

Abstract
High‐density lipoproteins (HDL) are well known for their atheroprotective function, mainly due to their ability to remove cell cholesterol and to exert antioxidant and anti‐inflammatory activities. Through the same mechanisms HDL could also affect the development and progression of tumors. Cancer cells need cholesterol to proliferate, especially in hormone‐dependent tumors, as prostate cancer (PCa). Aim of the study was to investigate the ability of HDL to modulate cholesterol content and metabolism in androgen receptor (AR)‐positive and AR‐null PCa cell lines and the consequences on cell proliferation. HDL inhibited colony formation of LNCaP and PC3 cells. HDL reduced cell cholesterol content and proliferation of LNCaP cells loaded with low‐density lipoproteins but were not effective on PC3 cells. Here, the expression of the ATP‐binding cassette transporter A1 (ABCA1) was markedly reduced due to proteasome degradation. Bortezomib, a proteasome inhibitor, restored ABCA1 expression and HDL ability to promote cholesterol removal from PC3; consequently, HDL inhibited the proliferation of PC3 cells induced by LDL only after bortezomib pre‐treatment. In conclusion, the antiproliferative activity of HDL on AR‐positive and AR‐null PCa cells also rely on cholesterol removal, a process in which the ABCA1 transporter plays a key role.
Keywords: ABCA1, cholesterol, lipoproteins, prostate cancer
Abbreviations
- ABC
ATP‐binding cassette transporter
- ApoA‐I
apolipoprotein A‐I
- AR
androgen receptor
- HDL
high‐density lipoproteins
- HMGCoAR
HMG coenzyme A reductase
- LDL
low‐density lipoproteins
- LRP1
LDL receptor related protein 1
- PCa
prostate cancer
- RCT
reverse cholesterol transport
- SR‐BI
scavenger receptor type BI
- SRD5A1
steroid 5α‐reductase 1
- SREBP2
sterol regulatory element‐binding protein 2
- TC
total cholesterol
- UC
unesterified cholesterol
- βMCD
β‐methylcyclodextrin
1. INTRODUCTION
High‐density lipoproteins (HDL) are well known for their protective role against the development and progression of atherosclerosis. 1 Their central role in the reverse transport of cholesterol from peripheral cells to the liver for excretion (RCT) is generally believed as the milestone of HDL‐mediated atheroprotection. 2 The first step of RCT is the efflux of cholesterol from cells to extracellular acceptors as HDL or their main protein component, apolipoprotein A‐I (apoA‐I), which can occur through different mechanisms: passive diffusion according to the cholesterol gradient (which may also include unknown transporters), facilitated diffusion through the scavenger receptor BI (SR‐BI), and active transport promoted by the ATP‐binding cassette transporters ABCA1 and ABCG1. 3 In addition, HDL play many other anti‐atherogenic activities: they possess a direct antioxidant capacity and exert multiple positive effects toward several cell types involved in the atherosclerotic process as endothelial and immune cells. 4
Interestingly, through the same mechanisms HDL could also affect the development and progression of tumors, by directly acting on cancer cells or by affecting tumor microenvironment. 5 HDL can target tumor cells since they generally express SR‐BI, and infiltrate tumors because of their relatively small size. Here, they might reduce oxidative stress, as we recently showed in prostate cancer cells, 6 inflammation or cholesterol content, thus affecting cancer cell proliferation. Cholesterol represents an attractive target since it is needed as a structural component of cell membranes, and it is involved in several cellular functions. 7 For example, the presence of cholesterol in lipid rafts regulates the activation of transmembrane receptors, including those for growth factors, and their downstream signaling pathways. 8 Moreover, cholesterol can be converted into biologically active molecules, including locally produced hormones. Thus, cholesterol could be particularly relevant for cell proliferation in hormone‐dependent tumors, as PCa. 9 Cancer cells can synthetize cholesterol through the mevalonate pathway, or they can take up lipoproteins, especially those containing apolipoprotein B, as low‐density lipoproteins (LDL). 10 On the contrary, HDL could act as cholesterol acceptors, thus reducing its cellular content. However, SR‐BI, which binds to apoA‐I on HDL and facilitates the passive diffusion of cholesterol, could promote its efflux or influx according to the concentration gradient between cell membranes and the extracellular acceptors. 3
PCa is the most common cancer in men and the fourth most common cancer overall. 11 Since the prostate is an androgen‐dependent organ, androgen deprivation therapy is effective for the management of PCa progression and dissemination. 12 However, a significant percentage of PCa cases can evolve to a castration‐resistant phenotype, for which few therapeutic options with limited efficacy are available. 13 Thus, novel strategies to achieve a direct cytotoxic effect on PCa cells or to make tumor cells more sensitive to the action of classical cytotoxic agents are required.
In the present study, the ability of HDL to modulate cholesterol content and metabolism in PCa cell lines and the consequences on cell proliferation were investigated. To this aim, androgen receptor (AR)‐positive and AR‐null cells lines were used, which express high amounts of SR‐BI, higher levels of ABCG1, but lower levels of ABCA1 compared to normal prostate epithelial cells. 6
2. EXPERIMENTAL PROCEDURES
2.1. Cell lines
Human AR‐positive and AR‐null PCa epithelial cell lines (LNCaP and PC3, respectively) and the SV40‐immortalized non‐tumorigenic human prostatic epithelial cell line PNT2 were obtained from ECACC (UK, Catalogue no. 89110211; 90112714; and 95012613). According to the manufacturer instructions, cell lines were cultured in RPMI 1640 supplemented with 5% (PC3) or 10% (LNCaP and PNT2) of fetal bovine serum (FBS), 2 mmol/lL‐glutamine, 0.1 U/ml penicillin, and 0.1 μg/ml streptomycin, at 37°C in a humidified atmosphere with 5% CO2. Cell media, supplements, and sterile disposables were from EuroClone (Italy).
