

Abstract
The shrinking of glaciers is among the most iconic consequences of climate change. Despite this, the downstream consequences for ecosystem processes and related microbiome structure and function remain poorly understood. Here, using a space‐for‐time substitution approach across 101 glacier‐fed streams (GFSs) from six major regions worldwide, we investigated how glacier shrinkage is likely to impact the organic matter (OM) decomposition rates of benthic biofilms. To do this, we measured the activities of five common extracellular enzymes and estimated decomposition rates by using enzyme allocation equations based on stoichiometry. We found decomposition rates to average 0.0129 (% d−1), and that decreases in glacier influence (estimated by percent glacier catchment coverage, turbidity, and a glacier index) accelerates decomposition rates. To explore mechanisms behind these relationships, we further compared decomposition rates with biofilm and stream water characteristics. We found that chlorophyll‐a, temperature, and stream water N:P together explained 61% of the variability in decomposition. Algal biomass, which is also increasing with glacier shrinkage, showed a particularly strong relationship with decomposition, likely indicating their importance in contributing labile organic compounds to these carbon‐poor habitats. We also found high relative abundances of chytrid fungi in GFS sediments, which putatively parasitize these algae, promoting decomposition through a fungal shunt. Exploring the biofilm microbiome, we then sought to identify bacterial phylogenetic clades significantly associated with decomposition, and found numerous positively (e.g., Saprospiraceae) and negatively (e.g., Nitrospira) related clades. Lastly, using metagenomics, we found evidence of different bacterial classes possessing different proportions of EEA‐encoding genes, potentially informing some of the microbial associations with decomposition rates. Our results, therefore, present new mechanistic insights into OM decomposition in GFSs by demonstrating that an algal‐based “green food web” is likely to increase in importance in the future and will promote important biogeochemical shifts in these streams as glaciers vanish.
Keywords: alpine biogeochemistry, carbon cycling, ecological stoichiometry, extracellular enzyme activity, microbial ecology
Glacier‐fed streams are among the fastest‐changing aquatic habitats worldwide, yet the functional response of resident microbial communities to glacier shrinkage remains minimally explored. Here, we investigated how reductions in glacier influence affect the organic matter decomposition rates of sediment biofilms residing in these vanishing habitats by surveying 101 glacier‐fed streams across six major regions globally. Using this space for time approach, we found that glacier shrinkage will accelerate decomposition rates, and corresponds to increases in benthic algal biomass and shifts in the sediment bacterial microbiome.

1. INTRODUCTION
The decomposition of organic matter (OM) by microorganisms is a major intermediary to the global carbon cycle. Microorganisms produce extracellular enzymes that degrade organic compounds, which can be metabolized to CO2 or CH4, or incorporated into microbial biomass and further into the food web (Battin et al., 2008; Sinsabaugh et al., 2009). Therefore, assessing controls on OM decomposition is critical to understand carbon fluxes through ecosystems and climate‐change impacts on this ecosystem process. Current understanding of OM decomposition in streams (Boyero et al., 2021; Rosemond et al., 2015; Tiegs et al., 2019) is rooted in early stream ecosystem science and biased toward the decomposition of terrestrial OM subsidies and its relevance for the “brown food web” (Webster & Benfield, 1986; Webster & Meyer, 1997). In this context, leaf litter bags and cotton strips that have been widely used as model substrates for OM degradation in running waters revealed latitudinal and altitudinal controls on OM decomposition rates (Boyero et al., 2021; Follstad Shah et al., 2017; Tiegs et al., 2019). Yet, such approaches fail to capture OM decomposition in high‐latitude and high‐altitude streams above the treeline, where terrestrial subsidies of vascular plant material rich in cellulose are scarce or even absent.
Foremost among the streams with little terrestrial OM subsidies are glacier‐fed streams (GFSs), which sustain flow in some of the world's largest fluvial networks and contribute to large‐scale carbon fluxes (Hood et al., 2015; Horgby et al., 2019; Singer et al., 2012). Climate‐induced shrinkage of mountain glaciers is altering the GFS environment at an unprecedented pace (Milner et al., 2017). How these alterations affect biodiversity in GFSs is becoming increasingly well understood (Cauvy‐Fraunié & Dangles, 2019; Jacobsen & Dangles, 2012; Wilhelm et al., 2013), but less so ecosystem processes (Elser et al., 2020; Milner et al., 2017). It has been speculated that primary production will increase in GFSs following peak glacier melt when environmental conditions (e.g., higher water clarity) become more beneficial to primary producers (Milner et al., 2017). This is an important consideration given that primary production is the primary energy source in GFSs, particularly in reaches close to the glacier that are largely devoid of terrestrial OM subsidies (Zah & Uehlinger, 2001). To date, we do not properly understand how glacier shrinkage may affect primary production, and how this in turn may affect the decomposition of OM in GFSs.
Fungi and bacteria are the main decomposers of OM in streams and rivers (Findlay, 2021; Hall & Meyer, 1998). The role of fungal hyphomycetes as early decomposers of stream OM, particularly leaf litter, is generally well documented in the aquatic literature (Gessner & Chauvet, 1994); yet, how bacterial diversity and community composition affect OM degradation, both in terrestrial and aquatic ecosystems, remains a subject of active debate (Glassman et al., 2018; Hayer et al., 2021). In GFSs, OM decomposition and its microbial ecology is only rudimentarily understood. A recent study using cotton strips as a surrogate for OM highlighted the relevance of fungal hyphomycetes for OM decomposition in GFSs (Fell et al., 2021), and further related hyphomycete biomass to relative glacier coverage to infer that glacier shrinkage will accelerate decomposition. Despite these insights, we are still missing a comprehensive and mechanistic understanding of the drivers of OM decomposition in GFSs, and how it may respond to glacier shrinkage.
Using a space‐for‐time substitution approach, we studied how glacier shrinkage affects the rate of OM decomposition associated with benthic biofilms from 101 GFSs, spanning six major regions worldwide. We inferred decomposition rates from measured extracellular enzymatic activities (EEAs) involved in carbon (C), nitrogen (N), and phosphorus (P) acquisition (Hill et al., 2017; Sinsabaugh & Moorhead, 1994). The advantage of this approach, compared with others (e.g., leaf litter packs and cotton strips), is that it intrinsically and directly links OM decomposition to the microorganisms. We further used amplicon sequencing (16S rRNA and 18S rRNA genes), as well as metagenomics, to study relationships between decomposition rates and microbiome structure and function.
Based on fundamental considerations of ecophysiological and stoichiometric controls on OM decomposition (Brown et al., 2004; Follstad Shah et al., 2017; Rosemond et al., 2015), we anticipated that decreasing glacier influence affects decomposition rates through elevated water temperatures and nutrient N:P ratios. Furthermore, given the largely missing subsidies of vascular plant material in GFSs, we expected primary producer biomass to be a major driver of decomposition rates. Finally, we expected to find distinct components of the microbiome to be associated with changes in decomposition rates. Our findings shed new light on the downstream impacts of glacier shrinkage by quantifying the relationship between glacier coverage and decomposition rates across a wide range of GFSs worldwide and by revealing the role of microbiome structure and function for this important ecosystem process.
2. MATERIALS AND METHODS
2.1. Sampling and field data
We sampled 101 GFSs from six major regions globally over 2019–2020, including the Southern Alps of New Zealand (n = 20), the Nuuk and Qeqertarsuaq (Disko Island) areas of Greenland (n = 10), the Ecuadorian Andes (n = 15), the Russian Caucasus (n = 19), the Lyngen Alps and Jotunheim Mountains of Norway (n = 10), and the European Alps (n = 27). The sampled GFSs spanned a gradient in latitude, elevation, glacier size, and catchment geology (Data S1). Sampling was conducted as previously described (Fodelianakis et al., 2021; Kohler et al., 2020). Briefly, samples were collected from two reaches of each GFS, with one near the glacier snout and the other located downstream (median: 434 m) before the confluence with another tributary. The presence of physical barriers (cliffs, waterfalls, etc.) prevented a standardized distance between the glacier and associated transects. At each reach, three patches were sampled for benthic sediments (top <5 cm of the streambed) and gently sieved to retain the sandy fraction (from 250 μm to 3.15 mm); all sampling devices were flame‐sterilized via blowtorch prior to use. All sediment aliquots were flash‐frozen with liquid nitrogen in the field, pending analysis of chlorophyll a, EEA, and DNA extraction before being stored at –80°C.
Stream water temperature, pH, electrical conductivity, and O2 concentrations were measured in situ with a SenTix® 940‐P, a TetraCon® 925‐P, and a FDO® 925‐P probe on a MultiLine® meter (WTW Xylem Analytics Germany), respectively. Turbidity was measured with a Turb® 430 IR portable turbidity meter (WTW Xylem Analytics Germany). Stream water samples were collected from both reaches (except for New Zealand, where only upstream reaches were sampled) for the measurement of major ions, nutrients, and dissolved organic carbon (DOC). Samples for nutrient analyses were collected by filtering water through pre‐washed Whatman GF/F filters into acid‐washed Nalgene HDPE bottles and were frozen as soon as possible. Nutrient concentrations were measured on a LaChat QuikChem 8500 flow injection analyzer and included nitrate (; QuikChem® Method 10‐107‐05‐1‐C), nitrite + nitrate ; 10‐107‐04‐1‐B), ammonium (; 10‐107‐06‐3‐D), and soluble reactive phosphorus (P‐SRP; 10‐115‐01‐1‐M). New Zealand nutrient chemistry was measured on the same instrument model, but using slightly different QuikChem® methods for the same chemical species (see Kohler et al., 2020). Inorganic forms of N were summed to make dissolved inorganic N (DIN). Values were normalized using the molar mass of each element before calculating molar N:P ratios of stream water nutrients (DIN:SRP). Lastly, DOC samples were also filtered through pre‐ashed Whatman GF/F filters, stored at 4°C in acid‐washed amber glass vials, and measured on a Sievers M9 TOC Analyser (GE). All concentrations are reported in ppb.
