

Abstract
Rare disease patients are more likely to receive a rapid molecular diagnosis nowadays thanks to the wide adoption of next‐generation sequencing. However, many cases remain undiagnosed even after exome or genome analysis, because the methods used missed the molecular cause in a known gene, or a novel causative gene could not be identified and/or confirmed. To address these challenges, the RD‐Connect Genome‐Phenome Analysis Platform (GPAP) facilitates the collation, discovery, sharing, and analysis of standardized genome‐phenome data within a collaborative environment. Authorized clinicians and researchers submit pseudonymised phenotypic profiles encoded using the Human Phenotype Ontology, and raw genomic data which is processed through a standardized pipeline. After an optional embargo period, the data are shared with other platform users, with the objective that similar cases in the system and queries from peers may help diagnose the case. Additionally, the platform enables bidirectional discovery of similar cases in other databases from the Matchmaker Exchange network. To facilitate genome‐phenome analysis and interpretation by clinical researchers, the RD‐Connect GPAP provides a powerful user‐friendly interface and leverages tens of information sources. As a result, the resource has already helped diagnose hundreds of rare disease patients and discover new disease causing genes.
Keywords: data sharing, data standardization, diagnostics, genome analysis, NGS, patient matchmaking, rare diseases
The RD‐Connect Genome‐Phenome Analysis Platform (GPAP) is a scalable and interoperable online system which facilitates the collation, analysis, interpretation and sharing of integrated genome‐phenome datasets, with a particular focus on RD case diagnosis and novel gene discovery. It is free to use for all noncommercial members of the rare disease research community.
1. INTRODUCTION
Recent estimates suggest that between 3.5% and 7% of the world's population is affected by a rare disease (RD) at some point in their lifetime, placing significant social, emotional, and economic burden on society (Nguengang Wakap et al., 2020; https://ec.europa.eu/health/non_communicable_diseases/rare_diseases_en). Over the last decade the advent of cost‐effective massively parallel shotgun DNA sequencing, often referred to as next generation sequencing, has been a major boon to the study of rare Mendelian diseases. The wide‐scale application of exome sequencing, and more recently genome sequencing, to large numbers of cases with similar phenotypes has allowed researchers to identify many novel disease genes and variants, and provide molecular diagnoses to thousands of affected individuals (e.g., Fitzgerald et al., 2015; Retterer et al., 2016; Stranneheim et al., 2021; Turnbull et al., 2018; Wright, McRae, et al., 2018).
Nevertheless, even in specialist clinics, the majority of RDs will only be encountered once, if ever, by primary care physicians, and the heterogeneous, and sometimes dynamic, nature of symptoms with which many RD patients present means that correct diagnosis of the underlying disease poses a significant challenge. Unfortunately, this often leads to patients and their families undergoing a diagnostic odyssey whereby they receive a number of possible diagnoses over the course of several years, before a definitive diagnosis is eventually reached. This can result in delayed and/or missed opportunities for counseling, prevention, and treatment (Boycott et al., 2017).
There are a variety of bottlenecks that impede rapid resolution of RD cases through the application of next generation sequencing. One is the collection of the data itself, which is time consuming and requires the work of many skilled individuals, from the medical practitioners who examine the patient, record their phenotype and extract samples, to the specialized laboratory staff who perform DNA sequencing, and the bioinformaticians who process the data to identify variants, supported by high performance computing infrastructures. The ensuing challenge is to identify the one or two disease‐causing variants out of the millions found in each human genome. This requires the blending of information from many different data sources to aid variant interpretation, ideally using user‐friendly systems that can efficiently help clinical researchers achieve this goal. As scientific knowledge constantly grows and methodology improves over time, it is beneficial to keep the phenotypic and genomic data properly organized to facilitate review of the data with manual or semi‐automated procedures on a regular basis. However, historically this task has been made difficult by the lack of widely adopted standardized formats and nomenclature for describing genomic and phenotypic data.
Furthermore, as many RDs are extremely rare, having an incidence of less than 1 per million (Nguengang Wakap et al., 2020), the means to locate other patients with similar phenotypes can greatly aid in reaching a diagnosis. Therefore, one way in which the pace of research can be accelerated, and hence the time to diagnosis for RD patients be reduced, is through the promotion of the sharing of standardized phenotypic descriptions and genomic data in line with FAIR (Findable, Accessible, Interoperable, Reusable) principles (Wilkinson et al., 2016). However, the sharing of RD patient data faces a number of barriers as outlined in Box 1.
Box 1. Barriers to sharing Rare Disease data.
RD data often remains effectively siloed, thus impeding research and diagnoses, for a number of reasons, which may differ for different groups of stakeholders (e.g., funders, clinicians, researchers, industry, etc.).
-
1)Motivational
-
A.Culture of protecting research data to reduce the chance of prior publication by competing peers.
-
B.Concerns about intellectual property that could be derived from the data by third parties.
-
C.Cost of making and maintaining the data interoperable, discoverable, and accessible.
-
D.Lack of incentives for sharing data.
-
E.Concerns regarding quality of one's own data.
-
A.
-
2)Logistical
-
A.Data may not be findable. Clinical researchers often do not know which other professionals have similar cases.
-
B.Clinicians lack the time and resources to fully describe their own cases in a standardized format which would facilitate sharing and automated analysis.
-
C.Language barriers ‐ RD case descriptions may be written in different languages, and thus time consuming and difficult to compare.
-
D.Relevant information may have to be compiled from different sources (e.g. electronic health records, project databases, local files, etc.) requiring time consuming effort.
-
A.
-
3)Technical
-
A.Data may not be discoverable and/or accessible. Relevant data is often stored across different servers/databases which are not freely accessible outwith the institution to which they belong.
-
B.Processes to request access to data may not be clear, or too cumbersome.
-
C.Data may not be interoperable. Where common standards are not widely implemented, even in cases where communication is permitted, data is often stored in incompatible formats which are not machine readable, e.g. free‐form patient descriptions in electronic health records.
-
D.Sequencing data is large in size, of the order of 10–100 Gigabytes of raw data per individual. Thus it requires highly specialized knowledge, tools, and computing resources to manage, process, analyze, interpret, store, and distribute.
-
A.
-
4)Ethical and Legal
-
A.Data may not be reusable for example, if there is not an appropriate legal basis for processing in place, such as informed consent, permitting such usage.
-
B.Patient data is inherently private, and thus sharing must be limited, and in line with national and international legal restrictions for example, the European General Data Protection Regulation (GDPR), to prevent undesired re‐identification.
-
A.
In recent years, there has been much effort within the RD community towards overcoming these barriers, in particular with respect to the standardization of methodology and terminology for recording phenotypic descriptions, and the responsible processing and sharing of genomic data. These ongoing efforts include initiatives such as the human phenotype ontology (HPO, Köhler et al., 2021), the orphanet rare disease ontology (ORDO, Nguengang Wakap et al., 2020), ClinVar (Landrum et al., 2020), the American College of Medical Genetics (ACMG) standards for variant interpretation (Richards et al., 2015), Matchmaker Exchange (MME, Philippakis et al., 2015; Boycott et al., 2022), Genomics England PanelApp (Martin et al., 2019), and work along many different lines within the Global Alliance for Genomics and Health (GA4GH). All these efforts, and many others, will hopefully enable “all people living with a RD to receive an accurate diagnosis, care, and available therapy within one year of coming to medical attention,” which is the ambitious vision of the International Rare Disease Research Consortium (IRDiRC) for 2027 (Austin et al., 2018).
The primary goal of the European Union FP7 funded RD‐Connect project was to accelerate RD research by connecting databases, patient registries, biobanks, and clinical bioinformatics data through an intuitive user‐friendly platform to simplify the analysis of RD cases within a collaborative framework (Lochmüller et al., 2018; Thompson et al., 2014). One of the key assets developed in this project to overcome many of the barriers highlighted in Box 1 was the RD‐Connect Genome‐Phenome Analysis Platform (RD‐Connect GPAP), which is now an IRDiRC recognized resource (Lochmüller et al., 2017). It was designed from the ground up with leadership from RD clinicians involved in diagnosis and gene discovery in their own patient cohorts, giving it a unique perspective focused on the bottlenecks they face in their own research and diagnostic practice. The platform is free to use for RD researchers and clinicians who have the appropriate legal basis, such as informed consent, to share their participants’ (affected individuals and relatives) genomic and phenotypic data with other registered users. The RD‐Connect GPAP has been used as the primary analysis tool in a number of large European projects and is involved in many ongoing projects and initiatives including Solve‐RD (https://cordis.europa.eu/project/id/779257), the European Joint Program on Rare Diseases (EJP‐RD, https://cordis.europa.eu/project/id/951724), the Beyond One Million Genomes project (B1MG, https://cordis.europa.eu/project/id/951724), ELIXIR (https://elixir‐europe.org/), Matchmaker Exchange (MME), and the Global Alliance for Genomics and Health (GA4GH).
Here, we describe in detail the RD‐Connect GPAP, a scalable and interoperable online system which facilitates the collation, analysis, interpretation, and sharing of integrated genome‐phenome datasets, with a particular focus on RD case diagnosis and novel gene discovery. We provide an overview of the different modules that comprise the RD‐Connect GPAP, and detail how it facilitates filtering and prioritization of variants based upon both the phenotypic information provided and the incorporation of a wide variety of openly accessible annotations and tools. Further, we describe how case data can be shared in a controlled manner to support internal and external patient matchmaking, and how case resolution is recorded within the platform, in line with the ACMG guidelines. Finally, we highlight how the RD‐Connect GPAP has been successfully used in a number of European projects resulting in the diagnosis of hundreds of RD cases.
2. METHODS
2.1. User registration and data submission
Access to the RD‐Connect GPAP is free to all noncommercial members of the RD research community. To become an authorized user of the RD‐Connect GPAP, Principal Investigators, or equivalent, must indicate why they wish to access the platform, and sign the RD‐Connect Adherence Agreement and Code of Conduct (available via https://rd‐connect.eu/forms‐and‐guides/). Applications are then forwarded to the RD‐Connect GPAP Data Access Committee which consists of four leaders in the field of RDs. Upon approval, the Principal Investigator may then request access credentials for other members of their group, each of whom must also agree to the Code of Conduct. Additionally, researchers from countries outside the EU may have to sign a Data Transfer Agreement and a Data Access Agreement. The whole process is largely based on ethics research conducted within the RD‐Connect project (Mascalzoni et al., 2016).