2.2. Lipoproteins
LDL (d = 1.020–1.063 g/ml) and HDL (d = 1.063–1.21 g/ml) were isolated by sequential ultracentrifugation from the plasma of healthy volunteers. 14 Lipoproteins were dialyzed against sterilized saline immediately before use. Their concentration is expressed as protein content, as measured by the method of Lowry. ApoA‐I was isolated from HDL by affinity chromatography, as previously described. 15
2.3. Colony formation assay
LNCaP and PC3 were seeded at 200 cells/well in six‐well plates. Cells were treated with HDL (0.5 mg/ml) for 48 h. Then, the medium was replenished, and cells were maintained in regular growth medium for 12 days. In some experiments with PC3, bortezomib 50 pM (Cell Signaling, MA) was added to HDL during the first 24 h of incubation. Fourteen days after seeding, cells were fixed in 100% cold methanol for 7 min. Then, cells were stained for 45 min with Giemsa dye solution (Sigma‐Aldrich, MO) diluted in water 1:20. Colonies formed by more than 50 cells were counted under an optical microscope.
2.4. Cell proliferation assay
LNCaP and PC3 cells were seeded (150.000 and 100.000 cells/well, respectively) in six‐well culture plates in RPMI containing 5%–10% FBS (see above). After 48 h, cells were moved to RPMI 1640 without phenol red containing 5% or 10% charcoal stripped FBS and allowed to proliferate for additional 48 h. Medium was then refreshed, and cells were treated with LDL (20 μg protein/ml) for 24 h and then with HDL for 48 h (0.5 mg protein/ml). The incubation with LDL was limited to 24 h to avoid the possible confounding effect of lipoprotein oxidation. 6 At the end of the experiment, cells were harvested and counted by Trypan blue exclusion in a Burker chamber. Cell growth was further analyzed by measuring ATP levels through the CellTiter‐Glo® Luminescent Cell Viability Assay (Promega Corporation, WI) and bromodeoxyuridine (BrdU) incorporation with the BrdU cell proliferation colorimetric assay (Abcam, Cambridge). Assays were performed according to the manufacturers' instructions in 96‐well culture plates with a seeding density of 2000 cells/well. Luminescence and absorbance at 450 nm were measured with the Synergy H1 multi‐mode reader equipped with the Gen5 software (BioTek, Agilent, CA).
2.5. Cell cholesterol content
LNCaP and PC3 cells were seeded at 100,000 cells/well in 12 well/plates. Cells were incubated for 1 h with LDL (50 μg/ml) or HDL (0.5 mg/ml) in RPMI1640, 2 mmol/ll‐glutamine, 0.1 U/ml penicillin, and 0.1 μg/ml streptomycin. In a separated set of experiments, cells were incubated with LDL (50 μg/ml) for 24 h. Then, cells were washed and exposed to HDL (0.5 mg/ml), apoA‐I (0.5 mg/ml), or β‐methylcyclodextrin (βMCD, 2.5 mM, Sigma‐Aldrich, MO) for 1 h. After washing with PBS, cells were lysed overnight in 1% sodium cholate and 10 U/ml of DNase (Sigma‐Aldrich, MO) at room temperature. Total cholesterol was measured by fluorescence using the Amplex Red Cholesterol Assay Kit (Sigma‐Aldrich, MO), according to the manufacturer instructions. For each sample, cholesterol concentration was normalized by the protein concentration of the total cell lysate, measured with the microBCA assay (ThermoFisher Scientific, IL). In some experiments, cholesterol esterase was not added to the Amplex Red reagent to selectively measure unesterified cholesterol.
2.6. Real‐time PCR
Confluent PNT2, LNCaP, and PC3 cells were harvested in Trizol Plus reagent (Life Technologies, CA), and RNA was extracted according to the manufacturer's instructions. cDNA was prepared by reverse transcription of 0.8 μg of total RNA using the iScript cDNA Synthesis kit (Bio‐Rad Laboratories, Hercules, CA). Amplification was carried out with the iTaq Universal SYBR Green Supermix in a MiniOpticon System (Bio‐Rad Laboratories, Hercules, CA). Expression was calculated using the ΔΔCt method and normalized to a housekeeping gene (GAPDH). Primers are reported in Table S1.
2.7. Western blotting
Cells were harvested in lysis buffer (20 mmol/L Tris, 4% SDS, 20% glycerol containing 1 mmol/L EDTA, 1 mmol/L sodium orthovanadate, 1 mmol/L NaF, 1 μg/ml leupeptin, 1 mmol/L benzamidine, 10 μg/ml soy trypsin inhibitor, 1 mmol/L PMSF, and 0.5 mmol/L DTT, pH 6.8). Cell debris was removed by centrifugation and protein concentration was determined by the microBCA assay (ThermoFisher Scientific, IL). Proteins were separated by SDS‐PAGE and then transferred on a nitrocellulose membrane. After saturation with 5% nonfat dried milk, membranes were incubated with antibodies against LDL‐receptor, LRP1, HMGCoA‐reductase, SR‐BI, p21, NF‐kB p65 or phosphorylated p65, and then with a horseradish peroxidase–conjugated secondary antibody. Bands were visualized by enhanced chemiluminescence (GE Healthcare, IL). Membranes were then stripped and reprobed with an antibody against α‐tubulin. Band densities were evaluated with a GS‐690 Imaging Densitometer and Multi‐Analyst software (Bio‐Rad Laboratories). Antibodies are listed in Table S2.
2.8. Silencing of SR‐BI
PC3 cells were seeded in 24‐well plates and transfected with 100 pmol of siRNA against either SR‐BI or noncoding (scrambled) siRNA for 48 h using the OptiMEM/Lipofectamine 2000 system (Life Technologies, Carlsbad, CA), according to the manufacturer's protocol. siGenome ON‐TARGETplus SMARTpool were used (GE Healthcare Dharmacon, CO). Cells were then washed with PBS and cholesterol assay was performed as described above. Silencing efficiency was evaluated by Western blotting for SR‐BI and α‐tubulin, as described above.
