

Abstract
MS is the most common autoimmune demyelinating disease of the CNS. For the past decades, several immunomodulatory disease‐modifying treatments with multiple presumed mechanisms of action have been developed, but MS remains an incurable disease. Whereas high efficacy, at least in early disease, corroborates underlying immunopathophysiology, there is profound heterogeneity in clinical presentation as well as immunophenotypes that may also vary over time. In addition, functional plasticity in the immune system as well as in the inflamed CNS further contributes to disease heterogeneity. In this review, we will highlight immune‐pathophysiological and associated clinical heterogeneity that may have an implication for more precise immunomodulatory therapeutic strategies in MS.
Keywords: precision medicine ⋅ biomarkers ⋅ brain barriers immunotherapy ⋅multiple sclerosis
Currently, MS diagnosis and therapy follow stereotype guidelines, where in a “try and error” approach patients are divided into treatment responders or nonresponders. In this review, we discuss different immunopathophysiological sources of heterogeneity; still an unmet medical need, ideally these sources of heterogeneity would be the basis for individualized disease management decisions.
Introduction
MS is a demyelinating neuroimmunological disorder affecting the CNS. Due to MS heterogeneity, MS immunotherapy should ideally be tailored to patient phenotypes and pathomechanisms. “Precise” immunotherapy is important to achieve the optimum benefit‐risk profile of a therapeutic substance in the individual patient, especially in view of the limited capacity for regeneration of the CNS which determines a certain “window of opportunity,” that is, prophylactic immunotherapy before irreversible tissue damage has accrued. Thus, MS is a highly attractive candidate for strategies to stratify therapeutic interventions according to underlying disease mechanisms for specific patient groups [1]. The growing spectrum of unanticipated severe adverse drug reactions associated with different immunotherapies further underscores the need for “precise” (or “individualized, personalized”) therapeutic approaches. In this review, we will discuss immunopathophysiological heterogeneity both as a major contributor and a challenge to precision medicine and emphasize current approaches to stratify disease phenotypes using disease markers.
Multiple sclerosis: Clinical and pathological heterogeneity
Among patients, MS is often described as the disease of “1000 faces.” This in part is owed to the multitude of symptoms that can occur in an unpredictable fashion, among others visual disturbance, motor impairment, bladder and sexual dysfunction as well as cognitive symptoms and fatigue. Given the many potential layers of impairment and the fact that far more than two million individuals are affected worldwide often during decisive years of their professional careers and social relationships, the burden of disease and socioeconomic implications are immense [2]. Despite the presumed multifactorial etiology of MS, the initial clinical manifestation in more than 80% of patients follows a relapsing‐remitting phenotype with relapses characterized by the acute occurrence of new or worsening of pre‐existing neurological symptoms [3]. After approximately one decade, the disease phenotype often transforms into a secondary progressive disease course with chronic accrual of disability; a subset of patients (10–15%) progresses from disease onset (primary progressive). The “disease of the 1000 faces” also refers to treatment response that is not readily predictable for an individual patient. With far more than a dozen immunotherapies approved, there is still no uniformly valid, evidence‐based treatment algorithm that would encompass even “simple” clinical situations, that is, choice among the different options in the first‐line therapy in a newly diagnosed patient. These considerations become even more complicated for sequential treatment (e.g. switch due to lack of efficacy, side‐effects), with vulnerable patient groups (children, elderly), or a stop of immunotherapy. One major contributor to variable treatment response appears to be pathophysiological heterogeneity. The current concept stipulates that MS is a chronic inflammatory CNS disease of autoimmune etiology, influenced by genetic and environmental factors [3]. Histopathologically defined active plaques in early MS are characterized by focal white matter demyelinating lesions with perivenular cellular infiltrates consisting mainly of CD8+ T cells, CD20+ B cells, and plasmablasts [4]. To some extent, perivascular cuffs, but rather the associated blood–brain barrier (BBB) impairment, can be detected by magnetic resonance imaging (MRI) with relatively distinct radiological appearance (“central vein sign”) [5]. BBB impairment detected by MRI (i.e. lesions enhanced with the paramagnetic contrast agent gadolinium) serves as a paraclinical marker of focal “disease activity.” Whereas these characteristics appear to be predominant in early relapsing MS, in progressive disease morphologically distinct, CNS‐compartimentalized inflammatory reactions can be identified [4]. Here, an accumulation of T and B cells can especially be observed in the meninges (spatially associated with cortical lesions) and perivascular spaces (associated with white matter lesions) with little infiltration into the lesion parenchyma and both are also characterized by microglial activation and macrophage infiltration [6]. Furthermore, subpial cortical demyelination [7], diffuse white matter injury [8], slow expansion of pre‐existing lesions [9, 10], and extensive brain atrophy [11] constitute histopathological features of progressive MS. In addition, albeit not visible by MRI imaging, histopathological studies on postmortem MS brain tissues have provided evidence for continued abnormalities of BBB function in inactive MS lesions as well as in normal‐appearing white matter during progressive MS [12, 13].
Robust treatment results of immunotherapy are especially seen in early relapsing MS; in chronic progressive disease, treatment response is limited which indicates that morphological heterogeneity of the inflammatory reaction may also translate into functional differences [3]. Traditionally, a two‐stage process of progression has been suggested, where an initial phase is predominantly driven by more focal inflammatory changes, followed by a secondary phase where neurodegenerative and chronic inflammatory mechanisms of the disease become more dominant without evident relapses. More recently, this view of temporally distinct processes was challenged by the demonstration of insidious accrual of disability already in patients classified as early relapsing remitting MS [14]. In addition, in post‐hoc analyses of clinical trials, most accumulation of disability was not associated with clinical relapses but resulted from a steady “progression independent of relapse activity (PIRA)” [15]. These data would rather argue for a continuum of focal and diffuse (compartimentalized) inflammatory and degenerative mechanisms that occur throughout the disease spectrum. However, the “progression independent of relapse activity [PIRA]” concept is mainly based on clinical outcome parameters that are prone to variability, and objective biomarkers that would be able to define or even predict these different phenotypes with presumed underlying pathomechanisms are only beginning to emerge [16].
Despite the broad range of potential pathophysiological mechanisms involved, currently approved therapies focus on relatively few presumed targets (i.e. effector T cells, B cells, immune cell trafficking into the CNS) [3, 17]. These therapies comprise substances with potentially pleiotropic mechanisms of action but also mAb with restricted targets [18]. The functions of key inflammatory elements are determined by complex interactions, for example, those associated with the disease stage and subtypes, the CNS region, or simply age‐dependent aspects, which all will influence treatment response.
Key inflammatory effectors and heterogenous stage‐/phase‐specific roles
In the following sections, we will discuss the disease‐specific role, functional heterogeneity, and also plasticity of the targets that may govern the heterogeneity of treatment response including the brain barriers, T cells, B cells, and myeloid cells.
The brain barriers
The endothelial BBB maintains CNS homeostasis by protecting the CNS from the constantly changing milieu in the blood stream. The BBB is established by brain microvascular endothelial cells, which inhibit free paracellular diffusion of water‐soluble molecules by complex tight junctions [19]. Combined with an extremely low vesicular activity inhibiting transcellular passage, the BBB establishes a physical permeability barrier [20]. At the same time, the BBB establishes a metabolic barrier, in which polarized expression of specific transporters and enzymes ensures that toxic metabolites are rapidly removed from the CNS. These transporters may also be relevant for (heterogenous) distribution of therapeutic agents [21]. Importantly, the biochemically unique BBB characteristics are not intrinsic to brain microvascular endothelial cells but rather rely on the continuous cross‐talk involving wnt/β‐catenin and hedgehog signaling pathways with pericytes and astrocytes surrounding the CNS microvessels [20, 22].
The BBB also controls immune cell entry into the CNS. In the absence of neuroinflammation, the BBB limits immune cell trafficking mainly to effector and central memory T cells, which can reach the CSF compartments, but are separated from the CNS parenchyma by the glia limitans, ensuring CNS immune surveillance [23]. Under neuroinflammatory conditions, additional immune cell subsets cross the altered BBB and eventually the glia limitans, inducing clinical signs associated with MS [24, 25]. Migration of T cells across the BBB is a multistep process regulated by the sequential interaction of different signaling and adhesion molecules on the BBB endothelium and the T cells [26]. Accounting for the extreme tightness of the BBB, T‐cell migration across the BBB is characterized by unique adaptations. CD4 T cells crawl for long distances against the direction of blood flow on the surface of the BBB endothelium in search for rare sites permissive for paracellular diapedesis preferentially through the tricellular endothelial junctions [27, 28, 29, 30]. During neuroinflammation, the BBB endothelium presents an altered phenotype, leading to the increased infiltration of presumably pathogenic CD4+ T cells (i.e. Th1 or Th17 cells) and other immune cell subsets, into the CNS. Interestingly, this is accompanied by increased numbers of T cells crossing the inflamed BBB endothelium preferentially via a transcellular pathway, albeit endothelial junctional integrity is impaired at this time [27, 28]. While interaction of α4β1‐integrins with endothelial VCAM‐1 mediates T‐cell arrest on the BBB under physiological flow, subsequent polarization and crawling of T cells against the direction of blood flow prior to their diapedesis is mediated by LFA‐1 engaging endothelial ICAM‐1 and ICAM‐2 [31, 32, 33]. Further molecules, such as ALCAM [34, 35], MCAM [36] and ninjuirin‐1, as well as the atypical chemokine receptor 1 [37] were proposed to participate in T‐cell migration across the BBB during EAE and MS [38, 39] and need further exploitation.