Data submission is undertaken in three steps. First, the user creates a phenotypic record in the bespoke application PhenoStore, describing the case and any family members for which genomic data is available. In the second step, the submitter must provide a small amount of essential metadata to allow data processing to be undertaken correctly. This metadata describes the type of experiment performed for example, genomic library preparation and sequencing strategy, and to which participant the data belongs. These first two steps can either be performed directly within a user‐friendly graphical user interface or via bulk upload using MS Excel templates. In the final step, high‐speed transfer of the raw sequencing data, in standard formats (FASTQ, BAM, or CRAM) is facilitated via use of an Aspera server, provided by the Spanish academic and research network RedIris (https://www.rediris.es/). Furthermore, the RD‐Connect GPAP team can broker, upon agreement, the transfer of raw and processed data to the European Genome‐Phenome Archive (EGA, Lappalainen et al., 2015) to make them available to the broader research community. Upon request, data submitted to the RD‐Connect GPAP may be placed under embargo for up to 6 months, during which time only members of the submitting group have access to the data. Following expiry of the embargo period, the genomic data and limited phenotypic data become visible to all platform users.
2.2. Genomic data processing
Submitted raw sequencing data is processed through a benchmarked standard analysis pipeline optimized for the processing of short‐read data produced by Illumina and MGI platforms, which is currently the only type of raw genomic data the platform accepts, largely as described in Laurie et al. (2016), with only minor variations in the protocol associated to the experimental methodology used in order to minimize batch effects. The current RD‐Connect processing pipeline uses BWA‐MEM (version 0.7.8; Li, 2013) for alignment to the hs37d5 version of the human genome and GATK HaplotypeCaller (v3.6) for the detection of single nucleotide variants and short insertions and deletions in line with the GATK Best Practices workflow (Poplin et al., 2018). In addition to short variant identification, all runs of homozygosity in excess of 500kb in length are identified (Matalonga et al., 2020), and read depth calculated across the target region of interest to provide a measure of data quality for each individual experiment. Before any changes to the core pipeline being implemented, the output of the updated pipeline is benchmarked for validation against the genome sequencing data for the NA12878 sample from NIST/GIAB following the guidelines of Krusche et al. (2019), before full reanalysis starting with the raw data of all experiments in RD‐Connect using the new pipeline, to maintain internal consistency.
All detected variants which are covered by both a minimum of eight sequencing reads and having a genotype quality determined by GATK of at least 20 are uploaded to the GPAP. Nevertheless, the default filter recommendations are set to a minimum coverage of 10 reads and a genotype quality of 30 as our benchmarking has shown that the proportion of false‐positive calls below a read depth of 10 increased rapidly. Furthermore, it is in the user's hands to determine the fraction of reads that should support a heterozygote or homozygote alternative variant call, with defaults of 0.2–0.8 for a heterozygote, and >0.95 for a homozygote. The internal allele frequency for all variant positions in RD‐Connect is also calculated and this can be applied as a filter in the same way as the variant frequencies from gnomAD and the 1000 genomes project. This internal allele frequency is also shown in the results view allowing easy identification and removal of any common alignment and variant calling artifacts from the results. Finally, coverage metrics, for example mean/median depth of coverage, and proportion of target region of interest covered by 10 or 20 reads etc. for each experiment are available in the Data Management module, thus providing users with a measure of the overall quality of the data for the experiments undergoing analysis.
2.3. RD‐Connect GPAP architecture
The RD‐Connect GPAP variant storage and processing system have been built on fully scalable technologies. gVCF files output by the analysis pipeline are stored on a Hadoop File System (https://hadoop.apache.org/docs/r1.2.1/hdfs_user_guide.html) and the extract, transform and load process is leveraged by Apache Spark (https://spark.apache.org/) and Hail (https://hail.is/). The output of this process is loaded into an Elasticsearch cluster (https://www.elastic.co/elasticsearch/) which offers low‐latency response time to user queries.
The core functionalities of the platform are provided by three distinct modules: Data Management, PhenoStore, and Data Analysis, each of which consists of a server and client, all being connected to the same Identity Provider (Keycloak, https://www.keycloak.org/). The PhenoStore server is implemented in Python/Flask (https://flask.palletsprojects.com/), the Data Analysis server in Scala/Play2 (https://www.playframework.com/), and the Data Management module written in Python/Django (https://www.djangoproject.com/). The clients are all single‐page application implementations written in JavaScript and React (https://reactjs.org/). In addition to these core modules, other microservice modules have been developed such as the MME and Beacon V1 (Fiume et al., 2019) servers.
The RD‐Connect GPAP is hosted by the data centre of the Centro Nacional de Análisis Genómico (CNAG‐CRG, Barcelona, Spain). For security purposes, the Unix cluster nodes are not connected to the internet and only communicate to the outside world via a controlled proxy server. The platform stands behind a firewall which permits outbound requests from the internal nodes only if the host destination is on a white list, meanwhile inbound requests are all validated by a reverse proxy which then derives the request to the relevant service. The server and client libraries are continuously updated to the latest versions to minimize security vulnerabilities, and platform security is audited periodically by an external company to test for vulnerabilities: firstly, black box testing against the platform APIs is undertaken, and secondly, a security engineer is given standard user credentials and they attempt to upscale their permissions to gain access to information for which they do not have credentials (gray box testing). Any minor vulnerabilities identified are then immediately patched.
2.4. Data standards
To enable interoperability and to overcome language barriers, the RD‐Connect GPAP has been designed to use widely adopted and machine readable international and community standards and ontologies whenever possible. Within the PhenoStore module, patient descriptions are recorded using HPO, ORPHA, and OMIM (Amberger et al., 2015) terminology. Phenotypic records can be exported to the GA4GH approved Phenopackets file format (https://github.com/phenopackets/phenopacket‐schema), and family trees in PLINK PED format (Purcell et al., 2007). Genomic alignments and variants are stored and transferred (e.g. to the EGA) in GA4GH approved BAM/CRAM, and gVCF/VCF formats, respectively (https://www.ga4gh.org/genomic‐data‐toolkit/). Biological annotations, available in the Data Analysis module are provided by Ensembl Variant Effect Predictor (McLaren et al., 2016), and supplemented with data from other human genomics community resources such as ClinVar, gnomAD (Karczewski et al., 2020), and PanelApp. Data discovery and sharing are facilitated through the implementation of GA4GH Beacon V1 and MME APIs.
3. RESULTS
3.1. RD‐Connect GPAP implementation
From the user's perspective the RD‐Connect GPAP consists of 3 core modules (Figure 1):
-
(1)
The Data Management module, which facilitates the upload of raw sequencing data, together with associated metadata required for processing.
-
(2)
PhenoStore, an application for the storage and sharing of phenotypic descriptions of all participants (affected individuals and relatives), which uses machine readable ontologies and vocabularies such as HPO, ORDO, and OMIM.
-
(3)
The Genomic Analysis module, where researchers/clinicians analyze their own cases, and can discover and access data from cases shared by other researchers, to identify disease‐causing variants.
Figure 1.
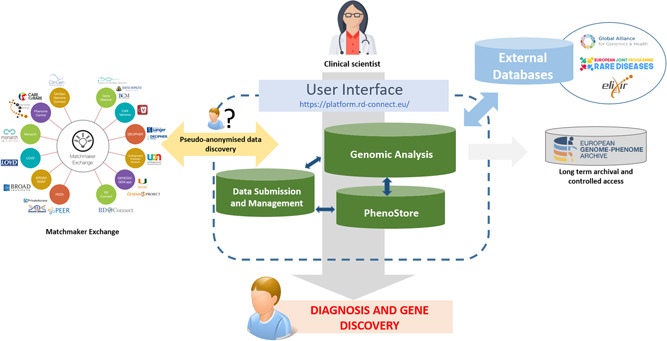
Flow of genome‐phenome data in the RD‐Connect Genome‐Phenome Analysis Platform. Clinical scientists submit their data to the RD‐Connect GPAP wherein it is processed through a standard analysis pipeline. The variants identified are returned to the user via a user‐friendly interface where they can undertake filtration and prioritization to diagnose their rare disease cases. This effort is supported by the integration of data from a large variety of external resources, and through live links via APIs to other resources. When an inconclusive but interesting result is found, patient matchmaking may be performed using the Matchmaker Exchange API to query other similar resources around the world
3.1.1. Data management
The Data Management module facilitates the upload of sequencing data and metadata and allows users to keep track of all data they have submitted. With this module, they can easily check for how many participants they have entered data into the system, which types of experiments are linked to these participants, which associated files were uploaded, and to which projects they pertain (Supp. Figure S1). They can also track the progress of their submission, data processing, and analysis status. Further, submitters can see which RD‐Connect GPAP users have made queries specifically on their genomic datasets.
3.1.2. PhenoStore
A requirement for collaboration in the RD‐Connect GPAP is that all submitted genomic data be accompanied by a corresponding pseudonymised phenotypic description of the individual from whom the sample was taken, which is recorded within the PhenoStore module developed primarily within the EJP‐RD. A deep phenotypic record for affected individuals can be generated using HPO terms, which describe observed patient signs and symptoms and, when informative, the absence of certain traits. HPO is a machine‐readable standardized vocabulary which aids in breaking language barriers, since each of the terms consists of a unique code linked to a description and additional information (e.g., synonyms, comments) which have been translated into several languages. HPO is continuously being updated, and new terms are added as necessary, and one of the successful mechanisms to update HPO, and broaden its usage, were workshops organized with disease experts from RD‐connect and Solve‐RD, among others (Kohler et al., 2021). Where the RD diagnosis is known at the time of submission, or upon case resolution, an ORPHA and/or OMIM identifier can be added to the record, together with a description of the causative variant(s). Each individual record is assigned to a specific family within the system, and a user‐friendly pedigree‐drawing tool facilitates the linking and addition of family members.
