

Abstract
The potential to use quantitative image analysis and artificial intelligence is one of the driving forces behind digital pathology. However, despite novel image analysis methods for pathology being described across many publications, few become widely adopted and many are not applied in more than a single study. The explanation is often straightforward: software implementing the method is simply not available, or is too complex, incomplete, or dataset‐dependent for others to use. The result is a disconnect between what seems already possible in digital pathology based upon the literature, and what actually is possible for anyone wishing to apply it using currently available software. This review begins by introducing the main approaches and techniques involved in analysing pathology images. I then examine the practical challenges inherent in taking algorithms beyond proof‐of‐concept, from both a user and developer perspective. I describe the need for a collaborative and multidisciplinary approach to developing and validating meaningful new algorithms, and argue that openness, implementation, and usability deserve more attention among digital pathology researchers. The review ends with a discussion about how digital pathology could benefit from interacting with and learning from the wider bioimage analysis community, particularly with regard to sharing data, software, and ideas. © 2022 The Author. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: digital pathology, computational pathology, image processing, image analysis, open science, software
Introduction
The growth of digital pathology and whole‐slide imaging have created the opportunity to extract more information from histological samples through image analysis. However, despite considerable progress over the last decade, digital pathology analysis remains difficult to employ in practice and much of its promise remains to be fulfilled.
The core ideas of image analysis are quite straightforward, although applying them is not. A digital image is composed of pixels. In pathology, most images are brightfield whole‐slide scans in RGB format: this means that each pixel comprises three numbers—usually 8‐bit integers in the range 0–255—that together represent the red, green, and blue components of the colour used to display the pixel. A typical whole‐slide image can therefore be thought of as a Width × Height × 3 array; the width and height often exceed 100,000 pixels each, so the raw data comprises billions of numbers. The challenge of analysis is to identify and interpret meaningful patterns within these numbers—and to do so in a way that is robust to variation from multifarious sources, including biology, tissue processing, staining, and scanning. Even a small study containing tens of images requires us to grapple with trillions of pixels, from which we often want to extract at most a few actionable insights per image.
The richness of histopathology imaging data means that a single whole‐slide scan affords a plethora of possibilities for analysis. Ideally, our choice would be driven by the precise question we want to answer from the image. In practice, we are limited by the tools at our disposal and how we use them. Most pathologists do not write software, while few algorithm and software developers know very much about pathology; if digital pathology software is to be useful, therefore, both sides need to be able to communicate effectively with one another. This involves having an accurate perception of the strengths and limitations of each discipline, focussing on areas where computational methods can have a meaningful impact.
Numerous recent digital pathology and artificial intelligence (AI) reviews provide an excellent overview of progress towards clinical applications [1, 2, 3, 4, 5, 6]. Here, I aim to offer a practical assessment of the use and development of digital pathology tools in research today. After distinguishing between approaches and techniques, I describe some of the main challenges faced by people who currently use digital pathology tools to analyse their data. This review ends with a discussion concerning how digital pathology could benefit from insights and practices from the broader field of bioimage analysis, particularly with regard to open data and software.
Approaches to digital pathology analysis
Digital pathology analysis can be broadly categorised into two main approaches: quantitative analysis and AI‐driven assessment. Although any specific application might require a combination of both, one approach usually dominates. Making this distinction can help to elucidate issues concerning how algorithms are developed and validated, and where a pathologist's knowledge fits into the digital system.
Quantitative analysis
Historically, pathology image analysis has focussed on some combination of detecting, classifying, counting, and/or quantifying specific ‘objects’ visible within the image. These objects may be of different kinds and identified at different scales. For example, determining whether a slide contains evidence of invasive primary tumour [7], or metastatic tumour [8], or Helicobacter pylori infection [9] are all primarily detection tasks. Often, what is detected needs to also be classified; for example, nuclei might be classified according to different cell types, and this used to determine metrics predictive of therapeutic response, such as the relative proportions of lymphocytes and tumour cells [10]. Alternatively, we might eschew cell detection in favour of classifying pixels directly as belonging to tumour epithelium, stroma, or other tissue types, and from this quantify the areas occupied by each tissue class (for example, to assess fat proportion [11], or the tumour‐stroma ratio [12, 13]). Each of the suggested analyses could be applied to the same whole‐slide scan of a tissue section, depending upon the questions we wish to address.
The common feature of such quantitative analysis is that it is concerned with assessing something that is well‐defined and visible. In principle, it replicates what a pathologist could ascertain by looking at a slide. A knowledgeable observer can determine whether the analysis has been performed appropriately by visualising what has been detected, classified, and quantified.
AI‐based assessment
The problems that may be addressed through quantitative analysis are often surrogates for what we really want to know. This includes questions concerning what diagnostic or prognostic information we can determine from the image, or to which treatment a specific patient is likely to respond.
Much recent work in digital pathology has focussed on demonstrating how such questions may be approached more directly—without explicitly detecting or quantifying specific features. An early example demonstrated that AI techniques could predict mutations in six commonly mutated genes in lung adenocarcinoma [14]. Similar strategies have since been applied to predict mutations for many more genes across a wide range of tissue types [15, 16, 17, 18, 19]. Other studies have shown that AI may be used to directly predict patient outcome from hematoxylin and eosin [H&E] slide scans [20, 21, 22, 23].
In these cases, the output is a prediction that is not based on directly measuring any particular feature or structure. This makes verifying the result more difficult, at least on a per‐image basis. Confidence needs to be earned through: (1) large‐scale validation studies using diverse datasets, and (2) the ability to visualise regions of the slide that contributed more or less strongly to the result. Such visualisations can reveal that the AI has learned to base its predictions of regions of the image that are already known to be clinically relevant [15, 16].
Hybrid approaches
While not all digital pathology applications fit neatly into the two categories above, we may still distinguish between outputs that are amenable to visual verification and those that are not. For example, numerous AI‐based methods have been developed for Gleason grading [24, 25]. Some include elements of object detection and quantification before the application of AI, whereas others operate more directly on the pixels without explicit detection; either way, a pathologist can judge the algorithm's performance by comparing the final generated gradings with their own assessments. This differs from an AI‐based approach to prostate cancer risk stratification [26] or cancer recurrence prediction [27], designed without recourse to any established grading system, which is less amenable to visual verification—and therefore perhaps less appealing to pathologists in terms of adoption.
Techniques for image analysis
Regardless of approach, the fundamental challenge of digital pathology remains the same: to uncover patterns among the pixels. This involves applying mathematical operations to the numbers in the input image, typically in a way that progressively transforms the image into a form in which the key features can be separated from everything else. While each individual operation may be straightforward, complexity ensues whenever hundreds—or even thousands—of such operations are combined into an algorithm that is applied to billions of pixels. Nevertheless, recognising the essential simplicity of building blocks used to construct digital pathology algorithms is central to understanding their strengths and predicting their limitations. In some cases, the success or failure of a complex analysis can come down to a single cutoff threshold applied at a key step.