Inhibition of immune cell trafficking across the BBB using natalizumab or sphingosine 1 phosphate receptor modulators (S1PR, currently approved: fingolimod, siponimod, ozanimod, ponesimod) has become a well‐established and successful therapeutic approach for the treatment of relapsing forms of MS [40, 41]. MS patients under treatment with an anti‐α4‐integrin subunit mAb (natalizumab) show strongly reduced counts of adaptive immune cells such as CD4 and CD8 T cells and B cells in their CSF [42, 43]. In addition, natalizumab reduces the number of DCs in perivascular CNS spaces [44]. Taken together, this supports the notion that natalizumab blocks CNS entry of both, adaptive and innate immune cell subsets, which may eventually interfere with CNS immune surveillance [45]. Although S1PR modulators limit the egress of T and B lymphocytes from the LNs, they reduce the numbers of T cells but not B cells in the CSF [43, 46]. This hypothesis has been supported by the observation that treatment with natalizumab but, to a lesser extent, also with fingolimod and dimethyl fumarate is associated with an increased risk of developing progressive multifocal leukoencephalopathy (PML), a rare but potentially fatal side‐effect caused by JC virus infection of glial cells [47]. As CD8 T cells are in charge of controlling CNS virus infections, development of PML is thought to be at least partly due to therapy‐mediated inhibition of CNS immune surveillance by virus‐specific CD8 T cells. Unfortunately, little is known about the molecular mechanisms involved in CD8 T‐cell trafficking across the BBB. While α4β1‐integrins are essential for CD8 T‐cell migration into the CNS during immune surveillance and neuroinflammation similar to CD4 T cells [38], there are distinct differences in the employment of the endothelial integrin ligands from the IgCAMs between CD8 and CD4 T cells [48, 49]. Thus, exploitation of the distinct trafficking signals available at the BBB for the different immune cell subsets is a prerequisite to develop therapies that specifically block CNS entry of pathogenic T cells while leaving those required for CNS immune surveillance untouched.
Importantly, serial MRI studies have indicated that BBB dysfunction may even precede CNS immune cell infiltration and myelin damage in MS [50, 51, 52]. This is supported by our recent observation that in vitro BBB models derived from human induced pluripotent stem cells from MS patients show functional impairments [53]. Thus, BBB dysfunction could be the underlying cause of MS pathogenesis in some patients, eventually leading to BBB breakdown later visible by MRI imaging and ensuing neuroinflammation. In addition, continuous BBB abnormalities may also be relevant in later disease stages, where histopathologically disrupted tight junctions or upregulated expression of adhesion molecules in brain microvessels were observed in inactive MS lesions as well as in normal‐appearing white matter of postmortem MS brain tissue [12, 13]. Therapeutic implications are unclear since natalizumab failed to reduce disability progression using a multicomponent endpoint in secondary progressive MS, but did reduce progression in subdomains [54]. Therapeutic tightening of the BBB may provide a novel treatment option for MS that has not yet been considered. In fact, tightening of the BBB is observed as an accompanying effect under several immunomodulatory MS therapies [18], underscoring the central role of BBB dysfunction in MS pathogenesis. As experimental tightening of the BBB ameliorates clinical signs of EAE, an animal model of MS [55], therapeutic tightening of the BBB for treating MS could be explored. This approach has been hampered until recently as studying the BBB of MS patients has been prohibited by a lack of access to the tissue from patients. Recent advancements in stem cell technology have, however, allowed the derivation of human and, thus, patient‐derived brain microvascular endothelial cells from human‐induced pluripotent stem cells [56, 57, 58]. hiPSC‐derived in vitro models of the BBB have proven successful to model BBB dysfunction in inheritable neurological disorders [59, 60, 61] as well as immune cell trafficking across the BBB in vitro [23]. Therefore, establishing hiPSC‐derived models of the neurovascular unit, in which all cell types are sourced from MS patients, may offer unprecedented opportunities to study somatic alterations at the level of the BBB contributing to the major hallmarks of MS pathogenesis, namely BBB dysfunction and increased immune cell entry into the CNS. In addition, these MS patient‐derived in vitro BBB models could serve as tools for developing therapeutic strategies for BBB stabilization and for testing adverse effects of disease‐modifying drugs in a personalized fashion.
Contribution of the dysfunction of other outer brain barriers, for example, the epithelial blood‐CSF barrier (BCSFB) surrounding the choroid plexuses or the arachnoid barrier establishing a BCSFB between the dura mater and the subarachnoid space [24] to MS pathogenesis has been largely ignored. More recent evidence for disturbance of the choroid plexus BCSFB in postmortem MS tissue [62] encourages development of MRI imaging sequences analyzing BCSFB function in MS patients. Similarly, dysfunction of the arachnoid barrier may contribute to the establishment of meningeal infiltrates, which are mainly comprised of B cells, possibly directly from the dura mater, which has been identified as a unique B‐cell niche [63, 64]; hypothetically this may allow to identify patients who respond particularly well to therapeutic targeting of B cells.
T cells
MS is considered a prototypic T cell‐mediated autoimmune disease based on MS‐ associated genetic variants (single nucleotide polymorphisms) that point to genes coding for molecules related to the activation or proliferation of T and other immune cells and due to observations made in the animal model EAE [3]. The classical EAE models rely on immunization with CNS‐derived autoantigens emulsified in potent immune adjuvants, thereby favoring the activation of CD4 T cells and the induction of autoantibodies. Both, γ‐IFN‐secreting Th1 and IL‐17‐ secreting Th17 cells (summarized in Ref. [65]) and also GM‐CSF producing CD4 T‐cell subsets [66] have been shown to play key pathogenic roles in triggering EAE.
Different CD4 T‐cell subsets differ in their effector functions and trafficking profiles. In contrast to Th1 cells, Th17 cells are a heterogenous group of T cells including a subgroup of Th1‐like Th17 cells expressing both Th1 and Th17 signature cytokines and chemokine receptors. It is these Th1‐like Th17 cells referred to as Th1* [67] or Th17.1 [68] that have recently been suggested to play a dominant role in MS pathogenesis. Therefore, studying the T‐cell profile of MS patients early in the disease may provide additional insights into potentially diverse disease triggers based on the observed alteration of distinct T‐cell subsets and provides a strong rationale to further explore more specific T‐cell‐targeted therapies in early MS. As an additional source of heterogeneity, studies in EAE have shown that Th17 and Th1 cells use different anatomical entry sites and distinct molecular mechanisms to reach the CNS. While Th17 cells preferentially enter the brain via the choroid plexus using the chemokine receptor CCR6 [69] and LFA‐1 [70], Th1 cells preferentially infiltrate the spinal cord and require α4β1‐integrins for CNS entry [65, 71]. In addition, a recent study has shown that the site of priming of autoaggressive T cells affects their ability to infiltrate CNS white and grey matter [72]. Exploring potential underlying T‐cell subset mediated pathomechanisms in individual MS patients may, thus, have implications for the likelihood of response to agents like natalizumab.
In addition to CD4 T cells, there is accumulating evidence for a role of CD8 T cells in the autoimmune CNS attack in MS [73]. Indeed, CD8 T cells accumulate within white matter lesions, where they often outnumber CD4 T cells and are found in close association with oligodendrocytes and demyelinated axons [6, 74, 75]. Furthermore, CD8 T cells harboring an effector memory phenotype were shown to accumulate in the inflamed CNS of MS patients, and these cells exhibit cytotoxic and inflammatory potential as they express granzyme B and IFN‐γ at high frequency [38, 76]. CD8 T cells were even detected in immune cell infiltrates in cortical demyelinating lesions, which are prevalent at early stages of MS [77], underscoring an involvement of CD8 T cells at early time points of MS pathogenesis. Analyses of TCR usage by CNS infiltrating T cells provided evidence for oligoclonal expansion of CD8 but not CD4 T cells within MS lesions [78, 79] and the CSF [80], which is a likely consequence of their local MHC class‐I‐dependent antigen‐driven activation. Furthermore, recent genetic studies showed a clear association between certain MHC class I alleles and MS susceptibility [81]. Finally, results from clinical trials further underscore the contribution of lymphocyte subsets other than CD4 T cells to MS pathogenesis [82, 83]. While anti‐inflammatory therapies globally targeting lymphocytes showed profound effects [84, 85], therapies specifically targeting CD4 T cells did not lead to clinical benefits in MS patients [86].
