

Abstract
Disease is a contributing factor to the decline of wildlife populations across the globe. Koalas, iconic yet declining Australian marsupials, are predominantly impacted by two pathogens, Chlamydia and koala retrovirus. Chlamydia is an obligate intracellular bacterium and one of the most widespread sexually transmitted infections in humans worldwide. In koalas, Chlamydia infections can present as asymptomatic or can cause a range of ocular and urogenital disease signs, such as conjunctivitis, cystitis and infertility. In this study, we looked at differences in response to Chlamydia in two northern populations of koalas using a targeted gene sequencing of 1209 immune genes in addition to genome‐wide reduced representation data. We identified two MHC Class I genes associated with Chlamydia disease progression as well as 25 single nucleotide polymorphisms across 17 genes that were associated with resolution of Chlamydia infection. These genes are involved in the innate immune response (TLR5) and defence (TLR5, IFNγ, SERPINE1, STAT2 and STX4). This study deepens our understanding of the role that genetics plays in disease progression in koalas and leads into future work that will use whole genome resequencing of a larger sample set to investigate in greater detail regions identified in this study. Elucidation of the role of host genetics in disease progression and resolution in koalas will directly contribute to better design of Chlamydia vaccines and management of koala populations which have recently been listed as “endangered.”
Keywords: Chlamydia, conservation genomics, GWAS, koala, wildlife disease
1. INTRODUCTION
Wildlife diseases are a major contributor to species’ declines across the globe, including chytridiomycosis in amphibians (Berger et al., 1998; Longcore et al., 1999), devil facial tumour disease (DFTD) in Tasmanian devils (Sarcophilus harissii) (Hawkins et al., 2006; Jones et al., 2007; McCallum, 2008; Pye et al., 2016), and sylvatic plague in black‐footed ferrets (Mustella nigripes) (Matchett et al., 2010; Williams et al., 1994). Koalas (Phascolarctos cinereus) are an iconic arboreal Australian marsupial occurring along the eastern coast of Australia. The species has suffered declines of ~24% over the past three generations (15–21 years) occurring across their entire range (Adams‐Hosking et al., 2016) and have recently been listed as endangered under the Australian Government's Environment Protection and Biodiversity Conservation Act 1999. Anthropogenic factors, including land clearing that results in habitat loss and fragmentation, vehicle strikes and dog attacks, are contributing to this decline (Beyer et al., 2018; Dique et al., 2003; Goldingay & Dobner, 2014; McAlpine et al., 2006; Rhodes et al., 2015). Disease is also a known threat. A 4‐year longitudinal monitoring study determined that predation was the cause of 49.5% of koala deaths, with disease the second biggest contributor at 28.9% with 62.1% of these attributed to Chlamydia (Beyer et al., 2018). With such a large proportion of koalas succumbing to disease pressures it is plausible that Chlamydia is acting as a selective pressure on koala populations. Two main pathogens, Chlamydia (Cockram & Jackson, 1974; Polkinghorne et al., 2013) and koala retrovirus (KoRV; Hanger et al., 2000), are contributing to the decline of koala populations. Koala population declines have been highest throughout the northern region of their range, with Queensland populations decreasing by 53% overall (Adams‐Hosking et al., 2016). High prevalence of Chlamydia infection in northern populations can reduce fertility and reproductive rates, impacting the ability of these populations to stabilize or increase (Polkinghorne et al., 2013; Rhodes et al., 2011).
Chlamydia are obligate intracellular bacteria which can cause ocular or genital tract disease in a range of hosts, including humans, mice (Mus musculus), great barred frogs (Mixophyes iteratus), chickens (Gallus gallus) and koalas (Horn, 2008). In the extracellular phase of the Chlamydia life cycle the bacterium exists as inactive particles called elementary bodies which are then phagocytosed into the cell where they become actively replicating reticulate bodies (Quigley & Timms, 2020; Zuck et al., 2017). This life cycle takes advantage of the host phagocytic machinery and there is potential that genes involved in phagocytosis may influence an individual's ability to clear an infection.
