

Abstract
Phosphotyrosine residues are essential functional switches in health and disease. Thus, phosphotyrosine biomimetics are crucial for the development of chemical tools and drug molecules. We report here the discovery and investigation of pentafluorophosphato amino acids as novel phosphotyrosine biomimetics. A mild acidic pentafluorination protocol was developed and two PF5‐amino acids were prepared and employed in peptide synthesis. Their structures, reactivities, and fluorine‐specific interactions were studied by NMR and IR spectroscopy, X‐ray diffraction, and in bioactivity assays. The mono‐anionic PF5 motif displayed an amphiphilic character binding to hydrophobic surfaces, to water molecules, and to protein‐binding sites, exploiting charge and H−F‐bonding interactions. The novel motifs bind 25‐ to 30‐fold stronger to the phosphotyrosine binding site of the protein tyrosine phosphatase PTP1B than the best current biomimetics, as rationalized by computational methods, including molecular dynamics simulations.
Keywords: Chemical Biology, Drug Development, Pentafluorophosphates, Phosphotyrosine Biomimetics, Protein Tyrosine Phosphatases
The more fluorine the better? Pentafluorophosphato amino acids A and B are stable new molecular entities showing surprisingly high binding affinities to the phosphotyrosine binding site of a protein tyrosine phosphatase—better than the current gold standard for phosphotyrosine mimetics. PF5 motifs display remarkable fluorine‐specific interactions and thus might enable a new class of potent tools for medicinal chemistry and chemical biology.
Phosphorylation of the amino acid L‐tyrosine is a key regulatory mechanism controlling the function of numerous proteins, governing cellular processes such as protein expression, cell division, development, mobility, and aging. [1] Aberrant activity of tyrosine kinases (TK) or protein tyrosine phosphatases (PTP) in biological systems is linked to diseases such as diabetes [2] and cancer, [3] and thus these proteins have been identified as pharmacologically relevant targets. [4] For a profound understanding of protein tyrosine phosphorylation chemical tools are required to bind, inhibit or manipulate phosphotyrosine binding sites without being prone to enzymatic cleavage or dephosphorylation. To date the most potent “gold standard” phosphotyrosine mimetic is 4‐phosphono‐difluoromethyl‐phenylalanine (PDFM‐Phe) 1 and numerous studies have demonstrated highly potent and selective inhibitors with this structure integrated in peptide sequences (Scheme 1A).[ 5 , 6 ] Phosphonic acids, however, are strong acids and form highly polar di‐anions resulting in low membrane permeability and thus inactivity in cells. [7]
Scheme 1.
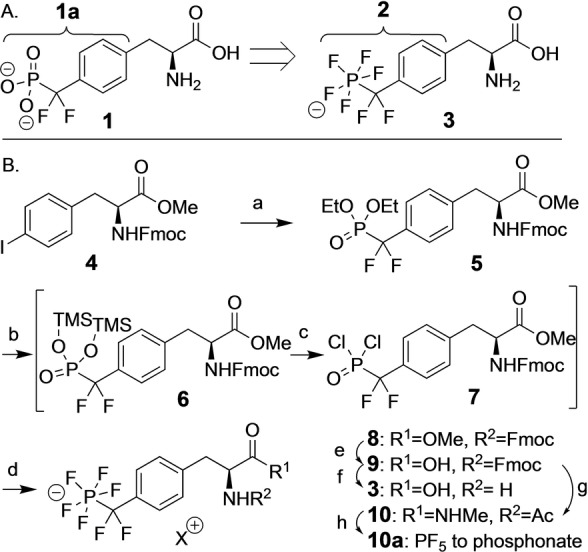
Design (A) and synthesis (B) of PFPDFM‐phenylalanine 3 as a potential phosphotyrosine mimetic. Reaction conditions: a) Cd, CuBr, BrCF2P(O)(OEt)2, DMF, 99 %; b) TMSBr (5 equiv), ACN; c) oxalylchloride (10 equiv), DMF (5 equiv); d) NMe4F (10 equiv) (68 % for b–d); e) bacillus licheniformis protease, 50 mM NH4HCO3 buffer, RT, o.n., 96 %; f) 20 % piperidine in ACN, RT, 8 h, 97 %; g) MeNH3Cl, TBTU, DIPEA, ACN, RT, 1 h, 95 %; 10 % piperidine in ACN, RT, 7 h, 96 %; Ac2O, DIPEA, ACN, RT, 4 h, quant.; h) 2 M HCl, 72 h, quant. TBTU=O‐(Benzotriazol‐1‐yl)‐N,N,N′,N′‐tetramethyluronium tetrafluoroborate, TMSBr=Trimethylsilylbromide, DIPEA=Di‐isopropyl‐ethylamine, RT=room temperature.