B cells
Although MS was traditionally considered as a predominantly T cell‐mediated autoimmune disease, B cells are now known to be key contributors to the pathogenesis of MS. B cells, plasmablasts, and plasma cells can be found in three different subcompartments of the CNS in patients with MS: the CSF, the parenchyma, and the meninges. The CSF of MS patients is characterized by the presence of oligoclonal bands in more than 90% of patients [87], and they also constitute a valuable biomarker in the diagnosis of MS [2]. The strong overlap observed between the Ig transcriptome of B cells and the corresponding Ig proteome in the CSF of MS patients demonstrated that the source of the antibodies giving rise to oligoclonal bands were clonally expanded intrathecal B cells [88]. The antigenic targets of B cells in MS patients remain unknown. Oligoclonal bands antibodies do not seem to be specific for CNS proteins but instead B‐cell responses are heterogeneous and include reactivity against ubiquitous self‐antigens released during tissue destruction [89]. The number of B cells is usually increased in the CSF of patients with MS and the B cell to other CSF immune cell ratio was found to influence MS disease severity and progression. In this context, a high B cell to monocyte ratio in the CSF was associated with more rapid MS progression, whereas a low ratio with a predominance of monocytes was observed in patients having slower progression [90]. B cells in the CSF of MS patients are largely class‐switched memory B cells and short‐lived plasmablasts [91, 92].
B cells can also be found in the brain parenchyma of MS patients. Autopsy tissue studies late in the disease course indicated that the classical deep white matter perivascular demyelinating lesions of MS patients typically exhibited few B cells and plasma cells compared to the greater abundance of myeloid cells and T cells [93]. However, studies conducted in MS patients early in the disease course revealed that the CNS lesions exhibited considerable numbers of B cells as well as plasma cells, in addition to T cells and myeloid cells [94]. Altogether, these findings raise the possibility that B cells and plasma cells may be a more common feature of early parenchymal lesions.
Ectopic follicle‐like aggregates containing B cells and plasma cells are found in the leptomeninges of patients with MS. Although these lymphoid structures were first described in patients with secondary progressive disease course [95], meningeal aggregates of B cells are also identified in patients with relapsing remitting MS and primary progressive MS [77, 96]. The importance of these findings stems from the potential causal relationship observed between meningeal B‐cell inflammation and cortical damage. In this context, patients with follicle‐like structures showed more pronounced subpial demyelination, microglia activation, and neurite loss [97] compared to those without follicle‐like aggregates. In addition, a gradient of neuronal loss from the pia mater to the white matter was observed in grey matter of patients with follicle‐like structures, suggesting the presence of soluble cytotoxic factors diffusing from the meninges into the grey matter [98]. The presence of ectopic follicle‐like structures has been associated with more aggressive clinical disease [97, 99]. More recently, the demonstration of a specific Th17‐cell subset that fosters a meningeal microenvironment that supports B‐cell tropism and development highlights complex interactions of the local immune network [100]. The remarkable clinical success of B cell‐depleting therapies has further underscored the importance of B cells in the pathogenesis of MS. Treatment of MS patients with rituximab and ocrelizumab results in near‐complete depletion of B cells in different blood and CNS compartments [101, 102] and is associated with significant reductions in inflammatory brain lesions and clinical relapses [103]. While both memory and naïve B cells are efficiently depleted by these therapies, plasmablasts and plasma cells, which do not express CD20, are not targeted by anti‐CD20 mABs, and hence, the clinical benefits of these therapies are not related with reductions in IgM and IgG antibody levels [103, 104]. However, the effect of B cell‐depleting therapies is modest in patients with progressive MS [105, 106], most likely reflecting the limited penetration of these drugs through the BBB and their difficult access to pathogenic meningeal B‐cell aggregates. This is highlighted by more recent data indicating a CNS‐specific, “private” source of meningeal B‐cells located in the dura mater that may or may not be potentially sourced from the nearby BM [63, 107].
Myeloid cells
The reactivation of infiltrating encephalitogenic T cells by self‐cognate antigens presented by innate immune cells at CNS barriers is considered to represent a crucial step in the pathogenesis of MS. Relevant cellular elements comprise the CNS resident microglia localized in the CNS parenchyma, CNS‐border‐associated macrophages, as well as circulating DCs and monocytes. Using an unbiased transcriptome approach of regionally distinct CNS‐resident and circulating myeloid cells, highly dynamic and complex changes of resident and recruited circulating myeloid cells during experimental disease can be demonstrated [108]. Thus, within the innate immune cell compartment, CNS‐resident macrophages undergo clonal expansion but are functionally dispensable for antigen presentation. In contrast, DCs and monocyte‐derived cells appear to be crucial for antigen presentation. Therefore, even among myeloid cells, there is a high degree of dynamic subtype specification dependent on neuroinflammatory disease stage and anatomical compartment. A high degree of plasticity is also observed when focusing solely on CNS resident microglia. Several (conditional) transgenic mouse models indicate disease phase‐specific roles of microglia with crucial disease‐promoting effector functions but also phagocytic clearance of tissue debris relevant in the recovery phase [109, 110, 111]. Necroptosis of proinflammatory microglia and repopulation to a regenerative state was demonstrated to drive remyelination in toxic and inflammatory CNS demyelination [112]. Therefore, targeting proinflammatory microglia may appear attractive both to modulate white matter inflammation and support remyelination. However, although considerable progress in the understanding of myeloid cell plasticity has been achieved, we are still not capable of exploiting functional and anatomical heterogeneity therapeutically [113, 114]. More recently, inhibitors of Bruton's tyrosine kinase (BTK), a key signaling element for both B cells and myeloid cells, demonstrated efficacy especially on MRI parameters of focal inflammation in phase II clinical trials [115, 116]. The hypothesis, that brain penetrant BTK‐inhibitors may also target chronic, microglia‐associated inflammation in chronic progressive MS is currently being explored in clinical phase III trials.
Biomarkers of disease heterogeneity and individualized treatment responses
Biomarkers are particularly relevant in disorders characterized by a high degree of heterogeneity, like MS, for which it is expected that biomarkers will fully capture the different aspects of disease heterogeneity and help in better disease diagnosis, stratification, and monitoring [117]. Among the different categories of biomarkers that have been described in MS, treatment response biomarkers constitute the cornerstone for the development of a personalized therapy in the disease. While treatment response biomarkers in the strict sense refer to those associated with the clinical response to therapies, the vast armamentarium of therapeutic options existing nowadays for MS patients certainly opens the potential benefits of this type of biomarkers to include guidance on dose adjustment, switch and escalation therapies, remission induction, safety monitoring and risk stratification. The ultimate goal of personalized therapy is to administer treatment to the patient who is going to respond to it and will not develop adverse drug effects. The clinical use of treatment response biomarkers may differ depending on the nature of biomarkers. Treatment response biomarkers of DNA or RNA nature can be identified by the use of omics technologies (this is the field of “pharmacogenomics”) such as genomics and transcriptomics. Ideally, these biomarkers can be determined before receiving treatment or during the first months of treatment, to make predictions of the response of each particular patient to the drug. After a follow‐up, patients will be classified according to clinical and/or radiological criteria, allowing to calculate the predictive accuracies of treatment response biomarkers by comparing the initial predictions made before treatment or in the first months of treatment with the real response phenotypes observed during follow‐up [118]. Unfortunately, there are no examples of treatment response biomarkers that are used in routine MS clinical practice.
A different scenario is observed for biomarkers of protein nature, which are more suitable for therapy monitoring, pharmacodynamics, and as surrogate endpoints [117]. A promising biomarker that is gradually being implemented into MS clinical practice for therapy monitoring is the blood neurofilament light subunit [119]. Blood neurofilament light levels are modified by the effect of MS therapies, and are associated with treatment response, and have the potential to be used as an informative endpoint in phase II clinical trials [120]. Interestingly, serum neurofilament levels have also been proposed as a biomarker for early identification of PML in MS patients, receiving treatment with natalizumab [121, 122].
Treatment of acute MS relapses: A paradigm for individualized therapy?
MS relapses lead to sustained residual disability in more than 40% of the patients [123]. Acute therapy follows a stereotypic approach with the administration of intravenous, high‐dose intravenous glucocorticosteroids (GC, methylprednisolone); relapses not responsive to GC can subsequently be treated using apheresis techniques (plasma exchange, immunoadsorption) [124]. Therapy in this situation is far from “personalized” with few, mainly clinical predictors of therapy response to GC or apheresis (Fig. 1). GC exert pleiotropic anti‐inflammatory effects, for example, by inhibiting secretion of proinflammatory mediators (Th1‐associated cytokines, chemokines, antibodies, nitric oxide), effects on activation and migration of different immune cells (T cells, macrophages, microglia), increase of CD25+, FoxP3+Treg cells, and effects on adhesion molecules and tight‐junction proteins in endothelial cells [125, 126]. Lack of response in a proportion of patients [127] may point to distinct pathomechanisms incompletely targeted by repeated and even dose escalated GC. Thus, in a cohort study of MS patients who had undergone brain biopsy, treatment response to apheresis in the context of steroid‐resistant relapse was associated with distinct histopathological characteristics [128]. Those patients, in which early demyelinating CNS‐lesions were characterized by the deposition of Ig and complement, responded relatively well to apheresis therapy. This was in contrast to patients with lesions characterized by changes reminiscent of primary oligodendrocyte damage [129]. Together with the concept of interindividual histopathological heterogeneity with intraindividually stable phenotypes, this potential interference of apheresis therapy with antibody‐ and complement‐mediated demyelination could hypothetically be an element of individualizing treatment algorithms [130, 131]. However, this concept is controversially discussed, and other confounding factors may be relevant in this highly selected cohort of patients that underwent brain biopsy, a procedure that is very rarely performed in the differential diagnosis of MS.