Conventional image processing
Conventional image processing involves explicitly defining the processing operations involved, typically drawn from a wide range of established techniques. For example, processing an H&E image often begins with stain separation by colour deconvolution [28]; this effectively recombines the red, green, and blue values for each pixel using a weighted sum characterised by the stain colour. This may be followed by a convolution: an image filtering operation that replaces each pixel by a weighted sum of the neighbouring pixels; the weights are defined by a convolution kernel, and different kernels result in output images that highlight different kinds of feature at different scales (e.g. filamental structures, edges, blobs of different sizes). Convolution is often applied to duplicates of the image using different kernels, then recombined by adding or subtracting corresponding pixels. Eventually, an image is produced wherein the pixel values corresponding to structures of interest can be separated from all other pixels by applying a threshold, thereby generating a binary image representing distinct objects that can be measured. Some additional operations may be needed (e.g. nonlinear filters, distance or watershed transforms) to adequately split clustered objects or refine boundaries. A link to an overview of these techniques is provided in the ‘Data availability statement’ section.
The digital pathology literature is replete with image processing publications at all levels: describing individual operations (e.g. new approaches to stain separation), combinations of operations for generic tasks (e.g. nucleus detection), and full algorithms devised for specific applications (e.g. Ki67 evaluation in breast cancer). In each case, the processing is hand‐crafted and deterministic. Core operations can be endlessly adjusted and recombined to construct different algorithms for different purposes.
Machine learning
Crafting robust image processing algorithms requires substantial effort and a good understanding of the data. It also takes imagination: the developer needs to guard against the many ways in which the algorithm might fail on unseen images, artefacts, and anomalies. In practice, this can never be entirely successful: it is hard to think of everything that might go wrong, and even recognised problems are hard to overcome.
This would be easier if the computer could exhibit human‐like intelligence, informed by example and experience. Such AI can be achieved (to an extent) by using the techniques of machine learning to train a model capable of making useful predictions on new data. Here, we will focus on supervised machine learning, whereby the model is trained to make predictions using labelled data with a defined target. This is in contrast to unsupervised approaches, which may be used to find clusters in data whenever labels are unavailable [29].
Developing a supervised machine‐learning algorithm for digital pathology requires inputs with associated labels, a model capable of making predictions from the inputs, and a loss function that computes an error between a prediction and a label. The goal during training is to iteratively refine the model until the loss between predictions and labels is minimised. The concept is very generic, and labels take different forms depending upon the task at hand. For example, regions annotated by a pathologist might be used to derive a label for every pixel of an image, and used to train a model that generates similar labels from new images that we convert to objects for quantitative analysis. Alternatively, an image may have a single associated label—perhaps based upon a pathologist's assessment, or other available data—and the model should also make a single prediction, such as mutational status or patient outcome.
Traditionally, machine‐learning models have been based on techniques such as random forests, support vector machines, and logistic regression [29, 30]. The algorithm developer chooses features from the image that could be relevant for prediction, and which will be provided as model inputs. These features are often the result of applying common image processing operations (e.g. convolutional filters with predefined kernels), although they might also be determined from objects previously detected in the image (e.g. the size, shape,and density of nuclei). Thus, image processing is still involved, but the developer does not explicitly define all the operations; rather, they tune the algorithm indirectly through the choice of training data, features, and model.
Deep learning refers to a subset of machine‐learning methods that have proven particularly powerful across multiple domains [1, 2]. For imaging applications, a deep‐learning model is frequently a type of convolutional neural network (CNN). As the name suggests, a CNN also relies upon convolution to generate features—however, the kernels themselves are learned during training. This has a profound impact upon what can be achieved. By learning hundreds of such convolution filters and applying them in combination with other nonlinear transforms and resizing operations, deep learning effectively frees us from the limits of human imagination in defining the input features. In practice, this makes it possible to identify far more complex or subtle patterns than could be found using other contemporary approaches—at a cost of requiring much more computational power.
Comparison of techniques
This very brief overview aims to demonstrate the overlaps between image processing, machine‐learning, and deep‐learning approaches to digital pathology analysis. Ultimately, all are applied to the pixel values of the image. Convolution—scaling and summing neighbouring pixel values—plays a starring role in each case.
An advantage of using conventional image processing exclusively to develop an algorithm is that the methods are well‐defined and tractable. The developer tunes performance by setting key parameters, such as filter sizes and thresholds. Simplicity is a virtue: an algorithm that uses a small number of intuitive parameters is easy to apply and adapt to work on new images, whereas a complicated, hand‐tuned algorithm is likely to be brittle and overfit to one dataset. However, simplicity is also a limiting factor: the complexity of pathology data means that image processing alone is often insufficient.
A benefit of traditional machine learning is that the developer can focus on higher‐level questions: instead of devising fixed rules, they can provide training data and labels representing the images that should be handled. Developing and applying traditional machine‐learning models can also be very fast: a model can be trained in a matter of seconds while interactively annotating an image, and progressively refined if required [13], although a more structured approach to model training across multiple images is usually preferable. However, even with extensive training the success will ultimately depend upon the usefulness of the input features, which may simply not be informative enough. Since key parameters are wrapped up inside the model, they cannot be readily tuned to work on new images. When the algorithm fails, we need to train a new model.
Most of the observations regarding traditional machine learning also apply to deep learning, with two important caveats. The first is that training a deep‐learning model from scratch is usually much slower: typically requiring several hours or more, depending upon the model, training data, and computational resources available—although this can be substantially reduced if the training can instead be applied to fine‐tune an existing model. The second is that the model performance is much less limited by available features, although it remains constrained by the available training data and definition of the loss function.
In practice, all techniques have strengths and weaknesses. Sophisticated analysis problems usually require elements of them all to be combined (Figure 1 ).
Figure 1.
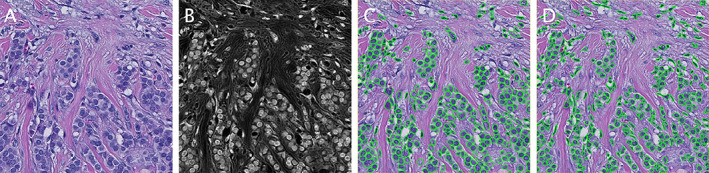
Nucleus segmentation using image processing, with and without deep learning. (A) Original H&E image. (B) Result of processing the image using colour deconvolution and image filtering to extract the haematoxylin information. (C) Segmented nuclei using a publicly available StarDist deep‐learning model trained for fluorescence data [44]. By using the processed image as input rather than the original, the model can achieve reasonable nucleus segmentation performance despite not being trained for H&E images. (D) Result of QuPath's built‐in cell detection using conventional image processing. The StarDist deep‐learning approach results in more regular contours and better handles densely‐packed regions, although the total number of nuclei detected are similar (319 and 331, respectively).
Analysis in practice
Anyone seeking to develop or apply digital pathology methods encounters many of the same challenges. Here I discuss some of the main difficulties, and consider ways they may be addressed.