Considering that phosphate binding sites are coated with a positively charged surface resulting from cationic arginine residues and from H‐bond donors, [8] we hypothesized that fragments containing fluorine atoms with negative partial charge might act as H‐bond acceptors and thus might be useful as phosphate mimetics. First aromatic fragments containing the pentafluoro‐phosphato‐difluoromethyl‐motif (PFPDFM) were prepared and the PFPDFM‐substituted benzene 2 was found to inhibit the phosphotyrosine phosphatase activity of PTP1B with low, millimolar affinity. [9] Synthesis and purification of amino acids containing the PFPDFM‐motif failed using the reported conditions with basic fluoride or anhydrous HF.[ 9 , 10 ] Thus, a new pentafluorination protocol needed to be developed. Here, we report the refined synthesis of the PFPDFM motif resulting in the unnatural amino acid 4‐pentafluoro‐phosphato‐difluoromethyl‐phenylalanine 3 PFPDFM‐Phe, Phe*, Scheme 1B) and investigate the (bio)physical, chemical and biochemical properties of the pentafluoro phosphate motif with a focus on the fluorine‐specific interactions exerted by it. Starting from O‐methyl N‐Fmoc‐4‐iodo‐phenylalanine 4 and subsequently di‐O‐ethyl‐phosphonato‐difluoromethyl derivative 5, the yield of the pentafluorination step toward 8 was raised from traces to almost 70 % under acidic conditions (Scheme 1B, b–d).
Using trimethylsilyl (TMS) bromide, 5 was converted to intermediary di‐O‐TMS‐phosphonate 6, which was transformed in situ to the di‐chloro‐phosphonate 7 with oxalyl chloride and DMF. Subsequently, fluorination with an excess of tetra‐methyl‐ammonium fluoride yielded compound 8, which was isolated after workup in aqueous buffer by reversed phase MPLC. Purity and structure of 8 was confirmed by high‐resolution mass spectrometry, 1H, 13C, 19F, and 31P NMR spectroscopy and by X‐ray diffraction of crystalized product. [11] In solution, the phosphorus (V) center was coordinated bipyramidally resulting in a doublet‐quintet splitting of the axial fluorine in the 19F spectrum. In the crystal, small deviations of bond angles between axial and equatorial fluorine atoms from 90°, and a slightly elongated axial P−F bond were observed (Figure 1, Supporting Information Figure S2, Supporting Information Table S2).
Figure 1.
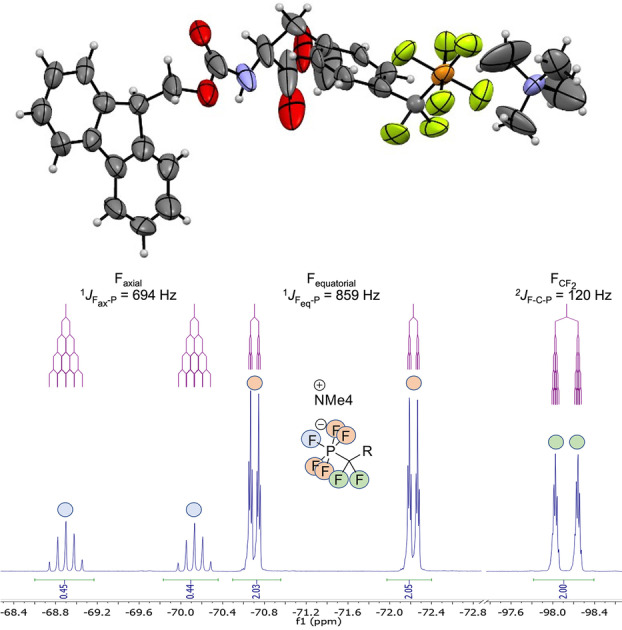
Crystal structure and 19F NMR spectrum of N‐Fmoc‐PFPDFM‐Phe‐OMe 8.