2.9. Immunofluorescence
PC3 cells were seeded at 80,000 cells/well in six‐well/plates containing sterilized slides. At 20%–50% confluence, cells were treated for 4 h with the following inhibitors: the proteasome inhibitors MG132 (50 μM) and bortezomib (5 nM), the lysosomal protease inhibitor chloroquine diphosphate (100 μM), the calpain inhibitor calpeptin (30 μg/ml) (Sigma‐Aldrich, St. Louis, MO). Then, cells were fixed in 100% cold methanol for 10 min or in 4% paraformaldehyde for 5 min at room temperature and incubated with 10% FBS in PBS for 20 min. Slides were incubated overnight at 4°C with primary antibodies against ABCA1 and α‐tubulin in PBS containing 0.1% Tween 20% and 1% BSA. After washing, cells were incubated with the fluorescent secondary antibodies anti‐mouse AlexaFluor‐488 and anti‐rabbit Rhodamine conjugated for 1 h at room temperature. Then, slides were rinsed with water and one drop of mounting medium containing DAPI (ThermoFisher Scientific, Waltham, MA) was added onto each slide. Images were captured with a Nikon A1 confocal microscope. Fluorescence intensities were measured with the ImageJ software and the ratio of ABCA1 and DAPI signals was calculated. Antibodies are listed in Table S2.
2.10. ABCA1 gene sequencing
Genomic DNA was extracted from PC3 cells by a standard procedure. 16 The promoter, the 50 exons and the splice junctions of the ABCA1 gene were amplified and sequenced on ABI PRISM 3130xl (Life Technologies), as reported previously. 17 The ABCA1 sequence variant nomenclature was designated according to the Human Genome Variation Society version 2016. 18
2.11. Statistical analysis
Data are expressed as mean and standard deviation (SD), if not otherwise stated. Group differences were evaluated by two‐sided one‐way ANOVA. The α level of significance was set at 0.05. Statistical analysis was performed with SigmaPlot 12.5 (Systat Software, Chicago, IL).
3. RESULTS
3.1. HDL reduced colony formation in LNCaP and PC3 cells
PCa cells were seeded at low density and allowed to growth for 14 days. A short treatment with HDL in the first 48 h resulted in a marked and long‐lasting inhibition of colony formation in both cell lines. Indeed, in LNCaP HDL reduced the number of colonies after 14 days by 74% ± 13% (p < 0.001, Figure 1A). In PC3 cells, HDL also significantly decreased colony formation by 49% ± 15% (p < 0.001, Figure 1B), even if to a lesser extent if compared to LNCaP.
FIGURE 1.
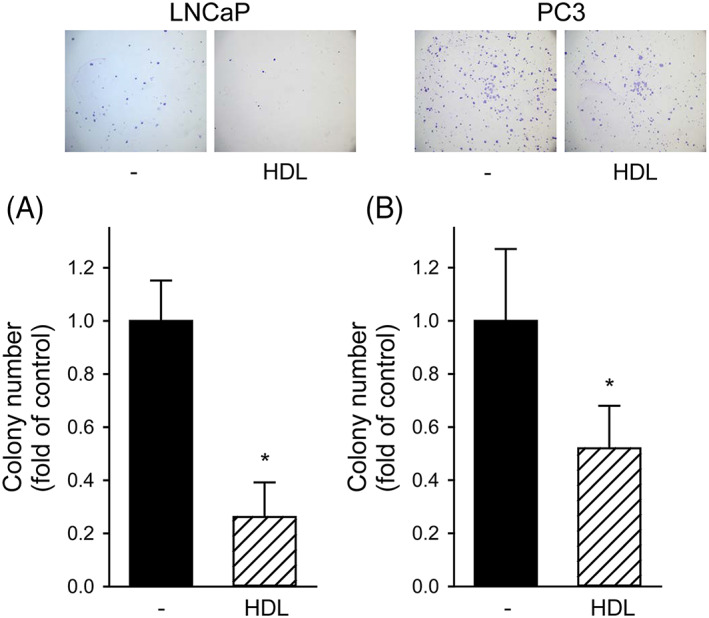
HDL inhibited colony formation in LNCaP and PC3 cells; LNCaP (Panel A) and PC3 (Panel B) cells were seeded at low density and allowed to grow in regular medium for 14 days (black bars). HDL were added at 0.5 mg/ml only in the first 48 h (open dashed bars). The number of colonies made by more than 50 cells was counted. Results are expressed as fold of untreated cells, mean ± SD, n = 18. *p < 0.05 versus untreated cells. Representative images are shown at the top
3.2. HDL inhibited LDL‐induced increase of cholesterol content and cell proliferation in LNCaP but not in PC3
We previously showed that the antioxidant effect of HDL is involved in their anti‐proliferative activity on both LNCaP and PC3 cells. 6 Here, the ability of HDL to reduce cell cholesterol content and the consequent impact on cell proliferation was assessed.
The incubation of LNCaP with LDL increased cell cholesterol content by 52% ± 16% (p < 0.001); the subsequent exposure to HDL significantly reduced cholesterol in LDL‐loaded cells by 24% ± 11% (p < 0.001, Figure 2A). Similar results were obtained when apoA‐I, the main protein component of HDL, was used. When cells were not preloaded with LDL, HDL did not affect cholesterol content (Figure 2A). The incubation of LNCaP with LDL resulted in a 22% ± 7% increase of cell number (p < 0.001), which was almost halved when HDL were also present (12% ± 5% p = 0.029 vs. LDL alone, Figure 2B), in agreement with the changes of cholesterol content. The modulation of cell proliferation was fully confirmed by ATP levels and BrdU incorporation‐based proliferation assays (Figure S1, Panels A and C).
FIGURE 2.