Generalisation and bias
Limited generalisation affects all digital pathology analysis, and has been described as ‘probably the single most important obstacle for wide‐scale implementation of [computational pathology] techniques in the clinic’ [1]. A seemingly successful algorithm can be thwarted in different—and often subtle—ways when confronted by data that differs appreciably from that used for development. For example, the top‐ranked deep‐learning algorithms in the CAMEYLON17 grand challenge for detecting lymph node metastases were all reported to struggle with benign areas that occurred rarely in the training set [31]. Another study reported improved robustness by using both a much larger training dataset and weaker annotations (i.e. slide‐level labels, rather than contours outlining individual metastases), but still acknowledged a drop in performance when slides used for training and testing were acquired from different sources, or using different scanners [7]. Subtle, subvisual changes in input images can also result in quite different predictions using some deep‐learning approaches, which can even be used as a form of ‘attack,’ whereby an image is deliberately manipulated to cause a different prediction [32].
One strategy to address this is to include more diverse training images from different sources, acquired using different scanners. A problem, however, is that this can introduce learnable hidden variables, and thereby batch effects [33]. For example, a study applying deep learning to melanoma slides from five institutions demonstrated that it was possible to learn information about slide origin, scanner type, patient age, and even (to a lesser extent) slide preparation date [34]. A similar study showed that site‐specific signatures are identifiable within the images in The Cancer Genome Atlas (TCGA), and these have a relationship to ethnicity [35]. Such effects may provide an inflated estimate of accuracy or a systematic bias if the model learns characteristics that correlate with the training labels due to the cohort makeup, rather than the disease.
Another way to broaden the training data is to augment it using image‐processing operations that deliberately introduce random variations (e.g. in resolution, rotation, colour, and sharpness), thereby pushing the model towards learning more informative features [36]. Alternatively, one can take the reverse approach of making the model inputs more standardised at the prediction stage by stain normalisation [37, 38]. These methods are not exclusive, and the best results may be achieved by both broadening the model training with diverse and augmented data, and then narrowing the input variation with stain normalisation [36].
Cell detection
The problem of generalisation is particularly clear in the continued struggle to accurately detect cells. This is a fundamental part of many pathology analysis workflows. Although few topics in bioimage analysis have received as much attention as nucleus segmentation, a review in 2012 describes how it remained unsolved after more than half a century of effort [39]. The last decade has seen substantial progress using deep learning, with hints that a single solution that handles most tissue, staining, and scanning variation is achievable [40, 41, 42, 43, 44]. Nevertheless, more work is needed devise accurate, robust, and computationally‐efficient cell detection methods that are incorporated into widely‐used software. This is particularly important because failures in cell segmentation are not randomly distributed, but rather tend to increase with specific morphologies.
Boundaries and cutoffs
A benefit of image analysis is that it enables us to quantitatively answer more detailed questions from images. An inconvenience is that, freed from the imprecision of human visual estimation but lacking the expert's intuition, we are pushed to define what should be assessed to a higher degree of exactness. This means imposing hard boundaries where none may exist. For example, the area of a tumour region may be precisely determined—but only if one accepts a precise boundary defining the tumour. Pathologists annotating tumour regions draw quite different contours, each of which may be justifiable for a particular purpose, but each of which would have a different area [45]. Accepting the lack of a definitive ground truth, one may argue for the value of digital pathology because it reduces human subjectivity and improves reproducibility. However, the reality is more complicated. The problem of limited generalisation means that the algorithm may produce one boundary for an image scanned using a particular scanner, but quite different boundaries if the same slide is scanned on a different scanner. The extent to which this occurs needs to be explored case‐by‐case, but one should avoid assuming a priori that a digital method will reduce variation.
Similarly, when analysing immunohistochemistry (IHC) images, a cell may be classified as ‘positive’ or ‘negative’ (or 1+, 2+, 3+) by applying fixed cutoff thresholds based upon a summary measurement of the pixel values within the cell. Given that measured staining intensity is effectively a continuous variable, the precise choice of threshold can have a considerable impact upon output metrics—such as positive percentage, H‐score [46, 47], or Allred score [48]—if a significant proportion of cells are measured close to the threshold value. At first glance, digital scoring still seems preferable to relying upon a pathologist's visual impression, since the digital cutoff can be strictly defined and fully reproducible. However, if the threshold is held constant but the image would have different colour characteristics—perhaps due to staining variations, tissue thickness, or the choice of scanner—the analysis would still yield different results. Locking the algorithm parameters is therefore not in itself sufficient for reproducible analysis, unless one can also lock all other preanalytical variables that will impact the pixel values (Figure 2 ).
Figure 2.
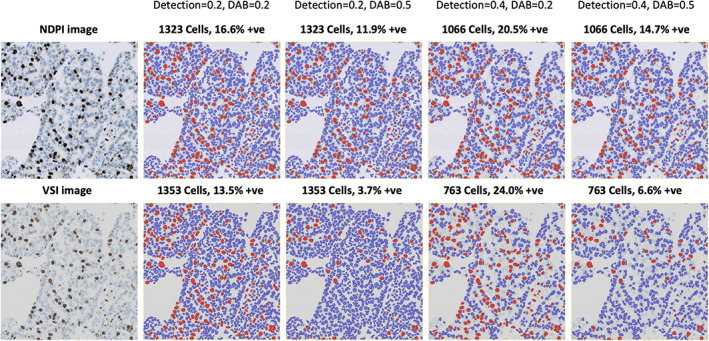
The impact of algorithm parameters and cutoff thresholds on images. QuPath's ‘positive cell detection’ command is used to determine Ki67 labelling indices for the same field of view, acquired using two different scanners. This conventional image‐processing algorithm uses multiple adjustable parameters, although here only the thresholds for nucleus detection and DAB positivity are varied. Horizontally adjacent images are from the same scanner, while vertically adjacent images are generated using the same thresholds. Detected nuclei are shown as red or blue, depending upon whether they are classified as positive or negative, respectively. Changing either threshold or the scanner can substantially change the results, although typically in predictable ways (e.g. a high detection threshold leads to negative nuclei being missed, and the labelling index is inflated; a high DAB threshold leads to positive nuclei being misclassified as negative, and the labelling index is reduced). Combining this knowledge with a careful evaluation of the markup images, it is possible for a user to identify and address many errors by adjusting algorithm parameters accordingly.
One might wish to mitigate this by avoiding hard thresholds, and using instead continuous measurements of staining intensity. While image analysis software can certainly derive numbers from such staining, these remain subject to imaging variations—and can easily be overinterpreted, e.g. if one ignores the biophysical properties of the diaminobenzidine (DAB) substrate [49]. Another tempting way to avoid the issue is to use machine learning to determine IHC positivity. This too requires caution, since it reduces tractability without necessarily improving results. For nuclear markers such as Ki67, we need to be careful that confounding features (e.g. nucleus size) cannot unduly influence any model prediction. Nevertheless, a machine‐learning approach may be justifiable for markers exhibiting complex staining patterns (e.g. PD‐L1), for which too much information is lost by simple summary metrics of staining intensity.