Reach coordinates were collected using a GPS (GPSMAPR 66s, GARMIN). Coordinates allowed the calculation of glaciological metrics, such as the distance to the glacier, glacier area, and percent glacierized catchment based on satellite data. Specifically, glacier area was defined as the total glacierized area within the catchment above the sampling point and was manually delineated from Sentinel‐2 imagery (Level 2a, March–April 2019, downloaded from https://scihub.copernicus.eu/) based on a catchment definition derived from the ASTER Global Digital Elevation Model (GDEM) Version 3 (NASA/Meti/Aist/Japan Spacesystems, and U.S./Japan Aster Science Team, 2019). Straight‐line distances to the termini were calculated from manually mapped terminus positions based on the same data sources and GPS coordinates. From these data, we calculated the “Glacial Index” (GI; Jacobsen & Dangles, 2012), where high values indicate a greater level of glacier influence than low values:
| (1) |
2.2. Biofilm biomass and enzymatic extracellular activity
Chlorophyll a was measured by adding 90% EtOH to weighed sediments (~2 g wet weight), extracted in a hot water bath (78°C) for ten min, and incubated in the dark at 4°C for 24 h. Following incubation, samples were vortexed, centrifuged (max. speed for 5 min), read in a plate reader at 436/680 excitation/emission, and sediment dried to normalize resulting values by the dry mass (DM) of sediment (µg chlorophyll a g−1 DM).
We measured potential activities of α‐1,4‐glucosidase (AG), β‐1,4‐glucosidase (BG), leucine aminopeptidase (LAP), β‐1,4‐N‐acetylglucosaminidase (NAG), and acid (alkaline) phosphatase (AP) using fluorescent 4‐methylumbelliferone (MUF) and 7‐Amino‐4‐methylcoumarin (AMC)‐linked substrates (4‐MUF‐α‐D‐glucoside, 4‐MUF‐β‐D‐glucoside, L‐Leucine‐7‐amino‐4‐AMC, 4‐MUF‐N‐acetyl‐β‐D‐glucosaminide, and 4‐MUF‐phosphate, respectively) as performed previously (Kohler et al., 2020). AG and BG acquire C, cleaving starch and cellulose, LAP and NAG acquire both N and C, cleaving peptides and chitin, while AP acquires P through the degradation of phosphomonoesters (Sinsabaugh et al., 2009). These extracellular enzymes are widely distributed across aquatic ecosystems where they have been extensively used to infer controls on microbial decomposition (Sinsabaugh et al., 2009; Sinsabaugh & Shah, 2012).
A substrate concentration of 0.3 µM and a 1.5 to 2 h incubation time were applied to all substrates based on preliminary work and were dissolved in artificial stream water with a pH adjusted to 7.5 (Kohler et al., 2020). Approximately 1 g (wet weight) of sediment was transferred to pre‐weighed centrifuge tubes, and 4 ml of MUF and AMC‐linked substrates were added chilled (4°C) to sediments incubated for 1.5–2 h in the dark on a shaker at 4°C. In addition to the assays, we incubated reference standards of MUF/AMC with controls for each sediment sample: a blank (artificial stream water only), a matrix control (sediment plus artificial stream water), a quench control (sediment plus MUF/AMC reference standard), and a deactivated control (sediment boiled for 30 min before adding substrate). Following incubation, 2 ml of glycine buffer (pH = 10.4) was quickly added to all tubes (2:1 sample:buffer vol:vol ratio) to raise pH and halt product generation. Tubes were vortexed, centrifuged, and the supernatant transferred to black 96‐well plates (Corning, flat bottom, polystyrene non‐binding surface) before fluorescences were read on a BioTek Synergy H1 high sensitivity plate reader at 365/455 excitation/emission wavelengths for MUF and 364/445 excitation/emission wavelengths for AMC. Following analyses, sediments were dried to a stable mass and weighed, and enzymatic activities (nmol h−1 g−1 DM) were calculated using equations in Kohler et al. (2020).
2.3. DNA extraction, amplification, and sequencing
Nucleic acids were extracted from ~5 g of sediment following a phenol‐chloroform‐based protocol modified for GFSs (Busi et al., 2020). Analysis of the prokaryotic microbial component was performed by amplifying the V3–V4 hypervariable region of the 16S rRNA gene using primers 341f (5′‐CCTACGGGNGGCWGCAG‐3′) and 785r (5′‐GACTACHVGGGTATCTAATCC‐3′; Klindworth et al., 2013). Similarly, the V4 loop of the eukaryotic 18S rRNA gene was amplified gene was amplified using the TAReuk454F––TAReukREV3 primers (Stoeck et al., 2010). While we acknowledge that other primers may be superior in cataloguing fungal diversity, such as the internal transcribed spacer (ITS) region, the 18S rRNA gene has been shown to adequately reflect fungal diversity at high taxonomic levels (e.g., order and higher, Yarza et al., 2017), which is sufficient for our purposes of providing class‐level or higher differences.
The KAPA HiFi DNA polymerase (Hot Start and Ready Mix formulation) was used in a 25‐µl‐amplification reaction containing 1x PCR buffer, 1 µM of each primer, 0.48 µg/µl BSA, and 1.0 µl of template DNA (concentration of ≤2–3 ng/µl). Amplification was performed in a Biometra Trio (Biometra) instrument and consisted of an initial denaturation at 95°C for 3 min, followed by 94°C for 30 s, 55°C for 30 s and 72°C for 30 s for 25 cycles followed by a final extension at 72°C for 5 min. Amplification was verified on a 1.5% agarose gel, libraries were prepared, and samples sequenced using a 300‐bp paired‐end protocol on the Illumina Miseq platform. Sequencing was performed partly in the Genomic Technologies Facility of the University of Lausanne and partly at the Biological Core Lab of the King Abdullah University of Science and Technology. 16S and 18S rRNA genes’ amplicon data were analyzed by using Trimmomatic (v.0.36, Bolger et al., 2014) for quality filtering and QIIME2 (v.2019.1, Bolyen et al., 2019) for sequence processing. DADA2 was used to create amplicon sequence variants (ASVs; Callahan et al., 2016), and the SILVA database (v138.1) was used for taxonomic classification (Quast et al., 2012). Singletons, chloroplasts, and mitochondria were removed prior to analysis from the 16S rRNA gene data set, and we removed singletons and all non‐eukaryotes from the 18S rRNA gene data set.
A subset (n = 50) of the samples were chosen for shotgun metagenomic sequencing (based on DNA concentration and successful library preparation) and included samples from all six regions (New Zealand, N = 18; European Alps, n = 6; Greenland, n = 2; Caucasus, n = 16; Ecuador, n = 3; Norway, n = 5; Data S1). Shotgun metagenomic libraries were prepared using the NEBNext Ultra II FS library kit. The libraries were prepared as described in Busi et al. (2021). Briefly, 50 ng of DNA was enzymatically fragmented for 12.5 min, followed by 6 PCR amplification cycles for constructing the libraries. The libraries were quantified using Qubit (Invitrogen), and the average insert size (~450 bp) was determined by quality assessment using the Bioanalyzer from Agilent. All the libraries were pooled and sequenced at the Functional Genomics Centre Zurich on a NovaSeq (Illumina) using a S4 flowcell.
Shotgun metagenomic samples were preprocessed using trim_galore with default parameters (Krueger, 2018) which uses a combination of cutadapt (Martin, 2011) and fastqc (Andrews, 2010) to check the quality and remove adapters and other unwanted sequences (e.g. containing primers, poly‐A tails) from the raw sequence files. Then, megahit (v1.2.9, Li et al., 2015) was used to assemble the reads into contigs per sample using the default parameters. ORFs were obtained by Prodigal with the meta options (v2.6.3, Hyatt et al., 2010) and then annotated with eggnog‐mapper using default parameters (v2.1.2, Huerta‐Cepas et al., 2017). The genes taxonomy was determined by using Kraken2 (v2.1.2, Wood & Salzberg, 2014) against the Kraken2 v2.0.9beta97 maxikraken database available at https://lomanlab.github.io/mockcommunity/mc_databases.html. To match genes with the laboratory‐measured EEA, we used Enzyme Commission numbers obtained from Sinsabaugh et al. (2009; AG = 3.2.1.20, BG = 3.2.1.21, NAG = 3.2.1.14, LAP = 3.4.11.1, and AP = 3.1.3.1). The enzymes were then dereplicated using cdhit‐est (Huang et al., 2010) by applying a global identity cutoff of 95% and a coverage of 80% for the shorter gene. The coverage of the different dereplicated enzymes among the samples was obtained with the coverM software (v0.6.1, Woodcroft, 2020) with the trimmed_mean method that removes the 5% of bases with highest coverage and 5% of bases with lowest coverage before averaging the coverage along the remaining base. The recA gene (K03553) was used to normalize the coverage between samples by dividing the number of reads from the EEA genes by the number of reads assigned to this prokaryotic single‐copy gene (Acinas et al., 2021).
2.4. Calculation of decomposition rate
We calculated decomposition rates from EEA based on equations described in Hill et al. (2017). As a first step, threshold elemental ratios (TERC:X) were calculated based on the equations formulated by Sinsabaugh et al. (2009) and modified by Tapia‐Torres et al. (2015):
| (2) |
| (3) |
where B C:X is the carbon to nutrient ratio of microbial cells (C:N = 8.6 and C:P = 60), according to Cleveland and Liptzin (2007), and n o and p o are scalers determined by finding the intercept between C‐acquiring (AG, BG, NAG, and LAP) and N‐ (NAG and LAP) and P‐acquiring (AP) EEA. Importantly, here and below, we have reorganized the equations to include LAP and NAG as both C‐ and N‐acquiring EEA. This is because LAP dominated enzymatic activity across all sites, which is presumably due to the high protein content of GFS dissolved OM, as previously reported (Singer et al., 2012). When carbon use efficiency (CUE) was calculated by using LAP and NAG as only N‐acquiring EEA (as outlined below), high values were obtained and were deemed unrealistic. Thus, we have modified our equations as recommended by Sinsabaugh and Shah (2012) for freshwater phytoplankton by also including LAP and NAG as C‐acquiring EEA in the model, with improved results. Lastly, scalers were calculated by using the entire data set so as not to induce differences as a function of geographic region.
The estimated TER values were next used to calculate CUE by using the equation described in Sinsabaugh et al. (2013, 2016):
| (4) |
where AX is the assimilation efficiency of N or P (0.9). Overall, CUE values averaged 0.23 (median 0.18) for our data set, indicating that our model performs reasonably compared with past meta‐studies (Sinsabaugh et al., 2013; Sinsabaugh & Shah, 2012). Decomposition rates (k, % d−1) were then calculated as formulated by Sinsabaugh and Moorhead (1994) and executed previously by Hill et al. (2017):
| (5) |
where each EEA value for a given site is normalized to the greatest value in the data set to remove scalar weighting, and EEAtotal is the sum of all normalized EEA for a given site.