Two main species of Chlamydia infect koalas: C. pneumoniae and C. pecorum, but C. pecorum is more prevalent and more commonly associated with disease (Horn, 2008; Polkinghorne et al., 2013). Clinical disease signs of Chlamydia infection are characterized by inflammatory and fibrotic lesions in the ocular tissues, such as conjunctivitis, and urinary and reproductive tracts, which in severe cases can cause infertility or death (Cockram & Jackson, 1974; McColl et al., 1984; Polkinghorne et al., 2013).
Disease occurs as a result of complex interactions between the host and the microbe. An individual's susceptibility to disease is due to a combination of factors, including sex, age, life history, nutrition, immune response and genetics (Casadevall & Pirofski, 1999, 2015; Godbout et al., 2020).
Koala populations show highly variable levels of Chlamydia infection and disease prevalence, with some populations having only 4% (Mount Lofty) of Chlamydia‐infected koalas showing disease signs and others with 71% (Brisbane) of infected koalas showing disease signs (Quigley & Timms, 2020). Studies have attempted to identify and quantify the drivers of Chlamydial disease progression, with both pathogen and host factors assessed, including: Chlamydia species, Chlamydia load (copies µl–1), Chlamydial major outer membrane protein gene ompA genotype, infection site, host age, sex and co‐infection with KoRV (Griffith et al., 2013; Legione et al., 2016; Quigley et al., 2018; Wan et al., 2011).
Recent studies have identified genetic associations with Chlamydia susceptibility. Lau et al. (2014) identified a single Major Histocompatibility Complex (MHC) Class II variant (DAβ*10) that was more prevalent in koalas with Chlamydia infection and a second MHC Class II variant (DBβ*04) associated with high Chlamydia‐hsp60 antibody levels. Quigley et al. (2018) identified six alleles of the marsupial MHC Class II DAβ that potentially contributed to urogenital disease. Robbins et al. (2020) identified an additional four MHC alleles from the Class II DCβ, DBβ, DAβ and Class I UC genes that were associated with disease progression. Cytokines (tumour necrosis factor α [TNF‐α] and interferon γ [IFNγ]) and interleukins (IL‐17A, IL‐4 and IL‐6; Maher et al., 2014; Mathew et al., 2013, 2014; Mathew, Pavasovic, et al., 2013; Quigley & Timms, 2020) have also previously been implicated in response to Chlamydia in koalas.
In this study, we aimed to measure levels of immune gene diversity at 1209 immune genes from 43 individuals in two koala populations from southeast Queensland. We used a target enrichment approach to target single nucleotide polymorphisms (SNPs) within single‐copy immune genes. Immunoglobulins, T cell receptors and NK receptors were not targeted due to their multicopy nature. We discovered 25 SNPs across 17 genes that were associated with resolution of Chlamydia infection. Additionally, we identified two MHC Class I genes with differences in haplotype frequencies between individuals that were able to resolve an infection and those that were not.
2. METHODS
2.1. Study populations and sample collection
Two koala populations were utilized for this study. The first population, in the Moreton Bay Region (MBR) (27.0946°S, 152.9206°E), is located in peri‐urban/urban koala habitat ~25 km north of Brisbane. The second population, at Old Hidden Vale (HV) (27.6594°S, 152.4672°E), is located ~70 km southwest of Brisbane. The two study sites are separated by the Brisbane Valley barrier (BVB; Johnson et al., 2018). These koala populations were part of ongoing population management programmes (Robbins et al., 2019, 2020). As part of these programmes, animals were subject to regular field monitoring and capture for comprehensive clinical examinations under anaesthesia and treatment of Chlamydia disease, if required (for detailed methods see Robbins et al., 2020). Briefly, at each clinical examination, swab samples were taken from ocular conjunctiva and urogenital sinus and blood was taken from the cephalic vein. Whole blood samples were stored in EDTA at −20°C prior to processing. Veterinarians also performed a physical examination, sonographic examination of the urogenital tract, and cytological examination of the blood and urine sediment.
Detailed clinical observations and diagnostic test results were also recorded for each koala.