There are, of course, other data‐driven and adaptive approaches to determining IHC positivity cutoffs [50, 51]. The key point is that image analysis using digital pathology can give a range of plausible results based upon the interaction of the algorithm, its parameters, and the data under consideration—including all preanalytical sources of variation. We should continually resist the ‘illusion of objectivity’ by recognising potential errors and limitations, and incorporating this knowledge into how we design algorithms and interpret their outputs [52].
Working across disciplines
These observations should not be interpreted as undermining the importance of digital pathology, nor suggesting that it cannot or does not reduce subjectivity; rather, they emphasise that it will not do so necessarily. Generalising is hard, there are many pitfalls, and validation details matter. Developing truly robust methods requires expertise from a range of disciplines—including pathology, histology, imaging, image processing, machine‐learning, statistics, and epidemiology—to come together. These problems are being tackled, including through increased attention being given to ‘explainable AI’ for digital pathology [53] and the creation of guidelines for the use of AI in clinical trials [54, 55]. Software and algorithm developers can help by documenting their design decisions, drawing attention to known weak points and key parameters, and providing visualisations that make it easier for others to understand what the analysis is really doing.
Detection and classification remain the most difficult (and error‐prone) steps for most quantitative analyses. False‐positives and ‐negatives occur even when using a deep‐learning approach—particularly in the presence of mimics or artefacts. On the other hand, counting and quantification are often trivial from a computational perspective. This largely represents an inversion of a human pathologist's skillset: an experienced pathologist may reliably distinguish the structures of interest while ignoring mimics and artefacts, but cannot feasibly count a million classified cells or precisely determine a 1‐mm2 hotspot by eye alone. This suggests that an optimal arrangement may combine the strengths of human and computer, and factor the limitations of algorithms into how they are used. One pragmatic solution for quantitative analysis is to require manual input when defining a region of interest, to steer the analysis away from challenging areas or artefacts. A proposed approach for AI‐based assessment is to use AI as a form of triage, prioritising sensitivity over specificity [7].
Implementation, accessibility, and openness
The difficulty of developing robust, fully‐automated analysis methods may partly explain why few digital pathology algorithms have moved beyond publications into becoming available to pathologists. However, algorithmic challenges are only one consideration in the creation of useful digital pathology tools.
Proof‐of‐concept versus usable methods
Research groups developing computational methods have traditionally published articles describing their own bespoke algorithms, validated on their own in‐house datasets. If code and data are shared at all, it is often ‘by reasonable request’. This presents a considerable barrier to anyone who might wish to test whether the method works on their own data: they need permission and support from the original authors, who may be unable or unwilling to provide it [56]. The alternative is to try to reimplement the method from the published description—but this inevitably involves considerable effort, and guesswork where the description is incomplete [56].
This closed, ‘share‐on‐demand‐(perhaps)’ approach assumes that the key research contribution of a new computational method is the idea described in the article—relegating implementation to a technical detail [57]. However, implementation within software is crucial for the method to be used by anyone, and real‐world use determines the true value of the idea [39, 58].
The situation is improving as open research becomes required by some funders and journals [59, 60]. ‘Open’ does, however, afford considerable room for interpretation [61]. Although more papers include code as supplementary material, what is publicly shared is seldom sufficient to replicate the analysis or apply the method to new data [56]. The data used to train an AI model and the AI model itself are rarely made available—meaning that any researcher wishing to test a published method must still replicate a large amount of the work in the original study to train their own model. This means it can take weeks or months of effort to fill in the gaps to find out whether an ostensibly ‘open’ method works or not. There is often little incentive for someone with the requisite knowledge to put in the effort, because it is time not spent developing the ‘new’ methods they may need to advance their own careers. This contributes to a scientific literature replete with novel algorithms and proofs of concept, but few usable implementations available to the wider community. Different groups reinvent similar techniques, and there is a substantial disconnect between what seems possible (based on the literature) and what is possible (based upon available software).
Complications of sharing
The reasons for this are understandable: sharing data and code is hard. On one side, whole‐slide images are large: making them available requires data storage and bandwidth, which come at a cost. The images are also typically saved in bespoke, scanner‐specific file formats that include additional labels and metadata; this makes them difficult to anonymise both fully and with confidence that nothing has been missed [62]. Sharing also raises important questions around how the images may be used, including whether it is permitted to develop commercial AI models based on them; these must be resolved, with clear license statements provided, for others to safely use the shared data [63, 64]. While each may be surmountable, this combination of technical, financial, legal, and ethical hurdles can make sharing raw images extremely difficult.
Sharing code is also not without risk and complication [65, 66, 67]. An obvious risk is that the code may simply not do what it should. Bugs are inevitable in any sophisticated software, and researchers who share their code expose themselves to criticism—even retraction—if their software is shown not to do what is described in the paper [68]. But even if it works as described, making software open involves taking on a lot of additional work and responsibility [69, 70]. Code intended for public consumption typically needs to be of a higher standard than in‐house code; one cannot make simplifying assumptions (e.g. only supporting image files from a single scanner), and must pay considerably more attention to code‐quality, portability and ease‐of‐installation. Appropriate software licenses should be chosen [71], which can involve securing agreement from a range of stakeholders with different priorities (including principal investigators, funders, innovation departments). Then, if the software has sufficient appeal and the authors desire to maximise its usefulness, the work is really only beginning: users require documentation and ongoing support, perhaps lasting far beyond the grant that originally funded the work [69].
Common datasets
The problem of sharing the data used to develop an algorithm can be partially circumvented by using common public datasets. Notably, the pathology slides from TCGA and The Cancer Imaging Archive (TCIA) have been widely used by the digital pathology community [15, 16, 23, 33, 72, 73, 74, 75]. This has limits: TCGA/TCIA images are not intended to be definitive and cannot be fully representative [75]—for example, the majority are of frozen sections in Aperio's SVS file format—and exclusive use of TCGA to train and validate algorithms can lead to overly‐optimistic performance assessments [35]. Nevertheless, TCGA/TCIA have proven valuable and will continue to play an important role in advancing the field, alongside new image repositories such as that being created through the IMI‐BIGPICTURE project [76].
Grand challenges
Irrespective of whether the data are public or not, whenever a research group develops, validates, and publishes their own algorithm, it is impossible to guard against the limitations of publication bias and multiple hypothesis testing—however inadvertent this may be—because the algorithm that is published will inevitably be the one that ‘worked’. Grand challenges provide a way to address this [4]. A grand challenge involves organisers releasing a labelled dataset, along with detailed information about assessment metrics. Groups then compete to develop algorithms using these data and metrics. Crucially, the organisers withhold additional labelled data used to rank algorithm performance. Grand challenges are often held in conjunction with conferences, where the best‐performing algorithm is unveiled and the methods described in a later publication. Prominent examples for pathology include the CAMELYON16 and CAMELYON17 challenges to detect lymph node metastasis [8, 31], the GlaS challenge for colon gland segmentation [77], the BACH challenge to classify diagnostically relevant regions in breast cancer biopsies [78], and the PANDA challenge for Gleason grading of prostate cancer [25].