2.5. Statistical analyses
To assess how the mean and variance of decomposition changes over glaciological (turbidity, glacier coverage, GI) and environmental gradients (chlorophyll a, stream water temperature and N:P), we used generalized additive models (GAMs) for location, scale, and shape (GAMLSS, Rigby et al., 2005). Furthermore, we built a model that included the sampled region (e.g., New Zealand, European Alps, etc.), sediment chlorophyll a, stream water N:P, and temperature as fixed effects. Chlorophyll a was chosen because it represents the primary OM source in GFSs, is generated in close proximity to the decomposers, and is also likely to be of considerably higher quality and quantity than the DOC in the water column. Nutrient concentrations have previously been linked to decomposition (Rosemond et al., 2015), and the N:P variable specifically was chosen because GFSs are generally thought to be N‐limited due to high levels of P loading from subglacial weathering (Ren et al., 2019). Therefore, a change in the balance of N to P may be influential for organisms in meeting elemental budgets. Lastly, temperature was included because organismal metabolic process rates, including decomposition, typically increase with temperature, per the metabolic theory of ecology (Brown et al., 2004). A GAM was chosen to model k because it allows nonlinear components for explanatory variables (i.e., chlorophyll a), and produced the lowest AIC values compared with other model structures. The global model formula was thus:
| (6) |
To investigate how decomposition was related to the microbiome, we used phylogenetic factorization (phylofactorization) to identify bacterial clades that explained significant variation in k (Washburne et al., 2017). Phylofactorization computes the isometric log‐ratio (ILR) abundances, y e, corresponding to each edge e in the phylogeny (to remove compositionality) and identifies edges maximizing an objective function quantifying a statistical pattern of interest. We were primarily interested in clades related to decomposition after controlling for region in order to account for possible region‐specific and/or biogeographical signals present in the data set. Thus, we first computed the linear model:
| (7) |
for each edge and chose the edges maximizing the t statistic of y, referred to as the “winning” edge. To control for the nested dependence of edges in the phylogeny (i.e., to ensure that a chain of descendant edges behind a lineage with the largest signal are not all chosen in sequence), phylofactorization cuts the tree at each identified edge, recalculates ILR abundances within sub‐trees, recomputes linear models for each edge, and repeats. Each iteration produces a p‐value from the t statistics of the winning y, and we control for multiple comparisons across all edges at each iteration using Holm’s sequentially rejective procedure for family‐wise error rates (FWER) at 5% as detailed in Washburne et al. (2019). We removed all samples with <10,000 total reads and all ASVs with <6 total reads from the 16S rRNA gene amplicon data set prior to phylofactorization. All statistics were performed using the R statistical environment (version 3.4.3, R Core Team, 2021).
3. RESULTS AND DISCUSSION
3.1. Decomposition rates across 101 glacier‐fed streams
Consistent with previous work in GFSs (Kohler et al., 2020), we found LAP to have the greatest activities among the five extracellular enzymes, followed by AP and BG (Figure S1b). This pattern of EEA contrasts with work published from soils where BG activity generally represents a larger proportion of the EEA pool (Sinsabaugh et al., 2008), likely due to the abundance of OM derived from vascular plants. Yet, the dominance of LAP has been previously observed for both freshwater planktonic and river biofilm communities (Sinsabaugh et al., 2010), likely due to proteins and aminopolysaccharides contributing a relatively large proportion of the total C available for production in these habitats (Sinsabaugh & Shah, 2012). Thus, given that LAP can be used to acquire both C and N, it provides a nutritionally and energetically efficient solution to using proteins; they likely constitute a significant resource in GFS biofilms due to the dominance of algal‐derived matter over terrestrial subsidies of vascular plant material (Zah & Uehlinger, 2001).
We used EEAs to calculate decomposition rates (% d−1; Sinsabaugh & Moorhead, 1994), which averaged 0.0129 (median: 0.0076; range: 0.0001 to 0.0773) across all 101 GFSs (Figure 1). These values are comparable with previous measurements from GFSs using cotton strips (mean: 0.0182; Fell et al., 2021) and from Alpine streams using leaf packs (mean: 0.0137 without mesh bags; Robinson & Gessner, 2000). Therefore, our enzymatic modeling approach is validated by this congruence between different methods. Strikingly, our calculated decomposition rates, encompassing patches, reaches, GFSs and mountain ranges, span the same range as those reported from streams and rivers draining across various biomes worldwide (Boyero et al., 2021; Tiegs et al., 2019). This finding is unexpected because the GFS environment (e.g., stream water temperature, pH) varies relatively little among regions (Table S1) and because autochthonous production consistently dominates the OM pool in GFSs. Therefore, we explored possible drivers of decomposition that are specific to GFS ecosystems, foremost glacier influence and related parameters.
FIGURE 1.
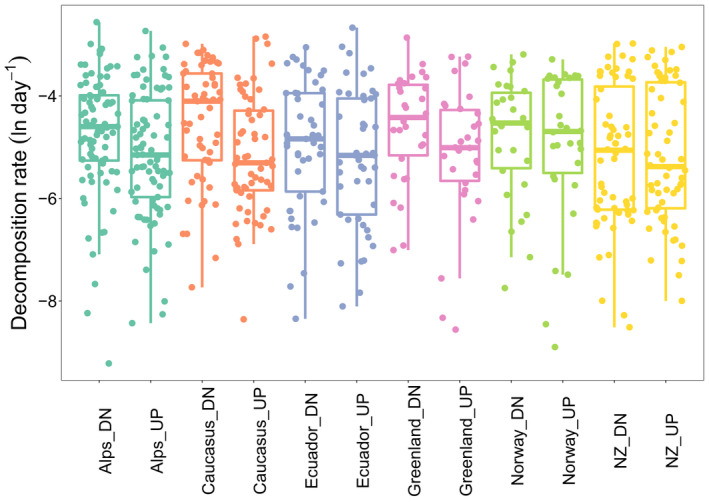
Decomposition rates exhibited a wide range of values across glacier‐fed streams, and were significantly greater at downstream (DN) versus upstream (UP) transects. We collected stream sediments from 101 glacier‐fed streams (GFSs) from six major global regions, including the European Alps (green, n = 27 GFSs), Russian Caucasus (orange, n = 19 GFSs), Ecuadorian Andes (blue, n = 15 GFSs), Greenland (violet, n = 10 GFSs), Norway (green, n = 10 GFSs), and New Zealand (yellow, n = 20 GFSs). At each GFS, two transects were sampled (up‐ and downstream), and at each transect three patches for a total of 606 individual measurements for decomposition rate, here expressed as natural log‐transformed % day−1
3.2. Decomposition rates increase with glacier shrinkage and primary producer biomass
The effects of glacier shrinkage on community ecology in GFSs are increasingly understood (Cauvy‐Fraunié & Dangles, 2019; Milner et al., 2017), less so on ecosystem processes (Elser et al., 2020). Capitalizing on our two‐reach sampling design (up‐ vs. downstream transects), we first created a generalized linear model including both transect and glacier as fixed effects, and found that upstream reaches have significantly lower decomposition rates than downstream (t = −6.110, p < .001, Figure 1). To further investigate how decomposition rates are related to glacier influence, and by using GAMLSS, we found that the average decomposition rate decreased with percent glacier coverage and GI (taking into account glacier area and distance from the glacier) across all GFSs, reaches, and patches, while the variance of decomposition increased across that same gradient (Figure 2a,b). Under the assumption that up‐ and downstream reaches, the GI, and percent glacier coverage substitute space for time (as used in past studies, Fell et al., 2021; Jacobsen & Dangles, 2012), these relationships would translate into an acceleration of OM decomposition in GFSs as glaciers recede. To explore this relationship further, we calculated the slope from the linear regression of un‐transformed decomposition rates versus percent glacier coverage and estimated that for every 1% reduction in glacier coverage in the catchment, the decomposition rate increases by 0.0091% d−1.
FIGURE 2.
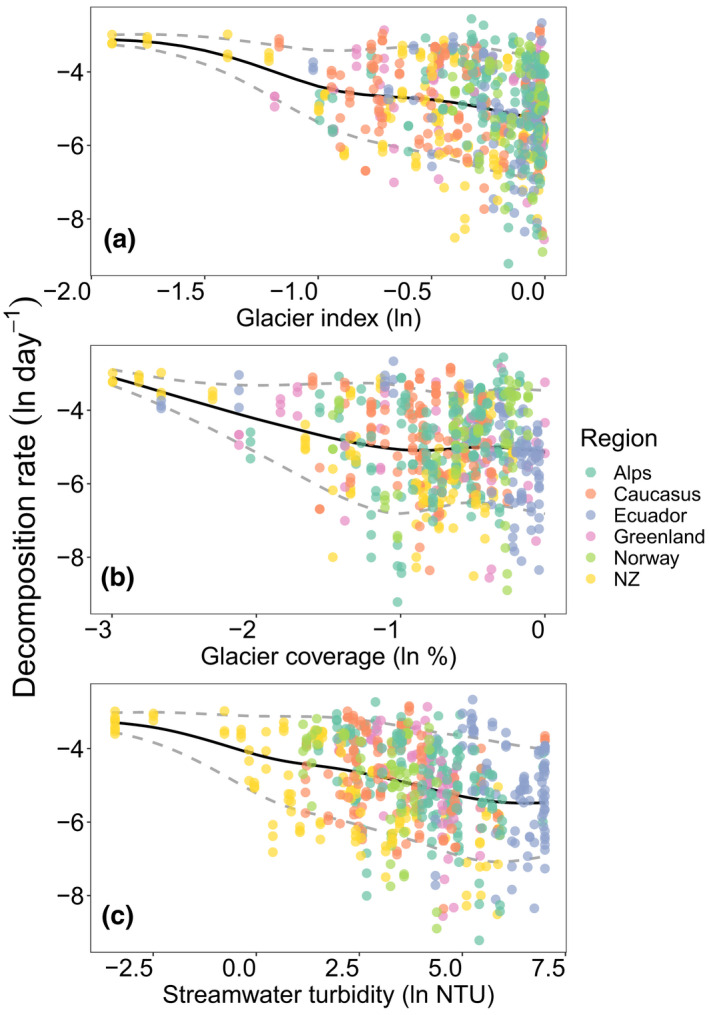
Decomposition rate increases with decreasing glacier influence using a space for time substitution. Decomposition rates in glacier‐fed streams were negatively associated with the (a) glacier index, (b) percent glacier coverage of catchments, and (c) stream water turbidity (all natural log‐transformed), suggesting that decomposition rates will increase with glacier shrinkage. Generalized additive models for location, scale and shape (GAMLSS) identified a significant decrease in the mean (black line), but an increase in variance (dashed gray line) in modeled decomposition rates with increases in all three variables (glacier index: μ, df = 3; t‐value = −21.47; p < 0.001; σ, df = 3; t‐value = 8.19; p < 0.001; percent glacier coverage: μ, df = 3; t‐value = −11.75; p < 0.001; σ, df = 3; t‐value = 5.88; p < 0.001; stream water turbidity: μ, df = 3; t‐value = −20.36; p < 0.001; σ, df = 3; t‐value = 6.06; p < 0.001)
Increasing variation in decomposition rates with glacier influence suggests that additional local factors not accounted for by these metrics are important for explaining decomposition. Indeed, it is likely that decomposition is not directly related to glacier influence, but instead related to the physical and chemical environment that changes with glacier influence. Stream water turbidity is such an environmental parameter, reflecting changes in suspended sediment concentrations largely driven by glacier extent, melt dynamics, and hydrological flow paths (Hodson & Ferguson, 1999; Swift et al., 2005). At the same time, turbidity attenuates light that reaches the benthic zone of the stream (Boix Canadell et al., 2021; Uehlinger et al., 2010) and induces physical abrasion of benthic biofilms (Francoeur & Biggs, 2006); hence, its effect on ecosystem processes is likely mediated by higher‐order glacier processes. This notion is supported by the negative relationship between stream water turbidity and decomposition (Figure 2c), which is similar to the previously described relationships between the glacier metrics (i.e., percent glacier coverage and GI) and decomposition rate (Figure 2a,b).