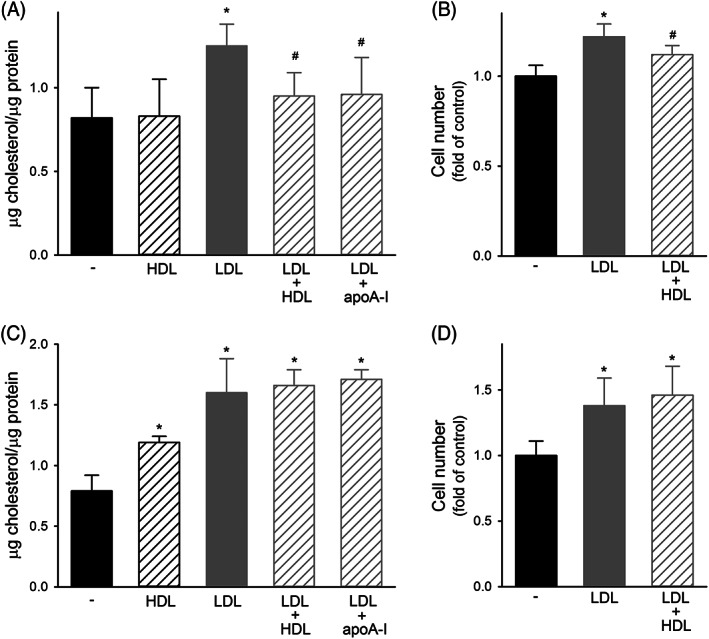
Effect of lipoproteins on cholesterol content and proliferation of PCa cell lines. Panels A and B, Effect on AR‐positive LNCaP. Panel A, Cholesterol content in untreated cells (black bars), or in cells incubated with (A) HDL at 0.5 mg/ml for 1 h (black dashed bars), (B) LDL at 50 μg/ml for 24 h (gray bars), (C) LDL at 50 μg/ml for 24 h followed by HDL or apoA‐I at 0.5 mg/ml for 1 h (gray dashed bars). Data are expressed as mean ± SD, n = 9. *p < 0.05 versus untreated cells, #p < 0.05 versus LDL. Panel B, Cell proliferation evaluated as cell number at 72 h in untreated cells (black bars) or in cells incubated with LDL at 20 μg/ml for the first 24 h followed (gray dashed bars) or not (gray bars) by HDL at 0.5 mg/ml for 48 h. Data are expressed as fold of untreated cells, mean ± SD, n = 12. *p < 0.05 versus untreated cells, #p < 0.05 versus LDL. Panels C and D, Effect on AR‐null PC3. Panel C, Cholesterol content in untreated cells (black bars), or in cells incubated with (A) HDL at 0.5 mg/ml for 1 h (black dashed bars), (B) LDL at 50 μg/ml for 24 h (gray bars), (C) LDL at 50 μg/ml for 24 h followed by HDL or apoA‐I at 0.5 mg/ml for 1 h (gray dashed bars). Data are expressed as mean ± SD, n = 9. *p < 0.05 versus untreated cells. Panel D, Cell proliferation evaluated as cell number at 72 h in untreated cells (black bars) or in cells incubated with LDL at 20 μg/ml for the first 24 h followed (gray dashed bars) or not (gray bars) by HDL at 0.5 mg/ml for 48 h. Data are expressed as fold of untreated cells, mean ± SD, n = 8. *p < 0.05 versus untreated cells
When the experiments were replicated in PC3 cells, very different results were obtained. First, HDL by themselves caused a significant increase of cell cholesterol content by 50% ± 7% (p < 0.001, Figure 2C). Second, the exposure to HDL or apoA‐I was not able to reduce the 102% ± 36% increase of cholesterol content induced by LDL (Figure 2C). Consistently, HDL were not able to inhibit the 38% ± 21% increase of PC3 cell number induced by LDL (Figure 2D). The lack of inhibition was confirmed by ATP levels and BrdU incorporation (Figure S1B,D).
3.3. Receptors for LDL are upregulated in LNCaP and PC3 cells
The LDL‐receptor (LDL‐R) and the LDL‐R related protein 1 (LRP1) are key for the binding and internalization of LDL into the cells. When compared to non‐tumor PNT2 cells, both LNCaP and PC3 showed a huge increase of mRNA and protein levels of LDL‐R and LRP1 (Figure 3, Panels A–D). Indeed, 15.1 ± 7.6‐fold and 16.0 ± 5.8‐fold increase of LDL‐R protein was observed in LNCaP and PC3 cells, respectively (p < 0.001 vs. PNT2). LRP1 protein levels were increased by 7.5 ± 2.8‐fold and 7.1 ± 4.4‐fold, as well (p = 0.013 and 0.008 vs. PNT2, respectively).
FIGURE 3.
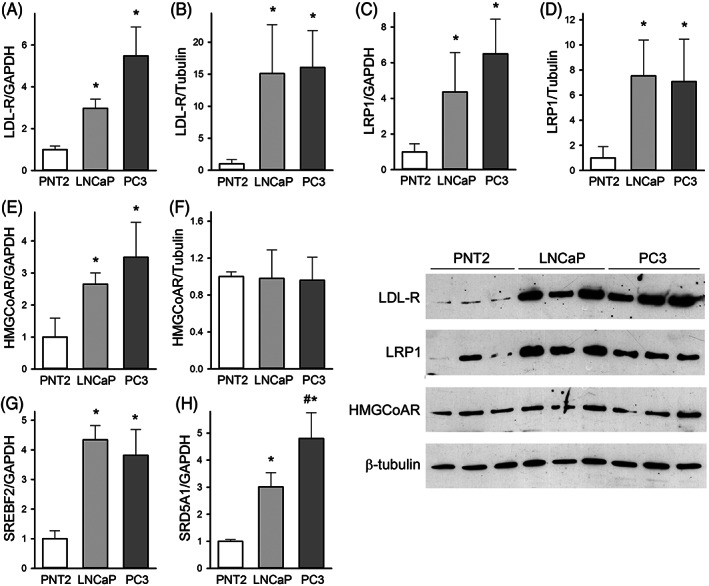
Gene and protein levels in PNT2, LNCaP, and PC3 cells; Real‐time PCR and Western blotting were used to assess the mRNA (Panels A, C, E, G, and H) or protein (Panels B, D, and F) levels of the LDL‐receptor (LDL‐R), LDL‐R related protein 1 (LRP1), 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase (HMGCoAR), sterol regulatory element binding transcription factor 2 (SREBF2), and steroid 5α‐reductase 1 (SRD5A1). Representative blots are shown on the right. Data are expressed as mean ± SD, n = 5 for mRNA and n = 6 for protein. *p < 0.05 versus non‐tumor PNT2, #p < 0.05 versus LNCaP
This increase is likely due to the upregulation of sterol regulatory element‐binding protein 2 (SREBP2), coded by the SREBF2 gene, which showed a 4.3 ± 0.5 and 3.8 ± 0.9‐fold increase in LNCaP and PC3 cells, respectively (p < 0.001 vs. PNT2, Figure 3G). 19 SREBP2 also regulates the expression of HMGCoA reductase (HMGCoAR), the rate limiting step of cholesterol synthesis. However, the 2.6 ± 0.3 to 3.5 ± 1.1‐fold increase in HMGCoAR mRNA levels observed in LNCaP and PC3 cells did not translate into significant changes in protein levels (Figure 3, Panel E–F), thus indicating that PCa cells mainly rely on lipoprotein uptake for their cholesterol needs. Consistently, the inhibition of HMGCoAR by lovastatin did not affect cell cholesterol content in PC3 (Figure S2).
