

Abstract
Sensitized triplet‐triplet annihilation upconversion (sTTA‐UC) mainly relies on precious metal complexes thanks to their high intersystem crossing (ISC) efficiencies, excited state energies, and lifetimes, while complexes of abundant first‐row transition metals are only rarely utilized and with often moderate UC quantum yields. [Cr(bpmp)2]3+ (bpmp=2,6‐bis(2‐pyridylmethyl)pyridine) containing earth‐abundant chromium possesses an absorption band suitable for green light excitation, a doublet excited state energy matching the triplet energy of 9,10‐diphenyl anthracene (DPA), a close to millisecond excited state lifetime, and high photostability. Combined ISC and doublet‐triplet energy transfer from excited [Cr(bpmp)2]3+ to DPA gives 3DPA with close‐to‐unity quantum yield. TTA of 3DPA furnishes green‐to‐blue UC with a quantum yield of 12.0 % (close to the theoretical maximum). Sterically less‐hindered anthracenes undergo a [4+4] cycloaddition with [Cr(bpmp)2]3+ and green light.
Keywords: Chromium, Dexter Energy Transfer, Doublet-Triplet Energy Transfer, Triplet-Triplet Annihilation, Upconversion
The molecular ruby [Cr(bpmp)2]3+ with an earth‐abundant chromium(III) central ion sensitizes green‐to‐blue triplet‐triplet annihilation upconversion with 9,10‐diphenylanthracene as the annihilator. The process involves a near‐unity energy transfer efficiency via an underexplored reaction pathway, an anti‐Stokes shift of 0.54 eV, and a maximum upconversion quantum yield of 12.0 %.

Introduction
Triplet‐triplet annihilation‐based sensitized photon upconversion (sTTA‐UC) converts low‐energy photons into anti‐Stokes‐shifted photons or allows demanding photochemical activation of substrates with low‐energy light. [1] Typically, a sTTA‐UC system consists of a strongly absorbing sensitizer with a long‐lived excited triplet state and a highly fluorescent organic annihilator. Photoexcitation of the sensitizer, which is often a transition metal complex with a precious metal, from its singlet ground state to a singlet excited state (frequently a singlet metal‐to‐ligand charge‐transfer state 1MLCT), is followed by intersystem crossing (ISC) to populate its long‐lived triplet state (often a 3MLCT state). The electronically excited triplet photosensitizer engages in collisional triplet‐triplet energy transfer (TTET) via a Dexter energy transfer mechanism with an acceptor possessing an appropriate triplet (T1) energy. Interaction of two triplet excited acceptors leads to triplet‐triplet annihilation (TTA), which yields one acceptor in its ground state S0 and the second one in its excited singlet state S1. The latter releases its energy by emitting delayed fluorescence at a shorter wavelength (higher energy) than the photons originally absorbed by the sensitizer.[ 1a , 1b , 1c , 1d , 1e , 1f , 1g , 1h , 1i ] Alternatively, this S1 state can be used for chemical transformations requiring more energy than a single low‐energy photon.[ 1j , 1k , 1l , 1m , 1n , 1o ]
| (1) |
Equation 1 [2] describes the dependence of the upconversion quantum yield (Φ UC) on the efficiencies of the involved processes, i.e., ISC of the sensitizer (Φ ISC), energy transfer (EnT; often TTET from 3MLCT states) from the excited sensitizer to the annihilator (Φ EnT), TTA of the annihilator (Φ TTA), and delayed annihilator fluorescence (Φ F). f equals the spin‐statistical factor for generating a singlet spin‐state via TTA. The combination of two acceptors in their T1 states in the TTA encounter complex generates nine spin states, namely one singlet, three triplet, and five quintet states. Depending on the energies of the annihilator's T1, T2, and S1 states, the theoretical maximum of the spin‐statistical factor f max amounts to 11.1 %, 40 % or 100 % for all channels (2E(T1)>E(S1); usually not accessible due to the too high energy of the quintet state), only singlet and triplet channels (2E(T1)>E(S1) and 2E(T1)>E(T2)), or only the singlet channel (E(T2)>2E(T1)>E(S1)) being open. [3] This leads to maximum UC quantum yields (Φ UC,max) of 5.55 %, 20 % or 50 %, respectively, for Φ ISC, Φ EnT, Φ TTA, and Φ F amounting to 100 %, taking the reaction stoichiometry into account.[ 2a , 4 ] The excited states of the most often used blue fluorescent annihilator 9,10‐diphenyl anthracene (DPA) allow both open singlet and triplet channels (Φ UC,max=20 % for f max=40 %).[ 3a , 5 ] Occasionally, Φ UC values >20 % were observed as well, [6] as other factors like spin dynamics and shapes of the excited singlet and triplet energy surfaces of the annihilator can also affect the overall UC efficiency.[ 3a , 7 ]
From the sensitizer perspective, both ISC and energy transfer—in most cases TTET—can be optimized. The ISC process is greatly accelerated in sensitizers with heavy metal ions, such as [Ru(bpy)3]2+ (bpy=2,2′‐bipyridine) and its derivatives. This explains the ubiquitous utilization of noble metal complexes in sTTA‐UC and other photochemical applications. Energy transfer processes from the excited sensitizer to the annihilator via the Dexter mechanism [8] require an overlap of the excited state wavefunction of the sensitizer and the annihilator's ground state wavefunction. Consequently, (triplet) charge‐transfer excited states with a wavefunction extending onto the ligands are particularly well suited for Dexter energy transfer. This lays the basis for exploiting complexes such as [Ru(bpy)3]2+ and its derivatives with long‐lived charge‐transfer states (mostly 3MLCT) in UC processes. [1h]
Most molecular triplet sensitizers featuring long‐lived excited charge‐transfer states contain precious second‐ or third‐row transition metals, e.g. RuII, IrIII, ReI, PtII, PdII or OsII. [9] Replacing precious metals with more earth‐abundant transition metals, in particular first‐row metals, opens non‐radiative decay pathways via low‐energy metal‐centered (MC) excited states. [10] This can reduce their excited state lifetimes. Notable exceptions are CuI 3MLCT and ZrIV 3LMCT sensitizers with 3d10 and 4d0 electron configurations that lack detrimental MC excited states,[ 6d , 11 ] as well as Cr0, MnI, and Mo0 triplet sensitizers with a d6 electron configuration in a strong ligand field.[ 1l , 12 ]
Fundamentally different from long‐lived 3MLCT (or 3LMCT) excited states are intraconfigurational MC excited states which are characterized by a spin‐flip within the lower‐energy d orbitals (t2g orbitals in octahedral symmetry). [13] Thanks to the spin‐forbidden and often Laporte‐forbidden character of these spin‐flip transitions, the excited state lifetimes can even reach milliseconds.[ 13a , 14 ] Certain CrIII complexes are called “molecular rubies” owing to their excited state landscapes resembling that of the oxidic mineral ruby, and they possess extraordinarily long excited state lifetimes and record photoluminescence quantum yields.[ 13a , 14 ] However, their excited state energies are very low.[ 13a , 14a , 14b , 14c , 14d , 14f , 15 ] This precludes their utilization in sTTA‐UC due to the mismatch with the triplet energies of typical annihilators like DPA. [1h] Moreover, their electronic structure renders efficient excitation at wavelength >470 nm barely feasible. For these CrIII complexes, energy transfer to triplet oxygen[ 14e , 16 ] or to lanthanide ions with low energy excited states has been successfully demonstrated as well as UC luminescence (UCL) using CrIII→ErIII energy transfer. [17] Energy transfer from YbIII to CrIII has also been reported to yield NIR‐to‐NIR upconverted photons. [18] Energy transfer from the π‐π* states of appended anthracenyl substituents to CrIII leads to quenching of the anthracene fluorescence and sensitized phosphorescence from the metal‐centered doublet state. [19] The reverse process of sensitizing the triplet state of anthracenes or other organic dyes by electronically excited CrIII complexes (doublet‐triplet energy transfer, DTET), relevant for sTTA‐UC, has not been observed so far due to the lack of CrIII complexes with suitably high doublet state energies and long excited state lifetimes.[ 13a , 14a , 14b , 14c , 14d , 15 ] The recently reported second generation molecular ruby [Cr(bpmp)2]3+ has a comparably high doublet energy (1.75 eV) and a high excited spin‐flip state lifetime in the low ms range (e.g., 1550 μs in H2O/HClO4) along with a comparably low‐energy absorption band at 462 nm (bpmp=2,6‐bis(2‐pyridylmethyl)pyridine). [14e] This bands tails up to about 550 nm, enabling excitation with green light. These photophysical properties recommend [Cr(bpmp)2]3+ as potential sensitizer in sTTA‐UC schemes.