Overall, decomposition rates depend on OM quantity and quality (Enríquez et al., 1993; Follstad Shah et al., 2017), temperature and stoichiometric constraints (Findlay, 2021; Rosemond et al., 2015; Sinsabaugh et al., 2009). DOC concentrations were low in the sampled streams (median = 150.35, range = 42.23–994 ppb; Table S1), which likely makes algae exudates the most important source of OM to GFS biofilms, which are in turn controlled by stream water turbidity. Therefore, to gain a more mechanistic understanding of decomposition in GFSs, we used a GAM to further explore variation in decomposition rate across all GFSs, reaches, and patches. Collectively, the deviance explained by the sampled region, chlorophyll a, stream water temperature, and the N:P ratio was 61.4% (Table S2). Most conspicuously, we found chlorophyll a to be a major driver of decomposition rate (Spearman’s r = 0.76, p < .001, Figure 3a). Stream water temperature (Spearman’s r = 0.24, p < .001) and N:P (Spearman's r = 0.15, p < .001) were also positively related with decomposition (Figure 3b,c); however, the magnitude of these correlations and the GAM effect sizes were relatively small compared with the relationship with chlorophyll a (Table S2).
FIGURE 3.
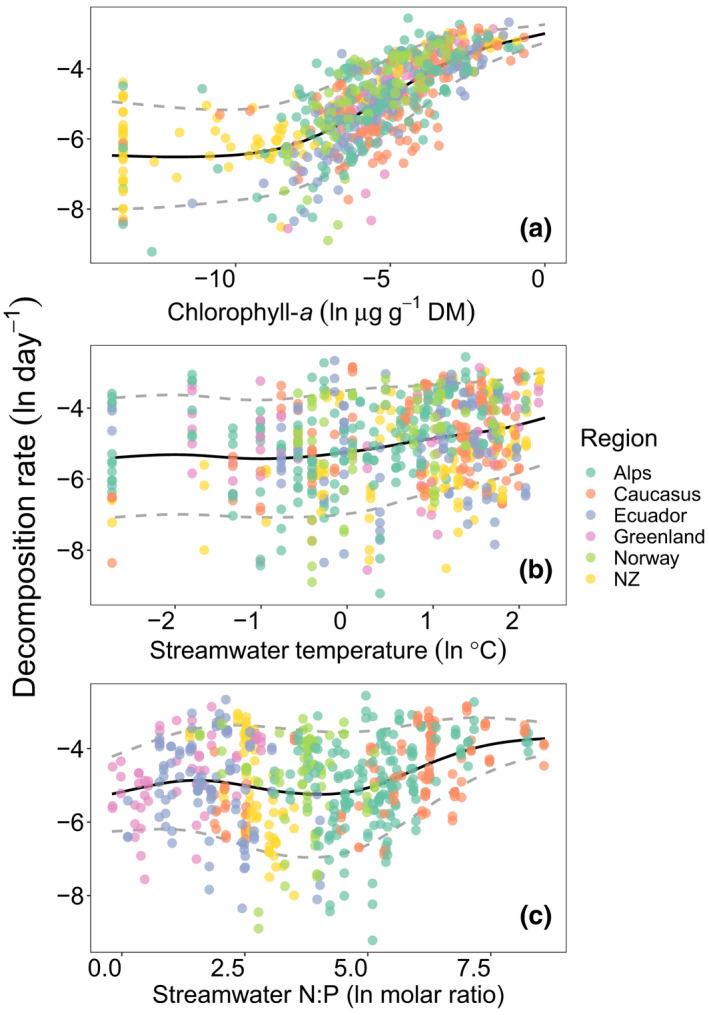
Decomposition in glacier‐fed streams is related to organic matter availability and subject to temperature and stoichiometric constraints. Chlorophyll a (a), stream water temperature (b), and stream water molar N:P ratios (c) all have significant positive univariate associations with modeled decomposition rates (all natural log‐transformed). For all three, GAMLSS identified a significant increase in the mean (black line), but an decrease in variance (dashed grey line) in modeled decomposition rates with increases in all three variables (chlorophyll a; μ, df = 3; t‐value = 26.61; p < .001; σ, df = 3; t‐value = −8.54; p < .001; temperature; μ, df = 3; t‐value = 5.93; p < .001; σ, df = 3; t‐value = −2.52; p < .001; stream water N:P; μ, df = 3; t‐value = 5.61; p < .001; σ, df = 3; t‐value = −3.56; p < .001)
Algae have been long recognized as a source of labile OM substrate (Enríquez et al., 1993) and exude macromolecules (e.g., carbohydrates), particularly when senescing, which can be readily decomposed by extracellular enzymes such as glucosidases (Espeland et al., 2001). In microbial biofilms, such enzymes act in close spatial proximity to the exuding algae, which is advantageous in a system characterized by high oligotrophy and continuous loss through advection (Battin et al., 2003, 2016). The efficient decomposition of the primary producers’ biomass by microorganisms is therefore a pivotal link to C and nutrient cycling in stream ecosystems. On the other hand, the lower effect size for stream water temperature in particular is likely attributable to the low variation in this parameter across and particularly within GFSs (temperature median = 2.35, range = 0 to 9.35°C; Table S1). It could also be argued that the decomposition of highly bioavailable algal‐derived OM is relatively insensitive to temperature compared with more recalcitrant OM (Davidson & Janssens, 2006; von Lützow & Kögel‐Knabner, 2009). Overall, temperature effects on OM decomposition in streams and rivers remain poorly understood compared with the terrestrial realm (Findlay, 2021).
3.3. Relating decomposition rates to the glacier‐fed stream microbiome
An enduring theme in ecosystem science has been the integration of biogeochemistry with microbial ecology. We used 18S rRNA and 16S rRNA gene amplicon sequencing to relate the composition of the biofilm microbiome to decomposition across all 101 GFSs. Based on relative abundances, we found that eukaryotes were dominated by photoautotrophs, particularly Ochrophytes (mean ± SD: 52.3% ± 24.3%), which includes the chrysophyte genus Hydrurus and diatoms, and green algae (Chlorophyta: 4.3% ± 6.3%; Charophyta: 2.5% ± 5.5%). The relative abundance of fungi (5.5% ± 5.3%) was low compared with the photoautotrophs (Figure S2b). Intriguingly, the overwhelming majority of fungal reads were from Chytridiomycetes (69.0% ± 22.3%) followed by Dikarya (8.9% ± 11.6%), which include the hyphomycetes Ascomycota and Basidiomycota (Figure S3).
Chytridiomycetes are known to parasitize algae, directly deriving OM from them rather than through decomposition, and potentially fostering the release of macromolecules through cell death and disruption of the cell walls (Senga et al., 2018). The relevance of these parasitic fungi for the production of a “fungal shunt” and related carbon fluxes is increasingly being recognized in aquatic ecosystems (Klawonn et al., 2021), including cryospheric ecosystems (Anesio et al., 2017; Brown et al., 2015). The apparent dominance of these parasitic fungi suggests that the majority of GFS fungi are primarily using algae‐derived OM (e.g., from Hydrurus) rather than allochthonous OM from glacier ice or terrestrial vegetation. As a result, the classical role of fungi as saprotrophic decomposers may be downplayed in GFSs in comparison with lower elevation/latitude systems. Our findings on the relative dominance of Chytridiomycetes contrasts those on the hyphomycetes associated with cotton strips used as model POM in GFSs (Fell et al., 2021). Cotton strips are largely composed of cellulose, a major compound of vascular plant material but largely absent from the upper reaches of GFSs. While we are aware that cellulose is an important component in the cell walls of green algae, it is not present in Hydrurus and diatoms (Lowe & LaLiberte, 2017), and green algae were a minority in the GFS photoautotrophic communities compared with the Ochrophyta (Figure S2b).
Within the bacterial realm of the microbiome, Gammaproteobacteria (42.8% ± 15.7%), Alphaproteobacteria (14.4% ± 5.7%), Bacteroidia (10.5% ± 5.6%), and Verrucomicrobiae (4.4% ± 3.8%) were the most abundant classes across all GFSs (Figure S2a). To investigate which clades within the microbiome covary with decomposition across all GFSs, we used phylofactorization (Washburne et al., 2017), accounting for biogeographical effects (see Methods, Table S3). Our analysis revealed clades with consensus taxonomies affiliated to Saprospiraceae (95 ASVs) within Bacteroidia, Fimbriiglobus (53 ASVs) within Gemmataceae, Acidobacteriae (256 ASVs), and Flavobacterium (50 ASVs) that were positively associated with increasing decomposition rate and chlorophyll a. While constituting relatively minor contributions (mean: 1.7%; range: 0%–5.8% of the total reads per sample) to the bacterial microbiome, all of these clades are known to include chemoorganotrophs (Kulichevskaya et al., 2017; McIlroy & Nielsen, 2014). We, therefore, suggest that these bacterial clades are involved in the decomposition of algal‐derived OM available in GFSs, potentially facilitated by, or in competition with, a fungal shunt as induced by the parasitic members of Chytridiomycetes.