Figure 1.
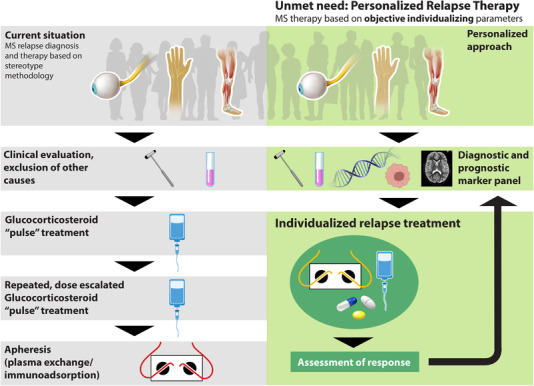
Unmet needs of personalized treatment approaches: MS relapse. (Left) MS standard treatment. With the occurrence of new or worsening symptoms of MS (e.g. visual, sensory, motor), further workup and therapy currently follow stereotypic flows and guidelines. Following neurological examination and exclusion of confounding factors (e.g. infections), high dose glucocorticosteroid “pulse therapy” is the current standard of care. If symptoms do not remit, a second, potentially dose‐escalated glucocorticosteroid pulse is administered. Apheresis techniques (plasma exchange/immunoadsorption) are employed in glucocorticosteroid‐resistant cases, not taking into account different sources of heterogeneity that lead to treatment nonresponse (“try and error”). (Right) MS personalized treatment. In a personalized approach, objective and prognostic markers on an individual level would be taken into account for a treatment decision. This marker profile would be informed by the mechanisms associated with heterogeneity in response to GC therapy as discussed in the text. Treatment could consist of different approaches (e.g. glucocorticosteroids, apheresis, new substances) also in combination. It is unclear, if treatment response in acute relapses varies over time, so marker profiles would potentially have to be individually updated to ensure response state during future relapses. Although some current findings will inform further research, at present no such marker profile exists.
GC resistance may involve additional or alternative dynamic mechanisms, for example, alterations of the GC receptor (GR) complex [132]. In MS, a different intracellular distribution of the GR and its molecular chaperone, hsp90 was demonstrated in GC‐resistant patients [133]. Alterations of GR isoform expression and DNA‐binding were reported in experimental MS models and in other non‐CNS disease models [134, 135]. Regulation by specific microRNAs, proteasomal degradation as well as specific ILs, which regulate transcription factors interfering with GR signaling may also play a role [136]. In an attempt to increase GC effects, synergistic effects of vitamin D and other mTOR inhibitors with GCs were demonstrated in an experimental model of MS [137]. Signaling via mTOR (mammalian/mechanistic target of rapamycin), an evolutionary highly conserved serine/threonine kinase crucial also during autoimmune neuroinflammation appears to be an important modulator of GC response [138, 139], and mTOR inhibition via vitamin D and also other specific mTOR inhibitors already in clinical use (e.g. everolimus) increased GR expression/GC‐mediated anti‐inflammatory effects [137]. Thus, if clinically validated, approaches to increase GC efficacy via enhancement of GR‐signaling may reduce the need for costly and risk‐prone apheresis procedures. As high‐dose GC therapy is also first‐line treatment in a variety of other neuroinflammatory diseases of different etiologies (e.g. CNS vasculitis, antibody‐mediated encephalitis), these approaches may also have implications beyond acute MS relapses [137]. Severe deficiency of serum vitamin D was associated with a higher likelihood of a GC‐resistant MS relapse. However, currently, there are no marker profiles that could guide “precise” choice of treatment in this context (Fig. 1) [140].
Conclusions and future perspectives
Despite major progress in the treatment of MS, there is an unmet need to tailor therapeutic approaches, which currently mainly rely on clinical, partly subjective parameters. Heterogeneity in pathomechanisms, changes over time also against the backdrop of aging not well appreciated by static scientific approaches and interindividual differences that govern pharmacodynamic and pharmacokinetic characteristics of pleiotropic substances are only some aspects that highlight the opportunities but also complexity of “individualized” therapeutic approaches (Fig. 2). More recent experimental findings underscoring that the site of T‐cell priming affects T‐cell trafficking and effector properties and thus immunopathology in the CNS open up the possibility of peripheral site‐directed treatment approaches (e.g. skin or gut) to modulate T‐cell pathophysiology at remote sites [72]. The challenges to further unravel pathophysiological heterogeneity and subsequent personalized treatment strategies in MS are high. Nevertheless, experience with related disorders, such as neuromyelitis spectrum disorders and more recently, also with myelin‐oligodendrocyte antibody disease has paved the way for further progress. Here, translational research into syndromes that clinically partially overlap with MS has elucidated distinct pathophysiological mechanisms, which has led to rapid advancement of specific therapeutic strategies [141].
Figure 2.
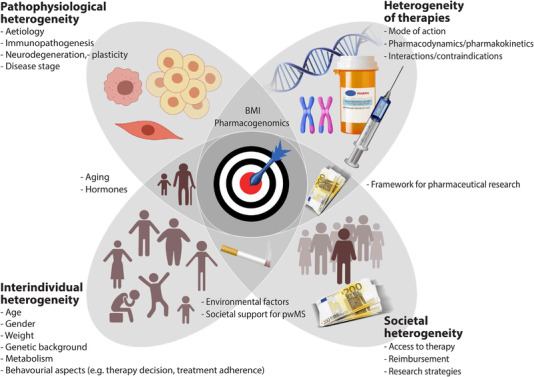
Factors that determine heterogeneity of treatment response in MS Individual factors (e.g. genetic background) combined with interindividual heterogeneity of immunopathophysiological changes as detailed in the text. Together they determine response to immunotherapy, which in itself harbors multiple sources of heterogeneity. Finally, societal aspects are relevant both in terms of MS pathophysiology (e.g. environmental risk factors), development of and access to treatment as well as support to cope with the disease. BMI, body mass index.
However, despite this progress in many fields we must not forget that for MS societal and political factors govern access to medication, and unforeseen developments such as the global COVID‐19 pandemia may at least temporally outweigh attempts to tailor therapy on an individual level.
Conflict of interest
BE received a grant from Biogen to study extended dosing of Natalizumab on T‐cell migration across the blood–brain barrier and a grant from CSL Behring to investigate the molecular underpinnings of blood brain barrier dysfunction in neurological disorders. BE is a co‐inventor on provisional US patent applications related to the EECM‐BMEC‐like cells (63/084980 and 63/185815). MC reports no conflict of interest in relation with this publication. AC has received speakers’/board honoraria from Actelion (Janssen/J&J), Almirall, Bayer, Biogen, Celgene (BMS), Genzyme, Merck KGaA (Darmstadt, Germany), Novartis, Roche, and Teva, all for hospital research funds and not related to this publication. He received research support from Biogen, Genzyme, and UCB, the European Union, and the Swiss National Foundation (SNF no. 310030_172952).
Author Contributions
All authors were involved in conceptualization, writing, and revising the context.
Abbreviations
- BBB
blood–brain barrier
- BCSFB
blood‐cerebrospinal fluid barrier
- BTK
Bruton's tyrosine kinase
- GC
glucocorticosteroids
- GR
GC receptor
- MRI
magnetic resonance imaging
- PML
progressive multifocal leukoencephalopathy
Acknowledgments
We thank our lab teams and clinical coworkers for their dedication. We thank people with MS that we have the privilege to counsel for their continuous support. Part of the work cited was funded by the Swiss National Fund (SNF no. 310030_172952) to AC.