2.2. Analysis for Chlamydia pecorum presence
Detection of Chlamydia pecorum followed methods used by Robbins et al. (2020). Briefly, ocular conjunctiva and urogenital tract swab samples were mixed with 500 µl of phosphate‐buffered saline and DNA was extracted from the suspension using a QIAamp DNA mini kit (Qiagen), according to the manufacturer's instructions. The extracted DNA was then used to screen for C. pecorum using a specific qPCR (quantitative polymerase chain reaction) assay that targets a 209‐bp region of the conserved gene CpecG_05739 (Jelocnik et al., 2017; Robbins et al., 2019), and Chlamydial plasmid DNA using a CDS5‐specifc qPCR (Phillips et al., 2018). A standard curve was generated for quantification of C. pecorum infection loads using a known concentration of C. pecorum genomic DNA. Samples were run in duplicate with positive and negative controls included in all qPCR assays. The qPCR and close veterinary observations were chosen for Chlamydia detection as these are more reliable than antibody tests in koalas (Hanger et al., 2013).
2.3. Koala study groupings
Based on the clinical observations and C. pecorum test results obtained during the monitoring period, koalas were divided into clinical groups for the purposes of this study. Study groups were created based on koalas that acquired a Chlamydial infection at some point during the monitoring period and either: (i) resolved that infection without medical intervention (resolvers, n = 12 koalas), or (ii) continued to carry C. pecorum for the remainder of the monitoring period and either did not develop clinical disease signs or developed clinical disease signs (nonresolvers, n = 31 koalas).
2.4. Probe design
We manually annotated MHC Class I and Class II in the koala genome (Johnson et al., 2018) using blast searches (Altschul et al., 1990) using previously identified marsupial MHC sequences. Manual inspection of all blast hits was used to identify start and stop codon sites and splicing locations between exons (Table S1). Exon sequences were then extracted from the genome assembly and submitted to Arbour Biosciences for bait development as described below. Genes to be targeted for enrichment were chosen using Gene Ontology (GO; Ashburner et al., 2000; Carbon et al., 2021) to generate a list of putative genes involved in “immune processes.” Broad criteria were used to include genes in order to minimize the risk of missing any genes which may impact the koala immune response, albeit indirectly. These genes were then searched within the koala genome annotation (Phascolarctos_cinereus.phaCin_unsw_v4.1.98.gff3; Johnson et al., 2018). Exon sequences for the target genes were extracted from the koala genome assembly (Koala phaCin_unsw_v4.1.fa) (Johnson et al., 2018) using bedtools version 2.29.2 (Quinlan & Hall, 2010) with a 40‐bp flanking region either side of each exon. The exon sequences were sent to Daicel Arbour Biosciences who designed 80‐bp RNA baits with a 40‐bp overlap between baits. Each bait was quality checked for blast (Altschul et al., 1990) specificity to the intended region of the koala genome (Johnson et al., 2018) at an estimated hybridization melting temperate (T m, °C) as well as GC content. Baits were filtered based on Arbour Biosciences moderate filtering criteria where a bait passed if: at most 10 blast hits between 62.5 and 65°C and two blast hits above 65°C, and fewer than two passing baits on each flank. Baits were also removed if they were more than 25% masked based on repeats in the mammalian database or if they were more than 25% masked based on repeats soft marked in the koala genome (Johnson et al., 2018). Any genes which had baits entirely filtered out of the probe set were searched on Web of Science with “gene name” and “Chlamydia,” and genes that had published examples of associations with Chlamydia disease were retained to capture all potential associations. The final bait set (see Results) was sent to Arbour Bioscience for synthesis.