The methyl ester of 8 was saponified by enzymatic cleavage with bacillus licheniformis protease followed by ion exchange on Amberlite yielding the sodium salt of Fmoc‐amino acid 9, the building block for solid phase peptide synthesis. 9 was further converted to the unprotected amino acid 3. Reaction with acetic anhydride and condensation with N‐methylamine using TBTU furnished the N‐acetyl‐N′‐methylamide 10.
Despite of its permanent negative charge, the PFPDFM motif displayed higher hydrophobicity than the phosphono‐difluoromethyl precursor 1. [12] The retention time of the pen‐tafluorinated amino acid 3 in reversed phase C18‐HPLC shifted relative to the respective phosphonate 1 by 2.2 min during a 6 min gradient of water and acetonitrile (3.8 min vs. 6 min), corresponding to an increase of from 66 % to 99 % of acetonitrile in the eluent mixture (Figure 2A). The amphiphilicity of the pentafluorophosphato residues was also reflected by the logarithmic partition coefficients (logP) of −0.23 and −0.73 for the tetramethyl ammonium salt of fragment 2 and for the sodium salt 10 measured in DCM/water, respectively. These logP values indicate that a significant portion of pentafluorophosphate salts is found in the organic phase, namely 37 % and 16 %, respectively. In contrast, phosphonates 1 and 10 a remained entirely in the water phase. The amphiphilic nature of the pentafluorophosphate was also reflected in the FTIR spectra. The difference ATR/FTIR spectrum of 2 dissolved in water, recorded against a pure water background, showed two characteristic water bands at 3630 cm−1 (O−H stretching mode) and at 1628 cm−1 (H−O−H bending mode) (Figure 2B). The peak position of the O−H stretching mode in the presence of 2 is higher and narrower as compared to the O−H stretching mode of bulk water (≈3340 cm−1, full width at half maximum (FWHM) ≈420 cm−1) and the corresponding bending mode also appears lower than that of bulk water (1640 cm−1). These values are characteristic of water molecules lacking one of the four typical hydrogen bonds of bulk water (so‐called dangling water). [13] The characteristic water bands do not appear in the IR spectrum of the phosphonate fragment 1 a suggesting that the pentafluorophosphato group forms a hydration shell with O−H−F hydrogen bonds and dangling water molecules.
Figure 2.
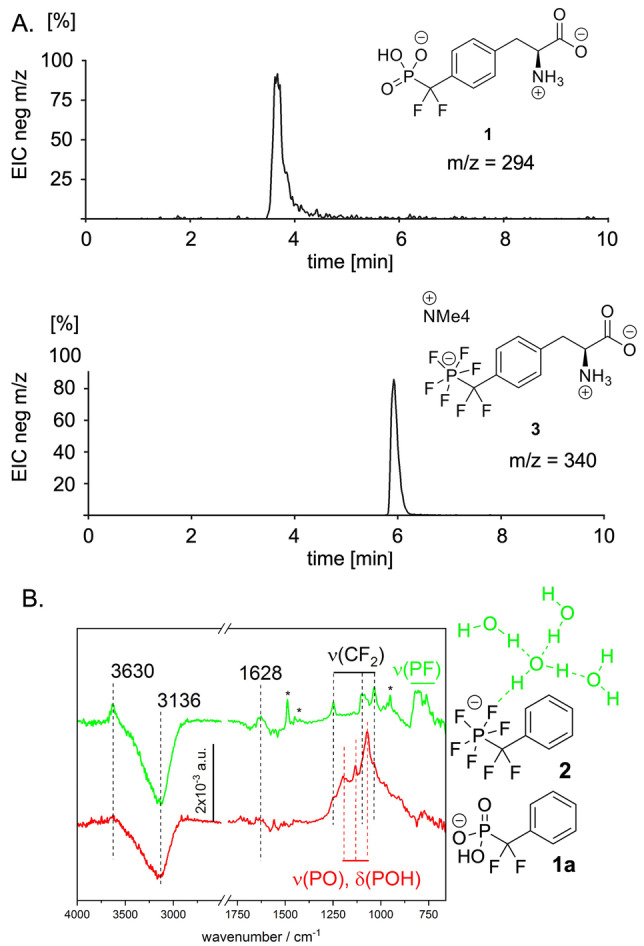
Amphiphilicity of the pentafluorophoshato‐difluormethyl (PFPDFM) motif in amino acid 3 and in 2. A. HPLC on RP‐18 silica of 1 and 3; B. FT‐ATR‐IR difference spectra of 1 a and 2 (10 mM) in comparison with the spectrum in water. Characteristic signals of the fluorinated fragments including dangling water signals (3630 and 1628 cm−1) are highlighted, full peak assignment and density functional theory (DFT) calculations in Supporting Information Table S3, Supporting Information Figures S3–S5.