Cholesterol can be used for the intracellular synthesis of dihydrotestosterone, as indicated by the increased expression of the steroid 5α‐reductase 1 (SRD5A1); its upregulation was proportional to the aggressiveness of the phenotype, varying from 3.0 ± 0.5‐fold in LNCaP to 4.8 ± 0.9‐fold in PC3 cells (p = 0.007 for LNCaP vs. PC3, Figure 3H).
3.4. ABCA1 protein is downregulated in PC3 cells
To understand why in PC3 cells cholesterol content was not reduced, but even increased by HDL, the possible involvement of SR‐BI was tested. When SR‐BI protein expression was reduced by 74.2% ± 7.3% through RNA interference, PC3 cell cholesterol content was not significantly increased after HDL exposure (p = 0.402), suggesting that SR‐BI could promote cholesterol influx from HDL to PC3 cells (Figure S3).
Next, to understand why HDL were not able to reduce cell cholesterol in LDL‐loaded cells, several hypotheses were investigated. Since the presence of unesterified cholesterol (UC) on cell membrane is needed for efflux, both unesterified and TC were measured in LNCaP and PC3 cells before and after loading with LDL. In untreated cells, the percentage of UC was comparable between the two cell lines; indeed, UC was 83% ± 1% of TC in LNCaP and 83% ± 0.5% in PC3. Similarly, after the incubation with LDL for 24 h, UC was 92% ± 4% in LNCaP and 84% ± 5% in PC3 (p = 0.09). Thus, no changes in the extent of cholesterol esterification were detected between the different cell lines. Furthermore, the availability of UC on PC3 cell membrane was confirmed by exposing LDL‐loaded cells to βMCD, which is a cholesterol acceptor through passive diffusion; as evident from Figure S2, βMCD completely abolished the increase of cell cholesterol content induced by LDL.
Then, the possible involvement of ABCA1 downregulation was investigated. ABCA1 mRNA levels were assessed by real‐time PCR, while ABCA1 protein signal on the cell surface was evaluated by immunofluorescence in non‐permeabilized cells (Figure 4). Consistent with our previous data, 6 ABCA1 mRNA and protein levels were significantly reduced in LNCaP if compared to non‐tumor PNT2. When PC3 cells were tested, the ABCA1 protein signal was also markedly reduced. However, its mRNA levels were increased by 5.9 ± 1.2‐fold in PC3 cells when compared to non‐tumor PNT2, thus indicating that the high levels of ABCA1 mRNA do not translate into ABCA1 protein on the cell membrane. To exclude the possibility that the lack of ABCA1 protein could be due to the presence of pathogenic mutations, the ABCA1 gene was sequenced, and only eight common polymorphisms were found (Table S3).
FIGURE 4.
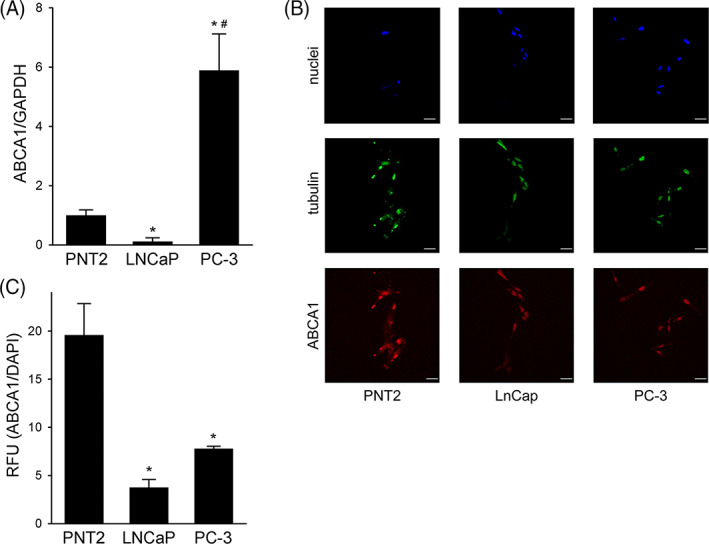
ABCA1 levels in PNT2, LNCaP, and PC3 cells; Panel A, ABCA1 mRNA levels by real‐time PCR. Data are expressed as mean ± SD, n = 3. *p < 0.05 versus PNT2, #p < 0.05 versus LNCaP. Panel B, Representative confocal immunofluorescence images for ABCA1 (red), tubulin (green), and DAPI (blue). Cells were fixed with 4% paraformaldehyde. Scale bar, 50 μm. Panel C, Quantitative assessment of ABCA1 fluorescence intensities. Data are expressed as ABCA1/DAPI signal ratio, mean ± SD, n = 3. *p < 0.05 versus PNT2
Finally, the possible intracellular degradation of newly synthetized ABCA1 protein was investigated. In particular, the impact of inhibitors of the 26S proteasome (MG132), of lysosomal proteases (chloroquine diphosphate) and of calpains (calpeptin) on ABCA1 protein levels was assessed. Only MG132 caused a significant upregulation of the ABCA1 signal (p = 0.010, Figure S4), thus indicating that ABCA1 is degraded by the proteasome in PC3 cells.