2.5. DNA extraction, library preparation and target capture
DNA was extracted from 200 µl of whole blood using the standard MagAttract HMW DNA kit (Qiagen) protocol with elution in 100 µl buffer AE. Quantity of DNA was determined by using a Nanodrop 2000 spectrophotometer (ThermoFisher Scientific) and quality was assessed through a 0.8% agarose TBE gel stained with SYBR Safe (Life Technologies), where 2 µl of DNA was stained with 4 µl of 10% loading dye (Bioline). DNA was separated alongside a 1‐kb size standard (Bioline) for 30 min at 90 V and bands were visualized using a Gel Doc XR + (Bio‐Rad) under ultraviolet light and images analysed with ImageLab (Bio‐Rad). DNA libraries were prepared using a Kapa Hyperprep Kit (Roche) following standard procedures. Briefly, 400 ng of DNA was cleaned using a 3× volume OF magnetic beads and eluted in 30 µl of buffer EB. Following this, 120 ng of DNA in 35 µl of EB was fragmented for 23 min at 37°C before A‐tailing, end repair and adapter ligation with KAPA unique dual indexed adapters (Roche). Libraries were then amplified in a T100 thermocycler (Bio‐Rad) with Illumina primers with an initial denaturation for 45 s at 98°C, then five cycles of denaturation at 98°C for 15 s, annealing at 60°C for 30 s and extension at 72°C for 30 s, and a final extension at 72°C for 1 min. Amplified libraries were then cleaned with a 1× bead volume and eluted in 15 µl of buffer EB. The quantity of the library was determined using a Nanodrop 2000 spectrophotometer (ThermoFisher) and average library size estimated on a 2100 Bioanalyzer instrument (Agilent). For target enrichment, eight samples were pooled in equimolar concentrations for a total of 500 ng of DNA. Hybridization of DNA to RNA baits occurred for 16 h at 63°C, following which the solution was washed with magnetic beads and a wash solution and eluted in 30 µl 0.05% TWEEN‐20 solution. Fifteen microlitres of the enriched library was amplified through 10 PCR cycles with conditions as listed above and a final 1× bead clean‐up and elution in 20 µl of buffer EB. Enriched libraries were quantified using a Qubit 2.0 fluorometer (Invitrogen). Confirmation of enrichment of intended gene regions was determined through copy number qPCR with two genes included that were present in the bait set (LOX and EOMES) and two genes as controls (ACRBP and CRCP) using primers designed for this study (Table S2). For qPCR, 2 µl of enriched library was diluted with 14 µl of EB and unenriched libraries diluted to similar concentration (~0.5 ng/µl), reactions were cycled on a CFX Connect Real‐Time PCR Detection System (Bio‐Rad) with initial denaturation at 95°C for 5 min, followed by 40 cycles at 95°C for 10 s, and then 55°C for 30 s, with images taken each cycle. This was followed by a melt curve from 50°C in 1°C increments every 5 s to 95°C. The copy number of each gene product was analysed on the CFX maestro Software version 1.0 (Bio‐Rad). All target reactions were then pooled equimolarly with a final concentration of 2 nm in 40 µl of buffer EB. Sequencing was done on an Illumina NovaSeq SP 2 × 150‐bp flowcell at The Ramaciotti Centre for Genomics (Kensington).
2.6. Bioinformatics
Sequencing reads were removed if their length was less than 50 bases and reads were trimmed if leading or trailing bases had a quality below 20 or if read quality was below 20 in a 4‐bp sliding window. Adapter sequences were removed using trimmomatic version 0.38 (Bolger et al., 2014). fastqc version 0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was run on trimmed reads and checked for read length and contamination. Trimmed reads were aligned to the koala reference genome (Johnson et al., 2018) using the Burrows Wheeler Aligner version 0.7.17 (bwa) mem function with default parameters (Li & Durbin, 2009) and duplicated reads marked with the picard version 2.21.9 mark duplicates function (http://broadinstitute.github.io/picard/). ngscat version 0.1 (Lopez‐Domingo et al., 2014) was run to assess coverage across target regions. The Genome Analysis Toolkit (gatk) version 4.1.9.0 (McKenna et al., 2010) “best practices” pipeline was run on aligned reads. Briefly, haplotypecaller was used to genotype individual samples over target regions, then genomicsdbimport was used to combine multiple g.vcf files to build a database of multisample variants, and finally genotypegvcfs was used to build multisample vcf files. Variant statistics were determined for each variant using the gatk VariantsToTable function and distributions of each statistic visualized in R version 4.0.2 (R Core Team, 2021; Figure S1) to determine filtering metrics. Variants were filtered through the gatk VariantFiltration function by; QD < 2, AN < 77.4, SOR > 3, FS > 60, MQ < 40, −2.5 < MQRankSum < 2.5, −2.5 < ReadPosRankSum < 2.5 and −3 < BaseQRankSum < 3. Following this, only biallelic SNPs were retained and alleles present at an MAF < 0.08 (to ensure an allele was seen in at least two individuals) were removed.
