10.1016/j.cell.2020.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondovcik, S.L. , Tamblyn, L. , McPherson, J.P. & Wells, P. (2012) Oxoguanine glycosylase 1 (OGG1) protects cells from DNA double‐Strand break damage following methylmercury (MeHg) exposure. Toxicological Sciences, 128, 272–283. [DOI] [PubMed] [Google Scholar]
- Panieri, E. & Santoro, M.M. (2016) ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death & Disease, 7, e2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish, M.C. , Chaim, I.A. , Nagel, Z.D. , Tannenbaum, S.R. , Samson, L.D. & Engelward, B.P. (2018) Nitric oxide induced S‐nitrosation causes base excision repair imbalance. DNA Repair, 68, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlolla, A. , Barnes, C. , Hackett, D. & Tchounwou, P. (2009a) Potassium dichromate induced cytotoxicity, genotoxicity and oxidative stress in human liver carcinoma (HepG2) cells. International Journal of Environmental Research and Public Health, 6, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlolla, A. , Barnes, C. , Yedjou, C. , Velma, V. & Tchounwou, P. (2009b) Oxidative stress, DNA damage, and antioxidant enzyme activity induced by hexavalent chromium in Sprague‐Dawley rats. Environmental Toxicology, 24, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, C. , Shikazono, N. , Thacker, J. & O'Neill, P. (2004) Enhanced mutagenic potential of 8‐oxo‐7,8‐dihydroguanine when present within a clustered DNA damage site. Nucleic Acids Research, 32, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning, T.M. (2017) Genotoxicity of ortho‐quinones: reactive oxygen species versus covalent modification. Toxicology Research, 6, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platel, A. , Nesslany, F. , Gervais, V. , Claude, N. & Marzin, D. (2011) Study of oxidative DNA damage in TK6 human lymphoblastoid cells by use of the thymidine kinase gene‐mutation assay and the in vitro modified comet assay: determination of no‐observed‐genotoxic‐effect‐levels. Mutation Research, 726, 151–159. [DOI] [PubMed] [Google Scholar]
- Platel, A. , Nesslany, F. , Gervais, V. & Marzin, D. (2009) Study of oxidative DNA damage in TK6 human lymphoblastoid cells by use of the in vitro micronucleus test: determination of no‐observed‐effect levels. Mutation Research, 678, 30–37. [DOI] [PubMed] [Google Scholar]
- Ramadan, A.M. & Suzuki, T. (2012) Detection of Genotoxicity of phenolic antioxidants, butylated hydroxyanisole and tert‐Butylhydroquinone in multiple mouse organs by the alkaline comet assay. Journal of American Science, 8, 722–727. [Google Scholar]
- Reliene, R. , Fischer, E. & Schiestl, R. (2004) Effect of N‐acetyl cysteine on oxidative DNA damage and the frequency of DNA deletions in Atm‐deficient mice. Cancer Research, 64, 5148–5153. [DOI] [PubMed] [Google Scholar]
- Renaudin, X. , Lee, M. , Shehata, M. , Surmann, E. & Venkitaraman, A.R. (2021) BRCA2 deficiency reveals that oxidative stress impairs RNaseH1 function to cripple mitochondrial DNA maintenance. Cell Reports, 36, 109478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva, L. , Pandiri, A.R. , Li, Y.R. , Droop, A. , Hewinson, J. , Quail, M.A. et al. (2020) The mutational signature profile of known and suspected human carcinogens in mice. Nature Genetics, 52, 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers, K. & McVey, M. (2016) Error‐prone repair of DNA double‐strand breaks. Journal of Cellular Physiology, 231, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszkowski, K. , Jozwicki, W. , Blaszczyk, P. , Mucha‐Malecka, A. & Siomek, A. (2011) Oxidative damage DNA: 8‐oxoGua and 8‐oxodG as molecular markers of cancer. Medical Science Monitor, 17, 329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, M.T. , Blasi, M.F. , Chiera, F. , Fortini, P. , Degan, P. , Macpherson, P. et al. (2004) The oxidized Deoxynucleoside triphosphate Pool is a significant contributor to genetic instability in mismatch repair‐deficient cells. Molecular and Cellular Biology, 24, 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydberg, B. , Cooper, B. , Cooper, P. , Holley, W. & Chatterjee, A. (2005) Dose‐dependent Misrejoining of radiation‐induced DNA double‐Strand breaks in human fibroblasts: experimental and theoretical study for high‐ and low‐LET radiation. Radiation Research, 163, 526–534. [DOI] [PubMed] [Google Scholar]