For projects focussing on a particular class of RD, customized templates are available in PhenoStore to accelerate data collation. In collaboration with Orphanet and members from several European Reference Networks (https://ec.europa.eu/health/ern_en), 20 templates based on the Genomics England RD data model (https://www.genomicsengland.co.uk/?wpdmdl=5500) have been implemented. These templates, which include diseases from all 24 European Reference Networks, promote data standardization and harmonization, by indicating which fields and HPO terms are most likely to be relevant for the appropriate RD class, though all HPOs remain available for selection. The templates may also include measurements that are not currently defined within the HPO, for example, the number of observed polyps in the case of tumor risk syndromes, or measurements of key molecular biomarkers. Together these provide a deep and meaningful description of the case which is easily understood by both human and machine alike. The latter is invaluable because it means that the phenotypic description can be directly applied by the user when undertaking variant prioritization within the Genomic Analysis module via the generation of real‐time in silico candidate gene panels through the integration of information from sources such as HPO, OMIM, DisGeNET (Piñero et al., 2020), and Exomiser (Smedley et al., 2015). Similarly, machine‐readable phenotypic descriptions are essential for patient matchmaking both within the RD‐Connect GPAP and worldwide via the MME network. Furthermore, data stored in PhenoStore can be exported to Phenopackets, a GA4GH open file standard for interoperability, transfer, and sharing of disease and phenotypic information. The RD‐Connect GPAP was one of the first testers and early adopters of Phenopackets, which are now extensively used within the Solve‐RD project as part of its data sharing and analysis strategy (manuscript in preparation).
3.1.3. Genomic analysis module
The Genomic Analysis module is the analytical core of the RD‐Connect GPAP, where researchers and clinicians analyze their own RD cases through the integration of the phenotypic information they have supplied, and the application of filters based on multiple annotations, tools and resources built into, or accessible from, the platform. The primary use case for the analysis is to discover disease‐causing variants in a submitted index case or family structure, and the system supports variant analysis in both a “diagnostic” paradigm (known variants or variants in known genes) and a “discovery” paradigm (potentially damaging variants in genes not previously associated with disease).
To begin an analysis, the user selects the samples they want to analyze and defines the mode of inheritance they wish to investigate. Following this, a range of filters can be applied within four broad categories: variant effect (McLaren et al., 2016) including presence in ClinVar; allele frequency described in control populations such as gnomAD and the 1000 Genomes Project (Genomes Project et al., 2015); predicted variant impact as determined by popular machine‐learning algorithms such as PolyPhen (Adzhubei et al., 2013) and CADD (Kircher et al., 2014); application of customized candidate gene lists, (see Table S1 for a detailed list of filtering options, and Figures S2 and S3 for screenshots). The user can add or remove filtering terms at will, and rerun their query, which will return updated results in seconds.
Summary results are displayed with color‐coding to highlight variants likely to be of particular interest, for example, variants recorded to be Likely Pathogenic or Pathogenic in ClinVar, and/or those predicted to be damaging by machine‐learning trained tools. To ease further exploration of variants of interest, hyperlinks to the appropriate record in popular genomic web servers are provided at both co‐ordinate and gene level for example, OMIM, Ensembl (Howe et al., 2021), gnomAD, the Human Gene Mutation database (Stenson et al., 2020), and the University of California at Santa Cruz Genome Browser (Kent et al., 2002). A full list of resources used in the platform is provided in Table S2.
Furthermore, the RD‐Connect GPAP provides fully detailed annotations for each variant in an extended results section, including the functional effect of the variant at the level of each Ensembl transcript, variant quality metrics, protein pathway, and interaction data, amongst others.
3.1.4. Advanced features
In addition to the more standard features described above, the RD‐Connect GPAP uniquely brings together information from a wide variety of sources which facilitate variant filtration, prioritization, and interpretation, beyond that provided by simple biological annotation. These include the ability to generate candidate gene lists on‐the‐fly via API connections to resources such as PanelApp, HPO, and DisGeNET. Long runs of homozygosity (>500kb) are identified in all experiments, and can be used as a filter within the platform, which is particularly useful when attempting to resolve cases in consanguineous progeny (Matalonga et al., 2020). Exomiser can be used to rank a shortlist of candidate variants using the HPO terms in the corresponding PhenoStore record, thus providing an accurate indication as to whether any variant is likely to be causal. Furthermore, hyperlinks to the appropriate page in resources which provide more detailed information for specific variants are provided, such as Varsome (Kopanos et al., 2019) for classification according to ACMG criteria. In combination, these features expedite the task of determining whether any variants returned by the initial filtering strategy are likely to be explanatory for the case under examination (Table 1). Tracking of the status of ongoing analysis of specific cases is also enabled, including time‐stamping, username recording, and the outcome of the analysis (e.g., solved case, negative case, variant under segregation analysis etc.). This information is accessible through the different RD‐Connect GPAP modules, facilitating case follow‐up and reporting.
Table 1.
Advanced features in the RD‐Connect Genome‐Phenome Analysis Platform for variant filtration, prioritization and interpretation
| Objective | Method | Tools/Resources |
|---|---|---|
| Variant filtration | Generate candidate gene lists on‐the‐fly via APIs | |
| Variant filtration | Identify variants in long runs of homozygosity | Runs of homozygosity >500 kb, 1 Mb, and 2 Mb in length are identified as described in Kancheva et al. (2016) |
| Variant filtration | Remove common variants | Filter by allele frequency from: |
| Variant prioritization | Score and rank list of candidate variants according to supplied HPO terms |
|
| Variant Iinterpretation | Hyperlinks to appropriate records in external resources |
|
| Variant interpretation | Classify according to ACMG Criteria | Link from variant to Varsome (Kopanos et al., 2019) |
| Search for variant in patient cohort | This internal search tool allows querying for specific genes or variants of interest across a cohort of accessible samples in the platform | |
| Tagging Variants | Users can tag variants in the platform and attribute a clinical significance, in accordance with the ACMG guidelines, for a specific patient and disorder. These tagged variants are visible to all other users and may be relevant for interpretation of their cases. | |
| Gene discovery | Internal data discovery |
|
| External data discovery |
Where initial analyses do not turn up any interesting variants from disease genes known to be associated with the phenotype of the case being investigated, the user may then shift to a gene discovery approach through more detailed investigation of variants that have passed the applied filters but are in unfamiliar genes. There are a number of features within the platform that facilitate this extended investigation. The simplest of these is the extended results view which indicates if genes are known to be involved in certain pathways, regulatory networks, or implicated in disease through annotations provided by databases such as Wikipathways, Reactome, and DisGeNet, and so on (see Table S2). A more advanced feature is the search‐across‐all tool which allows a user to search across all shared samples, or a user‐defined sub‐cohort of samples, to see if a particular variant, or a similar type of variant in the same gene is found in other affected individuals in the RD‐Connect GPAP, for example, a HIGH impact homozygous alternative variant in gene X. If such variants are commonly found in other cases, this would imply that the initial variant is less likely to be clinically relevant. However, if such variants are rarely found, but there is another case in the platform which has a somewhat similar phenotypic description, then this may suggest that the variant is worth pursuing further as it may represent a case of phenotypic expansion, or may indicate that one or other of the phenotypic descriptions is incomplete. When a good candidate is found in the latter case, the user has access to a contact button that sends a mail to the submitter of the newly identified case to initiate contact with both the user and the owner receiving a mail, as does the RD‐Connect helpdesk, allowing the researchers to compare notes and follow‐up at their own discretion. If the search‐across‐all proves fruitless, but a novel variant still appear highly interesting, then internal (within RD‐Connect) and external matchmaking is possible allowing the user to look for other cases, around the world, which have a similar phenotype and variants in the same gene (see Implementation of Matchmaker Exchange API section below).
3.1.5. Demonstration instance, training, and feedback
To provide potential new collaborators with the opportunity to test the system, and to facilitate training and workshops, a demonstration instance of the platform is available (https://playground.rd‐connect.eu/), which includes virtually all of the functionality available within the Genomic Analysis and PhenoStore modules. The genomic data therein comprises several trios based upon Illumina Platinum genome sequencing of the best‐characterized human genome in the world, the HapMap sample NA12878/HG001 and her parents, NA12891 and NA12892 (Eberle et al., 2017). The sequencing data were processed using the same analysis pipeline as for any other submitted sample but, before uploading the variants, causative variants from real cases were spiked into the gVCFs before upload to generate a variety of simulated RD trios. These are paired with corresponding simulated PhenoStore records with relevant HPO terms, allowing testers to attempt to diagnose these cases in the demo instance, as if they were their own RD cases. The demo instance has proven to be very useful for hands‐on training on the system itself and also for the human genomics community, as it has been used by thousands of attendees in over 30 events including students, researchers, healthcare providers and patients. We have recorded a number of the live webinars we have given, and furthermore have provided video tutorials on specific types of analyses that can be undertaken in the platform, all of which are available via the RD‐Connect YouTube channel (https://www.youtube.com/channel/UCwwcUPJZfyWGaW13Lvao7Ag).
3.2. Data sharing and collaboration in the RD‐Connect GPAP
An overarching goal of the RD‐Connect GPAP is to facilitate data sharing to promote rapid diagnosis. However, patient data is inherently sensitive in nature, and hence must be securely stored, and access controlled, in accordance with the consented use. Furthermore, all data sharing must be in compliance with the EU General Data Protection Regulation (GDPR, European Parliament and Council of European Union (2016) Regulation (EU) 2016/679; https://eur‐lex.europa.eu/legal‐content/EN/TXT/HTML/?uri=CELEX:32016R0679&from=EN), for which the RD‐Connect GPAP is regarded as a data controller.
To gain access to the RD‐Connect GPAP, interested parties must complete the registration form accessible via the platform home page (https://platform.rd‐connect.eu/), any queries regarding eligibility can be directed to the helpdesk (help@rd‐connect.eu). For users who do not wish to register, variant data can be queried in a limited fashion via the Beacon Network (https://beacon‐network.org/) thanks to the implementation of the GA4GH Beacon API. Newer versions of the Beacon API expected to be available in the near future may allow queries related to phenotypes and other clinical features. The variant and phenotypic data for all participants for whom matchmaking consent has been provided can be queried via partner APIs within the Matchmaker Exchange network.