2.7. RRS sequencing
Samples were also sequenced by RRS to investigate genome‐wide neutral diversity. RRS was performed using DArTseq (Diversity Arrays Technology PL; DArT). Sample DNA quality and quantity were assessed, as above, to ensure each sample met the minimum concentration of 50 ng/µl and volume of 20 µl required by DArT. The combination of frequent and infrequent cutting restriction enzymes (SphI and PstI, respectively) was used to prepare the genomic DNA, and sequencing of size‐selected amplified fragments was achieved with a HiSeq2500 using 77‐bp single‐end reads.
Reads were processed and genotypes called using our in‐house bioinformatic pipeline (Wright et al., 2019). Briefly, fastq files were quality checked with fastqc version 0.11.8, cleaned and trimmed using stacks version 2.0 process_radtags (Catchen et al., 2013) and aligned to the koala reference genome (Johnson et al., 2018) using bwa (Li & Durbin, 2009). SNPs were called using stacks ref_map pipeline to output one random SNP per locus, using a minimum call rate of 20% and maximum observed heterozygosity of 70%. Additional filtering was performed in R to retain loci with alleles sequenced to a minimum mean depth of 2.5, allelic coverage difference no greater than 80%, reproducibility (based on technical replicates performed by DArT) above 90%, MAF > 0.05 and removal of any potential sex‐linked loci (Wright et al., 2019).
2.8. Comparison of target enrichment and RRS
Exploratory principal coordinates analysis (PCoA) was performed using target enrichment and RRS variants (hereafter "immune gene" and "genome‐wide" SNPs, respectively) separately and plotted using adegenet (Jombart, 2008) in R to investigate genetic differences between koalas from HV and MBR. annovar version 20180416 (Wang et al., 2010) was used to characterize SNPs against the koala genome annotation (Johnson et al., 2018) as intergenic, intronic or exonic (nonsynonymous or synonymous). Heterozygosity of each animal was calculated using the genhet package (Coulon, 2010) in R. Average heterozygosity of individuals from each population (MBR and HV) was calculated separately for each data type and compared using an ANOVA.
2.9. Identifying candidate SNPs
Levels of inbreeding were calculated for each individual using plink (Purcell et al., 2007), with the mean F of each group (resolvers, nonresolvers) compared using an independent sample t test. An increased level of inbreeding results in a decrease in overall genetic diversity which may cause a loss of alleles responsible for disease resistance (Hedrick & Garcia‐Dorado, 2016). Second, a genome wide association study (GWAS) was performed using plink (Purcell et al., 2007) with both a chi‐square test and Fisher's exact test to identify any SNPs associated with the ability of a Chlamydia‐infected koala to resolve their infection (Figure S2). Any SNPs with a p‐value ≤ .001 were selected as significant SNPs (Batley et al., 2019, 2021). The basic GWAS is a commonly used approach that compares allele frequencies between case and control phenotypes and is used to identify loci associated with the trait of interest (Korte & Farlow, 2013); in this study we used both a chi‐squared test and Fisher's exact test to reduce the chance of false positives whilst maintaining the greatest number of candidate SNPs possible. Our GWAS was complemented with Weir and Cockerham's F ST test (Weir & Cockerham, 1984) using the ‐weir‐fst‐pop tool in vcftools version 0.1.14 (Danecek et al., 2011) and SNPs with an F ST more than 5 SD from the mean were said to be significant SNPs (Axelsson et al., 2013; Batley et al., 2021; Figure S2). We used Weir and Cockerham's F ST to identify regions of genetic differentiation between our resolvers and nonresolvers. These two methods work differently with a GWAS aiming to identify loci associated with a chosen phenotype (Korte & Farlow, 2013) whereas an F ST outlier identifies loci with allele frequency differences between groups than would be expected under drift alone (Lotterhos & Whitlock, 2014). Any SNP which was identified as significant in two or more of the three tests were chosen to be candidate SNPs to reduce false positives (Figure S2). The gene location of each candidate SNP was determined, and the GO slim for immunology annotation set in GOnet (Pomaznoy et al., 2018) was used to investigate key pathways that candidate genes were involved in. We identified any additional SNPs located within our candidate genes and determined whether those SNPs were synonymous or nonsynonymous using annovar version 20180416 (Wang et al., 2010).