We now report on unprecedented UC and photochemical cycloaddition reactions using the [Cr(bpmp)2]3+ sensitizer and green light excitation. The unique decisive DTET process from the excited CrIII complex to anthracene acceptors is probed by steady‐state and time‐resolved photoluminescence spectroscopy and excitation power density (P) dependent luminescence studies, complemented by nanosecond transient absorption (TA) spectroscopy.
Results and Discussion
The key idea is to identify a suitable CrIII based sensitizer which meets the sTTA‐UC requirements of a low excitation energy, a doublet excited state energy matching the energy of the DPA annihilator in its triplet state T1 (E T=1.72–1.77 eV; Figure 1a),[ 3a , 20 ] and a long excited state lifetime. To the best of our knowledge, out of the huge number of known CrIII complexes, only the CrIII complex [Cr(bpmp)2]3+ fulfills these requirements. [Cr(bpmp)2]3+ has an excitation energy of 2.33 eV (532 nm), a doublet excited state (2E/2T1) energy of 1.75 eV, an excited state lifetime of τ 0=890 μs independent of concentration (in deoxygenated DMF containing 0.1 M perchloric acid), and a high photostability (Figures 1a and 1b; Supporting Information, Figures S1, S2). [14e]
Figure 1.
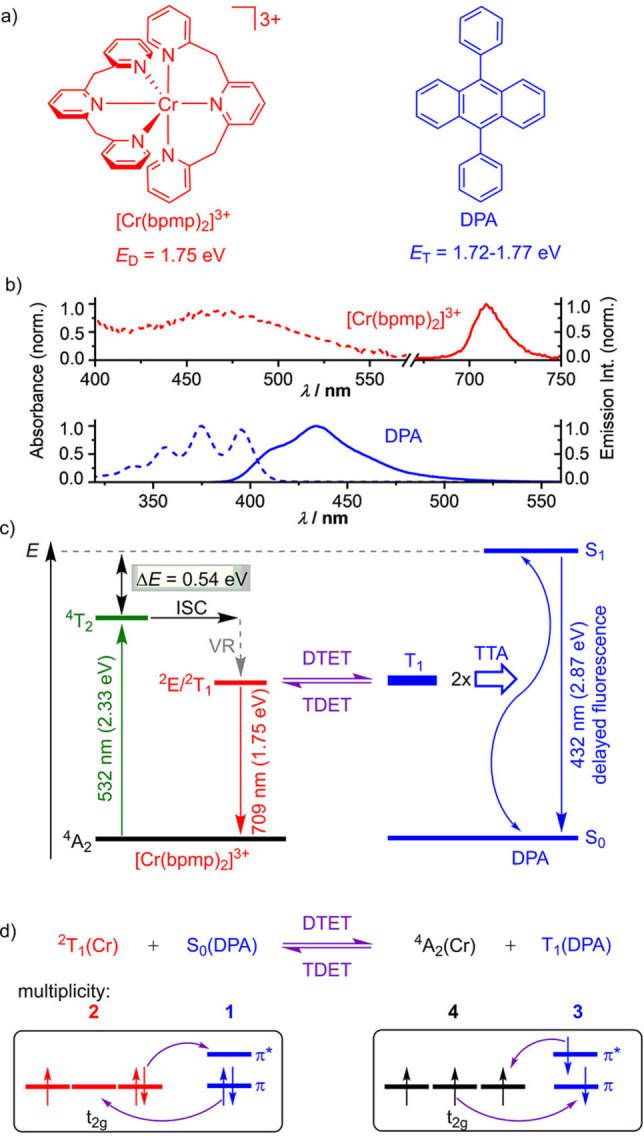
a) Structures and excited state energies of [Cr(bpmp)2]3+ and DPA, b) normalized absorption and emission spectra of [Cr(bpmp)2]3+ and DPA, c) Jablonski diagram illustrating the excitation of the sensitizer with green light, intersystem crossing (ISC) to its doublet states, vibrational relaxation (VR), doublet‐triplet energy transfer (DTET), the reverse process triplet‐doublet energy transfer (TDET), triplet‐triplet annihilation (TTA) and delayed fluorescence, and d) illustration of the Dexter energy transfer between the excited [Cr(bpmp)2]3+ sensitizer and ground state DPA (DTET) and the reverse process TDET using relevant microstates. The multiplicities (spin degeneracies) of the involved states are highlighted.
According to the intensity‐ and lifetime‐based Stern–Volmer studies, DPA quenches the [Cr(bpmp)2]3+ phosphorescence at 709 nm with K SV=5.7×104 M−1 and 5.0×104 M−1, respectively (Figure S3). The similar K SV values confirm dynamic quenching [21] of the 2E/2T1 states of [Cr(bpmp)2]3+ via DTET to DPA (Figure 1c)). The DTET rate constant k DTET=K SV/τ 0 amounts to (5.6–6.4)×107 M−1 s−1. This value is lower than the diffusion limit of bimolecular reactions in DMF (830×107 M−1 s−1) at 25 °C [22] but similar to the rates of self‐exchange reactions of CrIII complexes involving doublet‐doublet energy transfer. [23]
To visualize possible encounters of [Cr(bpmp)2]3+ and DPA and to estimate the distances involved in the Dexter‐type DTET processes with CrIII sensitizers, molecular models of anthracene as small DPA model and [Cr(bpmp)2]3+ in a solvent cage were optimized with Density Functional Theory calculations (CPCM(DMF)‐RI‐B3LYP‐D3BJ‐ZORA/def2‐TZVPP; Figure S4). These qualitative models, which do not consider counter ions and solvent molecules, do not support a strong and static interaction between [Cr(bpmp)2]3+ and anthracene in agreement with the dynamic quenching of the doublet states. The models of the exemplary optimized collisional encounter complexes suggest shortest distances of 5.8–9.5 Å between the CrIII ions to the centers of the anthracene rings. These distances are compatible with a Dexter mechanism [8] of the DTET process (Figure S4). Compared to sensitizers with triplet charge‐transfer states,[ 1l , 6d , 9 , 10 , 11 , 12 ] where the wavefunctions of the excited states extend onto the ligands, the metal‐localized nature of the CrIII spin‐flip states [13] allows only for comparably weak sensitizer‐annihilator orbital interactions in the encounter complexes. This—in addition to the small driving force for DTET (Figure 1c)—can explain the roughly 4.4–5.0 times lower energy transfer rate constant of [Cr(bpmp)2]3+/DPA as compared to, e.g. the triplet‐triplet energy transfer (TTET) rate constant k TTET of 28.2×107 M−1 s−1 reported by Castellano et al. for DPA and a CuI‐based 3MLCT sensitizer. [24]
The smaller k DTET value of the [Cr(bpmp)2]3+/DPA pair is more than compensated by the very long excited state lifetime τ 0=890 μs of [Cr(bpmp)2]3+. The DTET efficiencies Φ DTET=1−τ/τ 0 reach values of 84 % and >90 % for DPA concentrations of 100 μM and >300 μM (saturation), respectively. These values strongly exceed Φ DTET=69 % of the only other DTET process to an organic dye reported so far, that involved a luminescent doublet π‐radical sensitizer with τ 0 of only 27 ns [25] and DPA (5 mM), as well as Φ TTET=56 % of the classical [Ru(bpy)3]2+/DPA pair (τ 0=935 ns, Figure 2b). This underlines the importance of long sensitizer excited state lifetimes for these processes.