The chemical stability of the PFPDFM‐motif was investigated for compound 3 by HPLC‐MS and by 19F NMR spectroscopy. The pentafluorophosphate anion was stable in water from pH 2–12 at RT for 24 h. It tolerated organic bases like pyridine, 20 % piperidine, and 2 % 1,8‐diaza‐bicyclo[5,4,0]‐undec‐8‐en (DBU) in DMF or acetonitrile, reducing agents such as dithiothreitol (DTT) and Pd/C with hydrogen. No decomposition was observed by heating to 60 °C for one day, by sonification, and under typical condensation conditions for peptide synthesis using N,N′‐diisopropyl‐carbodiimide (DIC)/HOBt and TBTU/DIPEA. In contrast, both aqueous acid at pH<2 (0.1 M HCl or 0.1 % TFA) and non‐aqueous acid (10 % acetic acid in DCM or hexafluoro‐isopropanol (HFIP)) effected the hydrolysis of the PFPDFM motif to the monofluorophosphonate 11 (Scheme 2A). Monofluoro‐phosphonate 11 was stable at neutral pH and hydrolyzed slowly to the free phosphonate under acidic conditions, e.g. with 5 % perchloric acid over 3 d.
Scheme 2.

A) Conversion of amino acid 3 to products 11 and 12 (Reaction conditions to 12: poly‐HF‐pyridine, 1 % water, RT, 6 h). B) Fmoc‐based peptide synthesis employing the PFPDFM motif in amino acid 9. Reaction conditions: a) 20 % Piperidine/DMF, RT, 10 min (twice); b) 5 equiv Fmoc‐AA‐OH, 4.9 equiv TBTU, DIPEA, DMF, RT, 2 h, repeat a+b for each AA; c) Ac2O/pyridine (1 : 1), DMF, 1 h; d) dry poly‐HF‐pyridine, 10 % anisole, RT, 90 min; e) poly‐HF‐pyridine, 10 % anisole, 1 % water, RT, 6 h; f) 5 equiv 18, 4.9 equiv TBTU, HOBt, 10 equiv DIPEA, DMF, 2 h. HOBt=Hydroxybenzotriazole, Fmoc=9‐Fluorenyl‐methyl‐oxycarbonyl.
Lability of the PFPDFM motif under acidic conditions excluded the use of standard Fmoc or Boc strategies for solid phase peptide synthesis, even with very acid‐labile linkers such as 2‐chlorotrityl resin. As an alternative hydrogen fluoride was investigated for cleavage assuming that an excess of HF should protect the PF5‐residue from acidic decomposition. [13] Indeed, compounds 2 and 3 were stable when treated with dry pyridinium poly‐(hydrogen fluoride) (Olah's reagent). [14] In contrast, treatment with aqueous HF hydrolyzed the difluoromethyl position but not the pentafluorophosphato group, forming the novel amino acid 4‐(pentafluorophosphato‐carbonyl)‐phenylalanine (PFPC‐Phe) 12 in 87 % yield. Compound 12 is to our best knowledge the first example of an acyl‐pentafluorophosphate formed via hydrolysis of CF2 and was stable over a pH range from 2–12. Structurally related benzoyl phosphonates have been described as photoactive phosphotyrosine mimetics and have been employed in the photo‐crosslinking and photo‐deactivation of phosphotyrosine binding sites in proteins. [15] Amino acid 12 shows an n‐π* transition at 343 nm and irradiation in isopropanol/water (70 : 30) resulted in the photoconversion of the PF5 residue (Supporting InformationFigure S6).