2.10. MHC analysis
Single sample bam files (N = 43) were used as input to gatk version 4.1.9.0 (McKenna et al., 2010) to call variants occurring within annotated MHC genes in the koala genome (Johnson et al., 2018). HaplotypeCaller was used to call variants across scaffolds in the koala genome containing MHC genes, then GenomicsDBImport combined multiple g.vcf files and GenotypeGVCFs was used to build multisample vcf files. Variants within exons of MHC genes were phased using phase version 2.1.1 (Stephens & Scheet, 2005; Stephens et al., 2001), an allele was only considered real if it appeared twice within the data set. A Pearson's chi‐square test was used to test for differences between observed and expected MHC allele frequencies for animals able to resolve a Chlamydia infection and those unable to resolve an infection. As the number of MHC genes with variants investigated is small (23) and the nature of this study is exploratory, to identify as many variants as possible for further investigation we decided it was not necessary to correct for multiple testing (Bender & Lange, 2001). For the two MHC Class I genes significantly associated with being able to resolve a Chlamydia infection, we conducted AICC (corrected Akaike information criterion)‐based model selection following Grueber et al. (2013). We investigated the effects of heterozygosity at each gene as well as the effect of each allele on the ability to resolve an infection. Heterozygosity at each gene was coded as 0/1 (for homozygous and heterozygous, respectively) and each individual was coded as 1/0 for presence/absence of the allele in question. All models included heterozygosity as a factor. Each model was ranked based on AICC values and models ≥2 AICC from the base model were interpreted to be evidence for the given allele being able to influence the ability to resolve a Chlamydia infection (Grueber et al., 2013; Sepil et al., 2013).
2.10.1. Regulatory approvals
Koala management programmes were conducted under approvals issued by the Queensland Department of Agriculture and Fisheries (approvals CA 2012/03/597, CA 2013/09/719, CA 2014/06/777, CA 2015/03/852 and CA 2016/03/950), and work with koalas was authorized by scientific permits issued by the Queensland Department of Environment and Heritage Protection (approvals WISP 11525212, WISP 16125415, WISP 13661313, WITK 14173714, WISP 17273716 and WA 0008304). Swab samples were analysed under approval nos. AN/A/13/80 and AN/E/19/33 issued by the University of the Sunshine Coast Animal Ethics Committee.
3. RESULTS
3.1. Koala samples and Chlamydia pecorum status
Fourteen koalas from HV and 29 from MBR were included in the study, all of which were infected with C. pecorum during the monitoring period. Out of the 43 sampled koalas, 31 individuals were not able to resolve an infection (nonresolvers) and 12 individuals (resolvers) were able to resolve an infection. For RRS, 41 samples (resolvers = 11, nonresolvers = 30) had the required concentration and volume of DNA. Of the 12 resolvers, 11 were from MBR and one from HV; of the 31 non resolvers, 18 were from MBR and 13 from HV (Figure 1).
FIGURE 1.
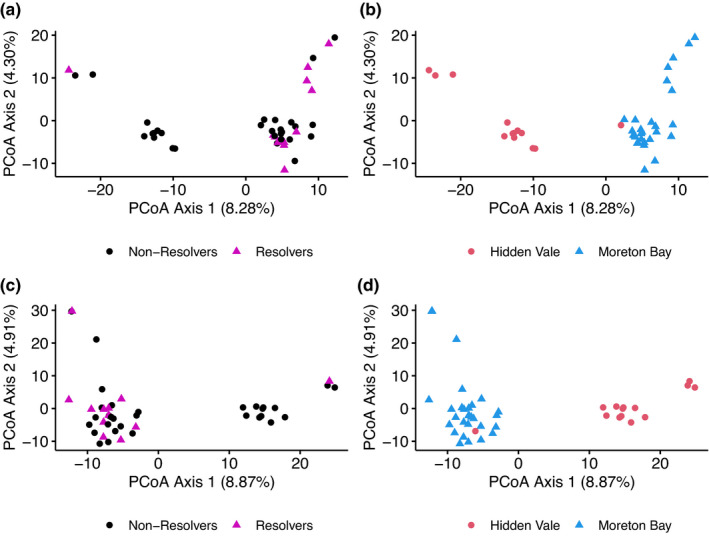