The RD‐Connect GPAP is designed to facilitate data sharing and foster collaboration between RD researchers across the world in a number of ways:
-
1)
Query parameters can be saved through the generation of unique URLs, thus allowing users to return to their analyses at any point in the future, and share results easily with other authorized users in the system.
-
2)
Variants of interest can be tagged and user interpretation applied, which is then visible to all other users. Furthermore, when a user confirms a variant as disease causing within the platform, a similar set of information to that required by ClinVar is requested to provide the necessary context and evidence.
-
3)
The search‐across‐all feature allows users who have identified a variant of interest in a certain gene to search across all genomic datasets available to them within the RD‐Connect GPAP to attempt to discover participants with similar or identical variants. If they identify such a variant, a contact button allows them to contact the submitter of the relevant participants to discuss the candidate variants further.
-
4)
A combinatorial query on the participants’ information (ORPHA and/or HPO terms, and assigned European Reference Network) allows definition of in‐silico cohorts of experiments to which the search‐across‐all feature can be applied to focus the query more clearly.
-
5)
The RD‐Connect GPAP is included in the Beacon Network (https://beacon‐network.org/) via the implementation of the GA4GH Beacon API v1, which permits queries to determine if a particular variant is found in at least one of the experiments in the RD‐Connect GPAP. Likewise, any interesting variant found in the RD‐Connect GPAP can be immediately queried in other beacons through the Beacon Network.
3.2.1. Implementation of matchmaker exchange API
The MME API (Buske et al., 2015) has been implemented in the RD‐Connect GPAP for internal and external matchmaking. Internal usage allows matchmaking only between data sets within the platform, empowering users to identify similar cases both in terms of phenotype and the variants they carry, thus facilitating confirmation of causality and case resolution. External usage currently permits bidirectional communication with four MME nodes: PhenomeCentral (Osmond et al, 2022), Decipher (Foreman et al., 2022), GeneMatcher (Hamosh et al., 2022), and MyGene2 (https://mygene2.org/MyGene2/).
In order for a case in the RD‐Connect GPAP to be discoverable through the MME API, the submitting clinical researcher must actively provide permission. There are currently 5,736 cases available for matchmaking within the system, with 821 unique genes tagged. When a query is received by the RD‐Connect GPAP node, matches are sought and a score between 0 and 1 is calculated. The score consists of the sum of a genomic component and a phenotypic component, each of which range between 0 and 0.5. To compute the genomic score, the genes from the query are compared with genes that have variants tagged in the system as “Pathogenic,” “Likely Pathogenic” or “Uncertain Significance.” If there is a match of at least one gene, the genomic score is set to 0.5, otherwise it is set to 0. For the phenotypic score, the HPO terms from the request are used to compute similarity with matchable participants from the RD‐Connect GPAP using the UI similarity algorithm (Guo et al., 2006), which is divided by 2 to yield a score between 0 and 0.5. If the sum of the genomic and phenotypic components is greater than 0.2, the participant PhenoStore ID is returned in the response to the querying node together with the score, the corresponding HPOs, the matched gene when appropriate, and an internal RD‐Connect GPAP email address so that the querying researcher may initiate contact if deemed relevant.
RD‐Connect GPAP users may only initiate matchmaking requests for samples which have been submitted by their group and which have been cleared for participation in MME. If matchmaking has been permitted, a button with the MME logo is visible in the Data Analysis module (see Figure S2). Upon clicking on the MME button a form is displayed which is automatically filled with the relevant information from the Genomic Analysis module and PhenoStore, including the HPOs from the participant and any genes for which a variant has been tagged. The user must specify to which MME node they wish to make a request, and may add other HPOs and genes if desired before sending the request. If the receiving node returns matching results, the user is presented with a list of matches which may include HPOs, a gene, or both, depending upon which node is returning a match, and a button to contact the data custodian of the potential match by email.
As further follow‐up is in the hands of the clinical researchers involved, we are often unaware as to whether a match was conclusive in providing a diagnosis for one or both cases involved, or whether the cases were found to be too dissimilar upon further investigation. Nevertheless, we are aware of a number of recent success stories. For example, matchmaking involving samples from the Consequitur project has led to the identification of 11 novel candidate genes in a variety of rare neurological cases presenting with a range of phenotypes including brain malformation, intellectual disability with epilepsy, Charcot‐Marie Tooth syndrome, myopathy, and ataxia (Kurul et al., 2021; Richard et al., 2021). While the Consequitur cases were Turkish in origin, the cases matched included individuals living in the UK and Egypt. We are also aware of successful matchmaking involving cases living as far apart as Australia and Holland, and a number of matches between Italy, Spain, and the UK.
3.3. Impact on rare disease diagnosis
Since its initial release in 2015, the RD‐Connect GPAP has been involved in the primary analysis and reanalysis of thousands of RD cases. Unfortunately, it is not possible to provide an accurate count of how many cases have been solved since the successful resolution of cases is not always updated in the system or communicated by the submitter. The total number of shared genome‐phenome datasets has grown steadily and now stands at over 22,000, including affected participants and relatives. Currently, the total number of affected participants is 13,676, of which 5,736 are discoverable through MME. The majority of submitted datasets have come from large European consortia projects, but in some cases, data from just a few sequenced individuals or trios from independent research groups have been uploaded and cases solved (e.g., Oktay et al., 2020; Owen et al., 2018; Permanyer et al., 2020; Topf et al., 2020; Yaramis et al., 2020). Before the initiation of the Solve‐RD project, the three largest projects which had used the system for primary analysis were NeurOmics (https://cordis.europa.eu/project/id/305121/reporting, n = 1,117 data sets), the Biobanking and Biomolecular Research Infrastructure—Large Prospective Cohorts (BBMRI‐LPC, https://cordis.europa.eu/project/id/313010/reporting) exome sequencing project (n = 757), and the Consequitur project (n = 626, Kurul et al., 2021). Currently, the Solve‐RD project is using the RD‐Connect GPAP as a key resource for the collection and reanalysis of 19,000 previously unsolved genome‐phenome datasets (Zurek et al., 2021). In addition, the RD‐Connect GPAP is further developed and made available to all European researchers through the EJP‐RD Virtual Platform, and also as an ELIXIR‐ES resource.
The EU FP7 NeurOmics project, funded in parallel to RD‐Connect, focussed on the investigation of cases of rare neuromuscular and neurodegenerative diseases, and was the first large project to upload genome‐phenome data to the RD‐Connect GPAP and use it as one of their analysis tools. This consisted of a total of 671 exome sequencing datasets, and 446 genome sequencing datasets, from a total of 375 families. By the end of the project in December 2017, 104 families (28%) had been solved, and over 100 new gene‐disease relationships described (Lochmüller et al., 2018).
The BBMRI‐LPC WES project was an open call offering exome sequencing for transnational European projects with the objective of diagnosing RD cases and enriching RD sample biobanks. Seventeen projects were selected focussing on different classes of RD for example, rare eye diseases, epileptic encephalopathies, mitochondrial disease, disorders of glycosylation. A total of 757 samples from 379 families were analyzed using the RD‐Connect GPAP, with in‐depth analysis undertaken by an in‐house clinical genomics expert and/or external domain experts, with variants of interest returned to the submitters for follow‐up and confirmation. To date 117 of 273 (43%) fully analyzed families have been solved (Ivanovski et al., 2020; Maini et al., 2018, 2021; Saredi et al., 2019; Urreizti et al., 2020).
The Consequitur project focussed on progeny from consanguineous marriages who have a RD. 626 samples from 190 families were analyzed using the RD‐Connect GPAP. Here the implementation of an algorithm to detect runs of homozygosity was particularly valuable as most causative variants in such families are likely to be found in regions identical by descent (Matalonga et al., 2020). To date, 137 families (72%) have been solved using the GPAP, with the majority of identified causative variants being found within long runs of homozygosity as expected, and further strong candidate variants are still undergoing follow‐up (Kurul et al., 2021).
One of the principal goals of the EU Horizon 2020 Solve‐RD project is to diagnose RD cases which have previously undergone exome sequencing and analysis, but remain unsolved, through systematic and extended reanalyses and, where resolution remains inconclusive, extending analyses to beyond the exome approaches, including short and long‐read genome sequencing, transcriptomics, metabolomics and so on. To this end, 19,000 genome‐phenome datasets are in the process of being collected, processed, and analyzed in the RD‐Connect GPAP. Programmatic reanalysis of the first ~4,000 families resulted in rapid resolution of 120 families (de Boer et al., 2021; Matalonga et al., 2021; Schüle et al., 2021; te Paske et al., 2021; Topf et al., 2021). These numbers, which have increased substantially since publication, were achieved through the identification of causative variants that had either been missed by the original variant filtering strategies undertaken by submitting centers, or were dismissed as being unimportant at the time due to a lack of supporting evidence, but for which further information has come to light linking the gene to the phenotype of the case in the intervening period. It is anticipated that further cases from this cohort will be resolved through regular repetition of this automated process once further new gene‐disease relationships are established by the RD community. The RD‐Connect GPAP is interoperable with the rest of the Solve‐RD data infrastructure, which includes additional components for data discovery, sharing, and analysis. Of note, all developments of general use which are triggered by the Solve‐RD project become available to all other RD‐Connect GPAP users.
4. DISCUSSION
The RD‐Connect GPAP has been facilitating collaborative analysis of genome‐phenome data since 2015 by enabling data discovery and sharing, with the aim of accelerating diagnosis and novel gene discovery. This has been possible thanks to collaboration within large EU funded projects and international initiatives, which have both fueled the development of the system and have shared thousands of datasets, but the platform is also used by many smaller research groups, such as those based in hospitals. Currently, the platform has 695 active users from 348 research groups across 185 different institutions located in 36 countries.
The powerful range of functionalities provided by the system, the sharing of data, and the expertize of registered peers, allow clinical researchers to enhance their analyses and/or to give a second chance to data for which previous analyses were inconclusive. In some cases, a good candidate variant has been found by the submitter shortly after commencing analysis. In other cases, the diagnosis has been achieved through patient matchmaking either within the platform or with other nodes from MME. In the latter cases, the patients are typically from different countries, making it unlikely that the referring clinicians would ever have come into contact with one another to discuss their cases were it not for the matchmaking mechanism.