Employing Rink amide linker on 1 % divinylbenzene (DVB)‐polystyrene resin 13 (0.34 mmol g−1), the dipeptide amide Ac‐Phe*‐Leu‐NH2 14 was synthesized as the first peptide containing the pentafluorophosphato residue (Scheme 2B). Coupling of Fmoc‐building block 9 succeeded after activation with TBTU. Following to the final Fmoc‐deprotection, the resin was N‐acetylated, washed, dried, and subsequently treated for 90 min with dry poly‐HF‐pyridine containing 10 % anisole. Products were washed off the resin with THF and the washing solution was neutralized with saturated aqueous sodium bicarbonate. After evaporation the resulting dipeptide 14 was isolated by reversed phase MPLC using a gradient of ammonium bicarbonate pH 7.5 and acetonitrile in a yield of 75 %. Addition of 1 % water to HF‐pyridine cleavage cocktail for 6 h furnished dipeptide 15 containing the pentafluorophosphato‐carbonyl (PFPC) residue instead of the PFPDFM‐group in 73 % yield after MPLC purification. Applying the water‐free protocol, hexapeptide amide Ac‐DADEF*L‐NH2 16 was prepared and isolated in 65 % yield (Scheme 2), representing the native autophosphorylation sequence of the epidermal growth factor (EGF) receptor and an established substrate of PTP1B. [16] Tripeptide mimetic 17 containing two PFPDFM residues was designed from a potent PTP1B inhibitor [17] carrying 4‐PFPDFM‐phenyl‐acetamide as N‐terminal cap. Synthesis of this peptide required the preparation of 4‐PFPDFM‐phenylacetic acid 18 starting from 4‐iodo‐phenylacetate in two steps. Peptide amide 17 could be obtained and isolated using the established protocol in a yield of 46 % (Scheme 2).
Amino acids and peptides were subsequently tested in an enzymatic assay of protein tyrosine phosphatase PTP1B using 6,8‐difluoro‐4‐methylumbelliferyl phosphate (DiFMUP) as the substrate. While phosphonate amino acid 1 displayed a K I value of 1.55 mM, pentafluorophosphato amino acid 3 showed a K I of 61 μM, indicating a 25‐fold increase of affinity (Figure 3A, Table 1). PF5‐amino acid 12 was an even stronger inhibitor of PTP1B with a K I of 52 μM, 30‐fold better than mimetic 1.
Figure 3.
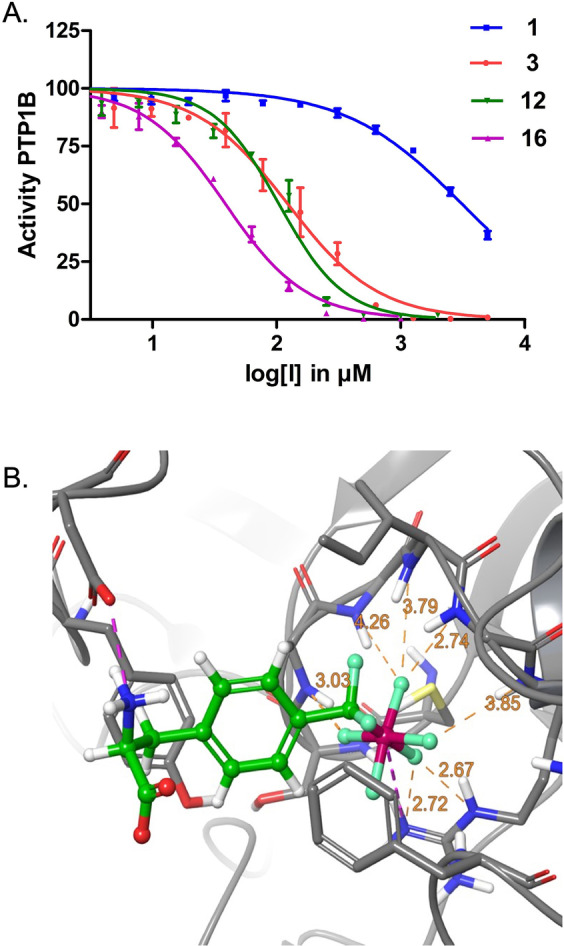
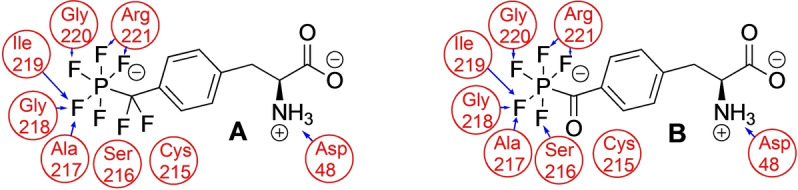
A) Inhibition of the protein tyrosine phosphatase PTP1B by the pentafluorophosphato‐phenylalanines 3 and 12, respectively, was 25‐fold and 30‐fold stronger than of the classical phosphono‐difluormethyl phenylalanine 1 and was further enhanced in peptide mimetics 15 and 16. B) Docking of 3 suggested the preferred binding pose in the phosphotyrosine binding pocket of PTP1B.