Figure 2.
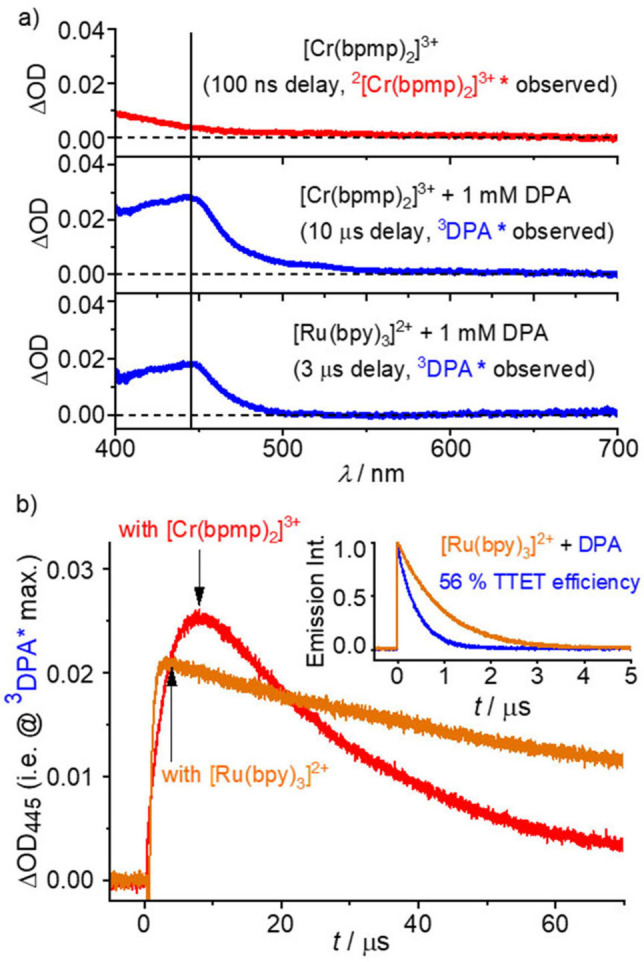
Transient absorption studies of the formation of 3DPA upon sensitizer excitation with a 532 nm laser in Ar‐saturated acidified DMF containing 1 mM of DPA. a) TA spectra of [Cr(bpmp)2]3+ (middle panel) and [Ru(bpy)3]2+ (bottom panel) in the presence of DPA integrated over 100 ns as well as control experiments with [Cr(bpmp)2]3+ (upper panel). b) Comparative TA traces monitoring the formation and decay of 3DPA with [Cr(bpmp)2]3+ and [Ru(bpy)3]2+ under these conditions. The measurement of the 3[Ru(bpy)3]2+ TTET quenching efficiency is shown in the inset. The concentrations of [Cr(bpmp)2]3+ and [Ru(bpy)3]2+ were adjusted to matching absorbances of 0.027±0.001 at 532 nm. The detection windows used for the measurement of the 3DPA spectra are indicated with black arrows.
Formation of the T1 state of DPA (3DPA) was assessed by nanosecond TA spectroscopy for higher concentrations of [Cr(bpmp)2]3+ and DPA than those used for the luminescence studies. This yields the characteristic and essentially solvent independent 445 nm 3DPA absorption band[ 1l , 22 , 26 ] (associated with electronic transitions from the long‐lived lowest triplet state to higher triplet states of DPA) 10 μs after the excitation pulse (Figure 2, Figure S5; 2 mM [Cr(bpmp)2]3+/1 mM DPA) and confirms DTET from the excited CrIII complex to DPA (Figure 2a, middle panel). The ISC process (Figure 1c) of the initially excited quartet state 4T2 to the doublet states 2E/2T1 and vibrational relaxation (VR, Figure 1c) are much faster according to reported fs transient absorption spectroscopic data of [Cr(bpmp)2]3+. [14e] In the absence of DPA (Figure 2a, top panel), the TA spectrum of the relaxed doublet state is observed 100 ns after the excitation pulse. Due to the large energy gap between 4T2 and 2E/2T1 states, back‐ISC is thermodynamically unfeasible (Figure 1b). To gain a deeper insight into the formation efficiency of the DPA triplet and to quantify the involved processes, we performed quantitative TA studies with the [Cr(bpmp)2]3+/DPA pair utilizing [Ru(bpy)3]2+ as a reference sensitizer. [27] As TTET from [Ru(bpy)3]2+ to anthracenes proceeds without side reactions as demonstrated by the characteristic 3DPA spectrum and the absence of DPA radical ion formation (indicated by the absence of additional absorption bands in the red spectral region in the lower panel of Figure 2a),[ 1j , 1l , 28 ] this system is a suitable actinometer in laser flash photolysis (LFP) studies. [27] Thereby, the efficiency of the combined ISC and DTET processes of the [Cr(bpmp)2]3+ sensitizer was determined to Φ ISC+DTET=(92±5) % using a TTET efficiency of [Ru(bpy)3]2+ of 56 % (Figure 2b; Supporting Information, Figure S6).
As shown in Figure 2b, the 3DPA decay in the presence of [Cr(bpmp)2]3+ is faster than that observed for [Ru(bpy)3]2+.ek; This suggests a back‐energy transfer process, namely triplet‐doublet energy transfer (TDET) from 3DPA to [Cr(bpmp)2]3+ in its 4A2 ground state which is thermodynamically feasible (Figure 1c). TDET manifests itself also in the time‐resolved luminescence spectra of [Cr(bpmp)2]3+ in the presence of DPA recorded with delays of 20 ns and 50 μs, revealing prompt and delayed [Cr(bpmp)2]3+ phosphorescence, respectively (Figure S7).
Beyond the small enthalpy difference between the 2E/2T1 manifold of [Cr(bpmp)2]3+ and the T1 state of DPA (Figure 1c), entropy aspects can be relevant for this equilibrium. [29] Assuming that i) only the lowest microstate of the 2E/2T1 manifold of [Cr(bpmp)2]3+ is significantly populated (a 2T1‐derived microstate; Figure 1d [14e] and ii) the spin conservation rules [30] do merely affect the kinetic feasibility and not the thermodynamics, only the spin degeneracies play a role (Figure 1d). The number of degenerate states of the [4A2(Cr)+T1(DPA)] combination (n=4×3=12) then exceeds the number of the [2T1(Cr)+S0(DPA)] combinations (n=2×1=2) (Figure 1c). This yields an upper limit for the entropic contribution of ΔS=R ln(12/2)=15 J mol−1 K−1, specific for CrIII doublet sensitizer/triplet annihilator pairs and the DTET process. This adds an amount of ≈4.5 kJ mol−1 to the driving force ΔG of the forward DTET at room temperature. In contrast, no entropic effects are expected for TTET with 3MLCT sensitizers such as [Ru(bpy)3]2+. Even without such electronic entropy effects (i.e. for conventional TTET reactions), uphill energy transfer processes can occur at room temperature. This has been recently observed for an IrIII 3MLCT sensitizer and benzothiophenes (triplet energy difference of 24 kJ mol−1). [31] The feasibility of this up‐hill process was attributed to the thermal population of vibrational or rotational levels of the excited sensitizer or ground‐state acceptor molecules which helped to overcome the thermodynamic energy gap. [31]
Consequently, [Cr(bpmp)2]3+ and DPA engage in an excited state equilibrium due to their long‐excited state lifetimes, similar excited state energies, and the available thermal energy to overcome small energy gaps. As the doublet states, which can be reached by direct excitation of [Cr(bpmp)2]3+ and by repopulation via TDET, form an excited‐state reservoir, no energy is lost. Hence, the TDET/DTET energy transfer pathways seem to barely affect the following TTA process (Figure 1c) and the overall UC process is very efficient despite the reduced lifetime of the 3DPA state (see below).