3.5. Bortezomib increased ABCA1 expression and restored HDL anti‐proliferative activity in PC3 cells
Bortezomib, the first approved proteasome inhibitor for cancer therapy, was able to increase ABCA1 expression in PC3 cells when used at the final concentration of 5 nM; indeed, ABCA1 signal raised from 2.08 ± 0.34 in untreated cells to 3.01 ± 0.53 in cells exposed to bortezomib (p = 0.025, Figure 5). The concentration of bortezomib was selected in order to increase ABCA1 without causing a direct anti‐proliferative effect on PC3. Indeed, bortezomib at 5 nM did not reduce cell number (Figure 5E and Figure S1) or colony formation (data not shown) in LDL‐treated cells. Consistently, bortezomib at 5 nM did not affect p21 expression, a well‐known inhibitor of cell cycle involved in the direct cytotoxic effect of bortezomib in PCa cells. 20 When tested at 10 and 15 nM, bortezomib caused a fivefold increase of p21 expression; on the contrary, a mild 1.46‐fold increase was detected at 5 nM (p = 0.53 vs. untreated cells) (Figure 5C). In addition, bortezomib at 5 nM did not affect NF‐kB activation, as shown by the preserved phosphorylated/total p65 ratio (Figure S5).
FIGURE 5.
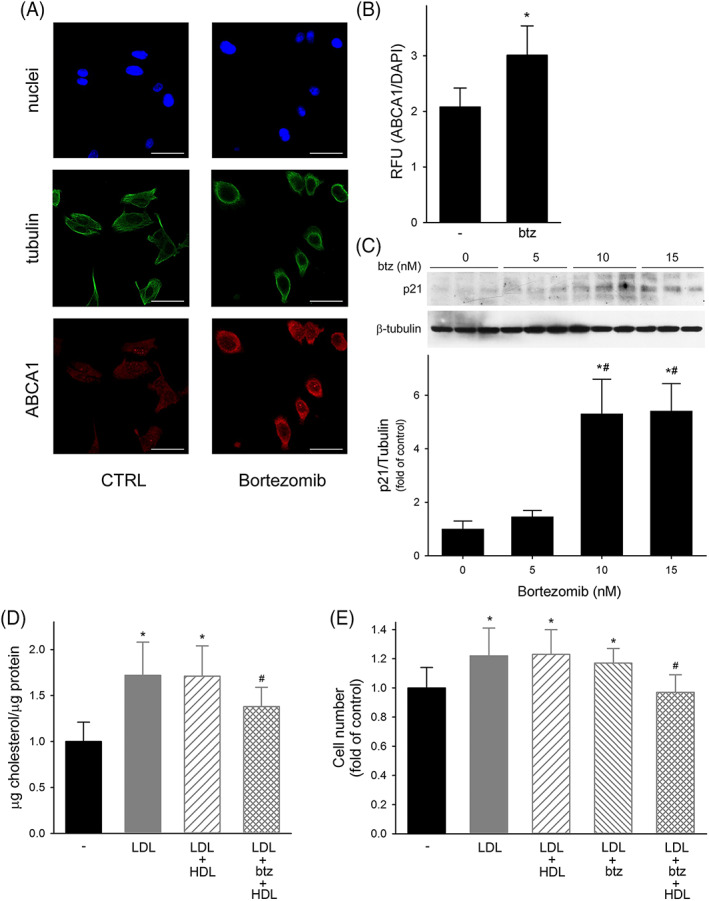
Effect of bortezomib on ABCA1 expression, cholesterol content, and proliferation in PC3 cells; Panel A, Representative confocal immunofluorescence images for ABCA1 (red), tubulin (green), and DAPI (blue). Scale bar, 50 μm. Panel B, Quantitative assessment of ABCA1 fluorescence intensities in untreated cells (−) and in cells incubated with 5 nM bortezomib (btz). Data are expressed as ABCA1/DAPI signal ratio, mean ± SD, n = 3. *p < 0.05 versus untreated cells. Panel C, Expression of p21 by Western blotting in PC3 cells incubated with increasing concentrations of bortezomib. Data are expressed as fold increase of untreated cells, n = 3. *p < 0.05 versus control, #p < 0.05 versus bortezomib 5 nm. Panel D, Cholesterol content in untreated cells (black bars), or in cells incubated with (A) LDL at 50 μg/ml for 24 h (gray bars), (B) LDL at 50 μg/ml for 24 h followed by HDL at 0.5 mg/ml for 1 h (gray dashed bars), or (C) LDL at 50 μg/ml plus bortezomib (btz) 5 nM for 24 h, followed by HDL at 0.5 mg/ml for 1 h (gray crossed bars). Data are expressed as mean ± SD, n = 9. *p < 0.05 versus untreated cells, #p < 0.05 versus LDL. Panel E, Cell proliferation evaluated as cell number at 72 h in untreated cells (black bars) or in cells incubated with (A) LDL at 20 μg/ml for 24 h (gray bars), (B) LDL at 20 μg/ml for the first 24 h followed by HDL at 0.5 mg/ml for 48 h (gray dashed bars), (C) LDL at 20 μg/ml plus bortezomib 5 nM for 24 h followed (gray crossed bars) or not (gray dashed bars) by HDL at 0.5 mg/ml for 48 h
When PC3 cells were pre‐treated with bortezomib, HDL were able to reduce the content of cholesterol in LDL‐loaded cells by 19.8% (Figure 5D). The restoration of ABCA1 expression and cholesterol efflux by bortezomib allowed HDL to completely abolish the increase of cell proliferation induced by LDL (Figure 5E and Figure S1). Consistently, the pretreatment with bortezomib significantly increased HDL ability to reduce PC3 colony formation from 49% ± 15% to 68% ± 8% (p = 0.002 for bortezomib plus HDL vs. HDL alone), similar to what observed in LNCaP.