Table 1.
Binding affinities of the phosphotyrosine mimetic amino acids and peptides 1, 3, 10, 12, and 14–17 to protein tyrosine phosphatase PTP1B calculated from enzyme inhibition assays.
|
Compound |
K I in μM[a] |
|---|---|
|
1 |
1555±183[b] |
|
3 |
61±8[c] |
|
10 |
[f] |
|
12 |
52±7[d] |
|
14 |
90±10[c] |
|
15 |
24±4,[b] 25±4,[c] 21±3[d] |
|
16 |
74±13,[e] 33±5,[c] 19±3[d] |
|
17 |
[f] |
[a] Assays were performed in triplicate with DiFMUP as a substrate (see Supporting Information part for raw data). Enzyme concentration was 1.5 nM, substrate concentration 67 μM, identical with the experimentally determined K M value of the substrate. IC50 values were converted into the corresponding K I values applying the Cheng‐Prusoff equation (with [S]=K M this results in IC50/2=KI ). [b] 0 % DMSO, [c] 2.5 %, [d] 5 % DMSO, [e] 2.5 %, stock diluted in buffer, [f] visible precipitate.
As protected amino acid 10 and tripeptide mimetic 17 precipitated in the assay, DMSO concentrations and dilution protocols were varied in the peptide assays. For peptide 15, identical K I‐values (24, 25, 21 μM) were recorded at 0, 2.5, and 5 % DMSO, while the apparent affinity of peptide 16 was raised significantly from 74 μM (2.5 % DMSO, dilution in buffer) to 33 μM (2.5 %, dilution in DMSO) and 19 μM (5 % DMSO). This observation corresponded with the higher polarity of 15 vs. 16 as shown in RP‐HPLC (Supporting Information Figure S16). While 15 was soluble in aqueous buffer, dynamic light scattering revealed the aggregation of peptide 16 in DMSO/buffer (Supporting Information Figures S12–S15) which reduced the apparent affinity to the target. Both peptides 15 and 16 showed stronger inhibition than the respective PF5 amino acids alone. These data suggest that the bioactivity of PF5‐containing compounds depends not only on their target‐binding but also on their physico‐chemical properties and formulation.
Docking studies of amino acids 1, 3, 10, and peptide 15 to PTP1B using the commercial docking software Glide[ 18 , 19 ] revealed binding poses in the binding pocket and key interactions between the amino acids and the protein (Figure 3B, Supporting Information Figures S14, S15, S20–S23). In the docked pose, the ligand was held in place by two salt bridges, one between the negative charge of the phosphate residue and the sidechain of Arg221 and the other between the positively charged, protonated alpha‐amino group and the negatively charged sidechain of Asp48. We derived forcefield parameters for amino acids 1 and 3 and were able to extensively sample the conformational flexibility of these ligands in the binding pocket through molecular dynamics (MD) simulations (see Supporting Information Figures S15–S19). While the salt bridge from the phosphate head group stayed in place throughout 100 ns of the MD simulation, in explicit solvent the backbone amine turned outward at the start of the simulation and preferred solvent exposure. The aromatic rings of 1 and 3, respectively, were close to the aromatic rings of Phe182 and Tyr46, however, π‐π interactions were not observed in the final docking poses and rarely found in the simulation snapshots. In the MD simulations, the backbone amide NH groups remained close to the fluorines of the PF5 moiety and for amino acids 217–221 the closest average N⋅⋅⋅F distance was found to be generally below 3 Å, which implied the continuous presence of N−H−F interactions.