Excitation of an oxygen‐free acidified DMF solution of [Cr(bpmp)2]3+ and DPA with a 532 nm laser leads to strong blue DPA fluorescence at 432 nm (Figure 3a). The UCL maximum is blue shifted by 0.54 eV relative to the 532 nm excitation (Figure 1b, Figure 3a). In the absence of [Cr(bpmp)2]3+, no DPA fluorescence occurs under these conditions (Figure 1b; Figure S9). The integrated UCL intensity I 400–500 exceeds the residual emission of [Cr(bpmp)2]3+ (I 680–750) by a factor of about 45 indicating an efficient TTA‐UC process. Even excitation at 532 nm with a significantly less intense xenon lamp (≈1 mW cm−2) leads to noticeable UCL of DPA (Figure S10).
Figure 3.
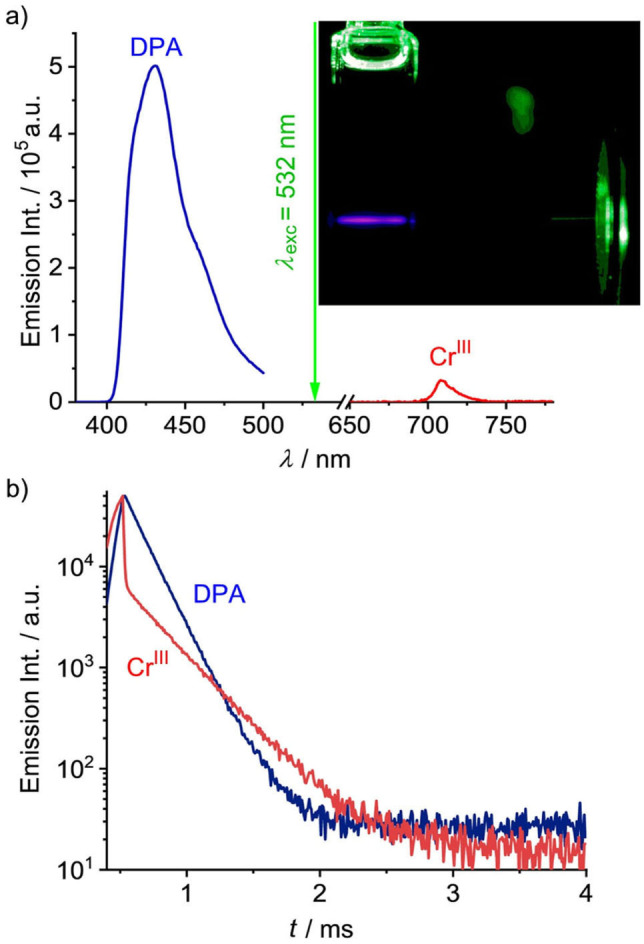
a) UCL spectrum (532 nm, cw, 1.5 W cm−2) and b) UCL decay (532 nm, 250 Hz, pulse width 500 μs) of [Cr(bpmp)2]3+/DPA (blue/red). The red emission trace in (b) corresponds to the concomitant [Cr(bpmp)2]3+ phosphorescence decay. Inset: Photograph of the sample under 532 nm laser excitation (laser power ≈40 mW). The sensitizer and acceptor concentrations in deoxygenated acidified DMF were 50 μM and 1 mM.
Increasing the DPA concentration enhances the UCL signal, which gradually approaches saturation (Figure S11). The fluorescence lifetime of directly excited DPA equals 6.6 ns, while the lifetime of the DPA fluorescence fed by green light excitation of [Cr(bpmp)2]3+ and DTET/TTA amounts to 162 μs (Figure 3b). The T1 state of DPA has a millisecond lifetime in the absence of non‐radiative processes. [32] Consequently, the long UC fluorescence lifetime of 162 μs confirms its delayed nature caused by the intermediate population of the long‐lived T1 state of DPA (Figure 1c).
Time‐resolved phosphorescence measurements of solutions of the [Cr(bpmp)2]3+/DPA pair (50 μM/1 mM; λ exc=532 nm, λ obs=709 nm) reveal biexponential decay kinetics with lifetimes τ 1=7 μs (15 %) and τ 2=303 μs (85 %), assigned to the prompt and delayed phosphorescence of [Cr(bpmp)2]3+, respectively (Figure 3b). The delayed signal arises from the doublet states of [Cr(bpmp)2]3+ which can be repopulated by TDET from the long‐lived T1 state of DPA (Figure 1c, d). An increase in the sensitizer concentration leads to a reduction in the delayed phosphorescence lifetime and an increase in its contribution to the luminescence decay (enlarged relative amplitude, see Supporting Information for details, Figure S8, Table S1). These data agree well with the assumed excited state equilibrium of [Cr(bpmp)2]3+ and DPA involving DTET/TDET processes (Figures 1c, d). [33] Under these conditions and using the lifetime of the prompt [Cr(bpmp)2]3+ phosphorescence τ 1 of 7 μs, the DTET yield amounts to Φ DTET=1−τ 1/τ 0=99.2 % in agreement with the high DPA triplet quantum yield determined by quantitative TA spectroscopy. Notably, these values imply a lower limit for the ISC efficiency of [Cr(bpmp)2]3+ on the order of 90 %. This high value is remarkable considering that chromium is a first‐row transition metal with a lower spin‐orbit coupling constant compared to precious metals such as ruthenium. [34]
Due to the biphotonic nature of sTTA‐UC, UCL from sensitizer/annihilator pairs nonlinearly depends on excitation power density and shows a slope factor (photonic order) of about two in the non‐saturated regime which approaches one upon saturation. Increasing the excitation power density of the 532 nm laser (cw, 1.5 W cm−2) drastically enhances the UCL of DPA (Figure 4a). The integrated UCL (I 400–500) of the [Cr(bpmp)2]3+/DPA pair shows a slope factor of 1.88 as expected for a biphotonic process. Yet, saturation is not reached up to power densities of 1.5 W cm−2 (Figure S12). To determine Φ UC for the saturated system, we utilized a more intense 520 nm laser (cw, ca. 8 W cm−2). Under these conditions, the integrated UCL nearly quadratically (slope factor of 1.91) depends on power density for power densities of up to about 2 W cm−2 and then starts to saturate as indicated by the linear slope of 1.03. The power density threshold value I th, which indicates the turning point from a biphotonic to a linear (monophotonic) process reflecting UC saturation, amounts to about 1.56 W cm−2 (Figure 4b). This value, which is relatively high compared to I th values of sTTA‐UC systems reported for systems utilizing ZnII, ZrIV, and CuI sensitizers,[ 6d , 11d , 35 ] is ascribed to the very small absorption of [Cr(bpmp)2]3+ above 520 nm (ϵ≈30–45 M−1 cm−1). [14e] This leaves room for improvement of the sTTA‐UC by further red shifting the absorption band the of the CrIII sensitizer and/or increasing its molar absorption coefficient for green light excitation. The relative determination of Φ UC (50 μM [Cr(bpmp)2]3+/1 mM DPA) give Φ UC=8.7 % for excitation with the 532 nm laser and Φ UC=(12.0±0.6) % for 520 nm excitation under saturated conditions (Figure 4c, Figure S12, S13). This value is amongst the highest UC efficiencies reported so far for sensitizers containing only earth‐abundant elements.[ 1l , 6d , 11d , 35a ]
Figure 4.