Grand challenges have proven extremely effective in galvanising the community—across both academia and industry—to work on solving a specific problem, with a consistent method of comparing performance. There are inevitably limitations, and impressive results in a challenge do not indicate that a problem has been ‘solved’ [31]. For example, the rankings do not necessarily reflect the algorithm that is ‘best’ in terms of generalisation and performance on most real‐world data, but rather only how algorithms fared using the specific test dataset and defined metrics [79]. Particularly when the same group may submit multiple algorithms, one can attempt to game the system or achieve a high score without necessarily developing a practical method that others may use. Nevertheless, grand challenges are open by design, and multiple groups competing in the challenge will be able to review and critique the data and metrics; this represents a major improvement over groups publishing incomparable work using their own in‐house data and assessment criteria. Recently, guidelines have been published that aim to improve the transparent reporting of challenges [80].
Open data and software
Much modern science depends upon Free and Open‐Source Software (FOSS), often referred to simply as ‘open software’. Users of FOSS tend to focus on the ‘free’ aspect in terms of ‘no financial cost’. However, proponents of open software use ‘free’ in a broader sense, referring also to the freedom to use the software for any purpose, to examine and modify the source code, and to redistribute both original and modified copies [71]. These elements are central to open software's unique contribution to research, enabling methods to not only be accessible to others, but to be interrogated, extended, enhanced, and shared by the wider community. Indeed, scanner vendors often provide image viewers that can be freely downloaded, but for which source code is not available, and numerous research publications provide software code only with restrictions (e.g. noncommercial use only); despite the lack of financial cost for the user, neither is FOSS.
Sharing digital pathology tools under a recognised FOSS license goes some way towards enabling them to be used freely by others. However, ‘software’ itself is a broad term: much software exists in the form of code libraries or scripts that lack the user‐friendly interface and visualisation capabilities required by nonprogrammers. Components such as these must be incorporated into larger software applications to be directly useful for pathologists. This larger application may or may not then be open itself; many companies integrate open‐source components into their proprietary software.
Developing, documenting, and supporting comprehensive software applications is complex, challenging, and time‐consuming [69, 81]. It is infeasible—and undesirable—for individual research groups to develop their own complete software applications simply to make their algorithms more usable. Fortunately, it is also not necessary. Several open software platforms exist that can solve the majority of the challenges around data management, visualisation, and interactivity while permitting others to add new algorithms and functionality for specific tasks.
The most important demonstration of this in the bioimaging community is ImageJ (NIH, Bethesda, MD, USA). Over more than a quarter of a century, ImageJ has dominated bioimage analysis and a recent review listed its direct precursor, NIH Image, as one of the ‘ten computer codes that transformed science’ [82]. Extensibility is key to ImageJ's continued success: when developers release new algorithms as ImageJ macros or plugins, users can install and run them, without needing to learn a whole new interface [83]. Several other bioimage analysis applications with a similar philosophy were later developed, including 3D Slicer [84], CellProfiler [85], and Icy [86].
All of these predate the growth of digital pathology, and none were designed to handle the specific challenges of whole‐slide image analysis. A few ImageJ plugins for pathology applications enable relatively straightforward processing techniques to low‐resolution images or cropped fields of view [87, 88]. Without full whole‐slide or AI support, these have limited use nowadays, but were nevertheless important in demonstrating the value of open methods being available for pathologists. More recently, several new open‐software applications have been developed with whole‐slide image analysis as a primary goal [89], including TMarker [90], Orbit [91], and QuPath [13] (desktop applications), and Cytomine [92] and the Digital Slide Archive [93] (web‐based platforms).
The importance of making research software that is open and flexible extends beyond enabling the analysis from a single paper to be reproduced. To take one example, QuPath's original publications had a narrow focus on colon and breast cancer biomarkers [13, 94], but the software has since been further developed and used in over 1,000 published journal articles across a wide range of diseases and applications. Openness means that the strengths and limitations of the tool can be freely explored, independently of the author's claims, and numerous studies have compared QuPath‐based analysis with both other software and with manual evaluation [95, 96, 97, 98, 99]. Groups using QuPath have made their scripts, extensions, and protocols available to aid reproducibility, in a way that could not be achieved without open software [10, 100, 101].
The value of openness applies not only to code and data. The Scientific Community Image Forum (https://image.sc) was established to help improve the practice of scientific image analysis [102]. It is now the primary discussion channel for more than 40 open‐software projects, including ImageJ, QuPath, Orbit, and Cytomine [69]. The forum aims to be inclusive and collaborative, with users from many disciplines contributing to discussions. Currently, image.sc contains over 200,000 posts across more than 28,000 different topics—each tagged and searchable—with more added each day. The forum acts both as a substitute for paid software support and as a venue for cross‐disciplinary discussions, while simultaneously providing community feedback directly to developers.
Outlook
The potential of deep learning for AI‐based assessment of clinical samples has been firmly established. Nevertheless, significant challenges remain concerning generalisation and validation if we are to see robust algorithms come into widespread clinical use [1]. These challenges cannot be adequately addressed by individual groups working solely with in‐house or public data. Rather, they will require long‐term projects incorporating insights from multiple disciplines, large and diverse datasets, and innovative approaches to training AI models at scale [62].
In the meantime, digital pathology analysis is already widely used. This is driven by the proliferation of whole‐slide scanners in research institutes, which has dramatically increased the number of scientists using histological samples for basic biology, preclinical, and veterinary studies. These data include H&E alongside other stains and IHC markers, across tissues, species, and image types—including multiplexed immunofluorescence and imaging mass cytometry data, the value of which can only be realised through computational analysis [103, 104]. The ubiquity brings concerns: digital pathology in no way changes the fact that biologists should engage with pathologists to properly evaluate such data [105, 106], but rather adds a need to understand the computational methods used as well. This is made difficult by a lack of pathologists, image analysis specialists, and capable software tools to meet the growing and varied demand [106, 107].
The growth of open science enables anyone with an interest in the field to contribute directly. This includes sharing data and code under open licenses where possible. Developers know that custom algorithms and AI‐models will often fail on new data, but this is no reason to withhold them: the limitations of the field need to be understood if they are to be addressed. In some cases, providing open software will enable more researchers to analyse their own data efficiently and effectively; in others, it may lead to finding that the software does not work as expected. Either outcome provides more value to the scientific community than a published method that lacks any usable implementation.
In the past, a pathologist engaging in digital pathology algorithm development might expect to spend hours laboriously outlining thousands of image regions, with no clear path leading to software they could later use. This should no longer be the case. Devising efficient annotation strategies is a computational problem that can be solved, while developers can make their algorithms accessible through open‐software platforms if they wish. Through initiatives like the Scientific Community Image Forum, pathologists can directly engage with algorithm and software developers, sharing their expertise and expressing their needs [102]. By understanding the strengths, challenges, and incentives of researchers and companies who develop digital pathology software, we can all contribute to a scientific culture that enables the field to advance more rapidly.