- Saki, M. & Prakash, A. (2017) DNA damage related crosstalk between the nucleus and mitochondria. Free Radical Biology & Medicine, 107, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky, C.J. , Ayyar, S. , Deem, A. , Chung, W.H. , Ira, G. & Malkova, A. (2015) Translesion polymerases drive microhomology‐mediated break‐induced replication leading to complex chromosomal rearrangements. Molecular Cell, 60, 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salk, J.J. & Kennedy, S.R. (2020) Next‐generation genotoxicity: using modern sequencing technologies to assess somatic mutagenesis and cancer risk. Environmental and Molecular Mutagenesis, 61, 135–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, J.C. , Allemang, A. , Bryce, S.M. , Custer, L. , Dearfield, K. , Dietz, Y. et al. (2020) Application of the adverse outcome pathway framework to genotoxic modes of action. Environmental and Molecular Mutagenesis, 61, 114–134. [DOI] [PubMed] [Google Scholar]
- Schipler, A. & Iliakis, G. (2013) DNA double‐strand–break complexity levels and their possible contributions to the probability for error‐prone processing and repair pathway choice. Nucleic Acids Research, 41, 7589–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, T. , Rangaswamy, S. & Izumi, T. (2014) Repair of oxidative DNA damage and cancer: recent progress in DNA base excision repair. Antioxidants & Redox Signaling, 20, 708–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedletska, Y. , Radicella, J.P. & Sage, E. (2013) Replication fork collapse is a major cause of the high mutation frequency at three‐base lesion clusters. Nucleic Acids Research, 41, 9339–9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol, J. , Shim, E.Y. & Lee, S.E. (2018) Microhomology‐mediated end joining: good, bad and ugly. Mutation Research, 809, 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafirovich, V. , Kropachev, K. , Anderson, T. , Li, Z. , Kolbanovskiy, M. , Martin, B. et al. (2016) Base and nucleotide excision repair of Oxidatively generated guanine lesions in DNA. The Journal of Biological Chemistry, 291, 5309–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, A. , Gray, K. , Figg, N. , Finigan, A. , Starks, L. & Bennett, M. (2018) Defective Base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation, 138, 1446–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, Z. , Oka, S. , Tsuchimoto, D. , Abolhassani, N. , Nomaru, H. , Sakumi, K. et al. (2012) 8‐Oxoguanine causes neurodegeneration during MUTYH‐mediated DNA base excision repair. The Journal of Clinical Investigation, 122, 4344–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, M. , Gruz, P. , Kamiya, H. , Masutani, C. , Xu, Y. , Usui, Y. et al. (2007) Efficient and erroneous incorporation of oxidized DNA precursors by human DNA polymerase η. The Biochemist, 46, 5512–5522. [DOI] [PubMed] [Google Scholar]
- Skipper, A. , Sims, J. , Yedjou, C. & Tchounwou, P. (2016) Cadmium chloride induces DNA damage and apoptosis of human liver carcinoma cells via oxidative stress. International Journal of Environmental Research and Public Health, 13, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M.T. , Guyton, K. , Gibbons, K. , Fritz, J. , Portier, C. , Rusyn, I. et al. (2016) Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environmental Health Perspectives, 124, 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , Harashima, H. & Kamiya, H. (2010) Effects of base excision repair proteins on mutagenesis by 8‐oxo‐7,8‐dihydroguanine (8‐hydroxyguanine) paired with cytosine and adenine. DNA Repair, 9, 542–550. [DOI] [PubMed] [Google Scholar]
- Suto, Y. , Akiyama, M. , Noda, T. & Hirai, M. (2015) Construction of a cytogenetic dose – response curve for low‐dose range gamma‐irradiation in human peripheral blood lymphocytes using three‐color FISH. Mutation Research ‐ Genetic Toxicology and Environmental Mutagenesis, 794, 32–38. [DOI] [PubMed] [Google Scholar]