To enable data discovery and data sharing, the RD‐Connect GPAP has addressed many of the barriers that traditionally block such activities, beginning with careful and ongoing evaluation of ethical and legal issues, which were initiated during the RD‐Connect project. This demonstrated that effective genome‐phenome data sharing across countries is possible if the proper procedures and measures are applied. To facilitate data entry and enable interoperability, the platform relies on widely adopted tools and standards for read alignment, variant detection and annotation, and the recording of phenotype descriptions and diseases. This is particularly relevant within the platform since the phenotypic information combined with the information from commonly used RD genomics resources can be directly applied to the objective of identifying disease‐causing variants. Taken together with the functionalities that address technical barriers by enabling users to analyze data without needing their own infrastructure or bioinformatics know‐how, the RD‐Connect GPAP helps clinicians and researchers to rapidly and accurately identify which variants are of potential interest. Furthermore, the PhenoStore module facilitates data transfer and sharing as records can be easily exported, by individual patient or in bulk, into Phenopackets, CSV, or PDF formats. The advantage of using Phenopackets is that the data can then be automatically used or imported by tools compliant with this GA4GH standard. As a result, phenotypic and genomics data can be re‐used within the RD‐Connect GPAP or beyond if it is shared within a consortium infrastructure, and/or submitted to a resource enabling controlled access such as the EGA. The Solve‐RD project provides a good demonstration of how data can be successfully shared and used within a consortium to undertake further analyses beyond those which are currently possible within the RD‐Connect GPAP (Zurek et al., 2021).
The RD‐Connect GPAP is undergoing continuous development thanks to participation in several initiatives, and in response to feedback from its users. In this sense, surveys are conducted in many of the workshops to ask the attendees about the system's utility and suggestions for improvement. In many cases, conversations with users help define the specifications of new features and, when deemed valuable, a few users are kindly asked to beta‐test the new developments before they are released. Current topics under evaluation and/or development include a new user interface, and the inclusion of results from other types of variants and mechanisms beyond single nucleotide variants and short insertions and deletions, such as copy number variants, short tandem repeats, mitochondrial heteroplasmy, and also through the implementation of features to facilitate the analysis and interpretation of the noncoding part of the genome, such as SQUIRLS for splicing variants (Danis et al., 2021). The identification of variants might be improved via re‐processing of the data with newer or updated tools on a more comprehensive genome reference. In a similar way, updated annotations and new discoveries may help diagnose historic cases. However, reinterpretation of all data becomes increasingly challenging as more data accumulates. Therefore, it is crucial to apply periodic programmatic reanalysis mechanisms (Machini et al., 2019; Wright, FitzPatrick, et al., 2018) as has already proven successful in Solve‐RD (Matalonga et al., 2021). Finally, to increase the chance of finding relevant data between resources, the RD‐Connect GPAP is exploring connections to additional MME nodes and, together with many other partners, is testing and further developing enhanced mechanisms of data discovery, which may allow more complex queries than those enabled with the current production versions of the Beacon and MME APIs. Some of these methods should facilitate the provision of aggregated or summary information to non‐registered users to let them know, for example, if there is data from individuals with a certain disease or phenotype (or even the number of available datasets of each kind).
We are constantly looking for novel methods to aid in RD diagnosis, and with this in mind we are evaluating how other –omics data (e.g., transcriptomics, proteomics, metabolomics, epigenomics, etc.) could be incorporated in a systematic and user‐friendly manner to help improve diagnostic yield. Similarly, as long‐read sequencing becomes more common, we will also develop workflows to accept and process such data. It would also be useful to expand connections to other types of data source, such as patient registries, biobanks and biomaterials resources, all of which are included, among others, within the EJP‐RD Virtual Platform for RD research of which the RD‐Connect GPAP is also a key resource.
In summary, the RD‐Connect GPAP has become a key resource in the EU for clinical research and has helped advance the field of genome‐phenome data discovery, sharing, and analysis. In the years to come, we expect it to grow and develop further, continuing to accelerate diagnosis and novel gene discovery and contributing to the IRDiRC Vision for 2027. In this sense, the most tangible impact of the RD‐Connect GPAP so far has been to the hundreds of patients, and their families, for whom it has contributed to ending their diagnostic odyssey.
CONFLICTS OF INTEREST
Orion Buske is the CEO of PhenoTips. Jules Jacobsen is a consultant to Congenica. All other authors declare no conflict of interest.
Supporting information
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
ACKNOWLEDGMENTS
The authors would like to thank Corina Antonescu, Giulia Babbi, Gareth Baynam, Matthew Bellgard, Rudy Benfredj, André Blavier, Niklas Blomberg, Gisèle Bonne, Kym Boycott, Anthony J. Brookes, Mike Brudno, Han G. Brunner, Kate Bushby, Alberto Calderone, Salvador Capella‐Gutierrez, Claudio Carta, Jessica X. Chong, Daniel Danis, Hugh Dawkins, Richarda de Voer, Sergiu Dumitriu, Kornelia Ellwanger, Teresinha Evangelista, Chris Evelo, Angelo Facchiano, Peter Fish, Julia Foreman, Mallory Freeberg, Antonio Fuentes, Laura Furlong, Marta Girdea, Ada Hamosh, Mark Hanauer, Nicoline Hoogerbrugge, Alexander Hosichen, Lawrence Hunter, Ben Hutton, Joseph Irwin, Thomas Keane, Bartha Knoppers, Finlay Macrae, Anna Marabotti, Ellen McDonagh, Per Nilsson, José Luis Oliveira, Annie Olry, Justin Paschall, Ernesto Picardi, Janet Piñero, Roberto Preste, Giuseppe Profiti, Heidi Rehm, Marco Roos, Ferran Sanz, Gary Saunders, Franz Schaefer, Rebecca Schüle, Serena Scollen, Nara Sobreira, Lincoln Stein, Matthis Synofzik, Domenica Taruscio, Coline Thomas, Alfonso Valencia, Gert‐Jan van Ommen, Angelo Varvara, Joanna Vella, Alain Verloes, Lisenka Vissers, Mary Wang, Petra Wilson, Luca Zalatnai, Birte Zurek, the ELIXIR Rare Disease Community members, and the RD‐Connect Principal Investigators and users. They have submitted data, analyzed it and reported back findings, provided expertize on diseases, phenotypes and genome analysis, enabled interoperability and connection of tools, tested the system, provided support, disseminated results, provided training, suggested improvements, and provided technical or conceptual advice and insightful discussions. We acknowledge the support of the developers of PhenoTips, which was used in the past by RD‐Connect and NeurOmics as the primary tool to collate phenotypic data. We would also like to thank the leaders and members of the Instituto Nacional de Bioinformática (INB) and ELIXIR for their support and collaboration throughout the years. RD‐Connect (RD‐Connect, an integrated platform connecting registries, biobanks, and clinical bioinformatics) received funding from the Seventh Framework (FP7) Programme of the European Union under grant agreement No 305444. Data were analyzed using the RD‐Connect GPAP, which received funding from EU projects Solve‐RD, EJP‐RD (grant numbers H2020 779257, H2020 825575), Instituto de Salud Carlos III (Grant numbers PT13/0001/0044, PT17/0009/0019; Instituto Nacional de Bioinformática, INB), ELIXIR‐EXCELERATE (Grant number EU H2020 #676559) and ELIXIR Implementation Studies (Remote real‐time visualization of human rare disease genomics data (RD‐Connect) stored at the EGA ELIXIR. 2017‐2018; ELIXIR IT‐2017‐INTEGRATION, Rare Disease Infrastructure ELIXIR, 2019‐2020 and the Beacon ELIXIR, 2019‐2021). The RD‐Connect GPAP has leveraged developments funded through project VEIS (001‐P‐001647 co‐financed by the European Regional Development Fund of the European Union in the framework of the Operational Program FEDER of Catalonia 2014‐2020 with the support of the Secretaria d'Universitats i Recerca del Departament d'Empresa i Coneixement de la Generalitat de Catalunya) and URD‐Cat (PERIS SLT002/16/00174, Departament de Salut, Generalitat de Catalunya). The research leading to these results has received funding from Consequitur (Newton Fund UK/Turkey, MR/N027302/1), BBMRI‐LPC (EU FP7 #313010), NeurOmics (EU FP7 #305121), the Economic Development Department of the Navarra Government (Grant number 001114112017), the European Reference Network for Rare Neurological Diseases (Project ID number 739510) and NIH, National Institute of Child Health and Human Development (1R01HD103805‐01). We acknowledge the support of the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) to the EMBL partnership, the Centro de Excelencia Severo Ochoa, and the CERCA Program/Generalitat de Catalunya. We also acknowledge the support of the Generalitat de Catalunya through Departament de Salut and Departament d'Empresa i Coneixement and Co‐financing by the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) with funds from the European Regional Development Fund (ERDF) corresponding to the 2014‐2020 Smart Growth Operating Program. HL receives support from the Canadian Institutes of Health Research (Foundation Grant FDN‐167281), the Canadian Institutes of Health Research and Muscular Dystrophy Canada (Network Catalyst Grant for NMD4C), the Canada Foundation for Innovation (CFI‐JELF 38412), and the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950‐232279).
Laurie, S. , Piscia, D. , Matalonga, L. , Corvó, A. , Fernández‐Callejo, M. , Garcia‐Linares, C. , Hernandez‐Ferrer, C. , Luengo, C. , Martínez, I. , Papakonstantinou, A. , Picó‐Amador, D. , Protassio, J. , Thompson, R. , Tonda, R. , Bayés, M. , Bullich, G. , Camps‐Puchadas, J. , Paramonov, I. , Trotta, J.‐R. , … Beltran, S. (2022). The RD‐Connect Genome‐Phenome Analysis Platform: Accelerating diagnosis, research, and gene discovery for rare diseases. Human Mutation, 43, 717–733. 10.1002/humu.24353
Steven Laurie and Davide Piscia contributed equally to this study.
DATA AVAILABILITY STATEMENT
As this study describes a software platform, there is no data to be made available. However, we confirm that free access to the platform will be granted to any bona fide researcher with an interest in rare diseases.