Rather unexpectedly, the majority of the clades that we uncovered were negatively associated with decomposition rate. Most prominent among them was a large clade including 3,244 ASVs with consensus taxonomies affiliated to Gammaproteobacteria, Alphaproteobacteria, and Chloroflexi (Figure 4). This clade included genera that we have previously found in GFSs, including Polaromonas, Methylotenera, Rhodoferax, Leptothrix, and Thiobacillus, some of which are known for chemolithoautotrophy (Fodelianakis et al., 2021). This is also true for the other clades negatively associated with decomposition rate; they include Nitrospira, Patescibacteria, Sulfurifustis, Veruccomicrobiota, Planctomycetales, and Omnitrophales, with several of them being likely chemolithoautotrophic and previously reported from proglacial environments (Dindhoria et al., 2021; Fodelianakis et al., 2021; Wilhelm et al., 2013).
FIGURE 4.
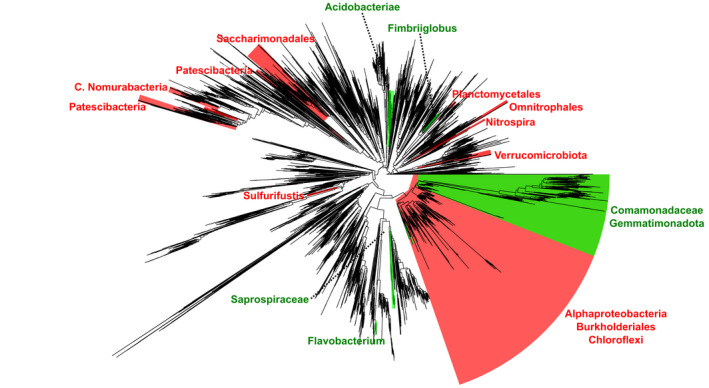
Multiple bacterial clades covary with decomposition rates in the sediment biofilm microbiome across 101 glacier‐fed streams. Phylogenetic tree showing significant positively (green) and negatively (red) associated phylogenetic clades with decomposition after controlling for the effect of the sampled region. Consensus taxonomies are shown next to each clade. For the large green clade on the right the outgroup was the adjacent large red clade. For all other clades, the outgroup was the rest of the phylogeny
Notably, three clades nested within the largest negatively associated clade were concomitantly increasing in abundance with increasing decomposition rates. The consensus taxonomy of the largest among them was affiliated to Comamonadaceae and Gemmatimonadota (1,427 ASVs) and the taxonomies of the other two to Burkholderiales (53 ASVs) and Ellin6067 (59 ASVs). The taxonomic similarities between the negatively associated clade and the nested clades could suggest ecological differences among phylogenetically close relative taxa as we previously found in GFSs in New Zealand (Fodelianakis et al., 2021). This could explain the contrasting patterns within the large phylogenetic clade that is negatively associated with decomposition rate and also agrees with our previous observation that, overall, taxa therein are decreasing both in diversity and in abundance with increasing sediment chlorophyll a content (Fodelianakis et al., 2021).
Because of the collinearity between decomposition rate and chlorophyll a (Figure 3a), we cannot unequivocally disentangle the effects of either on the bacterial clades. Nevertheless, our findings suggest that the microbiome transits from a “gray” to a “green” food web depending on primary production. In GFSs with sparse primary production, often related to high glacier influence with elevated turbidity and unstable channels, chemolithotrophy fuels a “gray” food web, similar to the subglacial environment (Boyd et al., 2014; Dieser et al., 2014; Hamilton et al., 2013). In fact, some of the clades identified by phylofactorization, such as members of Comamonadaceae, may be constituents of this “gray” food web rather than actually suppressing decomposition. Members of Comamonadaceae are found in abundance within subglacial sediments globally (Vinšová et al., 2022), and interestingly, are also known to oxidize carbon‐monoxide in glacier surface sediments (i.e., cryoconite, Franzetti et al., 2016). As glacier influence on GFSs diminishes, their environment becomes more favorable to benthic photoautotrophs, which increasingly sustain a “green” food web and thereby creates niches for bacterial decomposers (i.e., heterotrophs). We therefore suggest, considering the space‐for‐time substitution across our studied GFSs, that food webs in GFSs may become “greener” as glaciers shrink, providing evidence toward earlier predictions for GFS ecosystems (Milner et al., 2017).
3.4. Relating extracellular enzymatic activity to the glacier‐fed stream microbiome
While phylofactorization identified bacterial clades related to decomposition across GFSs, we acknowledge that it does not necessarily unveil causal relationships. Therefore, we took advantage of the fact that EEAs underlie microbial decomposition and used gene‐centric metagenomics to relate EEAs to bacterial groups across a subset of 50 GFSs (Data S1). Analyzing the metagenomes, we found that the number of gene copies, normalized by housekeeping genes, overall mirrored the patterns of measured EEA, with the greatest number of gene copies found for LAP and AP (Figure S1a). The interpretation of gene abundances from metagenomic data comes with assumptions, particularly the relationship between gene abundance and gene expression; however, the relationship between EEA and normalized gene copies was relatively robust (Figure S1c), indicating that gene abundances may be at least partially related with their expression and activity.
To identify bacteria putatively involved with EEA production, we assigned normalized gene copies to their respective taxonomy at the class level. Compared with the relative abundance based on the prokaryotic 16S rRNA gene amplicons, we found disproportionate amounts of the AP gene copies linked to Alphaproteobacteria, AG and BG gene copies linked to Bacteroidia, and LAP gene copies linked to Gammaproteobacteria (Figure 5). Gammaproteobacteria had the greatest relative abundances in the GFS biofilms (Figure S2a), and given that a disproportionate amount of the LAP gene copies could also be mapped to Gammaproteobacteria, these two observations help to explain the dominance of LAP within the total EEA pool. In contrast, Alphaproteobacteria generally dominate the soil microbiome (Delgado‐García et al., 2018), with lower contributions of Gammaproteobacteria (i.e., former Betaproteobacteria), and the soil EEA pool generally has greater proportions of BG activity than in GFSs (Sinsabaugh et al., 2008). Alphaproteobacteria were also present in the GFS biofilms, although at lower relative abundances than Gammaproteobacteria (Figure S2a), and a disproportionate number of BG gene copies were linked to this class. These findings on various bacterial classes differentially contributing to the gene abundance of extracellular enzymes and hence differentially involved with OM decomposition corroborates the notion of ecological coherence of high phylogenetic ranks (e.g., classes; Philippot et al., 2010).
FIGURE 5.
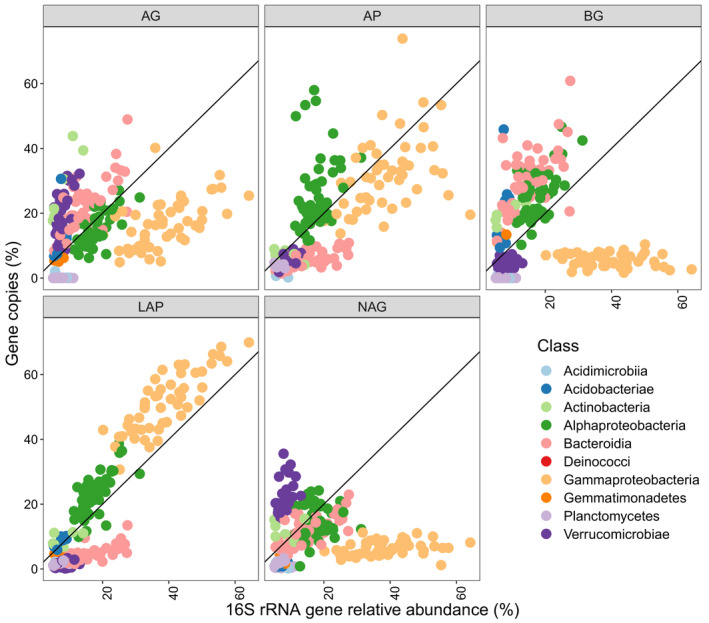
Bacterial classes differed in the number of gene copies for measured extracellular enzymatic activity that could be mapped through shotgun metagenomics. Relative abundance of the gene copies of a given enzyme mapped to bacterial classes are plotted against the relative abundance of a given bacterial class from 16S rRNA gene amplicon sequencing. A 1:1 line is provided for comparison. Extracellular enzymes include α‐1,4‐glucosidase, β‐1,4‐glucosidase, leucine aminopeptidase, β‐1,4‐N‐acetylglucosaminidase, and acid (alkaline) phosphatase
In the enzyme allocation model as used here (Equation 5), proportionately greater activities of C‐acquiring extracellular enzymes may lead to higher decomposition rates. Thus, groups disproportionately encoding for AG and BG, namely Bacteroidia and Alphaprotoebacteria, might disproportionately influence decomposition through their enzyme production. Indeed, Saprospiraceae, a member of Bacteroidia, showed a positive relationship with decomposition in the phylofactorization analysis, as did Acidobacteria. Meanwhile, Gammaproteobacteria showed an overall negative association with decomposition rate in the same analysis, and very few AG and BG gene copies could be traced to this class. Given the high prevalence of Gammaprotebacteria and the lower relative abundances of Bacteroidia and Alphaproteobacteria, it is, therefore, likely that modeled decomposition is disproportionately driven by bacterial clades constituting a minority of these bacterial communities.
While decomposition may be at least partially determined by the differential contributions of individual bacterial classes, variability in decomposition may also arise through the responses of individual taxa to the resource gradient (e.g., chlorophyll a). For example, LAP, despite being the dominant EEA, was less sensitive to changes in the chlorophyll a gradient (slope from regression = 0.25, R 2 = 0.19) than AG and BG, both of which strongly increased with increasing chlorophyll a (slopes = 0.51 and 0.55, R 2 = 0.41 and 0.54, respectively, Figure S4). These differences in response indicate that more AG and BG are produced per unit of increasing chlorophyll a than LAP, but whether these differences in activity are responses of individual taxa to changing environmental conditions via their enzyme production, community‐level shifts over the same gradient, or both, remains to be resolved.
To conclude, we have presented evidence that glacier shrinkage increases decomposition rates in 101 GFSs worldwide. Our findings highlight the role of primary production for decomposition, thereby expanding current understanding on stream OM decomposition, which has been largely based on ecosystems subsidized by terrestrial vascular plant material. Phylofactorization and metagenomics collectively allowed us to pinpoint bacterial clades that are putatively involved in decomposition. The potential existence of a fungal shunt mediating between primary producers and decomposition in GFSs seems plausible given that primary producers constitute the prime source of OM in GFSs but certainly warrants further investigations. Ultimately, our findings suggest that glacier shrinkage induces a shift of GFS ecosystem energetics from a “gray” food web sustained by chemolithoautotrophy to a “green” food web sustained by photoautotrophy.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
TJB conceived the project, and along with TJK, GM, HP, SF, PP, SBB, and PW designed the experiment. TJK, HP, MT, VS, MS, and MS conducted the fieldwork. TJK, HP, SF, PP, MT, VS, JB, MS, MS, SBB, RM, DD, and ND executed the laboratory work. TJK, GM, HP, SF, MB, and LE analyzed the data, and TJK, GM, HP, SF, LE, MB, SBB, PW, and TJB interpreted the results. TJK and TJB wrote the paper with editorial input from all authors.