(a) PCoA plot produced using genome‐wide neutral SNPs from 41 sequenced individuals, with individuals labelled by whether they were able to resolve a Chlamydia infection (resolvers—purple triangles) or not (nonresolvers—black circles). (b) PCoA plot produced using genome‐wide neutral SNPs from 41 sequenced individuals, with individuals labelled by study population, either Moreton Bay or Hidden Vale. (c) PCoA plot produced using biallelic SNPs from targeted immune genes in 43 sequenced individuals, with individuals labelled by whether they were able to resolve a Chlamydia infection (resolvers) or not (nonresolvers). (d) PCoA plot produced using biallelic SNPs from targeted immune genes in 43 sequenced individuals, with individuals labelled by study population, either Moreton Bay (blue triangles) or Hidden Vale (red circles)
3.2. Bait probe design
Our initial probe set consisted of 99,034 baits targeting 14,820 exonic sequences representing 1258 immune‐related genes (Table S3). We did not attempt to target immunoglobulin or T‐cell receptor genes as variation in these regions is largely generated through V(D)J recombination (Tonegawa, 1983; Ujvari & Belov, 2015). We identified 19 MHC Class I genes, including all 11 identified by Cheng et al. (2018), 14 of these with full‐length coding sequences (CDS), and we also found 16 MHC Class II for inclusion in our probe (Table 1, Figure 2). Based on filtering by Arbour Biosciences, 1140 baits failed the moderate filtering criteria, 5936 baits were more than 25% masked by the mammalian repeat database and 22,446 baits were more than 25% masked based on repeats in the koala genome. Manual screening of the removed baits resulted in 2151 bait sequences being reintroduced to the probe set. This process brought the final bait probe set to 73,397 sequences (Table S4) containing 13,783 exons from 1,209 immune‐related genes and covering 4,253,688 bp (or ~0.13% of the total genome).
TABLE 1.
Genomic coordinates of the 35 MHC genes we annotated as well as the number of haplotypes observed and p‐value from a chi‐squared test investigating allele frequency differences between individuals that resolved a Chlamydia infection and individuals that did not resolve an infection
| Gene | Scaffold | Strand | Start position | End position | No. of alleles | p |
|---|---|---|---|---|---|---|
| DAA | MSTS01000255.1 | − | 1,210,001 | 1,214,547 | 4 | .20 |
| DAB1 | MSTS01000401.1 | + | 78,815 | 85,317 | 0 | NA |
| DAB2 | MSTS01000401.1 | − | 145,139 | 153,948 | 15 | .06 |
| DAB3 | MSTS01000544.1 | + | 63,131 | 71,649 | 12 | .47 |
| DAB4 | MSTS01000840.1 | − | 12,395 | 23,791 | 2 | .89 |
| DAB5 | MSTS01000302.1 | + | 47,412 | 85,351 | 0 | NA |
| DBA1 | MSTS01000255.1 | − | 1,175,692 | 1,178,484 | 6 | .44 |
| DBA2 | MSTS01000255.1 | − | 1,141,123 | 1,143,891 | 0 | NA |
| DBA3 | MSTS01000255.1 | − | 1,071,439 | 1,074,196 | 5 | .22 |
| DBB1 | MSTS01000255.1 | + | 1,049,332 | 1,053,643 | 3 | .35 |
| DBB2 | MSTS01000255.1 | + | 1,071,439 | 1,074,196 | 4 | .21 |
| DBB3 | MSTS01000255.1 | + | 1,118,112 | 1,122,579 | 6 | .59 |
| DCA | MSTS01000255.1 | − | 1,414,232 | 1,417,702 | 0 | NA |
| DCB | MSTS01000255.1 | + | 1,420,849 | 1,430,221 | 4 | .16 |
| DMA | MSTS01000255.1 | + | 316,690 | 319,085 | 4 | .33 |
| DMB | MSTS01000255.1 | + | 340,447 | 343,862 | 6 | .37 |
| MHCI‐1(UI) | MSTS01000347.1 | + | 1,561,564 | 1,564,694 | 5 | .87 |
| MHCI‐10(UF) | MSTS01000255.1 | + | 986,710 | 989,373 | 4 | .20 |