Author contributions statement
PB wrote the manuscript.
Acknowledgements
The author thanks Dr Maurice Loughrey and Dr Andrew Janowczyk for their helpful comments and feedback during the writing of this article. The original whole‐slide images used in the preparation of Figures 1 and 2 are from the OpenSlide freely‐distributable test data (https://openslide.org).
For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.
No conflicts of interest were declared.
Data Availability Statement
A short overview of the image processing terms used in this review is available at https://petebankhead.github.io/2022-image-processing-overview. The images and scripts used in the preparation of figures for this article are available at https://github.com/petebankhead/2022-qupath-cell-detection.
References
- 1. van der Laak J, Litjens G, Ciompi F. Deep learning in histopathology: the path to the clinic. Nat Med 2021; 27: 775–784. [DOI] [PubMed] [Google Scholar]
- 2. Srinidhi CL, Ciga O, Martel AL. Deep neural network models for computational histopathology: a survey. Med Image Anal 2021; 67: 101813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cui M, Zhang DY. Artificial intelligence and computational pathology. Lab Invest 2021; 101: 412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Serag A, Ion‐Margineanu A, Qureshi H, et al. Translational AI and deep learning in diagnostic pathology. Front Med (Lausanne) 2019; 6: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rakha EA, Toss M, Shiino S, et al. Current and future applications of artificial intelligence in pathology: a clinical perspective. J Clin Pathol 2021; 74: 409–414. [DOI] [PubMed] [Google Scholar]
- 6. Bera K, Schalper KA, Rimm DL, et al. Artificial intelligence in digital pathology ‐ new tools for diagnosis and precision oncology. Nat Rev Clin Oncol 2019; 16: 703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campanella G, Hanna MG, Geneslaw L, et al. Clinical‐grade computational pathology using weakly supervised deep learning on whole slide images. Nat Med 2019; 25: 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ehteshami Bejnordi B, Veta M, Johannes van Diest P, et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA 2017; 318: 2199–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou S, Marklund H, Blaha O, et al. Deep learning assistance for the histopathologic diagnosis of Helicobacter pylori . Intell Based Med 2020; 1‐2: 100004. [Google Scholar]
- 10. Acs B, Ahmed FS, Gupta S, et al. An open source automated tumor infiltrating lymphocyte algorithm for prognosis in melanoma. Nat Commun 2019; 10: 5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marti‐Aguado D, Rodríguez‐Ortega A, Mestre‐Alagarda C, et al. Digital pathology: accurate technique for quantitative assessment of histological features in metabolic‐associated fatty liver disease. Aliment Pharmacol Ther 2021; 53: 160–171. [DOI] [PubMed] [Google Scholar]
- 12. Zhao K, Li Z, Yao S, et al. Artificial intelligence quantified tumour‐stroma ratio is an independent predictor for overall survival in resectable colorectal cancer. EBioMedicine 2020; 61: 103054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep 2017; 7: 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coudray N, Ocampo PS, Sakellaropoulos T, et al. Classification and mutation prediction from non‐small cell lung cancer histopathology images using deep learning. Nat Med 2018; 24: 1559–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kather JN, Heij LR, Grabsch HI, et al. Pan‐cancer image‐based detection of clinically actionable genetic alterations. Nat Cancer 2020; 1: 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fu Y, Jung AW, Torne RV, et al. Pan‐cancer computational histopathology reveals mutations, tumor composition and prognosis. Nat Cancer 2020; 1: 800–810. [DOI] [PubMed] [Google Scholar]
- 17. Bilal M, Raza SEA, Azam A, et al. Development and validation of a weakly supervised deep learning framework to predict the status of molecular pathways and key mutations in colorectal cancer from routine histology images: a retrospective study. Lancet Digit Health 2021; 3: e763–e772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loeffler CML, Gaisa NT, Muti HS, et al. Predicting mutational status of driver and suppressor genes directly from histopathology with deep learning: a systematic study across 23 solid tumor types. Front Genet 2022; 12: 806386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmauch B, Romagnoni A, Pronier E, et al. A deep learning model to predict RNA‐Seq expression of tumours from whole slide images. Nat Commun 2020; 11: 3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bychkov D, Linder N, Turkki R, et al. Deep learning based tissue analysis predicts outcome in colorectal cancer. Sci Rep 2018; 8: 3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skrede OJ, De Raedt S, Kleppe A, et al. Deep learning for prediction of colorectal cancer outcome: a discovery and validation study. Lancet 2020; 395: 350–360. [DOI] [PubMed] [Google Scholar]
- 22. Zadeh Shirazi A, Fornaciari E, Bagherian NS, et al. DeepSurvNet: deep survival convolutional network for brain cancer survival rate classification based on histopathological images. Med Biol Eng Comput 2020; 58: 1031–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wulczyn E, Steiner DF, Xu Z, et al. Deep learning‐based survival prediction for multiple cancer types using histopathology images. PLoS One 2020; 15: e0233678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Linkon AHM, Labib MM, Hasan T, et al. Deep learning in prostate cancer diagnosis and Gleason grading in histopathology images: an extensive study. Inform Med Unlocked 2021; 24: 100582. [Google Scholar]
- 25. Bulten W, Kartasalo K, Chen PHC, et al. Artificial intelligence for diagnosis and Gleason grading of prostate cancer: the PANDA challenge. Nat Med 2022; 28: 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xie W, Reder NP, Koyuncu C, et al. Prostate cancer risk stratification via nondestructive 3D pathology with deep learning‐assisted gland analysis. Cancer Res 2022; 82: 334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar N, Verma R, Arora A, et al. Convolutional neural networks for prostate cancer recurrence prediction. In Medical Imaging 2017: Digital Pathology (Vol. 10140), Gurcan MN, Tomaszewski JE (eds). SPIE, 2017; 106–117. [Google Scholar]
- 28. Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 2001; 23: 291–299. [PubMed] [Google Scholar]
- 29. Rashidi HH, Tran NK, Betts EV, et al. Artificial intelligence and machine learning in pathology: the present landscape of supervised methods. Acad Pathol 2019; 6: 2374289519873088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Komura D, Ishikawa S. Machine learning methods for histopathological image analysis. Comput Struct Biotechnol J 2018; 16: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bandi P, Geessink O, Manson Q, et al. From detection of individual metastases to classification of lymph node status at the patient level: the CAMELYON17 Challenge. IEEE Trans Med Imaging 2019; 38: 550–560. [DOI] [PubMed] [Google Scholar]
- 32. Bortsova G, González‐Gonzalo C, Wetstein SC, et al. Adversarial attack vulnerability of medical image analysis systems: unexplored factors. Med Image Anal 2021; 73: 102141. [DOI] [PubMed] [Google Scholar]
- 33. Janowczyk A, Zuo R, Gilmore H, et al. HistoQC: an open‐source quality control tool for digital pathology slides. JCO Clin Cancer Inform 2019; 3: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schmitt M, Maron RC, Hekler A, et al. Hidden variables in deep learning digital pathology and their potential to cause batch effects: prediction model study. J Med Internet Res 2021; 23: e23436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Howard FM, Dolezal J, Kochanny S, et al. The impact of site‐specific digital histology signatures on deep learning model accuracy and bias. Nat Commun 2021; 12: 4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tellez D, Litjens G, Bándi P, et al. Quantifying the effects of data augmentation and stain color normalization in convolutional neural networks for computational pathology. Med Image Anal 2019; 58: 101544. [DOI] [PubMed] [Google Scholar]
- 37. Anghel A, Stanisavljevic M, Andani S, et al. A high‐performance system for robust stain normalization of whole‐slide images in histopathology. Front Med (Lausanne) 2019; 6: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ciompi F, Geessink O, Bejnordi BE, et al. The importance of stain normalization in colorectal tissue classification with convolutional networks. In 2017 IEEE 14th International Symposium on Biomedical Imaging (ISBI 2017), 2017; 160–163.