REFERENCES
- Adzhubei, I. , Jordan, D. M. , & Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics, 7(suppl. 76), 7. 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger, J. S. , Bocchini, C. A. , Schiettecatte, F. , Scott, A. F. , & Hamosh, A. (2015). OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an Online catalog of human genes and genetic disorders. Nucleic Acids Research, 43, 789–798. 10.1093/nar/gku1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin, C. P. , Cutillo, C. M. , Lau, L. , Jonker, A. H. , Rath, A. , Julkowska, D. , Thomson, D. , Terry, S. F. , de Montleau, B. , Ardigò, D. , Hivert, V. , Boycott, K. M. , Baynam, G. , Kaufmann, P. , Taruscio, D. , Lochmüller, H. , Suematsu, M. , Incerti, C. , Draghia‐Akli, R. , … International Rare Diseases Research Consortium (2018). Future of rare diseases research 2017–2027: An IRDiRC perspective. Clinical and Translational Science, 11, 21–27. 10.1111/cts.12500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. M. , Azzariti, D. R. , Hamosh, A. , & Rehm, H. L. (2022). Seven years since the launch of the Matchmaker Exchange: The evolution of genomic matchmaking. Human Mutation. 10.1002/humu.24373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. M. , Rath, A. , Chong, J. X. , Hartley, T. , Alkuraya, F. S. , Baynam, G. , Brookes, A. J. , Brudno, M. , Carracedo, A. , den Dunnen, J. T. , Dyke, S. , Estivill, X. , Goldblatt, J. , Gonthier, C. , Groft, S. C. , Gut, I. , Hamosh, A. , Hieter, P. , Höhn, S. , … Lochmüller, H. (2017). International cooperation to enable the diagnosis of all rare genetic diseases. American Journal of Human Genetics, 100, 695–705. 10.1016/j.ajhg.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buske, O. J. , Schiettecatte, F. , Hutton, B. , Dumitriu, S. , Misyura, A. , Huang, L. , Hartley, T. , Girdea, M. , Sobreira, N. , Mungall, C. , & Brudno, M. (2015). The Matchmaker Exchange API: Automating patient matching through the exchange of structured phenotypic and genotypic profiles. Human Mutation, 36(10), 922–927. 10.1002/humu.22850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clima, R. , Preste, R. , Calabrese, C. , Diroma, M. A. , Santorsola, M. , Scioscia, G. , Simone, D. , Shen, L. , Gasparre, G. , & Attimonelli, M. (2017). HmtDB 2016: Data update, a better performing query system and human mitochondrial DNA haplogroup predictor. Nucleic Acids Research, 45(D1), 698. 10.1093/nar/gkw1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danis, D. , Jacobsen, J. O. B. , Carmody, L. C. , Gargano, M. A. , McMurry, J. A. , Hegde, A. , Haendel, M. A. , Valentini, G. , Smedley, D. , & Robinson, P. N. (2021). Interpretable prioritization of splice variants in diagnostic next‐generation sequencing. American Journal of Human Genetics, 108(9), 1564–1577. 10.1016/j.ajhg.2021.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, E. , Ockeloen, C. W. , Matalonga, L. , Horvath, R. , Rodenburg, R. J. , Coenen, M. J. H. , Janssen, M. , Henssen, D. , Gilissen, C. , Steyaert, W. , Paramonov, I. , Trimouille, A. , Kleefstra, T. , Verloes, A. , & Vissers, L. E. L. M. (2021). A MT‐TL1 variant identified by whole exome sequencing in an individual with intellectual disability, epilepsy, and spastic tetraparesis. European Journal of Human Genetics, 29, 1359–1368. 10.1038/s41431-021-00900-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders. (2015). Large‐scale discovery of novel genetic causes of developmental disorders. Nature, 519, 223–228. 10.1038/nature14135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle, M. A. , Fritzilas, E. , Krusche, P. , Källberg, M. , Moore, B. L. , Bekritsky, M. A. , Iqbal, Z. , Chuang, H. Y. , Humphray, S. J. , Halpern, A. L. , Kruglyak, S. , Margulies, E. H. , McVean, G. , & Bentley, D. R. (2017). A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three‐generation 17‐member pedigree. Genome Research, 27(1), 157–164. 10.1101/gr.210500.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat, A. , Jupe, S. , Matthews, L. , Sidiropoulos, K. , Gillespie, M. , Garapati, P. , Haw, R. , Jassal, B. , Korninger, F. , May, B. , Milacic, M. , Roca, C. D. , Rothfels, K. , Sevilla, C. , Shamovsky, V. , Shorser, S. , Varusai, T. , Viteri, G. , Weiser, J. , … D'Eustachio, P. (2018). The reactomepathway knowledgebase. Nucleic Acids Research, 46(D1):649. 10.1093/nar/gkx1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiume, M. , Cupak, M. , Keenan, S. , Rambla, J. , de la Torre, S. , Dyke, S. , Brookes, A. J. , Carey, K. , Lloyd, D. , Goodhand, P. , Haeussler, M. , Baudis, M. , Stockinger, H. , Dolman, L. , Lappalainen, I. , Törnroos, J. , Linden, M. , Spalding, J. D. , Ur‐Rehman, S. , … Scollen, S. (2019). Federated discovery and sharing of genomic data using Beacons. Nature Biotechnology, 37, 220–224. 10.1038/s41587-019-0046-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman, J., Brent, S., Perrett, D., Bevan, A. P., Hunt, S. E., Cunningham, F., Hurles, M. E., & Firth, H. V. (2022). DECIPHER: Supporting the interpretation and sharing of rare disease phenotype‐linked variant data to advance diagnosis and research. Human Mutation. 10.1002/humu.24340 [DOI] [PMC free article] [PubMed]
- Genomes Project, C. , Auton, A. , Brooks, L. D. , Durbin, R. M. , Garrison, E. P. , Kang, H. M. , Korbel, J. O. , Marchini, J. L. , Mccarthy, S. , Mcvean, G. A. , & Abecasis, G. R. (2015). A global reference for human genetic variation. Nature, 526, 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, X. , Liu, R. , Shriver, C. D. , Hu, H. , & Liebman, M. N. (2006). Assessing semantic similarity measures for the characterization of human regulatory pathways. Bioinformatics, 22(8), 967–973. 10.1093/bioinformatics/btl042 [DOI] [PubMed] [Google Scholar]
- Hamosh, A., Wohler, E., Martin, R., Griffith S., Rodrigues, E., Antonescu, C., Doheny, K. F., Valle, D., & Sobreira, N. (2022). The impact of GeneMatcher on international data sharing and collaboration. Human Mutation. 10.1002/humu.24350 [DOI] [PMC free article] [PubMed]
- Howe, K. L. , Achuthan, P. , Allen, J. , Allen, J. , Alvarez‐Jarreta, J. , Amode, M. R. , Armean, I. M. , Azov, A. G. , Bennett, R. , Bhai, J. , Billis, K. , Boddu, S. , Charkhchi, M. , Cummins, C. , da Rin Fioretto, L. , Davidson, C. , Dodiya, K. , El Houdaigui, B. , Fatima, R. , … Flicek, P. (2021). Ensembl 2021. Nucleic Acids Research, 49(D1), 884. 10.1093/nar/gkaa942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovski, I. , Caraffi, S. G. , Magnani, E. , Rosato, S. , Pollazzon, M. , Matalonga, L. , Piana, S. , Nicoli, D. , Baldo, C. , Bernasconi, S. , Frasoldati, A. , Zuffardi, O. , & Garavelli, L. (2020). Alazami syndrome: The first case of papillary thyroid carcinoma. Journal of Human Genetics, 65(2), 133–141. 10.1038/s10038-019-0682-5 [DOI] [PubMed] [Google Scholar]
- Kancheva, D. , Atkinson, D. , De Rijk, P. , Zimon, M. , Chamova, T. , Mitev, V. , Yaramis, A. , Maria Fabrizi, G. , Topaloglu, H. , Tournev, I. , Parma, Y. , Battaloglu, E. , Estrada‐Cuzcano, A. , & Jordanova, A. (2016). Novel mutations in genes causing hereditary spastic paraplegia and Charcot‐Marie‐Tooth neuropathy identified by an optimized protocol for homozygosity mapping based on whole‐exome sequencing. Genetics in Medicine, 18(6), 600–607. 10.1038/gim.2015.139 [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , England, E. M. , Seaby, E. G. , Kosmicki, J. A. , … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W. J. , Sugnet, C. W. , Furey, T. S. , Roskin, K. M. , Pringle, T. H. , Zahler, A. M. , & Haussler, A. D. (2002). The human genome browser at UCSC. Genome Research, 12(6), 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, S. , Gargano, M. , Matentzoglu, N. , Carmody, L. C. , Lewis‐Smith, D. , Vasilevsky, N. A. , Danis, D. , Balagura, G. , Baynam, G. , Brower, A. M. , Callahan, T. J. , Chute, C. G. , Est, J. L. , Galer, P. D. , Ganesan, S. , Griese, M. , Haimel, M. , Pazmandi, J. , Hanauer, M. , … Robinson, P. N. (2021). The human phenotype ontology in 2021. Nucleic Acids Research, 49(D1), 1207. 10.1093/nar/gkaa1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanos, C. , Tsiolkas, V. , Kouris, A. , Chapple, C. E. , Albarca Aguilera, M. , Meyer, R. , & Massouras, A. (2019). VarSome: The human genomic variant search engine. Bioinformatics, 35(11), 1978–1980. 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusche, P. , Trigg, L. , Boutros, P. C. , Mason, C. E. , De La Vega, F. M. , Moore, B. L. , Gonzalez‐Porta, M. , Eberle, M. A. , Tezak, Z. , Lababidi, S. , Truty, R. , Asimenos, G. , Funke, B. , Fleharty, M. , Chapman, B. A. , Salit, M. , & Zook, J. M. (2019). Best practices for benchmarking germline small‐variant calls in human genomes. Nature Biotechnology, 37, 555–560. 10.1038/s41587-019-0054-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurul, S. H. , Oktay, Y. , Töpf, A. , Szabó, N. Z. , Güngör, S. , Yaramis, A. , Sonmezler, E. , Matalonga, L. , Yis, U. , Schon, K. , Paramonov, I. , Kalafatcilar, İ. P. , Gao, F. , Rieger, A. , Arslan, N. , Yilmaz, E. , Ekinci, B. , Edem, P. P. , Aslan, M. , … Horvath, R. (2021). High diagnostic rate of trio exome sequencing in consanguineous families with neurogenetic diseases. Brain, 2021, awab395. 10.1093/brain/awab395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum, M. J. , Chitipiralla, S. , Brown, G. R. , Chen, C. , Gu, B. , Hart, J. , Hoffman, D. , Jang, W. , Kaur, K. , Liu, C. , Lyoshin, V. , Maddipatla, Z. , Maiti, R. , Mitchell, J. , O'Leary, N. , Riley, G. R. , Shi, W. , Zhou, G. , Schneider, V. , … Kattman, B. L. (2020). ClinVar: improvements to accessing data. Nucleic Acids Research, 48(D1), 835. 10.1093/nar/gkz972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen, I. , Almeida‐King, J. , Kumanduri, V. , Senf, A. , Spalding, J. D. , Ur‐Rehman, S. , Saunders, G. , Kandasamy, J. , Caccamo, M. , Leinonen, R. , Vaughan, B. , Laurent, T. , Rowland, F. , Marin‐Garcia, P. , Barker, J. , Jokinen, P. , Torres, A. C. , De Argila, J. R. , Llobet, O. M. , … Flicek, P. (2015). The European Genome‐phenome Archive of human data consented for biomedical research. Nature Genetics, 47, 692–695. 10.1038/ng.3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie, S. , Fernandez‐Callejo, M. , Marco‐Sola, S. , Trotta, J. R. , Camps, J. , Chacón, A. , Espinosa, A. , Gut, M. , Gut, I. , Heath, S. , & Beltran, S. (2016). From wet‐lab to variations: concordance and speed of bioinformatics pipelines for whole genome and whole exome sequencing. Human Mutation, 37, 1263–1271. 10.1002/humu.23114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. ArXiv Preprint ArXiv, 3. arXiv:1303.3997.