In summary, this work established the first synthesis and validation of pentafluorophosphato amino acids. High‐yielding pentafluorinations were developed as a one‐pot reaction under acidic conditions and yielded pentafluorophosphato‐difluoromethyl (PFPDFM) phenylalanine 3. Hydrolysis with aqueous HF provided the pentafluorophosphato‐carbonyl (PFPC) phenylalanine 12. Both PF5‐amino acids were successfully incorporated into peptides using HF for cleavage. The new chemical entities displayed remarkable structural, physico‐chemical, and biochemical properties resulting from fluorine‐specific interactions of the PF5‐anion. These include interactions of PF5 residues with water, hydrophobic surfaces, organic solvents, as well as aggregation events. As a result, PF5 amino acids bind 25 to 30 times stronger to the phosphotyrosine binding site of PTP1B than classical phosphonate biomimetics. Our studies demonstrate that developing improved PF5 ligands requires the optimization of protein‐ligand interactions but also of physico‐chemical properties and formulation of the molecules.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project‐ID 387284271—SFB 1349 (fluorine‐specific interactions, projects A3, A5, A6, and C5). M. A. received a graduate fellowship from the SFB 765. We thank Dr. Felix Bredendiek for analytical support, the work was supported by the DFG‐funded core facility BioSupraMol and the Zentraleinrichtung für Datenverarbeitung (ZEDAT) of Freie Universität Berlin. Open Access funding enabled and organized by Projekt DEAL.
M. Accorsi, M. Tiemann, L. Wehrhan, L. M. Finn, R. Cruz, M. Rautenberg, F. Emmerling, J. Heberle, B. G. Keller, J. Rademann, Angew. Chem. Int. Ed. 2022, 61, e202203579; Angew. Chem. 2022, 134, e202203579.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1.
- 1a. Hunter T., Cell 2000, 100, 113–127; [DOI] [PubMed] [Google Scholar]
- 1b. Ardito F., Giuliani M., Perrone D., Troiano G., Lo Muzio L., Int. J. Mol. Med. 2017, 40, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abdelsalam S. S., Korashy H. M., Zeidan A., Agouni A., Biomolecules 2019, 9, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Kostrzewa T., Styszko J., Gorska-Ponikowska M., Sledzinski T., Kuban-Jankowska A., Anticancer Res. 2019, 39, 3379–3384; [DOI] [PubMed] [Google Scholar]
- 3b. Singh V., Ram M., Kumar R., Prasad R., Roy B. K., Singh K. K., Protein J. 2017, 36, 1–6. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Cicirelli M. F., Tonks N. K., Diltz C. D., Weiel J. E., Fischer E. H., Krebs E. G., Proc. Natl. Acad. Sci. USA 1990, 87, 5514–5518; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Kraskouskaya D., Duodu E., Arpin C. C., Gunning P. T., Chem. Soc. Rev. 2013, 42, 3337–3370; [DOI] [PubMed] [Google Scholar]
- 4c. He R. J., Yu Z. H., Zhang R. Y., Zhang Z. Y., Acta Pharmacol. Sin. 2014, 35, 1227–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. T. R. Burke, Jr. , Smyth M. S., Otaka A., Nomizu M., Roller P. P., Wolf G., Case R., Shoelson S. E., Biochemistry 1994, 33, 6490–6494; [DOI] [PubMed] [Google Scholar]
- 5b. Shen I. K., Keng Y. F., Wu L., Guo X. L., Lawrence D. S., Zhang Z. Y., J. Biol. Chem. 2001, 276, 47311–47319; [DOI] [PubMed] [Google Scholar]
- 5c. Burke T., Curr. Top. Med. Chem. 2006, 6, 1465–1471; [DOI] [PubMed] [Google Scholar]
- 5d. Boutselis G., Yu X., Zhang Z. Y., Borch R. F., J. Med. Chem. 2007, 50, 856–864; [DOI] [PubMed] [Google Scholar]
- 5e. Zhang S., Zhang Z. Y., Drug Discovery Today 2007, 12, 373–381; [DOI] [PubMed] [Google Scholar]
- 5f. Köhn M., Meyer C., Synthesis 2011, 3255–3260; [Google Scholar]
- 5g. Meyer C., Hoeger B., Temmerman K., Tatarek-Nossol M., Pogenberg V., Bernhagen J., Wilmanns M., Kapurniotu A., Köhn M., ACS Chem. Biol. 2014, 9, 769–776; [DOI] [PubMed] [Google Scholar]