Open access funding provided by Inselspital Universitatsspital Bern.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1. Haendel, M.A. , Chute, C.G. and Robinson, P.N. , Classification, ontology, and precision medicine. N. Engl. J. Med. 2018. 379: 1452–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thompson, A.J. , Banwell, B.L. , Barkhof, F. , Carroll, W.M. , Coetzee, T. , Comi, G. , Correale, J. et al., Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018. 17: 162–173. [DOI] [PubMed] [Google Scholar]
- 3. Baecher‐Allan, C. , Kaskow, B.J. and Weiner, H.L. , Multiple sclerosis: mechanisms and immunotherapy. Neuron 2018. 97: 742–768. [DOI] [PubMed] [Google Scholar]
- 4. Lassmann, H. , Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front. Immunol. 2018. 9: 3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maggi, P. , Absinta, M. , Grammatico, M. , Vuolo, L. , Emmi, G. , Carlucci, G. , Spagni, G. et al., Central vein sign differentiates multiple sclerosis from central nervous system inflammatory vasculopathies. Ann. Neurol. 2018. 83: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frischer, J.M. , Bramow, S. , Dal‐Bianco, A. , Lucchinetti, C.F. , Rauschka, H. , Schmidbauer, M. , Laursen, H. et al., The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009. 132: 1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bø, L. , Vedeler, C.A. , Nyland, H.I. , Trapp, B.D. and Mørk, S.J. , Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J. Neuropathol. Exp. Neurol. 2003. 62: 723–732. [DOI] [PubMed] [Google Scholar]
- 8. Kutzelnigg, A. , Lucchinetti, C.F. , Stadelmann, C. , Brück, W. , Rauschka, H. , Bergmann, M. , Schmidbauer, M. et al., Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005. 128: 2705–2712. [DOI] [PubMed] [Google Scholar]
- 9. Sethi, V. , Nair, G. , Absinta, M. , Sati, P. , Venkataraman, A. , Ohayon, J. , Wu, T. et al., Slowly eroding lesions in multiple sclerosis. Mult. Scler. 2017. 23: 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dal‐Bianco, A. , Grabner, G. , Kronnerwetter, C. , Weber, M. , Höftberger, R. , Berger, T. , Auff, E. et al., Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol. 2017. 133: 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bermel, R.A. and Bakshi, R. , The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurol. 2006. 5: 158–170. [DOI] [PubMed] [Google Scholar]
- 12. McQuaid, S. , Cunnea, P. , McMahon, J. and Fitzgerald, U. , The effects of blood‐brain barrier disruption on glial cell function in multiple sclerosis. Biochem. Soc. Trans. 2009. 37: 329–331. [DOI] [PubMed] [Google Scholar]
- 13. Sospedra, M. and Martin, R. , Immunology of multiple sclerosis. Semin. Neurol. 2016. 36: 115–127. [DOI] [PubMed] [Google Scholar]
- 14. Cree, B.A.C. , Hollenbach, J.A. , Bove, R. , Kirkish, G. , Sacco, S. , Caverzasi, E. , Bischof, A. et al., Silent progression in disease activity‐free relapsing multiple sclerosis. Ann. Neurol. 2019. 85: 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kappos, L. , Wolinsky, J.S. , Giovannoni, G. , Arnold, D.L. , Wang, Q. , Bernasconi, C. , Model, F. et al., Contribution of relapse‐independent progression vs relapse‐associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 2020. 77: 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dal‐Bianco, A. , Grabner, G. , Kronnerwetter, C. , Weber, M. , Kornek, B. , Kasprian, G. , Berger, T. et al., Long‐term evolution of multiple sclerosis iron rim lesions in 7 T MRI. Brain 2021. 144: 833–847. [DOI] [PubMed] [Google Scholar]
- 17. Bar‐Or, A. and Li, R. , Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol. 2021. 20: 470–483. [DOI] [PubMed] [Google Scholar]
- 18. Martin, R. , Sospedra, M. , Rosito, M. and Engelhardt, B. , Current multiple sclerosis treatments have improved our understanding of MS autoimmune pathogenesis. Eur. J. Immunol. 2016. 46: 2078–2090. [DOI] [PubMed] [Google Scholar]
- 19. Tietz, S. and Engelhardt, B. , Brain barriers: crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015. 209: 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engelhardt, B. and Sorokin, L. , The blood‐brain and the blood‐cerebrospinal fluid barriers: function and dysfunction. Semin. Immunopathol. 2009. 31: 497–511. [DOI] [PubMed] [Google Scholar]
- 21. Thiele Née Schrewe, L. , Guse, K. , Tietz, S. , Remlinger, J. , Demir, S. , Pedreiturria, X. , Hoepner, R. et al., Functional relevance of the multi‐drug transporter abcg2 on teriflunomide therapy in an animal model of multiple sclerosis. J. Neuroinflamm. 2020. 17: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liebner, S. , Dijkhuizen, R.M. , Reiss, Y. , Plate, K.H. , Agalliu, D. and Constantin, G. , Functional morphology of the blood‐brain barrier in health and disease. Acta Neuropathol. 2018. 135: 311–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nishihara, H. and Engelhardt, B. , Brain barriers and multiple sclerosis: novel treatment approaches from a brain barriers perspective. Handb. Exp. Pharmacol. 2020. 10.1007/164_2020_407 [DOI] [PubMed] [Google Scholar]
- 24. Engelhardt, B. , Vajkoczy, P. and Weller, R.O. , The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017. 18: 123–131. [DOI] [PubMed] [Google Scholar]
- 25. Owens, T. , Bechmann, I. and Engelhardt, B. , Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008. 67: 1113–1121. [DOI] [PubMed] [Google Scholar]
- 26. Engelhardt, B. and Ransohoff, R.M. , Capture, crawl, cross: the T cell code to breach the blood‐brain barriers. Trends Immunol. 2012. 33: 579–589. [DOI] [PubMed] [Google Scholar]
- 27. Abadier, M. , Haghayegh Jahromi, N. , Cardoso Alves, L. , Boscacci, R. , Vestweber, D. , Barnum, S. , Deutsch, U. et al., Cell surface levels of endothelial ICAM‐1 influence the transcellular or paracellular T‐cell diapedesis across the blood‐brain barrier. Eur. J. Immunol. 2015. 45: 1043–1058. [DOI] [PubMed] [Google Scholar]
- 28. Lutz, S.E. , Smith, J.R. , Kim, D.H. , Olson, C.V.L. , Ellefsen, K. , Bates, J.M. , Gandhi, S.P. et al., Caveolin1 is required for th1 cell infiltration, but not tight junction remodeling, at the blood‐brain barrier in autoimmune neuroinflammation. Cell Rep. 2017. 21: 2104–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bartholomäus, I. , Kawakami, N. , Odoardi, F. , Schläger, C. , Miljkovic, D. , Ellwart, J.W. , Klinkert, W.E.F. et al., Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 2009. 462: 94–98. [DOI] [PubMed] [Google Scholar]
- 30. Castro Dias, M. , Odriozola Quesada, A. , Soldati, S. , Bösch, F. , Gruber, I. , Hildbrand, T. , Sönmez, D. et al., Brain endothelial tricellular junctions as novel sites for T cell diapedesis across the blood‐brain barrier. J. Cell Sci. 2021. 134: jcs253880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vajkoczy, P. , Laschinger, M. and Engelhardt, B. , Alpha4‐integrin‐VCAM‐1 binding mediates G protein‐independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J. Clin. Invest. 2001. 108: 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steiner, O. , Coisne, C. , Cecchelli, R. , Boscacci, R. , Deutsch, U. , Engelhardt, B. and Lyck, R. , Differential roles for endothelial ICAM‐1, ICAM‐2, and VCAM‐1 in shear‐resistant T cell arrest, polarization, and directed crawling on blood‐brain barrier endothelium. J. Immunol. 2010. 185: 4846–4855. [DOI] [PubMed] [Google Scholar]
- 33. Lyck, R. and Engelhardt, B. , Going against the tide–how encephalitogenic T cells breach the blood‐brain barrier. J. Vasc. Res. 2012. 49: 497–509. [DOI] [PubMed] [Google Scholar]
- 34. Cayrol, R. , Wosik, K. , Berard, J.L. , Dodelet‐Devillers, A. , Ifergan, I. , Kebir, H. , Haqqani, A.S. et al., Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat. Immunol. 2008. 9: 137–145. [DOI] [PubMed] [Google Scholar]
- 35. Lyck, R. , Lecuyer, M.A. , Abadier, M. , Wyss, C.B. , Matti, C. , Rosito, M. , Enzmann, G. et al., ALCAM (CD166) is involved in extravasation of monocytes rather than T cells across the blood‐brain barrier. J. Cereb. Blood Flow Metab. 2017. 37:2894‐2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Larochelle, C. , Cayrol, R. , Kebir, H. , Alvarez, J.I. , Lécuyer, M.‐.A. , Ifergan, I. , Viel, É. et al., Melanoma cell adhesion molecule identifies encephalitogenic T lymphocytes and promotes their recruitment to the central nervous system. Brain 2012. 135: 2906–2924. [DOI] [PubMed] [Google Scholar]