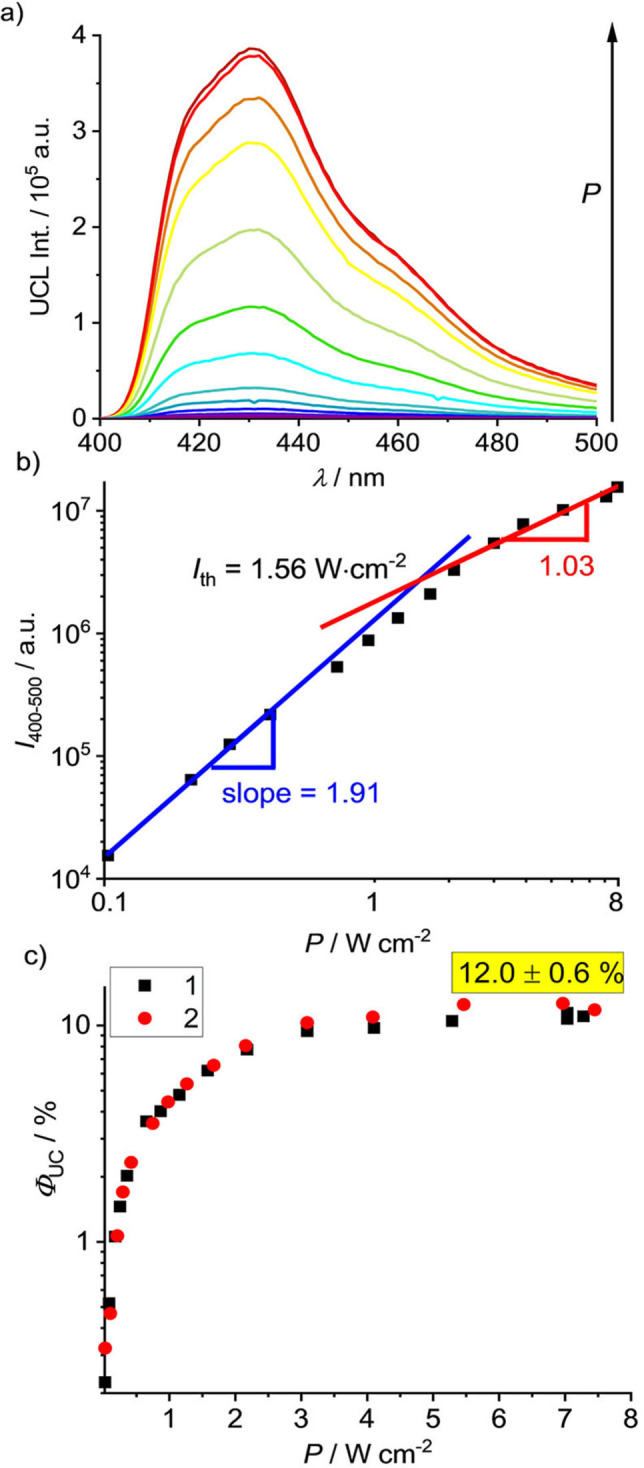
a) UCL spectra of [Cr(bpmp)2]3+/DPA (50 μM/1 mM in deoxygenated acidified DMF) as a function of excitation power density of a 520 nm laser (cw, ≈8 W cm−2), b) excitation power density dependence of the integrated UCL (I 400–500) of DPA, c) relatively determined Φ UC of the [Cr(bpmp)2]3+/DPA pair as a function of excitation power density of the 520 nm laser; the red and black symbols denote two independent experiments.
With Φ F of DPA of 82.8 % (measured in diluted, acidified DMF solution), Φ DTET+ISC of about 90 % and a spin‐statistical factor for DPA of f=40 %, Equation (1) gives an upper limit of the sTTA‐UC quantum yield Φ UC of about 15 %. This value is close to the actual Φ UC=(12.0±0.6) % achieved with [Cr(bpmp)2]3+/DPA. The slight discrepancy is probably due to optical losses [2b] resulting from the higher DPA concentration. In addition to this relatively high sTTA‐UC efficiency, the excellent photostability of [Cr(bpmp)2]3+ and DPA enable a constant UCL intensity under continuous laser illumination at 532 nm for more than two hours (Figure S14).
Sterically less‐hindered anthracene derivatives form [4+4] cycloaddition products under UV illumination by the reaction of an anthracene in its S1 state with a ground state anthracene. [36] Alternative to direct excitation with UV light, the S1 state of these anthracenes can also be populated via sTTA‐UC, e.g., by using [Ru(bpy)3]2+ derivatives as sensitizer and excitation with a 457.9 nm laser, yielding the respective anthracene dimer. [1i] To demonstrate that the [Cr(bpmp)2]3+ sensitizer can also initiate photochemical reactions via sTTA‐UC, anthracene‐9‐propionic acid (APA), 9‐anthracene carboxylic acid (ACA), and anthracene (An) were illuminated with 532 nm light in the presence of [Cr(bpmp)2]3+ in acidified deoxygenated DMF. All annihilators quench the [Cr(bpmp)2]3+ emission as expected from their T1 energies, which are similar to that of DPA (Figure S15). UCL is observed in the steady‐state and time‐resolved spectra, showing the characteristic UC dependence on P (Figures S16–S21). Yet, the Φ UC values obtained for APA, ACA, and An are much lower than Φ UC of [Cr(bpmp)2]3+/DPA (Figure S22, Table S2). Photolysis at 532 nm in the presence of [Cr(bpmp)2]3+ leads to a decrease of the absorption bands and UCL of the anthracenes (Figures S23, S24). Exemplary for the [Cr(bpmp)2]3+/An pair, anthracene photodimerization was confirmed by proton NMR spectroscopy. The 1H NMR spectra of [Cr(bpmp)2]3+/An in oxygen‐free acidified d7‐DMF obtained during photolysis with a 525 nm Kessil LED indicated the formation of the [4+4] dimer on the basis of the increasing characteristic 1H NMR resonance at δ=4.47 ppm for the bridgehead protons of the anthracene dimer (Figure S25). [1i]
Conclusion
The CrIII complex [Cr(bpmp)2]3+ can replace classical 3MLCT sensitizers, in particular [Ru(bpy)3]2+ derivatives, in sensitized triplet‐triplet annihilation upconconversion (sTTA‐UC) processes with 9,10‐diphenyl anthracene (DPA) as the annihilator generating upconverted blue photons from green photons. The key to success for efficient sTTA‐UC with [Cr(bpmp)2]3+ lies in its comparably low excitation energy (2.33 eV), its relatively high doublet state energy (1.75 eV), its high photostability, and especially in its long doublet state lifetime of 890 μs. The latter can overcompensate the comparably lower doublet‐triplet energy transfer rate, leading to high energy transfer efficiencies close to unity, thereby even outperforming [Ru(bpy)3]2+. This high energy transfer efficiency furnishes a UC quantum yield Φ UC of (12.0±0.6) %. For the sterically less‐hindered anthracene derivatives anthracene‐9‐propionic acid, 9‐anthracene carboxylic acid, and anthracene, the [Cr(bpmp)2]3+ sensitizer enables a [4+4] cycloaddition with green light that traditionally requires UV light.