- Lochmüller, H. , Badowska, D. M. , Thompson, R. , Knoers, N. V. , Aartsma‐Rus, A. , Gut, I. , Wood, L. , Harmuth, T. , Durudas, A. , Graessner, H. , & Schaefer, F. (2018). RD‐Connect, NeurOmics and EURenOmics: Collaborative European initiative for rare diseases. European Journal of Human Genetics, 26, 778–785. 10.1038/s41431-018-0115-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochmüller, H. , Le Cam, Y. , Jonker, A. H. , Lau, L. P. , Baynam, G. , Kaufmann, P. , Lasko, P. , Dawkins, H. J. , Austin, C. P. , & Boycott, K. M. (2017). “IRDiRC Recognized Resources”: A new mechanism to support scientists to conduct efficient, high‐quality research for rare diseases. European Journal of Human Genetics, 25, 162–165. 10.1038/ejhg.2016.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machini, K. , Ceyhan‐Birsoy, O. , Azzariti, D. R. , Sharma, H. , Rossetti, P. , Mahanta, L. , Hutchinson, L. , McLaughlin, H. , Green, R. C. , Lebo, M. , & Rehm, H. L. (2019). Analyzing and reanalyzing the genome: Findings from the MedSeq Project. American Journal of Human Genetics, 105(1), 177–188. 10.1016/j.ajhg.2019.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini, I. , Caraffi, S. G. , Peluso, F. , Valeri, L. , Nicoli, D. , Laurie, S. , Baldo, C. , Zuffardi, O. , & Garavelli, L. (2021). Clinical manifestations in a girl with naa10‐related syndrome and genotype–phenotype correlation in females. Genes, 12(6), 900. 10.3390/genes12060900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini, I. , Farnetti, E. , Caraffi, S. G. , Ivanovski, I. , De Bernardi, M. L. , Gelmini, C. , Pollazzon, M. , Rosato, S. , Laurie, S. , Matalonga, L. , Baldo, C. , & Garavelli, L. (2018). A novel CCND2 mutation in a previously reported case of megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus. Neuropediatrics, 49, 222–224. 10.1055/s-0038-1641722 [DOI] [PubMed] [Google Scholar]
- Martin, A. R. , Williams, E. , Foulger, R. E. , Leigh, S. , Daugherty, L. C. , Niblock, O. , Leong, I. , Smith, K. R. , Gerasimenko, O. , Haraldsdottir, E. , Thomas, E. , Scott, R. H. , Baple, E. , Tucci, A. , Brittain, H. , de Burca, A. , Ibañez, K. , Kasperaviciute, D. , Smedley, D. , … McDonagh, E. M. (2019). PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nature Genetics, 51, 1560–1565. 10.1038/s41588-019-0528-2 [DOI] [PubMed] [Google Scholar]
- Mascalzoni, D. , Dove, E. S. , Rubinstein, Y. , Dawkins, H. J. S. , Kole, A. , McCormack, P. , Woods, S. , Riess, O. , Schaefer, F. , Lochmüller, H. , Knoppers, B. M. , & Hansson, M. (2016). Erratum: international Charter of principles for sharing bio‐specimens and data. European Journal of Human Genetics, 24(7), 1096. 10.1038/ejhg.2015.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalonga, L. , Hernández‐Ferrer, C. , Piscia, D. , Solve‐RD SNV‐Indel Working, G , Schüle, R. , Synofzik, M. , Töpf, A. , Vissers, L. , de Voer, R. , Solve‐Rd, D. , Solve‐Rd, D. , Solve‐Rd, D. , Solve‐Rd, D. , Tonda, R. , Laurie, S. , Fernandez‐Callejo, M. , Picó, D. , Garcia‐Linares, C. , Papakonstantinou, A. , … Solve‐RD, C. (2021). Solving patients with rare diseases through programmatic reanalysis of genome‐phenome data. European Journal of Human Genetics, 29, 1337–1347. 10.1038/s41431-021-00852-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalonga, L. , Laurie, S. , Papakonstantinou, A. , Piscia, D. , Mereu, E. , Bullich, G. , Thompson, R. , Horvath, R. , Pérez‐Jurado, L. , Riess, O. , Gut, I. , van Ommen, G.‐J. , Lochmüller, H. , & Beltran, S. (2020). Improved diagnosis of rare disease patients through systematic detection of runs of homozygosity. The Journal of Molecular Diagnostics, 22, 1205–1215. 10.1016/j.jmoldx.2020.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren, W. , Gil, L. , Hunt, S. E. , Riat, H. S. , Ritchie, G. R. S. , Thormann, A. , Flicek, P. , & Cunningham, F. (2016). The ensembl variant effect predictor. Genome Biology, 17(1), 122. 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguengang Wakap, S. , Lambert, D. M. , Olry, A. , Rodwell, C. , Gueydan, C. , Lanneau, V. , Murphy, D. , Le Cam, Y. , & Rath, A. (2020). Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. European Journal of Human Genetics, 28(2), 165–173. 10.1038/s41431-019-0508-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oktay, Y. , Güngör, S. , Zeltner, L. , Wiethoff, S. , Schöls, L. , Sonmezler, E. , Yilmaz, E. , Munro, B. , Bender, B. , Kernstock, C. , Kaemereit, S. , Liepelt, I. , Töpf, A. , Yis, U. , Laurie, S. , Yaramis, A. , Zuchner, S. , Hiz, S. , Lochmüller, H. , & Schüle, R. (2020). Confirmation of TACO1 as a leigh syndrome disease gene in two additional families. Journal of Neuromuscular Diseases, 7(3), 301–308. 10.3233/JND-200510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmond, M. , Hartley, T. , Johnstone, B. , Andjic, S. , Girdea, M. , Gillespie, M. , Buske, O. , Dumitriu, S. , Koltunova, V. , Ramani, A. , Boycott, K. M. , & Brudno, M. (2022). PhenomeCentral: 7 years of rare disease matchmaking. Human Mutation. 10.1002/humu.24348 [DOI] [PubMed] [Google Scholar]
- Owen, D. , Töpf, A. , Preethish‐Kumar, V. , Lorenzoni, P. J. , Vroling, B. , Scola, R. H. , Dias‐Tosta, E. , Geraldo, A. , Polavarapu, K. , Nashi, S. , Cox, D. , Evangelista, T. , Dawson, J. , Thompson, R. , Senderek, J. , Laurie, S. , Beltran, S. , Gut, M. , Gut, I. , & Nalini, A. (2018). Recessive variants of MuSK are associated with late onset CMS and predominant limb girdle weakness. American Journal of Medical Genetics, Part A, 176, 1594–1601. 10.1002/ajmg.a.38707 [DOI] [PubMed] [Google Scholar]
- Permanyer, E. , Laurie, S. , Blasco‐Lucas, A. , Maldonado, G. , Amador‐Catalan, A. , Ferrer‐Curriu, G. , Fuste, B. , Perez, M. L. , Gonzalez‐Alujas, T. , Beltran, S. , Comas‐Riu, J. , Bardají, A. , Evangelista, A. , & Galiñanes, M. (2020). A single nucleotide deletion resulting in a frameshift in exon 4 of TAB2 is associated with a polyvalvular syndrome. European Journal of Medical Genetics, 63, 103854. 10.1016/j.ejmg.2020.103854 [DOI] [PubMed] [Google Scholar]
- Philippakis, A. A. , Azzariti, D. R. , Beltran, S. , Brookes, A. J. , Brownstein, C. A. , Brudno, M. , Brunner, H. G. , Buske, O. J. , Carey, K. , Doll, C. , Dumitriu, S. , Dyke, S. O. , den Dunnen, J. T. , Firth, H. V. , Gibbs, R. A. , Girdea, M. , Gonzalez, M. , Haendel, M. A. , Hamosh, A. ,, … Rehm, H. L. (2015). The matchmaker exchange: A platform for rare disease gene discovery. Human Mutation, 36, 915–921. 10.1002/humu.22858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñero, J. , Ramírez‐Anguita, J. M. , Saüch‐Pitarch, J. , Ronzano, F. , Centeno, E. , Sanz, F. , & Furlong, L. I. (2020). The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Research, 48, 845. 10.1093/nar/gkz1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplin, R. , Ruano‐Rubio, V. , DePristo, M. A. , Fennell, T. J. , Carneiro, M. O. , Van der Auwera, G. A. , Kling, D. E. , Gauthier, L. D. , Levy‐Moonshine, A. , Roazen, D. , Shakir, K. , Thibault, J. , Chandran, S. , Whelan, C. , Lek, M. , Gabriel, S. , Daly, M. J. , Neale, B. , MacArthur, D. G. , & Banks, E. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv, 1, 201178–201190. 10.1101/201178 [DOI] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M. A. R. , Bender, D. , Maller, J. , Sklar, P. , De Bakker, P. I. W. , Daly, M. J. , & Sham, P. C. (2007). PLINK: A tool set for whole‐genome association and population‐based linkage analyses. American Journal of Human Genetics, 81(3), 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer, K. , Juusola, J. , Cho, M. T. , Vitazka, P. , Millan, F. , Gibellini, F. , Vertino‐Bell, A. , Smaoui, N. , Neidich, J. , Monaghan, K. G. , McKnight, D. , Bai, R. , Suchy, S. , Friedman, B. , Tahiliani, J. , Pineda‐Alvarez, D. , Richard, G. , Brandt, T. , Haverfield, E. , & Chung, W. K. (2016). Clinical application of whole‐exome sequencing across clinical indications. Genetics in Medicine, 18(7), 696–704. 