- 37. Minten, C. , Alt, C. , Gentner, M. , Frei, E. , Deutsch, U. , Lyck, R. , Schaeren‐Wiemers, N. et al., DARC shuttles inflammatory chemokines across the blood‐brain barrier during autoimmune central nervous system inflammation. Brain 2014. 137: 1454–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ifergan, I. , Kebir, H. , Terouz, S. , Alvarez, J.I. , Lécuyer, M.‐.A. , Gendron, S. , Bourbonnière, L. et al., Role of Ninjurin‐1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann. Neurol. 2011. 70: 751–763. [DOI] [PubMed] [Google Scholar]
- 39. Odoardi, F. , Sie, C. , Streyl, K. , Ulaganathan, V.K. , Schläger, C. , Lodygin, D. , Heckelsmiller, K. et al., T cells become licensed in the lung to enter the central nervous system. Nature 2012. 488: 675–679. [DOI] [PubMed] [Google Scholar]
- 40. Khoy, K. , Mariotte, D. , Defer, G. , Petit, G. , Toutirais, O. and Le Mauff, B. , Natalizumab in multiple sclerosis treatment: from biological effects to immune monitoring. Front. Immunol. 2020. 11: 549842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bordet, R. , Camu, W. , De Seze, J. , Laplaud, D.A. , Ouallet, J.C. and Thouvenot, E. , Mechanism of action of s1p receptor modulators in multiple sclerosis: the double requirement. Rev. Neurol. (Paris) 2020. 176: 100–112. [DOI] [PubMed] [Google Scholar]
- 42. Alvermann, S. , Hennig, C. , Stuve, O. , Wiendl, H. and Stangel, M. , Immunophenotyping of cerebrospinal fluid cells in multiple sclerosis: in search of biomarkers. JAMA Neurol. 2014. 71: 905–912. [DOI] [PubMed] [Google Scholar]
- 43. Kowarik, M.C. , Astling, D. , Lepennetier, G. , Ritchie, A. , Hemmer, B. , Owens, G.P. and Bennett, J.L. , Differential effects of fingolimod and natalizumab on B cell repertoires in multiple sclerosis patients. Neurotherapeutics 2021. 18: 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manouchehri, N. , Hussain, R.Z. , Cravens, P.D. , Esaulova, E. , Artyomov, M.N. , Edelson, B.T. , Wu, G.F. et al., CD11c(+)CD88(+)CD317(+) myeloid cells are critical mediators of persistent CNS autoimmunity. Proc. Natl. Acad. Sci. U. S. A. 2021. 118: e2014492118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stüve, O. , Marra, C.M. , Bar‐Or, A. , Niino, M. , Cravens, P.D. , Cepok, S. , Frohman, E.M. et al., Altered CD4+/CD8+ T‐cell ratios in cerebrospinal fluid of natalizumab‐treated patients with multiple sclerosis. Arch. Neurol. 2006. 63: 1383–1387. [DOI] [PubMed] [Google Scholar]
- 46. Mathias, A. , Perriot, S. , Canales, M. , Blatti, C. , Gaubicher, C. , Schluep, M. , Engelhardt, B. et al., Impaired T‐cell migration to the CNS under fingolimod and dimethyl fumarate. Neurol. Neuroimmunol. Neuroinflamm. 2017. 4: e401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wenning, W. , Haghikia, A. , Laubenberger, J. , Clifford, D.B. , Behrens, P.F. , Chan, A. and Gold, R. , Treatment of progressive multifocal leukoencephalopathy associated with natalizumab. N. Engl. J. Med. 2009. 361: 1075–1080. [DOI] [PubMed] [Google Scholar]
- 48. Martin‐Blondel, G. , Pignolet, B. , Tietz, S. , Yshii, L. , Gebauer, C. , Perinat, T. , Van Weddingen, I. et al., Migration of encephalitogenic CD8 T cells into the central nervous system is dependent on the alpha4beta1‐integrin. Eur. J. Immunol. 2015. 45: 3302–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rudolph, H. , Klopstein, A. , Gruber, I. , Blatti, C. , Lyck, R. , Engelhardt, B. , Postarrest stalling rather than crawling favors CD8(+) over CD4(+) T‐cell migration across the blood‐brain barrier under flow in vitro. Eur. J. Immunol. 2016. 46: 2187–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Filippi, M. , Rocca, M.A. , Martino, G. , Horsfield, M.A. , Comi, G. , Magnetization transfer changes in the normal appearing white matter precede the appearance of enhancing lesions in patients with multiple sclerosis. Ann. Neurol. 1998. 43: 809–814. [DOI] [PubMed] [Google Scholar]
- 51. Werring, D.J. , Brassat, D. , Droogan, A.G. , Clark, C.A. , Symms, M.R. , Barker, G.J. , MacManus, D.G. et al., The pathogenesis of lesions and normal‐appearing white matter changes in multiple sclerosis: a serial diffusion MRI study. Brain 2000. 123: 1667–1676. [DOI] [PubMed] [Google Scholar]
- 52. Goodkin, D.E. , Rooney, W.D. , Sloan, R. , Bacchetti, P. , Gee, L. , Vermathen, M. , Waubant, E. et al., A serial study of new MS lesions and the white matter from which they arise. Neurology 1998. 51: 1689–1697. [DOI] [PubMed] [Google Scholar]
- 53. Nishihara, H. , Perriot, S. , Gastfriend, B.D. , Steinfort, M. , Cibien, C. , Soldati, S. , Matsuo, K. et al., Intrinsic blood‐brain barrier dysfunction contributes to multiple sclerosis pathogenesis. Brain 2022. 27: awac019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kapoor, R. , Ho, P.‐R. , Campbell, N. , Chang, I. , Deykin, A. , Forrestal, F. , Lucas, N. et al., Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double‐blind, placebo‐controlled trial with an open‐label extension. Lancet Neurol. 2018. 17: 405–415. [DOI] [PubMed] [Google Scholar]
- 55. Pfeiffer, F. , Schäfer, J. , Lyck, R. , Makrides, V. , Brunner, S. , Schaeren‐Wiemers, N. , Deutsch, U. et al., Claudin‐1 induced sealing of blood‐brain barrier tight junctions ameliorates chronic experimental autoimmune encephalomyelitis. Acta Neuropathol. 2011. 122: 601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stebbins, M.J. , Wilson, H.K. , Canfield, S.G. , Qian, T. , Palecek, S.P. and Shusta, E.V. , Differentiation and characterization of human pluripotent stem cell‐derived brain microvascular endothelial cells. Methods 2016. 101: 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lippmann, E.S. , Azarin, S.M. , Kay, J.E. , Nessler, R.A. , Wilson, H.K. , Al‐Ahmad, A. , Palecek, S.P. et al., Derivation of blood‐brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 2012. 30: 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qian, T. , Maguire, S.E. , Canfield, S.G. , Bao, X. , Olson, W.R. , Shusta, E.V. and Palecek, S.P. , Directed differentiation of human pluripotent stem cells to blood‐brain barrier endothelial cells. Sci. Adv. 2017. 3: e1701679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lim, R.G. , Quan, C. , Reyes‐Ortiz, A.M. , Lutz, S.E. , Kedaigle, A.J. , Gipson, T.A. , Wu, J. et al., Huntington's disease iPSC‐derived brain microvascular endothelial cells reveal WNT‐mediated angiogenic and blood‐brain barrier deficits. Cell Rep. 2017. 19: 1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vatine, G.D. , Al‐Ahmad, A. , Barriga, B.K. , Svendsen, S. , Salim, A. , Garcia, L. , Garcia, V.J. et al., Modeling psychomotor retardation using iPSCs from MCT8‐deficient patients indicates a prominent role for the blood‐brain barrier. Cell Stem Cell 2017. 20: 831–843.e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vatine, G.D. , Barrile, R. , Workman, M.J. , Sances, S. , Barriga, B.K. , Rahnama, M. , Barthakur, S. et al., Human iPSC‐derived blood‐brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell 2019. 24: 995–1005.e1006. [DOI] [PubMed] [Google Scholar]
- 62. Kooij, G. , Kopplin, K. , Blasig, R. , Stuiver, M. , Koning, N. , Goverse, G. , Van Der Pol, S.M.A. et al., Disturbed function of the blood‐cerebrospinal fluid barrier aggravates neuro‐inflammation. Acta Neuropathol. 2014. 128: 267–277. [DOI] [PubMed] [Google Scholar]
- 63. Brioschi, S. , Wang, W.‐L. , Peng, V. , Wang, M. , Shchukina, I. , Greenberg, Z.J. , Bando, J.K. et al., Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science 2021. 373: eabf9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schafflick, D. , Wolbert, J. , Heming, M. , Thomas, C. , Hartlehnert, M. , Börsch, A.‐L. , Ricci, A. et al., Single‐cell profiling of CNS border compartment leukocytes reveals that B cells and their progenitors reside in non‐diseased meninges. Nat. Neurosci. 2021. 24: 1225–1234. [DOI] [PubMed] [Google Scholar]
- 65. Pierson, E. , Simmons, S.B. , Castelli, L. and Goverman, J.M. , Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol. Rev. 2012. 248: 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Codarri, L. , Gyülvészi, G. , Tosevski, V. , Hesske, L. , Fontana, A. , Magnenat, L. , Suter, T. et al., RORγt drives production of the cytokine GM‐CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011. 12: 560–567. [DOI] [PubMed] [Google Scholar]
- 67. Zielinski, C.E. , Mele, F. , Aschenbrenner, D. , Jarrossay, D. , Ronchi, F. , Gattorno, M. , Monticelli, S. et al., Pathogen‐induced human TH17 cells produce IFN‐gamma or IL‐10 and are regulated by IL‐1beta. Nature 2012. 484: 514–518. [DOI] [PubMed] [Google Scholar]