Overall, this proof‐of‐concept study paves the way for novel sTTA‐UC photosensitizers based on earth‐abundant metal ions using long‐lived spin‐flip excited states instead of the traditionally employed precious metal sensitizers relying on charge‐transfer excited states. To further lower the excitation power density threshold of sTTA‐UC, on‐going studies aim for increasing the absorptivity of the CrIII sensitizers in the visible region.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (RE 1203/23‐2, HE 2778/10‐2, KE 2313/3‐1). Parts of this research were conducted using the supercomputer Elwetritsch and advisory services offered by the TU Kaiserslautern (https://elwe.rhrk.uni‐kl.de) which is a member of the Allianz für Hochleistungsrechnen Rheinland‐Pfalz (AHRP). We thank Dr. Dipl.‐Ing. Yi You from Division Instrumental Analytics of BAM (Berlin, Germany) for taking the photographs of the UC samples with 532 nm laser and Dipl.‐Ing. (FH) Arne Güttler for technical support for the UCL measurements. C.K. is grateful to the Chemical Industry Funds for a Liebig Fellowship. W.R.K. is grateful to the Chemical Industry Funds for a Kekulé Fellowship. Open Access funding enabled and organized by Projekt DEAL.
C. Wang, F. Reichenauer, W. R. Kitzmann, C. Kerzig, K. Heinze, U. Resch-Genger, Angew. Chem. Int. Ed. 2022, 61, e202202238; Angew. Chem. 2022, 134, e202202238.
Contributor Information
Prof. Dr. Christoph Kerzig, Email: ckerzig@uni-mainz.de.
Prof. Dr. Katja Heinze, Email: katja.heinze@uni-mainz.de.
Dr. Ute Resch‐Genger, Email: ute.resch@bam.de.
Data Availability Statement
All experimental data, procedures for data analysis and pertinent data sets are provided in the Sup. Inform. document. Further raw data sets are available from the authors upon reasonable request.
References
- 1.
- 1a. Parker C. A., Hatchard C. G., Proc. R. Soc. London Ser. A 1962, 269, 574–584; [Google Scholar]
- 1b. Parker C. A., Hatchard C. G., Proc. Chem. Soc. London 1962, 386–387; [Google Scholar]
- 1c. Parker C. A., Hatchard C. G., Joyce T. A., Nature 1965, 205, 1282–1284; [Google Scholar]
- 1d. Bharmoria P., Bildirir H., Moth-Poulsen K., Chem. Soc. Rev. 2020, 49, 6529–6554; [DOI] [PubMed] [Google Scholar]
- 1e. Gray V., Dzebo D., Abrahamsson M., Albinsson B., Moth-Poulsen K., Phys. Chem. Chem. Phys. 2014, 16, 10345–10352; [DOI] [PubMed] [Google Scholar]
- 1f. Richards B. S., Hudry D., Busko D., Turshatov A., Howard I. A., Chem. Rev. 2021, 121, 9165–9195; [DOI] [PubMed] [Google Scholar]
- 1g. Gray V., Moth-Poulsen K., Albinsson B., Abrahamsson M., Coord. Chem. Rev. 2018, 362, 54–71; [Google Scholar]
- 1h. Singh-Rachford T. N., Castellano F. N., Coord. Chem. Rev. 2010, 254, 2560–2573; [Google Scholar]
- 1i. Islangulov R. R., Castellano F. N., Angew. Chem. Int. Ed. 2006, 45, 5957–5959; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6103–6105; [Google Scholar]
- 1j. Kerzig C., Wenger O. S., Chem. Sci. 2018, 9, 6670–6678; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1k. Pfund B., Steffen D. M., Schreier M. R., Bertrams M. S., Ye C., Borjesson K., Wenger O. S., Kerzig C., J. Am. Chem. Soc. 2020, 142, 10468–10476; [DOI] [PubMed] [Google Scholar]
- 1l. Bilger J. B., Kerzig C., Larsen C. B., Wenger O. S., J. Am. Chem. Soc. 2021, 143, 1651–1663; [DOI] [PubMed] [Google Scholar]
- 1m. Castellanos-Soriano J., Herrera-Luna J. C., Díaz Díaz D., Jiménez M. C., Pérez-Ruiz R., Org. Chem. Front. 2020, 7, 1709–1716; [Google Scholar]
- 1n. Majek M., Faltermeier U., Dick B., Perez-Ruiz R., Jacobi von Wangelin A., Chem. Eur. J. 2015, 21, 15496–15501; [DOI] [PubMed] [Google Scholar]
- 1o. Ravetz B. D., Pun A. B., Churchill E. M., Congreve D. N., Rovis T., Campos L. M., Nature 2019, 565, 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Bossanyi D. G., Sasaki Y., Wang S., Chekulaev D., Kimizuka N., Yanai N., Clark J., JACS Au 2021, 1, 2188–2201; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Zhou Y., Castellano F. N., Schmidt T. W., Hanson K., ACS Energy Lett. 2020, 5, 2322–2326. [Google Scholar]
- 3.
- 3a. Gray V., Dreos A., Erhart P., Albinsson B., Moth-Poulsen K., Abrahamsson M., Phys. Chem. Chem. Phys. 2017, 19, 10931–10939; [DOI] [PubMed] [Google Scholar]
- 3b. Wang X., Tom R., Liu X., Congreve D. N., Marom N., J. Mater. Chem. C 2020, 8, 10816–10824; [Google Scholar]
- 3c. Schmidt T. W., Castellano F. N., J. Phys. Chem. Lett. 2014, 5, 4062–4072. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Cheng Y. Y., Fückel B., Khoury T., Clady R. G. C. R., Tayebjee M. J. Y., Ekins-Daukes N. J., Crossley M. J., Schmidt T. W., J. Phys. Chem. Lett. 2010, 1, 1795–1799; [Google Scholar]
- 4b. Saltiel J., March G. R., Smothers W. K., Stout S. A., Charlton J. L., J. Am. Chem. Soc. 1981, 103, 7159–7164. [Google Scholar]
- 5. Monguzzi A., Tubino R., Hoseinkhani S., Campione M., Meinardi F., Phys. Chem. Chem. Phys. 2012, 14, 4322–4332. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Fan C., Wei L., Niu T., Rao M., Cheng G., Chruma J. J., Wu W., Yang C., J. Am. Chem. Soc. 2019, 141, 15070–15077; [DOI] [PubMed] [Google Scholar]
- 6b. Ogawa T., Yanai N., Monguzzi A., Kimizuka N., Sci. Rep. 2015, 5, 10882; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Dzebo D., Moth-Poulsen K., Albinsson B., Photochem. Photobiol. Sci. 2017, 16, 1327–1334; [DOI] [PubMed] [Google Scholar]
- 6d. Yang M., Sheykhi S., Zhang Y., Milsmann C., Castellano F. N., Chem. Sci. 2021, 12, 9069–9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yokoyama K., Wakikawa Y., Miura T., Fujimori J., Ito F., Ikoma T., J. Phys. Chem. B 2015, 119, 15901–15908. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Skourtis S. S., Liu C., Antoniou P., Virshup A. M., Beratan D. N., Proc. Natl. Acad. Sci. USA 2016, 113, 8115–8120; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Scandola F., Indelli M. T., Chiorboli C., Bignozzi C. A. in Photoinduced Electron and Energy Transfer in Polynuclear Complexes, Vol. 158 (Ed. Mattay J.), Springer, Berlin, 1990; [Google Scholar]
- 8c. Balzani V., Bergamini G., Campagna S., Puntoriero F. in Photochemistry and Photophysics of Coordination Compounds: Overview and General Concepts., Vol. 280 (Eds.: Balzani V., Campagna S.), Springer, Berlin, 2007, pp. 1–36. [Google Scholar]
- 9.