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Richard, E. M. , Bakhtiari, S. , Marsh, A. , Kaiyrzhanov, R. , Wagner, M. , Shetty, S. , Pagnozzi, A. , Nordlie, S. M. , Guida, B. S. , Cornejo, P. , Magee, H. , Liu, J. , Norton, B. Y. , Webster, R. I. , Worgan, L. , Hakonarson, H. , Li, J. , Guo, Y. , Jain, M. , … Kruer, M. C. (2021). Bi‐allelic variants in SPATA5L1 lead to intellectual disability, spastic‐dystonic cerebral palsy, epilepsy, and hearing loss. American Journal of Human Genetics, 108(10), 2006–2016. 10.1016/j.ajhg.2021.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saredi, S. , Gibertini, S. , Matalonga, L. , Farina, L. , Ardissone, A. , Moroni, I. , & Mora, M. (2019). Exome sequencing detects compound heterozygous nonsense LAMA2 mutations in two siblings with atypical phenotype and nearly normal brain MRI. Neuromuscular Disorders, 29(5), 376–380. 10.1016/j.nmd.2019.04.001 [DOI] [PubMed] [Google Scholar]
- Schüle, R. , Timmann, D. , Erasmus, C. E. , Reichbauer, J. , Wayand, M. , van de Warrenburg, B. , Schöls, L. , Wilke, C. , Bevot, A. , Zuchner, S. , Beltran, S. , Laurie, S. , Matalonga, L. , Graessner, H. , & Synofzik, M. (2021). Solving unsolved rare neurological diseases—a Solve‐RD viewpoint. European Journal of Human Genetics, 29, 1332–1336. 10.1038/s41431-021-00901-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley, D. , Jacobsen, J. O. B. , Jäger, M. , Köhler, S. , Holtgrewe, M. , Schubach, M. , Siragusa, E. , Zemojtel, T. , Buske, O. J. , Washington, N. L. , Bone, W. P. , Haendel, M. A. , & Robinson, P. N. (2015). Next‐generation diagnostics and disease‐gene discovery with the Exomiser. Nature Protocols, 10, 2004–2015. 10.1038/nprot.2015.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Chapman, M. , Evans, K. , Azevedo, L. , Hayden, M. , Heywood, S. , Millar, D. S. , Phillips, A. D. , & Cooper, D. N. (2020). The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Human Genetics, 139, 1197–1207. 10.1007/s00439-020-02199-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranneheim, H. , Lagerstedt‐Robinson, K. , Magnusson, M. , Kvarnung, M. , Nilsson, D. , Lesko, N. , Engvall, M. , Anderlid, B. M. , Arnell, H. , Johansson, C. B. , Barbaro, M. , Björck, E. , Bruhn, H. , Eisfeldt, J. , Freyer, C. , Grigelioniene, G. , Gustavsson, P. , Hammarsjö, A. , Hellström‐Pigg, M. , … Wedell, A. (2021). Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Medicine, 13(1), 40. 10.1186/s13073-021-00855-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Paske, I. , Garcia‐Pelaez, J. , Sommer, A. K. , Matalonga, L. , Starzynska, T. , Jakubowska, A. , Solve‐Rd‐Genturis, g , van der Post, R. S. , Lubinski, J. , Oliveira, C. , Hoogerbrugge, N. , & de Voer, R. M. (2021). A mosaic PIK3CA variant in a young adult with diffuse gastric cancer: Case report. European Journal of Human Genetics, 29, 1354–1358. 10.1038/s41431-021-00853-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, R. , Johnston, L. , Taruscio, D. , Monaco, L. , Béroud, C. , Gut, I. G. , Hansson, M. G. , T., Hoen, P. B. A. , Patrinos, G. P. , Dawkins, H. , Ensini, M. , Zatloukal, K. , Koubi, D. , Heslop, E. , Paschall, J. E. , Posada, M. , Robinson, P. N. , Bushby, K. , & Lochmüller, H. (2014). RD‐connect: An integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research. Journal of General Internal Medicine, 29(Suppl 3), 780–787. 10.1007/s11606-014-2908-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Töpf, A. , Oktay, Y. , Balaraju, S. , Yilmaz, E. , Sonmezler, E. , Yis, U. , Laurie, S. , Thompson, R. , Roos, A. , MacArthur, D. G. , Yaramis, A. , Güngör, S. , Lochmüller, H. , Hiz, S. , & Horvath, R. (2020). Severe neurodevelopmental disease caused by a homozygous TLK2 variant. European Journal of Human Genetics, 28, 383–387. 10.1038/s41431-019-0519-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Töpf, A. , Pyle, A. , Griffin, H. , Matalonga, L. , Schon, K. , Solve‐RD SNV‐indel working, g , Solve‐Rd, D. , Sickmann, A. , Schara‐Schmidt, U. , Hentschel, A. , Chinnery, P. F. , Kölbel, H. , Roos, A. , & Horvath, R. (2021). Exome reanalysis and proteomic profiling identified TRIP4 as a novel cause of cerebellar hypoplasia and spinal muscular atrophy (PCH1). European Journal of Human Genetics, 29(9), 1348–1353. 10.1038/s41431-021-00851-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull, C. , Scott, R. H. , Thomas, E. , Jones, L. , Murugaesu, N. , Pretty, F. B. , Halai, D. , Baple, E. , Craig, C. , Hamblin, A. , Henderson, S. , Patch, C. , O'Neill, A. , Devereau, A. , Smith, K. , Martin, A. R. , Sosinsky, A. , McDonagh, E. M. , Sultana, R. , … Genomes, P. (2018). The 100 000 Genomes Project: Bringing whole genome sequencing to the NHS. BMJ, 361, 1687. 10.1136/bmj.k1687 [DOI] [PubMed] [Google Scholar]
- Urreizti, R. , Lopez‐Martin, E. , Martinez‐Monseny, A. , Pujadas, M. , Castilla‐Vallmanya, L. , Pérez‐Jurado, L. A. , Serrano, M. , Natera‐De Benito, D. , Martínez‐Delgado, B. , Posada‐De‐La‐Paz, M. , Alonso, J. , Marin‐Reina, P. , O'Callaghan, M. , Grinberg, D. , Bermejo‐Sánchez, E. , & Balcells, S. (2020). Five new cases of syndromic intellectual disability due to KAT6A mutations: Widening the molecular and clinical spectrum. Orphanet Journal of Rare Diseases, 15(1), 44. 10.1186/s13023-020-1317-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, M. , Dumontier, M. , & Aalbersberg, I. (2016). The FAIR guiding principles for scientific data management and stewardship. Scientific Data, 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, C. F. , FitzPatrick, D. R. , & Firth, H. V. (2018). Paediatric genomics: Diagnosing rare disease in children. Nature Reviews Genetics, 19, 253–268. 10.1038/nrg.2017.116 [DOI] [PubMed] [Google Scholar]
- Wright, C. F. , McRae, J. F. , Clayton, S. , Gallone, G. , Aitken, S. , FitzGerald, T. W. , Jones, P. , Prigmore, E. , Rajan, D. , Lord, J. , Sifrim, A. , Kelsell, R. , Parker, M. J. , Barrett, J. C. , Hurles, M. E. , FitzPatrick, D. R. , & Firth, H. V. (2018). Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome‐wide data in 1,133 families with developmental disorders. Genetics in Medicine, 20(10), 1216–1223. 10.1038/gim.2017.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaramis, A. , Lochmüller, H. , Töpf, A. , Sonmezler, E. , Yilmaz, E. , Hiz, S. , Yis, U. , Gungor, S. , Polat, A. I. , Edem, P. , Beltran, S. , Laurie, S. , Yaramis, A. , Horvath, R. , & Oktay, Y. (2020). COL4A1 ‐related autosomal recessive encephalopathy in 2 Turkish children. Neurology: Genetics, 6, 392. 10.1212/NXG.0000000000000392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurek, B. , Ellwanger, K. , Vissers, L. , Schüle, R. , Synofzik, M. , Töpf, A. , de Voer, R. M. , Laurie, S. , Matalonga, L. , Gilissen, C. , Ossowski, S. , 't Hoen, P. , Vitobello, A. , Schulze‐Hentrich, J. M. , Riess, O. , Brunner, H. G. , Brookes, A. J. , Rath, A. , Bonne, G. , … Solve‐RD, C (2021). Solve‐RD: Systematic pan‐European data sharing and collaborative analysis to solve rare diseases. European Journal of Human Genetics, 29, 1325–1331. 10.1038/s41431-021-00859-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.
Supporting Information.
Supporting Information.
Supporting Information.
Data Availability Statement
As this study describes a software platform, there is no data to be made available. However, we confirm that free access to the platform will be granted to any bona fide researcher with an interest in rare diseases.