4. DISCUSSION
The main finding of this work is that HDL are able to inhibit LDL‐induced cell proliferation by reducing cholesterol content in PCa cell lines. These findings support a role for cholesterol in PCa progression, making the modulation of cell cholesterol homeostasis an attractive target for the development of novel therapeutic strategies aimed at starving cancer cells. The expression of proteins involved in cholesterol homeostasis and the experiments performed with lovastatin indicate that PCa cell lines mainly rely on the uptake of lipoproteins, with the concomitant inhibition of cholesterol efflux, rather than on intracellular cholesterol synthesis to meet their cholesterol needs. These data further support the inhibition of lipoprotein endocytosis, for example, by silencing the LDL‐R, rather than of the mevalonate pathway by statins as a starving strategy against cancer. 21 , 22 , 23 Moreover, the progressive increase of SRD5A1 according to the aggressiveness of the phenotype, with PC3 cells showing the highest levels, further supports the ability of PCa cells in advanced stages of the disease to synthesize androgens from cholesterol, as previously shown. 24
Interestingly, HDL were not able to modulate cholesterol content in AR‐null PC3, but only in AR‐positive LNCaP. Since ABCG1 expression is even increased in both cell lines, 6 we focused on the ABCA1 transporter, showing its degradation by the proteasome; proteasome inhibition, by restoring ABCA1 levels, rescued HDL ability to modulate cholesterol and LDL‐induced cell proliferation. Since the mechanisms responsible for the progression of PCa from androgen‐dependent to androgen‐resistant phenotype are unclear, our findings provide insights on ABCA1 and, more generally, on cholesterol homeostasis as possible players in this transition. Our data also confirmed the downregulation of ABCA1 expression in the LNCaP cell line, in which the reduction of both mRNA and protein levels is consistent with the hypermethylation at the ABCA1 promoter 25 ; however, in LNCaP, the residual ABCA1 expression was sufficient to ensure cholesterol efflux. Interestingly, ABCA1 promoter hypermethylation is frequent in intermediate‐ to high‐grade PCa, and ABCA1 levels were shown to be inversely correlated with Gleason score. 25
Proteasome inhibitors are used for the treatment of multiple myelomas and of other plasma cell malignancies. Bortezomib, a dipeptide boronic acid derivative, was the first approved proteasome inhibitor and showed a potent antitumor activity; however, its use is limited by a significant toxicity, especially peripheral neuropathy, and by the development of resistance. 26 For these reasons, second‐generation inhibitors were developed, as carfilzomib and ixazomib; however, carfilzomib has a significant myocardial toxicity as well, while neuropathy is still observed with ixazomib. 27 Proteasome inhibitors can directly elicit apoptosis of cancer cells by altering cell cycle control and by promoting the accumulation of unfolded and misfolded proteins. Interestingly, bortezomib was also shown to be effective in PCa cells, with a p21‐dependent mechanism, and in human prostate tumor xenografts 20 , 28 ; consequently, its use in patients with PCa is under investigation. 29 Our findings extend the current knowledge on the antitumoral effect of bortezomib by suggesting a role for the increased ABCA1 expression as an additional mechanism in PCa cells.
Even if HDL were not able to remove cholesterol from AR‐null PC3 cells without bortezomib pre‐treatment, they significantly reduced colony formation, thus indicating that HDL can affect cell proliferation through other mechanisms. We previously showed the antioxidant effect of HDL on both LNCaP and PC3 cells, 6 but future studies are needed to explore additional antiproliferative mechanisms of HDL. In this regard, the modulation of inflammation or of other metabolic pathways should be taken into account. From a translational point of view, might HDL‐raising agents be used for the prevention and the treatment of cancer? The epidemiological link between plasma levels of HDL‐C and the risk of cancer is still debated, since results from retrospective, prospective or Mendelian randomization‐based studies performed to date gave opposite results. 5 In addition, no studies were performed to formally assess whether the treatment with HDL‐raising agents as niacin, fibrates or CETP inhibitors, is associated with a reduced risk of cancer or to a better prognosis. In addition, the picture is further complicated since several conditions are associated with changes in HDL composition which can significantly impair their functions, 30 likely including cancer or the consequent therapeutic interventions to manage it. In this context, pursuing the preservation or the improvement of HDL function might be a better strategy than the elevation of HDL levels. Finally, HDL are also under development as delivery agents for classical and innovative drugs to increase their tumor‐targeting, thanks to the high expression of SR‐BI in many cancer cells. Indeed, due to their biocompatibility, long circulating time, and loading capacity, HDL were able to increase the intracellular concentration and, consequently, the cytotoxic activity of several drugs as paclitaxel or doxorubicin in different tumor types. 5 When compared with other delivery strategies, as our and other evidence in the literature suggests, HDL could have the advantage to exert an antitumor activity per se.
The present study has two main limitations. The first is the lack of an in vivo validation of HDL antiproliferative effect. In addition, even if bortezomib alone did not exert a cytotoxic effect and did not modulate p21 expression or NF‐kB activation at the low dose we used, it is not possible to exclude the involvement of other mechanisms beyond ABCA1 in the reduction of cell proliferation after treatment with bortezomib and HDL, including a role for other non‐proteasome proteases whose activity is also modulated by bortezomib. 26
AUTHOR CONTRIBUTIONS
Alice Ossoli performed the experiments, data curation, and visualization. Eleonora Giorgio, Federica Cetti, Claudio Rabacchi, and Matteo Pedrelli performed the experiments and data curation. Massimiliano Ruscica, Patrizia Tarugi, and Paolo Parini reviewed and supervised the study. Monica Gomaraschi designed and supervised the study, analyzed the data, and wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Appendix S1
ACKNOWLEDGEMENT
This work was supported by an intramural grant for young researchers of Università degli Studi di Milano to Monica Gomaraschi (Linea 2, azione A). Open Access Funding provided by Universita degli Studi di Milano within the CRUI‐CARE Agreement.