- 9a. Islangulov R. R., Kozlov D. V., Castellano F. N., Chem. Commun. 2005, 3776–3778; [DOI] [PubMed] [Google Scholar]
- 9b. Gharaati S., Wang C., Förster C., Weigert F., Resch-Genger U., Heinze K., Chem. Eur. J. 2020, 26, 1003–1007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Zhao W., Castellano F. N., J. Phys. Chem. A 2006, 110, 11440–11445; [DOI] [PubMed] [Google Scholar]
- 9d. Wei Y., Zheng M., Chen L., Zhou X., Liu S., Dalton Trans. 2019, 48, 11763–11771; [DOI] [PubMed] [Google Scholar]
- 9e. Elgar C. E., Otaif H. Y., Zhang X., Zhao J., Horton P. N., Coles S. J., Beames J. M., Pope S. J. A., Chem. Eur. J. 2021, 27, 3427–3439; [DOI] [PubMed] [Google Scholar]
- 9f. Wu W., Ji S., Wu W., Shao J., Guo H., James T. D., Zhao J., Chem. Eur. J. 2012, 18, 4953–4964; [DOI] [PubMed] [Google Scholar]
- 9g. Yi X., Zhao J., Sun J., Guo S., Zhang H., Dalton Trans. 2013, 42, 2062–2074; [DOI] [PubMed] [Google Scholar]
- 9h. Singh-Rachford T. N., Castellano F. N., J. Phys. Chem. Lett. 2010, 1, 195–200; [Google Scholar]
- 9i. Yakutkin V., Aleshchenkov S., Chernov S., Miteva T., Nelles G., Cheprakov A., Baluschev S., Chem. Eur. J. 2008, 14, 9846–9850; [DOI] [PubMed] [Google Scholar]
- 9j. Singh-Rachford T. N., Castellano F. N., Inorg. Chem. 2009, 48, 2541–2548; [DOI] [PubMed] [Google Scholar]
- 9k. Amemori S., Sasaki Y., Yanai N., Kimizuka N., J. Am. Chem. Soc. 2016, 138, 8702–8705; [DOI] [PubMed] [Google Scholar]
- 9l. Sasaki Y., Amemori S., Kouno H., Yanai N., Kimizuka N., J. Mater. Chem. C 2017, 5, 5063–5067. [Google Scholar]
- 10.
- 10a. Förster C., Heinze K., Chem. Soc. Rev. 2020, 49, 1057–1070; [DOI] [PubMed] [Google Scholar]
- 10b. McCusker J. K., Science 2019, 363, 484–488. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Garakyaraghi S., Crapps P. D., McCusker C. E., Castellano F. N., Inorg. Chem. 2016, 55, 10628–10636; [DOI] [PubMed] [Google Scholar]
- 11b. Garakyaraghi S., Danilov E. O., McCusker C. E., Castellano F. N., J. Phys. Chem. A 2015, 119, 3181–3193; [DOI] [PubMed] [Google Scholar]
- 11c. McCusker C. E., Castellano F. N., Inorg. Chem. 2015, 54, 6035–6042; [DOI] [PubMed] [Google Scholar]
- 11d. Zhang Y., Petersen J. L., Milsmann C., J. Am. Chem. Soc. 2016, 138, 13115–13118; [DOI] [PubMed] [Google Scholar]
- 11e. Zhang Y., Lee T. S., Petersen J. L., Milsmann C., J. Am. Chem. Soc. 2018, 140, 5934–5947; [DOI] [PubMed] [Google Scholar]
- 11f. Zhang Y., Lee T. S., Favale J. M., Leary D. C., Petersen J. L., Scholes G. D., Castellano F. N., Milsmann C., Nat. Chem. 2020, 12, 345–352. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Büldt L. A., Wenger O. S., Angew. Chem. Int. Ed. 2017, 56, 5676–5682; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5770–5776; [Google Scholar]
- 12b. Büldt L. A., Guo X., Vogel R., Prescimone A., Wenger O. S., J. Am. Chem. Soc. 2017, 139, 985–992; [DOI] [PubMed] [Google Scholar]
- 12c. Herr P., Kerzig C., Larsen C. B., Häussinger D., Wenger O. S., Nat. Chem. 2021, 13, 956–962; [DOI] [PubMed] [Google Scholar]
- 12d. Heinze K., Nat. Chem. 2021, 13, 926–928; [DOI] [PubMed] [Google Scholar]
- 12e. Büldt L. A., Guo X., Prescimone A., Wenger O. S., Angew. Chem. Int. Ed. 2016, 55, 11247–11250; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11413–11417; [Google Scholar]
- 12f. Herr P., Glaser F., Büldt L. A., Larsen C. B., Wenger O. S., J. Am. Chem. Soc. 2019, 141, 14394–14402. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Kitzmann W. R., Moll J., Heinze K., Photochem. Photobiol. Sci. 2022, 10.1007/s43630-022-00186-3; [DOI] [PubMed] [Google Scholar]
- 13b. Otto S., Dorn M., Förster C., Bauer M., Seitz M., Heinze K., Coord. Chem. Rev. 2018, 359, 102–111; [Google Scholar]
- 13c. Scattergood P. A. in Recent advances in chromium coordination chemistry: luminescent materials and photocatalysis, Vol. 43 Royal Society of Chemistry, Cambridge, 2020, pp. 1–34. [Google Scholar]
- 14.
- 14a. Otto S., Grabolle M., Förster C., Kreitner C., Resch-Genger U., Heinze K., Angew. Chem. Int. Ed. 2015, 54, 11572–11576; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11735–11739; [Google Scholar]
- 14b. Wang C., Otto S., Dorn M., Kreidt E., Lebon J., Srsan L., Di Martino-Fumo P., Gerhards M., Resch-Genger U., Seitz M., Heinze K., Angew. Chem. Int. Ed. 2018, 57, 1112–1116; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1125–1130; [Google Scholar]
- 14c. Treiling S., Wang C., Förster C., Reichenauer F., Kalmbach J., Boden P., Harris J. P., Carrella L. M., Rentschler E., Resch-Genger U., Reber C., Seitz M., Gerhards M., Heinze K., Angew. Chem. Int. Ed. 2019, 58, 18075–18085; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18243–18253; [Google Scholar]
- 14d. Jiménez J.-R., Doistau B., Cruz C. M., Besnard C., Cuerva J. M., Campaña A. G., Piguet C., J. Am. Chem. Soc. 2019, 141, 13244–13252; [DOI] [PubMed] [Google Scholar]
- 14e. Reichenauer F., Wang C., Förster C., Boden P., Ugur N., Báez-Cruz R., Kalmbach J., Carrella L. M., Rentschler E., Ramanan C., Niedner-Schatteburg G., Gerhards M., Seitz M., Resch-Genger U., Heinze K., J. Am. Chem. Soc. 2021, 143, 11843–11855; [DOI] [PubMed] [Google Scholar]
- 14f. Wang C., Kitzmann W. R., Weigert F., Förster C., Wang X., Heinze K., Resch-Genger U., ChemPhotoChem 2022, 10.1002/cptc.202100296. [DOI] [Google Scholar]
- 15. Sinha N., Jimenez J. R., Pfund B., Prescimone A., Piguet C., Wenger O. S., Angew. Chem. Int. Ed. 2021, 60, 23722–23728; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 23915–23921. [Google Scholar]
- 16.