- 39. Meijering E. Cell segmentation: 50 years down the road. IEEE Signal Process Mag 2012; 29: 140–145. [Google Scholar]
- 40. Caicedo JC, Goodman A, Karhohs KW, et al. Nucleus segmentation across imaging experiments: the 2018 Data Science Bowl. Nat Methods 2019; 16: 1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Greenwald NF, Miller G, Moen E, et al. Whole‐cell segmentation of tissue images with human‐level performance using large‐scale data annotation and deep learning. Nat Biotechnol 2022; 40: 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Graham S, Vu QD, Raza SEA, et al. Hover‐Net: simultaneous segmentation and classification of nuclei in multi‐tissue histology images. Med Image Anal 2019; 58: 101563. [DOI] [PubMed] [Google Scholar]
- 43. Stringer C, Wang T, Michaelos M, et al. Cellpose: a generalist algorithm for cellular segmentation. Nat Methods 2021; 18: 100–106. [DOI] [PubMed] [Google Scholar]
- 44. Schmidt U, Weigert M, Broaddus C, et al. Cell detection with star‐convex polygons. In Medical Image Computing and Computer Assisted Intervention—MICCAI 2018, Frangi AF, Schnabel JA, Davatzikos C, et al. (eds). Springer International Publishing: Cham, 2018; 265–273. [Google Scholar]
- 45. Hamilton PW, Wang Y, Boyd C, et al. Automated tumor analysis for molecular profiling in lung cancer. Oncotarget 2015; 6: 27938–27952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McCarty KS Jr, Szabo E, Flowers JL, et al. Use of a monoclonal anti‐estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Res 1986; 46(8 Suppl): 4244s–4248s. [PubMed] [Google Scholar]
- 47. Goulding H, Pinder S, Cannon P, et al. A new immunohistochemical antibody for the assessment of estrogen receptor status on routine formalin‐fixed tissue samples. Hum Pathol 1995; 26: 291–294. [DOI] [PubMed] [Google Scholar]
- 48. Harvey JM, Clark GM, Osborne CK, et al. Estrogen receptor status by immunohistochemistry is superior to the ligand‐binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol 1999; 17: 1474–1481. [DOI] [PubMed] [Google Scholar]
- 49. Rimm DL. What brown cannot do for you. Nat Biotechnol 2006; 24: 914–916. [DOI] [PubMed] [Google Scholar]
- 50. Korzynska A, Roszkowiak L, Lopez C, et al. Validation of various adaptive threshold methods of segmentation applied to follicular lymphoma digital images stained with 3,3’‐Diaminobenzidine&Haematoxylin. Diagn Pathol 2013; 8: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kerby A, Graham N, Wallworth R, et al. Development of dynamic image analysis methods to measure vascularisation and syncytial nuclear aggregates in human placenta. Placenta 2022; 120: 65–72. [DOI] [PubMed] [Google Scholar]
- 52. Coveney PV, Highfield RR. When we can trust computers (and when we can't). Philos Trans A Math Phys Eng Sci 2021; 379: 20200067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Border SP, Sarder P. From what to why, the growing need for a focus shift toward Explainability of AI in digital pathology. Front Physiol 2022; 12: 821217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cruz Rivera S, Liu X, Chan AW, et al. Guidelines for clinical trial protocols for interventions involving artificial intelligence: the SPIRIT‐AI extension. Lancet Digit Health 2020; 2: e549–e560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ibrahim H, Liu X, Rivera SC, et al. Reporting guidelines for clinical trials of artificial intelligence interventions: the SPIRIT‐AI and CONSORT‐AI guidelines. Trials 2021; 22: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stodden V, Seiler J, Ma Z. An empirical analysis of journal policy effectiveness for computational reproducibility. Proc Natl Acad Sci U S A 2018; 115: 2584–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Benureau FCY, Rougier NP. Re‐run, repeat, reproduce, reuse, replicate: transforming code into scientific contributions. Front Neuroinform 2018; 11: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eglen SJ, Marwick B, Halchenko YO, et al. Toward standard practices for sharing computer code and programs in neuroscience. Nat Neurosci 2017; 20: 770–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wilkinson MD, Dumontier M, Aalbersberg IJ, et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data 2016; 3: 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cadwallader L, Papin JA, Mac Gabhann F, et al. Collaborating with our community to increase code sharing. PLoS Comput Biol 2021; 17: e1008867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Couture JL, Blake RE, McDonald G, et al. A funder‐imposed data publication requirement seldom inspired data sharing. PLoS One 2018; 13: e0199789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lu MY, Chen RJ, Kong D, et al. Federated learning for computational pathology on gigapixel whole slide images. Med Image Anal 2022; 76: 102298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilson G, Bryan J, Cranston K, et al. Good enough practices in scientific computing. PLoS Comput Biol 2017; 13: e1005510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Coulter C, McKay F, Hallowell N, et al. Understanding the ethical and legal considerations of Digital Pathology. J Pathol Clin Res 2022; 8: 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Barnes N. Publish your computer code: it is good enough. Nature 2010; 467: 753. [DOI] [PubMed] [Google Scholar]
- 66. Ince DC, Hatton L, Graham‐Cumming J. The case for open computer programs. Nature 2012; 482: 485–488. [DOI] [PubMed] [Google Scholar]
- 67. Alston JM, Rick JA. A beginner's guide to conducting reproducible research. Bull Ecol Soc Am 2021; 102: e01801. [Google Scholar]
- 68. Perkel JM. How to fix your scientific coding errors. Nature 2022; 602: 172–173. [DOI] [PubMed] [Google Scholar]
- 69. Levet F, Carpenter AE, Eliceiri KW, et al. Developing open‐source software for bioimage analysis: opportunities and challenges. F1000Res 2021; 10: 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nowogrodzki A. How to support open‐source software and stay sane. Nature 2019; 571: 133–134. [DOI] [PubMed] [Google Scholar]
- 71. Fortunato L, Galassi M. The case for free and open source software in research and scholarship. Philos Trans A Math Phys Eng Sci 2021; 379: 20200079. [DOI] [PubMed] [Google Scholar]
- 72. Clark K, Vendt B, Smith K, et al. The Cancer Imaging Archive (TCIA): maintaining and operating a public information repository. J Digit Imaging 2013; 26: 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sirinukunwattana K, Domingo E, Richman SD, et al. Image‐based consensus molecular subtype (imCMS) classification of colorectal cancer using deep learning. Gut 2021; 70: 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Saltz J, Gupta R, Hou L, et al. Spatial organization and molecular correlation of tumor‐infiltrating lymphocytes using deep learning on pathology images. Cell Rep 2018; 23: 181–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cooper LA, Demicco EG, Saltz JH, et al. PanCancer insights from The Cancer Genome Atlas: the pathologist's perspective. J Pathol 2018; 244: 512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Moulin P, Grünberg K, Barale‐Thomas E, et al. IMI—Bigpicture: a central repository for digital pathology. Toxicol Pathol 2021; 49: 711–713. [DOI] [PubMed] [Google Scholar]