- 5h. Liao H., Pei D., Org. Biomol. Chem. 2017, 15, 9595–9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For a recent review on phosphotyrosine mimetics: Makukhin N., Ciulli A., RSC Med. Chem. 2021, 12, 8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Kornberg R. D., McNamee M. G., McConnell H. M., Proc. Natl. Acad. Sci. USA 1972, 69, 1508–1513; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Xie L., Lee S. Y., Andersen J. N., Waters S., Shen K., Guo X. L., Moller N. P., Olefsky J. M., Lawrence D. S., Zhang Z. Y., Biochemistry 2003, 42, 12792–12804; [DOI] [PubMed] [Google Scholar]
- 7c. Wiemer A. J., Wiemer D. F., Top. Curr. Chem. 2015, 360, 115–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Sun J. P., Wu L., Fedorov A. A., Almo S. C., Zhang Z. Y., J. Biol. Chem. 2003, 278, 33392–33399; [DOI] [PubMed] [Google Scholar]
- 8b. Zhao D., Sun L., Zhong S., Mol. Diversity 2021, 10.1007/s11030-021-10323-2. [DOI] [PubMed] [Google Scholar]
- 9. Wagner S., Accorsi M., Rademann J., Chem. Eur. J. 2017, 23, 15387–15395. [DOI] [PubMed] [Google Scholar]
- 10.Preparation of few simple pentafluorophosphates has been described before via disproportionation under harsh conditions not tolerating functional groups like esters and carbamates:
- 10a. Pavlenko N. V., Babadzhanova L. A., Gerus I. I., Yagupolskii Y. L., Tyrra W., Naumann D., Eur. J. Inorg. Chem. 2007, 1501–1507; [Google Scholar]
- 10b. Tian C., Nie W., Borzov M. V., Su P., Organometallics 2012, 31, 1751–1760; [Google Scholar]
- 10c. Guzyr O. I., Zasukha S. V., Vlasenko Y. G., Chernega A. N., Rozhenko A. B., Shermolovich Y. G., Eur. J. Inorg. Chem. 2013, 4154–4158; [Google Scholar]
- 10d. Ignatyev N., Barthen P., Koppe K., Frank W., WO 2016/074757A, 2016.
- 11.Deposition Number 2157920 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 12. Hoffmann W., Langenhan J., Huhmann S., Moschner J., Chang R., Accorsi M., Seo J., Rademann J., Meijer G., Koksch B., Bowers M. T., von Helden G., Pagel K., Angew. Chem. Int. Ed. 2019, 58, 8216–8220; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8300–8304. [Google Scholar]
- 13.
- 13a. Ben-Amotz D., J. Am. Chem. Soc. 2019, 141, 10569–10580; [DOI] [PubMed] [Google Scholar]
- 13b. Scatena L. F., Brown M. G., Richmond G. L., Science 2001, 292, 908–912; [DOI] [PubMed] [Google Scholar]
- 13c. Perera P. N., Fega K. R., Lawerence C., Sundstrom E. J., Tomlinson-Philips J., Ben-Amotz D., Proc. Natl. Acad. Sci. USA 2009, 106, 12230–12234; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Bonn M., Nagata Y., Backus E. H. G., Angew. Chem. Int. Ed. 2015, 54, 5560–5576; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5652–5669. [Google Scholar]
- 14. Matsuura S., Niu C.-H., Cohen J. S., J. Chem. Soc. Chem. Commun. 1976, 451–452. [Google Scholar]
- 15.
- 15a. Horatscheck A., Wagner S., Ortwein J., Lisurek M., Kim B. G., Beligny S., Schütz A., Rademann J., Angew. Chem. Int. Ed. 2012, 51, 9441–9447; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9577–9583; [Google Scholar]
- 15b. Wagner S., Schütz A., Rademann J., Bioorg. Med. Chem. 2015, 23, 2839–2847; [DOI] [PubMed] [Google Scholar]
- 15c. Wong E. L., Nawrotzky E., Arkona C., Kim B. G., Beligny S., Wang X., Wagner S., Lisurek M., Carstanjen D., Rademann J., Nat. Commun. 2019, 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jia Z., Barford D., Flint A. J., Tonks N. K., Science 1995, 268, 1754–1758. [DOI] [PubMed] [Google Scholar]
- 17. Sun J.-P., Fedorov A. A., Lee S.-Y., Guo X.-L., Shen K., Lawrence D. S., Almo S. C., Zhang Z.-Y., J. Biol. Chem. 2003, 278, 12406–12414. [DOI] [PubMed] [Google Scholar]
- 18. Lu C., et al., J. Chem. Theory Comput. 2021, 17, 4291–4300. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Friesner R. A. et al., J. Med. Chem. 2004, 47, 1739–1749; [DOI] [PubMed] [Google Scholar]
- 19b. Friesner R. A. et al., J. Med. Chem. 2006, 49, 6177–6196; [DOI] [PubMed] [Google Scholar]
- 19c. Halgren T. A. et al., J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.