- 16a. Otto S., Nauth A. M., Ermilov E., Scholz N., Friedrich A., Resch-Genger U., Lochbrunner S., Opatz T., Heinze K., ChemPhotoChem 2017, 1, 344–349; [Google Scholar]
- 16b. Wang C., Otto S., Dorn M., Heinze K., Resch-Genger U., Anal. Chem. 2019, 91, 2337–2344. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Lazarides T., Davies G. M., Adams H., Sabatini C., Barigelletti F., Barbieri A., Pope S. J., Faulkner S., Ward M. D., Photochem. Photobiol. Sci. 2007, 6, 1152–1157; [DOI] [PubMed] [Google Scholar]
- 17b. Ward M. D., Coord. Chem. Rev. 2010, 254, 2634–2642; [Google Scholar]
- 17c. Aboshyan-Sorgho L., Cantuel M., Petoud S., Hauser A., Piguet C., Coord. Chem. Rev. 2012, 256, 1644–1663; [Google Scholar]
- 17d. Aboshyan-Sorgho L., Nozary H., Aebischer A., Bünzli J. C., Morgantini P. Y., Kittilstved K. R., Hauser A., Eliseeva S. V., Petoud S., Piguet C., J. Am. Chem. Soc. 2012, 134, 12675–12684; [DOI] [PubMed] [Google Scholar]
- 17e. Aboshyan-Sorgho L., Besnard C., Pattison P., Kittilstved K. R., Aebischer A., Bünzli J. C., Hauser A., Piguet C., Angew. Chem. Int. Ed. 2011, 50, 4108–4112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4194–4198; [Google Scholar]
- 17f. Suffren Y., Zare D., Eliseeva S. V., Guénée L., Nozary H., Lathion T., Aboshyan-Sorgho L., Petoud S., Hauser A., Piguet C., J. Phys. Chem. C 2013, 117, 26957–26963. [Google Scholar]
- 18. Kalmbach J., Wang C., You Y., Förster C., Schubert H., Heinze K., Resch-Genger U., Seitz M., Angew. Chem. Int. Ed. 2020, 59, 18804–18808; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18966–18970. [Google Scholar]
- 19. DeRosa F., Bu X., Pohaku K., Ford P. C., Inorg. Chem. 2005, 44, 4166–4174. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Faulkner L. R., Bard A. J., J. Am. Chem. Soc. 1969, 91, 209–210; [Google Scholar]
- 20b. Serevičius T., Komskis R., Adomėnas P., Adomėnienė O., Kreiza G., Jankauskas V., Kazlauskas K., Miasojedovas A., Jankus V., Monkman A., Juršėnas S., J. Phys. Chem. C 2017, 121, 8515–8524; [Google Scholar]
- 20c. Gray V., Dzebo D., Lundin A., Alborzpour J., Abrahamsson M., Albinsson B., Moth-Poulsen K., J. Mater. Chem. C 2015, 3, 11111–11121. [Google Scholar]
- 21. Arias-Rotondo D. M., McCusker J. K., Chem. Soc. Rev. 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- 22. Montalti M., Credi A., Prodi L., Gandolfi M. T., Handbook of Photochemistry, CRC Taylor & Francis, Boca Raton, 2006,. [Google Scholar]
- 23.
- 23a. Vagnini M. T., Rutledge W. C., Wagenknecht P. S., Inorg. Chem. 2010, 49, 833–838; [DOI] [PubMed] [Google Scholar]
- 23b. Wagenknecht P. S., Kane-Maguire N. A. P., Speece D. G., Helwic N., Inorg. Chem. 2002, 41, 1229–1235. [DOI] [PubMed] [Google Scholar]
- 24. Rosko M. C., Wells K. A., Hauke C. E., Castellano F. N., Inorg. Chem. 2021, 60, 8394–8403. [DOI] [PubMed] [Google Scholar]
- 25. Han J., Jiang Y., Obolda A., Duan P., Li F., Liu M., J. Phys. Chem. Lett. 2017, 8, 5865–5870. [DOI] [PubMed] [Google Scholar]
- 26. Chattopadhyay S. K., Kumar C. V., Das P. K., Chem. Phys. Lett. 1983, 98, 250–254. [Google Scholar]
- 27.
- 27a. Müller P., Brettel K., Photochem. Photobiol. Sci. 2012, 11, 632–636; [DOI] [PubMed] [Google Scholar]
- 27b. Bertrams M. S., Kerzig C., Chem. Commun. 2021, 57, 6752–6755. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. El Roz K. A., Castellano F. N., Chem. Commun. 2017, 53, 11705–11708; [DOI] [PubMed] [Google Scholar]
- 28b. Yoshimura A., Hoffman M. Z., Sun H., J. Photochem. Photobiol. A 1993, 70, 29–33. [Google Scholar]
- 29. Isokuortti J., Allu S. R., Efimov A., Vuorimaa-Laukkanen E., Tkachenko N. V., Vinogradov S. A., Laaksonen T., Durandin N. A., J. Phys. Chem. Lett. 2020, 11, 318–324. [DOI] [PubMed] [Google Scholar]
- 30. Guo D., Knight T. E., McCusker J. K., Science 2011, 334, 1684–1687. [DOI] [PubMed] [Google Scholar]
- 31. Strieth-Kalthoff F., Henkel C., Teders M., Kahnt A., Knolle W., Gómez-Suárez A., Dirian K., Alex W., Bergander K., Daniliuc C. G., Abel B., Guldi D. M., Glorius F., Chem 2019, 5, 2183–2194. [Google Scholar]
- 32. Olesund A., Gray V., Martensson J., Albinsson B., J. Am. Chem. Soc. 2021, 143, 5745–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.
- 33a. Meroni D., Monguzzi A., Meinardi F., J. Chem. Phys. 2020, 153, 114302; [DOI] [PubMed] [Google Scholar]
- 33b. Perego J., Pedrini J., Bezuidenhout C. X., Sozzani P. E., Meinardi F., Bracco S., Comotti A., Monguzzi A., Adv. Mater. 2019, 31, 1903309; [DOI] [PubMed] [Google Scholar]
- 33c. Zähringer T. J. B., Bertrams M.-S., Kerzig C., J. Mater. Chem. C 2022, 10, 4568–4573. [Google Scholar]
- 34.
- 34a. Penfold T. J., Gindensperger E., Daniel C., Marian C. M., Chem. Rev. 2018, 118, 6975–7025; [DOI] [PubMed] [Google Scholar]
- 34b. Marian C. M., WIREs Comput. Mol. Sci. 2012, 2, 187–203; [Google Scholar]
- 34c. Dunn T. M., Trans. Faraday Soc. 1961, 57, 1441–1444. [Google Scholar]
- 35.
- 35a. Durandin N. A., Isokuortti J., Efimov A., Vuorimaa-Laukkanen E., Tkachenko N. V., Laaksonen T., Chem. Commun. 2018, 54, 14029–14032; [DOI] [PubMed] [Google Scholar]
- 35b. Durandin N. A., Isokuortti J., Efimov A., Vuorimaa-Laukkanen E., Tkachenko N. V., Laaksonen T., J. Phys. Chem. C 2019, 123, 22865–22872. [Google Scholar]
- 36. Klán P., Wirz J., Photochemistry of Organic Compounds: From Concepts to Practice, Wiley, Hoboken, 2009, pp. 46–60. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
All experimental data, procedures for data analysis and pertinent data sets are provided in the Sup. Inform. document. Further raw data sets are available from the authors upon reasonable request.