- 68. Van Langelaar, J. , Van Der Vuurst De Vries, R.M. , Janssen, M. , Wierenga‐Wolf, A.F. , Spilt, I.M. , Siepman, T.A. , Dankers, W. et al., T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain 2018. 141: 1334–1349. [DOI] [PubMed] [Google Scholar]
- 69. Reboldi, A. , Coisne, C. , Baumjohann, D. , Benvenuto, F. , Bottinelli, D. , Lira, S. , Uccelli, A. et al., C‐C chemokine receptor 6‐regulated entry of TH‐17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009. 10: 514–523. [DOI] [PubMed] [Google Scholar]
- 70. Rothhammer, V. , Heink, S. , Petermann, F. , Srivastava, R. , Claussen, M.C. , Hemmer, B. and Korn, T. , Th17 lymphocytes traffic to the central nervous system independently of alpha4 integrin expression during EAE. J. Exp. Med. 2011. 208: 2465–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Glatigny, S. , Duhen, R. , Oukka, M. and Bettelli, E. , Cutting edge: loss of alpha4 integrin expression differentially affects the homing of Th1 and Th17 cells. J. Immunol. 2011. 187: 6176–6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hiltensperger, M. , Beltrán, E. , Kant, R. , Tyystjärvi, S. , Lepennetier, G. , Domínguez Moreno, H. , Bauer, I.J. et al., Skin and gut imprinted helper T cell subsets exhibit distinct functional phenotypes in central nervous system autoimmunity. Nat. Immunol. 2021. 22: 880–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Friese, M.A. and Fugger, L. , Autoreactive CD8+ T cells in multiple sclerosis: a new target for therapy? Brain 2005. 128: 1747–1763. [DOI] [PubMed] [Google Scholar]
- 74. Liblau, R.S. , Wong, F.S. , Mars, L.T. and Santamaria, P. , Autoreactive CD8 T cells in organ‐specific autoimmunity: emerging targets for therapeutic intervention. Immunity 2002. 17: 1–6. [DOI] [PubMed] [Google Scholar]
- 75. Neumann, H. , Medana, I.M. , Bauer, J. and Lassmann, H. , Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002. 25: 313–319. [DOI] [PubMed] [Google Scholar]
- 76. Van Nierop, G.P. , Van Luijn, M.M. , Michels, S.S. , Melief, M.‐J. , Janssen, M. , Langerak, A.W. , Ouwendijk, W.J.D. et al., Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. 2017. 134: 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lucchinetti, C.F. , Popescu, B.F.G. , Bunyan, R.F. , Moll, N.M. , Roemer, S.F. , Lassmann, H. , Brück, W. et al., Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 2011. 365: 2188–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Babbe, H. , Roers, A. , Waisman, A. , Lassmann, H. , Goebels, N. , Hohlfeld, R. , Friese, M. et al., Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000. 192: 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Junker, A. , Ivanidze, J. , Malotka, J. , Eiglmeier, I. , Lassmann, H. , Wekerle, H. , Meinl, E. et al., Multiple sclerosis: T‐cell receptor expression in distinct brain regions. Brain 2007. 130: 2789–2799. [DOI] [PubMed] [Google Scholar]
- 80. Jacobsen, M. , Cepok, S. , Quak, E. , Happel, M. , Gaber, R. , Ziegler, A. , Schock, S. et al., Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain 2002. 125: 538–550. [DOI] [PubMed] [Google Scholar]
- 81. Mars, L.T. , Saikali, P. , Liblau, R.S. and Arbour, N. , Contribution of CD8 T lymphocytes to the immuno‐pathogenesis of multiple sclerosis and its animal models. Biochim. Biophys. Acta 2011. 1812: 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hohlfeld, R. , Dornmair, K. , Meinl, E. and Wekerle, H. , The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol. 2015.15:198–209. [DOI] [PubMed] [Google Scholar]
- 83. Hohlfeld, R. , Dornmair, K. , Meinl, E. and Wekerle, H. , The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse‐translational research. Lancet Neurol. 2016. 15: 317–331. [DOI] [PubMed] [Google Scholar]
- 84. Singer, B.A. , Parenteral treatment of multiple sclerosis: the advent of monoclonal antibodies. Semin. Neurol. 2016. 36: 140–147. [DOI] [PubMed] [Google Scholar]
- 85. Steinman, L. and Zamvil, S.S. , Beginning of the end of two‐stage theory purporting that inflammation then degeneration explains pathogenesis of progressive multiple sclerosis. Curr. Opin. Neurol. 2016. 29: 340–344. [DOI] [PubMed] [Google Scholar]
- 86. Saxena, A. , Martin‐Blondel, G. , Mars, L.T. and Liblau, R.S. , Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett. 2011. 585: 3758–3763. [DOI] [PubMed] [Google Scholar]
- 87. Link, H. and Huang, Y.M. , Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J. Neuroimmunol. 2006. 180: 17–28. [DOI] [PubMed] [Google Scholar]
- 88. Obermeier, B. , Mentele, R. , Malotka, J. , Kellermann, J. , Kümpfel, T. , Wekerle, H. , Lottspeich, F. et al., Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 2008. 14: 688–693. [DOI] [PubMed] [Google Scholar]
- 89. Brändle, S.M. , Obermeier, B. , Senel, M. , Bruder, J. , Mentele, R. , Khademi, M. , Olsson, T. et al., Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self‐proteins. Proc. Natl. Acad. Sci. U. S. A. 2016. 113: 7864–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cepok, S. , Jacobsen, M. , Schock, S. , Omer, B. , Jaekel, S. , Böddeker, I. , Oertel, W.H. et al., Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. Brain 2001. 124: 2169–2176. [DOI] [PubMed] [Google Scholar]
- 91. Cepok, S. , Rosche, B. , Grummel, V. , Vogel, F. , Zhou, D. , Sayn, J. , Sommer, N. et al., Short‐lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain 2005. 128: 1667–1676. [DOI] [PubMed] [Google Scholar]
- 92. Cepok, S. , Von Geldern, G. , Grummel, V. , Hochgesand, S. , Celik, H. , Hartung, H. and Hemmer, B. Accumulation of class switched IgD‐IgM‐memory B cells in the cerebrospinal fluid during neuroinflammation. J. Neuroimmunol. 2006. 180: 33–39. [DOI] [PubMed] [Google Scholar]
- 93. Lassmann, H. , Brück, W. and Lucchinetti, C.F. , The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007. 17: 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Barnett, M.H. , Parratt, J.D. , Cho, E.S. and Prineas, J.W. , Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann. Neurol. 2009. 65: 32–46. [DOI] [PubMed] [Google Scholar]
- 95. Serafini, B. , Rosicarelli, B. , Magliozzi, R. , Stigliano, E. , Aloisi, F. , Detection of ectopic B‐cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004. 14: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Choi, S.R. , Howell, O.W. , Carassiti, D. , Magliozzi, R. , Gveric, D. , Muraro, P.A. , Nicholas, R. et al., Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012. 135: 2925–2937. [DOI] [PubMed] [Google Scholar]
- 97. Magliozzi, R. , Howell, O. , Vora, A. , Serafini, B. , Nicholas, R. , Puopolo, M. , Reynolds, R. et al., Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007. 130: 1089–1104. [DOI] [PubMed] [Google Scholar]
- 98. Magliozzi, R. , Howell, O.W. , Reeves, C. , Roncaroli, F. , Nicholas, R. , Serafini, B. , Aloisi, F. et al., A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010. 68: 477–493. [DOI] [PubMed] [Google Scholar]
- 99. Howell, O.W. , Reeves, C.A. , Nicholas, R. , Carassiti, D. , Radotra, B. , Gentleman, S.M. , Serafini, B. et al., Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011. 134: 2755–2771. [DOI] [PubMed] [Google Scholar]
- 100. Hartlehnert, M. , Börsch, A.‐L. , Li, X. , Burmeister, M. , Gerwien, H. , Schafflick, D. , Heming, M. et al., Bcl6 controls meningeal Th17‐B cell interaction in murine neuroinflammation. Proc. Natl. Acad. Sci. U. S. A. 2021. 118: e2023174118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cross, A.H. , Stark, J.L. , Lauber, J. , Ramsbottom, M.J. and Lyons, J.A. , Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 2006. 180: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. del Pilar Martin, M. , Cravens, P.D. , Winger, R. , Kieseier, B.C. , Cepok, S. , Eagar, T.N. , Zamvil, S.S. et al., Depletion of B lymphocytes from cerebral perivascular spaces by rituximab. Arch. Neurol. 2009. 66: 1016–1020. [DOI] [PubMed] [Google Scholar]
- 103. Hauser, S.L. , Waubant, E. , Arnold, D.L. , Vollmer, T. , Antel, J. , Fox, R.J. , Bar‐Or, A. et al., B‐cell depletion with rituximab in relapsing‐remitting multiple sclerosis. N. Engl. J. Med. 2008. 358: 676–688. [DOI] [PubMed] [Google Scholar]
- 104. Hauser, S.L. , Bar‐Or, A. , Comi, G. , Giovannoni, G. , Hartung, H.‐P. , Hemmer, B. , Lublin, F. et al., Ocrelizumab versus interferon Beta‐1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017. 376: 221–234. [DOI] [PubMed] [Google Scholar]
- 105. Hawker, K. , O'connor, P. , Freedman, M.S. , Calabresi, P.A. , Antel, J. , Simon, J. , Hauser, S. et al., Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double‐blind placebo‐controlled multicenter trial. Ann. Neurol. 2009. 66: 460–471. [DOI] [PubMed] [Google Scholar]