Supporting information
Supplementary Material
Data S1
Data S2
ACKNOWLEDGMENTS
We thank the many students and technicians that assisted us with lab work and other analyses, including Emmy Marie Oppliger, Eline Grégoire, Ben Therrien, Maxwell Bergström, Florian Bielser, Dilan Resch, and Thierry Demierre. We also thank Alex Washburne for his help with the phylofactorization analysis and comments on an early draft of the manuscript. Finally, we are grateful to Scott Hotaling and one anonymous reviewer for their comments which greatly improved the manuscript. Open access funding provided by Ecole Polytechnique Federale de Lausanne.
Kohler, T. J. , Fodelianakis, S. , Michoud, G. , Ezzat, L. , Bourquin, M. , Peter, H. , Busi, S. B. , Pramateftaki, P. , Deluigi, N. , Styllas, M. , Tolosano, M. , de Staercke, V. , Schön, M. , Brandani, J. , Marasco, R. , Daffonchio, D. , Wilmes, P. , & Battin, T. J. (2022). Glacier shrinkage will accelerate downstream decomposition of organic matter and alters microbiome structure and function. Global Change Biology, 28, 3846–3859. 10.1111/gcb.16169
Funding information
This research was supported by The NOMIS Foundation project “Vanishing Glaciers” to TJB. SBB was supported by the Synergia grant (CRSII5_180241: Swiss National Science Foundation) to TJB. PW is supported by the Luxembourg National Research Fund (FNR; PRIDE17/11823097). DD acknowledges the financial support of King Abdullah University and Technology (KAUST) through the baseline research fund.
Contributor Information
Tyler J. Kohler, Email: tyler.j.kohler@gmail.com.
Tom J. Battin, Email: tom.battin@epfl.ch.
DATA AVAILABILITY STATEMENT
All shotgun metagenomic sequence data are archived under the NCBI project number PRJNA733707, while New Zealand 16S samples are under the accession number PRJEB40567 in the European Nucleotide Archive, and the rest of the 16S and 18S amplicon data are under project PRJNA781406. Supplementary data, including stream characteristics and functional data, are available in the Zenodo data repository at https://doi.org/10.5281/zenodo.6367068. Code to reproduce the analyses are available at https://github.com/michoug/EEA_GlacierStream.
REFERENCES
- Acinas, S. G. , Sánchez, P. , Salazar, G. , Cornejo‐Castillo, F. M. , Sebastián, M. , Logares, R. , Royo‐Llonch, M. , Paoli, L. , Sunagawa, S. , Hingamp, P. , Ogata, H. , Lima‐Mendez, G. , Roux, S. , González, J. M. , Arrieta, J. M. , Alam, I. S. , Kamau, A. , Bowler, C. , Raes, J. , … Gasol, J. M. (2021). Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities. Communications Biology, 4(1). 10.1038/S42003-021-02112-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC a quality control tool for high throughput sequence data. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Anesio, A. M. , Lutz, S. , Chrismas, N. A. M. , & Benning, L. G. (2017). The microbiome of glaciers and ice sheets. npj Biofilms and Microbiomes, 3(1). 10.1038/s41522-017-0019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battin, T. J. , Besemer, K. , Bengtsson, M. M. , Romani, A. M. , & Packmann, A. I. (2016). The ecology and biogeochemistry of stream biofilms. Nature Reviews Microbiology, 14(4), 251–263. 10.1038/nrmicro.2016.15 [DOI] [PubMed] [Google Scholar]
- Battin, T. J. , Kaplan, L. A. , Findlay, S. , Hopkinson, C. S. , Marti, E. , Packman, A. I. , Newbold, J. D. , & Sabater, F. (2008). Biophysical controls on organic carbon fluxes in fluvial networks. Nature Geoscience, 1(2), 95–100. 10.1038/NGEO101 [DOI] [Google Scholar]
- Battin, T. J. , Kaplan, L. A. , Newbold, J. D. , & Hansen, C. M. E. (2003). Contributions of microbial biofilms to ecosystem processes in stream mesocosms. Nature, 426(6965), 439–442. 10.1038/nature02152 [DOI] [PubMed] [Google Scholar]
- Boix Canadell, M. , Gómez‐Gener, L. , Ulseth, A. J. , Clémençon, M. , Lane, S. N. , & Battin, T. J. (2021). Regimes of primary production and their drivers in Alpine streams. Freshwater Biology, 66(8), 1449–1463. 10.1111/FWB.13730 [DOI] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/BIOINFORMATICS/BTU170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen, E. , Rideout, J. R. , Dillon, M. R. , Bokulich, N. A. , Abnet, C. C. , Al‐Ghalith, G. A. , Alexander, H. , Alm, E. J. , Arumugam, M. , Asnicar, F. , Bai, Y. , Bisanz, J. E. , Bittinger, K. , Brejnrod, A. , Brislawn, C. J. , Brown, C. T. , Callahan, B. J. , Caraballo‐Rodríguez, A. M. , Chase, J. , … Caporaso, J. G. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37(8), 852–857. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd, E. S. , Hamilton, T. L. , Havig, J. R. , Skidmore, M. L. , & Shock, E. L. (2014). Chemolithotrophic primary production in a subglacial ecosystem. Applied and Environmental Microbiology, 80(19), 6146–6153. 10.1128/AEM.01956-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyero, L. , Pérez, J. , López‐Rojo, N. , Tonin, A. M. , Correa‐Araneda, F. , Pearson, R. G. , Bosch, J. , Albariño, R. J. , Anbalagan, S. , Barmuta, L. A. , Beesley, L. , Burdon, F. J. , Caliman, A. , Callisto, M. , Campbell, I. C. , Cardinale, B. J. , Casas, J. J. , Chará‐Serna, A. M. , Ciapała, S. , … Yule, C. M. (2021). Latitude dictates plant diversity effects on instream decomposition. Science Advances, 7(13). 10.1126/SCIADV.ABE7860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J. H. , Gillooly, J. F. , Allen, A. P. , Savage, V. M. , & West, G. B. (2004). Toward a metabolic theory of ecology. Ecology, 85(7), 1771–1789. 10.1890/03-9000 [DOI] [Google Scholar]
- Brown, S. P. , Olson, B. J. S. C. , & Jumpponen, A. (2015). Fungi and algae co‐occur in snow: An issue of shared habitat or algal facilitation of heterotrophs? Arctic, Antarctic, and Alpine Research, 47(4), 729–749. 10.1657/AAAR0014-071 [DOI] [Google Scholar]
- Busi, S. B. , Bourquin, M. , Fodelianakis, S. , Kohler, J. , Peter, H. , Pramateftaki, P. , Styllas, M. , Tolosano, M. , Staercke, D. , de Nies, L. , Marasco, R. , Daffonchio, D. , Wilmes, P. , Battin, T. J. , Group, S. E. , Biofilm, S. , Abdullah, K. , & Arabia, S. (2021). Genomic and metabolic adaptations of biofilms to ecological windows of opportunities in glacier‐fed streams. BioRxiv, 10.1101/2021.10.07.463499 [DOI] [PMC free article] [PubMed]
- Busi, S. B. , Pramateftaki, P. , Brandani, J. , Fodelianakis, S. , Peter, H. , Halder, R. , Wilmes, P. , & Battin, T. J. (2020). Optimised biomolecular extraction for metagenomic analysis of microbial biofilms from high‐mountain streams. PeerJ, 8, 1–20. 10.7717/peerj.9973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B. J. , Sankaran, K. , Fukuyama, J. A. , McMurdie, P. J. , & Holmes, S. P. (2016). Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Research, 5(3), 1492. 10.12688/f1000research.8986.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauvy‐Fraunié, S. , & Dangles, O. (2019). A global synthesis of biodiversity responses to glacier retreat. Nature Ecology and Evolution, 3(12), 1675–1685. 10.1038/s41559-019-1042-8 [DOI] [PubMed] [Google Scholar]
- Cleveland, C. C. , & Liptzin, D. (2007). C:N:P stoichiometry in soil: Is there a “Redfield ratio” for the microbial biomass? Biogeochemistry, 85(3), 235–252. 10.1007/s10533-007-9132-0 [DOI] [Google Scholar]
- Davidson, E. A. , & Janssens, I. A. (2006). Temperature sensitivity of soil carbon decomposition and feedbacks to climate change. Nature, 440(7081), 165–173. 10.1038/NATURE04514 [DOI] [PubMed] [Google Scholar]
- Delgado‐García, M. , Contreras‐Ramos, S. M. , Rodríguez, J. A. , Mateos‐Díaz, J. C. , Aguilar, C. N. , & Camacho‐Ruíz, R. M. (2018). Isolation of halophilic bacteria associated with saline and alkaline‐sodic soils by culture dependent approach. Heliyon, 4(11), e00954. 10.1016/j.heliyon.2018.e00954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieser, M. , Broemsen, E. L. J. E. , Cameron, K. A. , King, G. M. , Achberger, A. , Choquette, K. , Hagedorn, B. , Sletten, R. , Junge, K. , & Christner, B. C. (2014). Molecular and biogeochemical evidence for methane cycling beneath the western margin of the Greenland Ice Sheet. ISME Journal, 8(11), 2305–2316. 10.1038/ISMEJ.2014.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dindhoria, K. , Kumar, S. , & Kumar, R. (2021). Taxonomic and functional analysis of proglacial water bodies of Triloknath glacier ecosystem from North‐Western Himalayas. Ecological Informatics, 64. 10.1016/J.ECOINF.2021.101365 [DOI] [Google Scholar]
- Elser, J. J. , Wu, C. , González, A. L. , Shain, D. H. , Smith, H. J. , Sommaruga, R. , Williamson, C. E. , Brahney, J. , Hotaling, S. , Vanderwall, J. , Yu, J. , Aizen, V. , Aizen, E. , Battin, T. J. , Camassa, R. , Feng, X. , Jiang, H. , Lu, L. , Qu, J. J. , … Saros, J. E. (2020). Key rules of life and the fading cryosphere: Impacts in alpine lakes and streams. Global Change Biology, 26(12), 6644–6656. 10.1111/gcb.15362 [DOI] [PubMed] [Google Scholar]