- 77. Sirinukunwattana K, Pluim JPW, Chen H, et al. Gland segmentation in colon histology images: the glas challenge contest. Med Image Anal 2017; 35: 489–502. [DOI] [PubMed] [Google Scholar]
- 78. Aresta G, Araújo T, Kwok S, et al. BACH: grand challenge on breast cancer histology images. Med Image Anal 2019; 56: 122–139. [DOI] [PubMed] [Google Scholar]
- 79. Maier‐Hein L, Eisenmann M, Reinke A, et al. Why rankings of biomedical image analysis competitions should be interpreted with care. Nat Commun 2018; 9: 5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maier‐Hein L, Reinke A, Kozubek M, et al. BIAS: transparent reporting of biomedical image analysis challenges. Med Image Anal 2020; 66: 101796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Carpenter AE, Kamentsky L, Eliceiri KW. A call for bioimaging software usability. Nat Methods 2012; 9: 666–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Perkel JM. Ten computer codes that transformed science. Nature 2021; 589: 344–348. [DOI] [PubMed] [Google Scholar]
- 83. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fedorov A, Beichel R, Kalpathy‐Cramer J, et al. 3D Slicer as an image computing platform for the Quantitative Imaging Network. Magn Reson Imaging 2012; 30: 1323–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006; 7: R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. de Chaumont F, Dallongeville S, Olivo‐Marin JC. ICY: A new open‐source community image processing software. In 2011 IEEE International Symposium on Biomedical Imaging: From Nano to Macro. 2011; 234–237.
- 87. Tuominen VJ, Ruotoistenmäki S, Viitanen A, et al. ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki‐67. Breast Cancer Res 2010; 12: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tuominen VJ, Tolonen TT, Isola J. ImmunoMembrane: a publicly available web application for digital image analysis of HER2 immunohistochemistry. Histopathology 2012; 60: 758–767. [DOI] [PubMed] [Google Scholar]
- 89. Marée R. Open practices and resources for collaborative digital pathology. Front Med (Lausanne) 2019; 6: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schüffler PJ, Fuchs TJ, Ong CS, et al. TMARKER: a free software toolkit for histopathological cell counting and staining estimation. J Pathol Inform 2013; 4: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Stritt M, Stalder AK, Vezzali E. Orbit Image Analysis: an open‐source whole slide image analysis tool. PLoS Comput Biol 2020; 16: e1007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rubens U, Hoyoux R, Vanosmael L, et al. Cytomine: toward an open and collaborative software platform for digital pathology bridged to molecular investigations. Proteomics Clin Appl 2019; 13: e1800057. [DOI] [PubMed] [Google Scholar]
- 93. Gutman DA, Khalilia M, Lee S, et al. The digital slide archive: a software platform for management, integration, and analysis of histology for cancer research. Cancer Res 2017; 77: e75–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bankhead P, Fernández JA, McArt DG, et al. Integrated tumor identification and automated scoring minimizes pathologist involvement and provides new insights to key biomarkers in breast cancer. Lab Invest 2018; 98: 15–26. [DOI] [PubMed] [Google Scholar]
- 95. Acs B, Pelekanou V, Bai Y, et al. Ki67 reproducibility using digital image analysis: an inter‐platform and inter‐operator study. Lab Invest 2019; 99: 107–117. [DOI] [PubMed] [Google Scholar]
- 96. Ribeiro GP, Endringer DC, De Andrade TU, et al. Comparison between two programs for image analysis, machine learning and subsequent classification. Tissue Cell 2019; 58: 12–16. [DOI] [PubMed] [Google Scholar]
- 97. Maguire AS, Woodie LN, Judd RL, et al. Whole‐slide image analysis outperforms micrograph acquisition for adipocyte size quantification. Adipocyte 2020; 9: 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hein AL, Mukherjee M, Talmon GA, et al. QuPath digital immunohistochemical analysis of placental tissue. J Pathol Inform 2021; 12: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Berben L, Wildiers H, Marcelis L, et al. Computerised scoring protocol for identification and quantification of different immune cell populations in breast tumour regions by the use of QuPath software. Histopathology 2020; 77: 79–91. [DOI] [PubMed] [Google Scholar]
- 100. Tratwal J, Bekri D, Boussema C, et al. MarrowQuant across aging and aplasia: a digital pathology workflow for quantification of bone marrow compartments in histological sections. Front Endocrinol (Lausanne) 2020; 11: 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Apaolaza PS, Petropoulou P‐I, Rodriguez‐Calvo T. Whole‐slide image analysis of human pancreas samples to elucidate the immunopathogenesis of type 1 diabetes using the QuPath software. Front Mol Biosci 2021; 8: 689799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rueden CT, Ackerman J, Arena ET, et al. Scientific Community Image Forum: a discussion forum for scientific image software. PLoS Biol 2019; 17: e3000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Schapiro D, Yapp C, Sokolov A, et al. MITI minimum information guidelines for highly multiplexed tissue images. Nat Methods 2022; 19: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lazcano R, Rojas F, Laberiano C, et al. Pathology quality control for multiplex immunofluorescence and image analysis assessment in longitudinal studies. Front Mol Biosci 2021; 8: 661222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ward JM, Schofield PN, Sundberg JP. Reproducibility of histopathological findings in experimental pathology of the mouse: a sorry tail. Lab Anim (NY) 2017; 46: 146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ince TA, Ward JM, Valli VE, et al. Do‐it‐yourself (DIY) pathology. Nat Biotechnol 2008; 26: 978–979; discussion 979. [DOI] [PubMed] [Google Scholar]
- 107. Jamali N, Dobson ET, Eliceiri KW, et al. 2020 BioImage Analysis Survey: community experiences and needs for the future. Biol Imaging 2022; 1: e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A short overview of the image processing terms used in this review is available at https://petebankhead.github.io/2022-image-processing-overview. The images and scripts used in the preparation of figures for this article are available at https://github.com/petebankhead/2022-qupath-cell-detection.