- Swenberg, J. , Lu, K. , Moeller, B. , Gao, L. , Upton, P. , Nakamura, J. et al. (2011) Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicological Sciences, 120, S130–S145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart, D. , Fredrickson, S. , Gadkari, V. & Suo, Z. (2014) Mutagenic potential of 8‐Oxo‐7,8‐dihydro‐2′‐deoxyguanosine bypass catalyzed by human Y‐family DNA polymerases. Chemical Research in Toxicology, 27, 931–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi‐Miyajima, J. , Yagi, T. & Takebe, H. (1993) Analysis of mutations caused by DNA double‐strand breaks produced by a restriction enzyme in shuttle vector plasmids propagated in ataxia telangiectasia cells. Mutation Research, 294, 317–323. [DOI] [PubMed] [Google Scholar]
- Trenz, K. , Landgraf, J. & Speit, G. (2003) Mutagen sensitivity of human lymphoblastoid cells with a BRCA1 mutation. Breast Cancer Research and Treatment, 78, 69–79. [DOI] [PubMed] [Google Scholar]
- Turner, H.C. , Shuryak, I. , Taveras, M. , Bertucci, A. , Perrier, J.R. , Chen, C. et al. (2015) Effect of dose rate on residual c‐H2AX levels and frequency of micronuclei in X‐irradiated mouse lymphocytes. Radiation Research, 183, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzortzki, E.G. , Dimakou, K. , Neofytou, E. , Tsikritsaki, K. , Samara, K. , Avgousti, M. et al. (2012) Oxidative DNA damage and somatic mutations. Chest, 141, 1243–1250. [DOI] [PubMed] [Google Scholar]
- Unfried, K. , Schurkes, C. & Abel, J. (2002) Distinct Spectrum of mutations induced by crocidolite Asbestos: clue for 8‐Hydroxydeoxyguanosine‐dependent mutagenesis in vivo. Cancer Research, 62, 104. [PubMed] [Google Scholar]
- Villeneuve, D. , Crump, D. , Garcia‐Reyero, N. , Hecker, M. , Hutchinson, T. , LaLone, C. et al. (2014) Adverse outcome pathway (AOP) development I: strategies and principles. Toxicological Sciences, 142, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Duan, Q. , Wang, T. , Ahmed, M. , Zhang, N. , Li, Y. et al. (2015) Mitochondrial respiratory chain inhibitors involved in ROS production induced by acute high concentrations of iodide and the effects of SOD as a protective factor. Oxidative Medicine and Cellular Longevity, 2015, 217670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , Li, C. , Qiao, P. , Xue, Y. , Zheng, X. , Chen, H. et al. (2018) OGG1‐initiated base excision repair exacerbates oxidative stress‐induced parthanatos. Cell Death and Disease, 9, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidinger, A. & Kozlov, A. (2015) Biological activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules, 5, 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker, A. , Schaich, M. , Smith, M.S. , Flynn, T. & Freudenthal, B. (2017) Base excision repair of oxidative DNA damage: from mechanism to disease. Frontiers in Bioscience, 22, 1493–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, M. , Zhang, Z. & Che, W. (2008) Suppression of a DNA base excision repair gene, hOGG1, increases bleomycin sensitivity of human lung cancer cell line. Toxicology and Applied Pharmacology, 228, 395–402. [DOI] [PubMed] [Google Scholar]
- Yang, N. , Chaudry, A. & Wallace, S. (2006) Base excision repair by hNTH1 and hOGG1: a two edged sword in the processing of DNA damage in gamma‐irradiated human cells. DNA Repair, 5, 43–51. [DOI] [PubMed] [Google Scholar]
- Yang, N. , Galick, H. & Wallace, S. (2004) Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair, 3, 1323–1334. [DOI] [PubMed] [Google Scholar]
- Yauk, C.L. , Lambert, I.B. , Meek, M.E. , Douglas, G.R. & Marchetti, F. (2015) Development of the adverse outcome pathway "alkylation of DNA in male premeiotic germ cells leading to heritable mutations" using the OECD's users' handbook supplement. Environmental and Molecular Mutagenesis, 56, 724–750. [DOI] [PubMed] [Google Scholar]
- Yu, H. , Venkatarangan, L. , Wishnok, J. & Tannenbaum, S.R. (2005) Quantitation of four guanine oxidation products from reaction of DNA with varying doses of Peroxynitrite. Chemical Research in Toxicology, 18, 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Taghizadeh, K. & Dedon, P. (2005) Chemical and biological evidence for base Propenals as the major source of the endogenous M1dG adduct in cellular DNA. The Journal of Biological Chemistry, 280, 25377–25382. [DOI] [PubMed] [Google Scholar]