- Enríquez, S. , Duarte, C. M. , & Sand‐Jensen, K. (1993). Patterns in decomposition rates among photosynthetic organisms: The importance of detritus C:N:P content. Oecologia, 94(4), 457–471. 10.1007/BF00566960 [DOI] [PubMed] [Google Scholar]
- Espeland, E. M. , Francoeur, S. N. , & Wetzel, R. G. (2001). Influence of algal photosynthesis on biofilm bacterial production and associated glucosidase and xylosidase activities. Microbial Ecology, 42(4), 524–530. 10.1007/s00248-001-1022-8 [DOI] [PubMed] [Google Scholar]
- Fell, S. C. , Carrivick, J. L. , Cauvy‐Fraunié, S. , Crespo‐Pérez, V. , Hood, E. , Randall, K. C. , Nicholass, K. J. M. , Tiegs, S. D. , Dumbrell, A. J. , & Brown, L. E. (2021). Fungal decomposition of river organic matter accelerated by decreasing glacier cover. Nature Climate Change, 11(4), 349–353. 10.1038/s41558-021-01004-x [DOI] [Google Scholar]
- Findlay, S. E. G. (2021). Organic matter decomposition. In K. C. Weathers, D. L. Strayer, & G. E. Likens (Eds.), Fundamentals of ecosystem science (2nd ed., pp. 81–102). Academic Press. 10.1016/B978-0-12-812762-9.00004-6 [DOI] [Google Scholar]
- Fodelianakis, S. , Washburne, A. D. , Bourquin, M. , Pramateftaki, P. , Kohler, T. J. , Styllas, M. , Tolosano, M. , De Staercke, V. , Schön, M. , Busi, S. B. , Brandani, J. , Wilmes, P. , Peter, H. , & Battin, T. J. (2021). Microdiversity characterizes prevalent phylogenetic clades in the glacier‐fed stream microbiome. ISME Journal. 10.1038/S41396-021-01106-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follstad Shah, J. J. , Kominoski, J. S. , Ardón, M. , Dodds, W. K. , Gessner, M. O. , Griffiths, N. A. , Hawkins, C. P. , Johnson, S. L. , Lecerf, A. , LeRoy, C. J. , Manning, D. W. P. , Rosemond, A. D. , Sinsabaugh, R. L. , Swan, C. M. , Webster, J. R. , & Zeglin, L. H. (2017). Global synthesis of the temperature sensitivity of leaf litter breakdown in streams and rivers. Global Change Biology, 23(8), 3064–3075. 10.1111/GCB.13609 [DOI] [PubMed] [Google Scholar]
- Francoeur, S. N. , & Biggs, B. J. F. (2006). Short‐term effects of elevated velocity and sediment abrasion on benthic algal communities. Hydrobiologia, 561(1), 59–69. 10.1007/s10750-005-1604-4 [DOI] [Google Scholar]
- Franzetti, A. , Tagliaferri, I. , Gandolfi, I. , Bestetti, G. , Minora, U. , Mayer, C. , Azzoni, R. S. , Diolaiuti, G. , Smiraglia, C. , & Ambrosini, R. (2016). Light‐dependent microbial metabolisms drive carbon fluxes on glacier surfaces. The ISME Journal, 10(12), 2984–2988. 10.1038/ismej.2016.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessner, M. O. , & Chauvet, E. (1994). Importance of stream microfungi in controlling breakdown rates of leaf litter. Ecology, 75(6), 1807–1817. 10.2307/1939639 [DOI] [Google Scholar]
- Glassman, S. I. , Weihe, C. , Li, J. , Albright, M. B. N. , Looby, C. I. , Martiny, A. C. , Treseder, K. K. , Allison, S. D. , & Martiny, J. B. H. (2018). Decomposition responses to climate depend on microbial community composition. Proceedings of the National Academy of Sciences of the United States of America, 115(47), 11994–11999. 10.1073/PNAS.1811269115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, R. O. , & Meyer, J. L. (1998). The trophic significance of bacteria in a detritus‐based stream food web. Ecology, 79(6), 1995–2012. 10.1890/0012-9658(1998)079%5B1995:TTSOBI%5D2.0.CO;2 [DOI] [Google Scholar]
- Hamilton, T. L. , Peters, J. W. , Skidmore, M. L. , & Boyd, E. S. (2013). Molecular evidence for an active endogenous microbiome beneath glacial ice. ISME Journal, 7(7), 1402–1412. 10.1038/ISMEJ.2013.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer, M. , Wymore, A. S. , Hungate, B. A. , Schwartz, E. , Koch, B. J. , & Marks, J. C. (2021). Microbes on decomposing litter in streams: Entering on the leaf or colonizing in the water? ISME Journal. 10.1038/S41396-021-01114-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, B. H. , Elonen, C. M. , Herlihy, A. T. , Jicha, T. M. , & Mitchell, R. M. (2017). A synoptic survey of microbial respiration, organic matter decomposition, and carbon efflux in U.S. streams and rivers. Limnology and Oceanography, 62, S147–S159. 10.1002/lno.10583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson, A. J. , & Ferguson, R. I. (1999). Fluvial suspended sediment transport from cold and warm‐based glaciers in Svalbard. Earth Surface Processes and Landforms, 24(11), 957–974. 10.1002/(SICI)1096-9837(199910)24:11<957:AID-ESP19>3.0.CO;2-J [DOI] [Google Scholar]
- Hood, E. , Battin, T. J. , Fellman, J. , O’Neel, S. , & Spencer, R. G. M. (2015). Storage and release of organic carbon from glaciers and ice sheets. Nature Geoscience, 8(2), 91–96. 10.1038/ngeo2331 [DOI] [Google Scholar]
- Horgby, Å. , Segatto, P. L. L. , Bertuzzo, E. , Lauerwald, R. , Lehner, B. , Ulseth, A. J. J. , Vennemann, T. W. W. , & Battin, T. J. J. (2019). Unexpected large evasion fluxes of carbon dioxide from turbulent streams draining the world’s mountains. Nature Communications, 10(1), 4888. 10.1038/s41467-019-12905-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. , Niu, B. , Gao, Y. , Fu, L. , & Li, W. (2010). CD‐HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics, 26(5), 680–682. 10.1093/BIOINFORMATICS/BTQ003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta‐Cepas, J. , Forslund, K. , Coelho, L. P. , Szklarczyk, D. , Jensen, L. J. , Von Mering, C. , & Bork, P. (2017). Fast genome‐wide functional annotation through orthology assignment by eggNOG‐mapper. Molecular Biology and Evolution, 34(8), 2115–2122. 10.1093/MOLBEV/MSX148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.‐L. , LoCascio, P. F. , Land, M. L. , Larimer, F. W. , & Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11(1), 119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen, D. , & Dangles, O. (2012). Environmental harshness and global richness patterns in glacier‐fed streams. Global Ecology and Biogeography, 21(6), 647–656. 10.1111/J.1466-8238.2011.00699.X [DOI] [Google Scholar]
- Klawonn, I. , van den Wyngaert, S. , Parada, A. E. E. , Arandia‐Gorostidi, N. , Whitehouse, M. J. J. , Grossart, H.‐P.‐P. , & Dekas, A. E. E. (2021). Characterizing the “fungal shunt”: Parasitic fungi on diatoms affect carbon flow and bacterial communities in aquatic microbial food webs. Proceedings of the National Academy of Sciences of the United States of America, 118(23), e2102225118. 10.1073/pnas.2102225118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , & Glöckner, F. O. O. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Research, 41(1), e1. 10.1093/nar/gks808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler, T. J. , Peter, H. , Fodelianakis, S. , Pramateftaki, P. , Styllas, M. , Tolosano, M. , de Staercke, V. , Schön, M. , Busi, S. B. , Wilmes, P. , Washburne, A. , & Battin, T. J. (2020). Patterns and drivers of extracellular enzyme activity in New Zealand glacier‐fed streams. Frontiers in Microbiology, 11(November). 10.3389/fmicb.2020.591465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, F. (2018). Trim Galore . Retrieved from https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- Kulichevskaya, I. S. , Ivanova, A. A. , Baulina, O. I. , Rijpstra, W. I. C. , Sinninghe Damsté, J. S. , & Dedysh, S. N. (2017). Fimbriiglobus ruber gen. Nov., sp. nov., a gemmata‐like planctomycete from Sphagnum peat bog and the proposal of gemmataceae fam. nov. International Journal of Systematic and Evolutionary Microbiology, 67(2), 218–224. 10.1099/IJSEM.0.001598 [DOI] [PubMed] [Google Scholar]
- Li, D. , Liu, C.‐M. , Luo, R. , Sadakane, K. , & Lam, T.‐W. (2015). MEGAHIT: An ultra‐fast single‐node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, 31(10), 1674–1676. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- Lowe, R. L. , & LaLiberte, G. D. (2017). Benthic stream algae: Distribution and structure. Methods in Stream Ecology: Third Edition, 1, 193–221. 10.1016/B978-0-12-416558-8.00011-1 [DOI] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.Journal, 17(1), 10. 10.14806/EJ.17.1.200 [DOI] [Google Scholar]
- McIlroy, S. J. , & Nielsen, P. H. (2014). The family saprospiraceae. The prokaryotes: Other major lineages of bacteria and the Archaea, 9783642389542, 863–889. 10.1007/978-3-642-38954-2_138 [DOI]
- Milner, A. M. , Khamis, K. , Battin, T. J. , Brittain, J. E. , Barrand, N. E. , Füreder, L. , Cauvy‐Fraunié, S. , Gíslason, G. M. , Jacobsen, D. , Hannah, D. M. , Hodson, A. J. , Hood, E. , Lencioni, V. , Ólafsson, J. S. , Robinson, C. T. , Tranter, M. , & Brown, L. E. (2017). Glacier shrinkage driving global changes in downstream systems. Proceedings of the National Academy of Sciences of the United States of America, 114(37), 9770–9778. 10.1073/pnas.1619807114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NASA/Meti/AIST/Japan Space Systems, and U.S./Japan ASTER Science Team . (2019). ASTER Global Digital Elevation Model V003 [Data set]. NASA EOSDIS Land Processes DAAC. 10.5067/ASTER/ASTGTM.003 [DOI]