- 106. Montalban, X. , Hauser, S.L. , Kappos, L. , Arnold, D.L. , Bar‐Or, A. , Comi, G. , De Seze, J. et al., Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017. 376: 209–220. [DOI] [PubMed] [Google Scholar]
- 107. Engelhardt, B. , Private immune protection at the border of the central nervous system. Nature 2021. 596: 38–40. [DOI] [PubMed] [Google Scholar]
- 108. Jordão, M.J.C. , Sankowski, R. , Brendecke, S.M. , Sagar, Locatelli, G. , Tai, Y.‐H. , Tay, T. L. et al., Single‐cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019. 363: eaat7554. [DOI] [PubMed] [Google Scholar]
- 109. Heppner, F.L. , Greter, M. , Marino, D. , Falsig, J. , Raivich, G. , Hövelmeyer, N. , Waisman, A. et al., Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 2005. 11: 146–152. [DOI] [PubMed] [Google Scholar]
- 110. Neumann, H. , Kotter, M.R. and Franklin, R.J. , Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 2009. 132: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yamasaki, R. , Lu, H. , Butovsky, O. , Ohno, N. , Rietsch, A.M. , Cialic, R. , Wu, P.M. et al., Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014. 211: 1533–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lloyd, A.F. , Davies, C.L. , Holloway, R.K. , Labrak, Y. , Ireland, G. , Carradori, D. , Dillenburg, A. et al., Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 2019. 22: 1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wang, J. , Wang, J. , Wang, J. , Yang, B. , Weng, Q. and He, Q. , Targeting microglia and macrophages: a potential treatment strategy for multiple sclerosis. Front. Pharmacol. 2019. 10: 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Voet, S. , Prinz, M. and van Loo, G. , Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med. 2019. 25: 112–123. [DOI] [PubMed] [Google Scholar]
- 115. Montalban, X. , Arnold, D.L. , Weber, M.S. , Staikov, I. , Piasecka‐Stryczynska, K. , Willmer, J. , Martin, E.C. et al., Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 2019. 380: 2406–2417. [DOI] [PubMed] [Google Scholar]
- 116. Reich, D.S. , Arnold, D.L. , Vermersch, P. , Bar‐Or, A. , Fox, R.J. , Matta, A. , Turner, T. et al., Safety and efficacy of tolebrutinib, an oral brain‐penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2021. 20: 729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Comabella, M. and Montalban, X. , Body fluid biomarkers in multiple sclerosis. Lancet Neurol. 2014. 13: 113–126. [DOI] [PubMed] [Google Scholar]
- 118. Chan, A. , Pirmohamed, M. and Comabella, M. , Pharmacogenomics in neurology: current state and future steps. Ann. Neurol. 2011. 70: 684–697. [DOI] [PubMed] [Google Scholar]
- 119. Delcoigne, B. , Manouchehrinia, A. , Barro, C. , Benkert, P. , Michalak, Z. , Kappos, L. , Leppert, D. et al., Blood neurofilament light levels segregate treatment effects in multiple sclerosis. Neurology 2020. 94: e1201–e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sormani, M.P. , Haering, D.A. , Kropshofer, H. , Leppert, D. , Kundu, U. , Barro, C. , Kappos, L. et al., Blood neurofilament light as a potential endpoint in Phase 2 studies in MS. Ann. Clin. Transl. Neurol. 2019. 6: 1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dalla Costa, G. , Martinelli, V. , Moiola, L. , Sangalli, F. , Colombo, B. , Finardi, A. , Cinque, P. et al., Serum neurofilaments increase at progressive multifocal leukoencephalopathy onset in natalizumab‐treated multiple sclerosis patients. Ann. Neurol. 2019. 85: 606–610. [DOI] [PubMed] [Google Scholar]
- 122. Fissolo, N. , Pignolet, B. , Rio, J. , Vermersch, P. , Ruet, A. , Desèze, J. , Labauge, P. et al., Serum neurofilament levels and PML risk in patients with multiple sclerosis treated with natalizumab. Neurol. Neuroimmunol. Neuroinflamm. 2021. 8: e1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lublin, F.D. , Baier, M. and Cutter, G. , Effect of relapses on development of residual deficit in multiple sclerosis. Neurology 2003. 61: 1528–1532. [DOI] [PubMed] [Google Scholar]
- 124. Bevan, C. and Gelfand, J.M. , Therapeutic management of severe relapses in multiple sclerosis. Curr. Treat. Options Neurol. 2015. 17: 345. [DOI] [PubMed] [Google Scholar]
- 125. Schweingruber, N. , Reichardt, S.D. , Lühder, F. and Reichardt, H.M. , Mechanisms of glucocorticoids in the control of neuroinflammation. J. Neuroendocrinol. 2012. 24: 174–182. [DOI] [PubMed] [Google Scholar]
- 126. Schweingruber, N. , Fischer, H.J. , Fischer, L. , Van Den Brandt, J. , Karabinskaya, A. , Labi, V. , Villunger, A. et al., Chemokine‐mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. 2014. 127: 713–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Nickerson, M. and Marrie, R.A. , The multiple sclerosis relapse experience: patient‐reported outcomes from the North American Research Committee on Multiple Sclerosis (NARCOMS) Registry. BMC Neurol. 2013. 13: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Stork, L. , Ellenberger, D. , Beißbarth, T. , Friede, T. , Lucchinetti, C.F. , Brück, W. and Metz, I. , Differences in the reponses to apheresis therapy of patients with 3 histopathologically classified immunopathological patterns of multiple sclerosis. JAMA Neurol. 2018. 75: 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Aboul‐Enein, F. , Rauschka, H. , Kornek, B. , Stadelmann, C. , Stefferl, A. , Brück, W. , Lucchinetti, C. et al., Preferential loss of myelin‐associated glycoprotein reflects hypoxia‐like white matter damage in stroke and inflammatory brain diseases. J. Neuropathol. Exp. Neurol. 2003. 62: 25–33. [DOI] [PubMed] [Google Scholar]
- 130. Lucchinetti, C. , Brück, W. , Parisi, J. , Scheithauer, B. , Rodriguez, M. and Lassmann, H. , Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 2000. 47: 707–717. [DOI] [PubMed] [Google Scholar]
- 131. Fox, R.J. , Tissue markers for acute multiple sclerosis treatment response: a step toward personalized medicine. JAMA Neurol. 2018. 75: 406–407. [DOI] [PubMed] [Google Scholar]
- 132. Barnes, P.J. and Adcock, I.M. , Glucocorticoid resistance in inflammatory diseases. Lancet 2009. 373: 1905–1917. [DOI] [PubMed] [Google Scholar]
- 133. Matysiak, M. , Makosa, B. , Walczak, A. and Selmaj, K. , Patients with multiple sclerosis resisted to glucocorticoid therapy: abnormal expression of heat‐shock protein 90 in glucocorticoid receptor complex. Mult. Scler. 2008. 14: 919–926. [DOI] [PubMed] [Google Scholar]
- 134. Gold, S.M. , Sasidhar, M.V. , Lagishetty, V. , Spence, R.D. , Umeda, E. , Ziehn, M.O. , Krieger, T. et al., Dynamic development of glucocorticoid resistance during autoimmune neuroinflammation. J. Clin. Endocrinol. Metab. 2012. 97: E1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Du, J. , Li, M. , Zhang, D. , Zhu, X. , Zhang, W. , Gu, W. , Feng, Y. et al., Flow cytometry analysis of glucocorticoid receptor expression and binding in steroid‐sensitive and steroid‐resistant patients with systemic lupus erythematosus. Arthritis Res. Ther. 2009. 11: R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kleinschnitz, C. , Blecharz, K. , Kahles, T. , Schwarz, T. , Kraft, P. , Göbel, K. , Meuth, S.G. et al., Glucocorticoid insensitivity at the hypoxic blood‐brain barrier can be reversed by inhibition of the proteasome. Stroke 2011. 42: 1081–1089. [DOI] [PubMed] [Google Scholar]
- 137. Hoepner, R. , Bagnoud, M. , Pistor, M. , Salmen, A. , Briner, M. , Synn, H. , Schrewe, L. et al., Vitamin D increases glucocorticoid efficacy via inhibition of mTORC1 in experimental models of multiple sclerosis. Acta Neuropathol. 2019. 138: 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Delgoffe, G.M. , Pollizzi, K.N. , Waickman, A.T. , Heikamp, E. , Meyers, D.J. , Horton, M.R. , Xiao, B. et al., The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011. 12: 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Crino, P.B. , The mTOR signalling cascade: paving new roads to cure neurological disease. Nat. Rev. Neurol. 2016. 12: 379–392. [DOI] [PubMed] [Google Scholar]
- 140. Gili‐Kovács, J. , Hoepner, R. , Salmen, A. , Bagnoud, M. , Gold, R. , Chan, A. and Briner, M. ,An algorithm using clinical data to predict the optimal individual glucocorticoid dosage to treat multiple sclerosis relapses. Ther. Adv. Neurol. Disord. 2021. 14: 17562864211020074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Pittock, S.J. , Zekeridou, A. and Weinshenker, B.G. , Hope for patients with neuromyelitis optica spectrum disorders: from mechanisms to trials. Nat. Rev. Neurol. 2021. 17: 759–773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.