Ossoli A, Giorgio E, Cetti F, Ruscica M, Rabacchi C, Tarugi P, et al. HDL‐mediated reduction of cholesterol content inhibits the proliferation of prostate cancer cells induced by LDL: Role of ABCA1 and proteasome inhibition. BioFactors. 2022;48(3):707–717. 10.1002/biof.1845
Funding information Università degli Studi di Milano, Grant/Award Number: 2017 ‐ Linea 2 Azione A
DATA AVAILABILITY STATEMENT
Data are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Rohatgi A, Westerterp M, von Eckardstein A, Remaley A, Rye KA. HDL in the 21st century: a multifunctional roadmap for future HDL research. Circulation. 2021;143:2293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006;113:2548–55. [DOI] [PubMed] [Google Scholar]
- 3. Rothblat GH, Phillips MC. High‐density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21:229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Calabresi L, Gomaraschi M, Franceschini G. Endothelial protection by high‐density lipoproteins: from bench to bedside. Arterioscler Thromb Vasc Biol. 2003;23:1724–31. [DOI] [PubMed] [Google Scholar]
- 5. Ossoli A, Wolska A, Remaley AT, Gomaraschi M. High‐density lipoproteins: a promising tool against cancer. Biochim Biophys Acta Mol Cell Biol Lipids. 2022;1867:159068. [DOI] [PubMed] [Google Scholar]
- 6. Ruscica M, Botta M, Ferri N, Giorgio E, Macchi C, Franceschini G, et al. High density lipoproteins inhibit oxidative stress‐induced prostate cancer cell proliferation. Sci Rep. 2018;8:2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gomaraschi M. Role of lipoproteins in the microenvironment of hormone‐dependent cancers. Trends Endocrinol Metab. 2020;31:256–68. [DOI] [PubMed] [Google Scholar]
- 8. Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–67. [DOI] [PubMed] [Google Scholar]
- 9. Krycer JR, Brown AJ. Cholesterol accumulation in prostate cancer: a classic observation from a modern perspective. Biochim Biophys Acta. 2013;1835:219–29. [DOI] [PubMed] [Google Scholar]
- 10. Menard JA, Cerezo‐Magaña M, Belting M. Functional role of extracellular vesicles and lipoproteins in the tumour microenvironment. Philos Trans R Soc Lond B Biol Sci. 2018;373:20160480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 12. Guo C, Yeh S, Niu Y, Li G, Zheng J, Li L, et al. Targeting androgen receptor versus targeting androgens to suppress castration resistant prostate cancer. Cancer Lett. 2017;397:133–43. [DOI] [PubMed] [Google Scholar]
- 13. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Lalla OF, Gofman JW. Ultracentrifugal analysis of serum lipoproteins. Methods Biochem Anal. 1954;1:459–78. [DOI] [PubMed] [Google Scholar]
- 15. Franceschini G, Vecchio G, Gianfranceschi G, Magani D, Sirtori CR. Apolipoprotein AIMilano. Accelerated binding and dissociation from lipids of a human apolipoprotein variant. J Biol Chem. 1985;260:16321–5. [PubMed] [Google Scholar]
- 16. Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 17. Altilia S, Pisciotta L, Garuti R, Tarugi P, Cantafora A, Calabresi L, et al. Abnormal splicing of ABCA1 pre‐mRNA in Tangier disease due to a IVS2 +5G>C mutation in ABCA1 gene. J Lipid Res. 2003;44:254–64. [DOI] [PubMed] [Google Scholar]
- 18. den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan‐Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–9. [DOI] [PubMed] [Google Scholar]
- 19. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lashinger LM, Zhu K, Williams SA, Shrader M, Dinney CP, McConkey DJ. Bortezomib abolishes tumor necrosis factor‐related apoptosis‐inducing ligand resistance via a p21‐dependent mechanism in human bladder and prostate cancer cells. Cancer Res. 2005;65:4902–8. [DOI] [PubMed] [Google Scholar]
- 21. Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 2015;112:2473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gallagher EJ, Zelenko Z, Neel BA, Antoniou IM, Rajan L, Kase N, et al. Elevated tumor LDLR expression accelerates LDL cholesterol‐mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene. 2017;36:6462–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caro‐Maldonado A, Camacho L, Zabala‐Letona A, Torrano V, Fernández‐Ruiz S, Zamacola‐Bascaran K, et al. Low‐dose statin treatment increases prostate cancer aggressiveness. Oncotarget. 2017;9:1494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dillard PR, Lin M‐F, Khan SA. Androgen‐independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Mol Cell Endocrinol. 2008;295:115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee BH, Taylor MG, Robinet P, Smith JD, Schweitzer J, Sehayek E, et al. Dysregulation of cholesterol homeostasis in human prostate cancer through loss of ABCA1. Cancer Res. 2013;73:1211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J, Fang Y, Fan RA, Kirk CJ. Proteasome inhibitors and their pharmacokinetics, pharmacodynamics, and metabolism. Int J Mol Sci. 2021;22:11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yan W, Wu Z, Zhang Y, Hong D, Dong X, Liu L, et al. The molecular and cellular insight into the toxicology of bortezomib‐induced peripheral neuropathy. Biomed Pharmacother. 2021;142:112068. [DOI] [PubMed] [Google Scholar]
- 28. Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ. Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol Cancer Ther. 2003;2:835–43. [PubMed] [Google Scholar]
- 29. Voutsadakis IA, Papandreou CN. The ubiquitin‐proteasome system in prostate cancer and its transition to castration resistance. Urol Oncol. 2012;30:752–61. [DOI] [PubMed] [Google Scholar]
- 30. Ossoli A, Pavanello C, Giorgio E, Calabresi L, Gomaraschi M. Dysfunctional HDL as a therapeutic target for atherosclerosis prevention. Curr Med Chem. 2019;26:1610–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Data are available from the corresponding author upon reasonable request.