- Philippot, L. , Andersson, S. G. E. , Battin, T. J. , Prosser, J. I. , Schimel, J. P. , Whitman, W. B. , & Hallin, S. (2010). The ecological coherence of high bacterial taxonomic ranks. Nature Reviews Microbiology, 8(7), 523–529. 10.1038/nrmicro2367 [DOI] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , Peplies, J. , & Glöckner, F. O. (2012). The SILVA ribosomal RNA gene database project: Improved data processing and web‐based tools. Nucleic Acids Research, 41(D1), D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing. Retrieved from https://www.r‐project.org/ [Google Scholar]
- Ren, Z. , Martyniuk, N. , Oleksy, I. A. , Swain, A. , & Hotaling, S. (2019). Ecological stoichiometry of the mountain cryosphere. Frontiers in Ecology and Evolution, 7(Sep). 10.3389/fevo.2019.00360 [DOI] [Google Scholar]
- Rigby, R. A. , Stasinopoulos, D. M. , & Lane, P. W. (2005). Generalized additive models for location, scale and shape. Journal of the Royal Statistical Society. Series C: Applied Statistics, 54(3), 507–554. 10.1111/J.1467-9876.2005.00510.X [DOI] [Google Scholar]
- Robinson, C. T. , & Gessner, M. O. (2000). Nutrient addition accelerates leaf breakdown in an alpine springbrook. Oecologia, 122(2), 258–263. 10.1007/PL00008854 [DOI] [PubMed] [Google Scholar]
- Rosemond, A. D. , Benstead, J. P. , Bumpers, P. M. , Gulis, V. , Kominoski, J. S. , Manning, D. W. P. , Suberkropp, K. , & Wallace, J. B. (2015). Experimental nutrient additions accelerate terrestrial carbon loss from stream ecosystems. Science, 347(6226), 1142–1145. 10.1126/SCIENCE.AAA1958 [DOI] [PubMed] [Google Scholar]
- Senga, Y. , Yabe, S. , Nakamura, T. , & Kagami, M. (2018). Influence of parasitic chytrids on the quantity and quality of algal dissolved organic matter (AOM). Water Research, 145, 346–353. 10.1016/J.WATRES.2018.08.037 [DOI] [PubMed] [Google Scholar]
- Singer, G. A. , Fasching, C. , Wilhelm, L. , Niggemann, J. , Steier, P. , Dittmar, T. , & Battin, T. J. (2012). Biogeochemically diverse organic matter in Alpine glaciers and its downstream fate. Nature Geoscience, 5(10), 710–714. 10.1038/NGEO1581 [DOI] [Google Scholar]
- Sinsabaugh, R. L. , Hill, B. H. , & Follstad Shah, J. J. (2009). Ecoenzymatic stoichiometry of microbial organic nutrient acquisition in soil and sediment. Nature, 462(7274), 795–798. 10.1038/nature08632 [DOI] [PubMed] [Google Scholar]
- Sinsabaugh, R. L. , Lauber, C. L. , Weintraub, M. N. , Ahmed, B. , Allison, S. D. , Crenshaw, C. , Contosta, A. R. , Cusack, D. , Frey, S. , Gallo, M. E. , Gartner, T. B. , Hobbie, S. E. , Holland, K. , Keeler, B. L. , Powers, J. S. , Stursova, M. , Takacs‐Vesbach, C. , Waldrop, M. P. , Wallenstein, M. D. , … Zeglin, L. H. (2008). Stoichiometry of soil enzyme activity at global scale. Ecology Letters, 11(11), 1252–1264. 10.1111/j.1461-0248.2008.01245.x [DOI] [PubMed] [Google Scholar]
- Sinsabaugh, R. L. , Manzoni, S. , Moorhead, D. L. , & Richter, A. (2013). Carbon use efficiency of microbial communities: Stoichiometry, methodology and modelling. Ecology Letters, 16(7), 930–939. 10.1111/ele.12113 [DOI] [PubMed] [Google Scholar]
- Sinsabaugh, R. L. , & Moorhead, D. L. (1994). Resource allocation to extracellular enzyme production: A model for nitrogen and phosphorus control of litter decomposition. Soil Biology and Biochemistry, 26(10), 1305–1311. 10.1016/0038-0717(94)90211-9 [DOI] [Google Scholar]
- Sinsabaugh, R. L. , & Shah, J. J. F. (2012). Ecoenzymatic stoichiometry and ecological theory. Annual Review of Ecology, Evolution, and Systematics, 43, 313–343. 10.1146/annurev-ecolsys-071112-124414 [DOI] [Google Scholar]
- Sinsabaugh, R. L. , Turner, B. L. , Talbot, J. M. , Waring, B. G. , Powers, J. S. , Kuske, C. R. , Moorhead, D. L. , & Shah, J. J. F. (2016). Stoichiometry of microbial carbon use efficiency in soils. Ecological Monographs, 86(2), 172–189. 10.1890/15-2110.1 [DOI] [Google Scholar]
- Sinsabaugh, R. L. , van Horn, D. J. , Shah, J. J. F. , & Findlay, S. (2010). Ecoenzymatic stoichiometry in relation to productivity for freshwater biofilm and plankton communities. Microbial Ecology, 60(4), 885–893. 10.1007/s00248-010-9696-4 [DOI] [PubMed] [Google Scholar]
- Stoeck, T. , Bass, D. , Nebel, M. , Christen, R. , Jones, M. D. M. , Breiner, H. W. , & Richards, T. A. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Molecular Ecology, 19(Suppl. 1), 21–31. 10.1111/J.1365-294X.2009.04480.X [DOI] [PubMed] [Google Scholar]
- Swift, D. A. , Nienow, P. W. , & Hoey, T. B. (2005). Basal sediment evacuation by subglacial meltwater: Suspended sediment transport from Haut Glacier d’Arolla, Switzerland. Earth Surface Processes and Landforms, 30(7), 867–883. 10.1002/ESP.1197 [DOI] [Google Scholar]
- Tapia‐Torres, Y. , Elser, J. J. , Souza, V. , & García‐Oliva, F. (2015). Ecoenzymatic stoichiometry at the extremes: How microbes cope in an ultra‐oligotrophic desert soil. Soil Biology and Biochemistry, 87, 34–42. 10.1016/j.soilbio.2015.04.007 [DOI] [Google Scholar]
- Tiegs, S. D. , Costello, D. M. , Isken, M. W. , Woodward, G. , McIntyre, P. B. , Gessner, M. O. , Chauvet, E. , Griffiths, N. A. , Flecker, A. S. , Acuña, V. , Albariño, R. , Allen, D. C. , Alonso, C. , Andino, P. , Arango, C. , Aroviita, J. , Barbosa, M. V. M. , Barmuta, L. A. , Baxter, C. V. , … Zwart, J. A. (2019). Global patterns and drivers of ecosystem functioning in rivers and riparian zones. Science Advances, 5(1), eaav0486. 10.1126/sciadv.aav0486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehlinger, U. , Robinson, C. T. , Hieber, M. , & Zah, R. (2010). The physico‐chemical habitat template for periphyton in alpine glacial streams under a changing climate. Hydrobiologia, 657(1), 107–121. 10.1007/s10750-009-9963-x [DOI] [Google Scholar]
- Vinšová, P. , Kohler, T. J. , Simpson, M. J. , Hajdas, I. , Yde, J. C. , Falteisek, L. , Žárský, J. D. , Yuan, T. , Tejnecký, V. , Mercl, F. , Hood, E. , & Stibal, M. (2022). The biogeochemical legacy of arctic subglacial sediments exposed by glacier retreat. Global Biogeochemical Cycles, 36(3). 10.1029/2021GB007126 [DOI] [Google Scholar]
- von Lützow, M. , & Kögel‐Knabner, I. (2009). Temperature sensitivity of soil organic matter decomposition—What do we know? Biology and Fertility of Soils, 46(1), 1–15. 10.1007/s00374-009-0413-8 [DOI] [Google Scholar]
- Washburne, A. D. , Silverman, J. D. , Leff, J. W. , Bennett, D. J. , Darcy, J. L. , Mukherjee, S. , Fierer, N. , & David, L. A. (2017). Phylogenetic factorization of compositional data yields lineage‐level associations in microbiome datasets. PeerJ, 5(2), e2969. 10.7717/peerj.2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburne, A. D. , Silverman, J. D. , Morton, J. T. , Becker, D. J. , Crowley, D. , Mukherjee, S. , David, L. A. , & Plowright, R. K. (2019). Phylofactorization: A graph partitioning algorithm to identify phylogenetic scales of ecological data. Ecological Monographs, 89(2), e01353. 10.1002/ecm.1353 [DOI] [Google Scholar]
- Webster, J. R. , & Benfield, E. F. (1986). Vascular plant breakdown in freshwater ecosystems. Annual Review of Ecology and Systematics, 17, 567–594. 10.1146/ANNUREV.ES.17.110186.003031 [DOI] [Google Scholar]
- Webster, J. R. , & Meyer, J. L. (1997). Organic matter budgets for streams: A synthesis. Journal of the North American Benthological Society, 16(1), 141–161. 10.2307/1468247 [DOI] [Google Scholar]
- Wilhelm, L. , Singer, G. A. , Fasching, C. , Battin, T. J. , & Besemer, K. (2013). Microbial biodiversity in glacier‐fed streams. ISME Journal, 7(8), 1651–1660. 10.1038/ismej.2013.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, D. E. , & Salzberg, S. L. (2014). Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biology, 15(3), R46. 10.1186/gb-2014-15-3-r46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcroft, B. J. (2020). CoverM . Retrieved from https://github.com/wwood/CoverM
- Yarza, P. , Yilmaz, P. , Panzer, K. , Glöckner, F. O. , & Reich, M. (2017). A phylogenetic framework for the kingdom Fungi based on 18S rRNA gene sequences. Marine Genomics, 36, 33–39. 10.1016/j.margen.2017.05.009 [DOI] [PubMed] [Google Scholar]
- Zah, R. , & Uehlinger, U. (2001). Particulate organic matter inputs to a glacial stream ecosystem in the Swiss Alps. Freshwater Biology, 46(12), 1597–1608. 10.1046/J.1365-2427.2001.00847.X [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data S1
Data S2
Data Availability Statement
All shotgun metagenomic sequence data are archived under the NCBI project number PRJNA733707, while New Zealand 16S samples are under the accession number PRJEB40567 in the European Nucleotide Archive, and the rest of the 16S and 18S amplicon data are under project PRJNA781406. Supplementary data, including stream characteristics and functional data, are available in the Zenodo data repository at https://doi.org/10.5281/zenodo.6367068. Code to reproduce the analyses are available at https://github.com/michoug/EEA_GlacierStream.