| MHCI‐11 | MSTS01000314.1 | + | 2,329,060 | 2,332,457 | 0 | NA |
| MHCI‐12(UH) | MSTS01000129.1 | − | 3,854,947 | 3,858,138 | 6 | .85 |
| MHCI‐13(UG) | MSTS01000129.1 | − | 3,816,284 | 3,819,564 | 6 | .29 |
| MHCI‐14 | MSTS01000778.1 | − | 11,798 | 15,205 | 0 | NA |
| MHCI‐15(UJ) | MSTS01000454.1 | − | 94,471 | 97,231 | 3 | .46 |
| MHCI‐16 | MSTS01001120.1 | − | 62 | 3,236 | 0 | NA |
| MHCI‐17 | MSTS01000129.1 | + | 3,931,562 | 3,934,737 | 0 | NA |
| MHCI‐18 | MSTS01000381.1 | − | 34,572 | 37,792 | 0 | NA |
| MHCI‐19(UB) | MSTS01000381.1 | − | 11,690 | 15,571 | 8 | .38 |
| MHCI‐2‐partial(UD) | MSTS01000347.1 | + | 1,588,258 | 1,590,606 | 4 | .63 |
| MHCI‐3‐partial | MSTS01000347.1 | + | 1,624,069 | 1,626,296 | 0 | NA |
| MHCI‐4(UA) | MSTS01000263.1 | − | 3,139,825 | 3,143,043 | 17 | .04 a |
| MHCI‐5(UK) | MSTS01000255.1 | + | 400,493 | 407,225 | 3 | .17 |
| MHCI‐6‐partial | MSTS01000255.1 | + | 548,482 | 551,594 | 0 | NA |
| MHCI‐7 | MSTS01000255.1 | + | 618,950 | 622,135 | 0 | NA |
| MHCI‐8(UC) | MSTS01000255.1 | + | 764,362 | 767,048 | 4 | .03 a |
| MHCI‐9(UE) | MSTS01000255.1 | + | 839,055 | 841,733 | 9 | .37 |
A significant difference between allele frequencies of individuals that resolved a Chlamydia infection and those that did not.
FIGURE 2.

Genomic location of the 19 MHC Class I and 16 MHC Class II genes annotated, with yellow bars indicating which genes were included in our target probe set. Created with BioRender.com
3.3. Genetic variants
Sequencing of the koala genomic fragments enriched from the bait probe screening resulted in 907,793,964 total raw reads, ranging from 5,369,327 to 69,984,213 per individual, at an average coverage over the target regions of 226.87 (59.4–652.4). After processing and filtering, 19,310 immune gene variants were detected, of which 14,921 were biallelic SNPs (Table 2). There was a total of 132,416,470 raw sequences from DArT; two samples did not have the required quantity for sequencing. Prior to filtering, there were 23,408 SNPs, and after filtering, 8,801 (genome‐wide) SNPs remained with 1.90% of these falling in coding regions (Table 2).
TABLE 2.
Breakdown of the number of variants detected in immune genes and across the genome
| Immune gene | Genome‐wide | |
|---|---|---|
| Unfiltered variants | 24,425 | 23,408 |
| Filtered variants | 19,310 | 8,801 |
| SNPs | 15,048 | 8,801 |
| INDEL | 4,062 | NA |
| Biallelic SNPs | 14,921 | 8,801 |
| Nonsynonymous | 2,953 | 64 |
| Synonymous | 3,110 | 103 |
Immune gene SNPs were detected via target enrichment and genome‐wide SNPs were detected via DArT.
3.4. Comparison of immune gene and genome‐wide SNPs
Exploratory population analysis using immune gene SNPs and genome‐wide SNPs produced similar structuring within PCoA plots, indicating that genome‐wide SNPs and targeted functional SNPs show similar population structuring and that each type of data is adequate to elucidate population trends (Figure 1). In contrast, comparison of heterozygosity between the two data types showed immune gene SNPs are significantly more homozygous than genome‐wide SNPs (Figure 3; F = 97.31 df =1, p = 1.56 × 10−15). As intended, a higher proportion of our immune gene SNPs fell within coding regions of the genome, compared to genome‐wide SNPs (40.6% vs. 1.9%).
FIGURE 3.
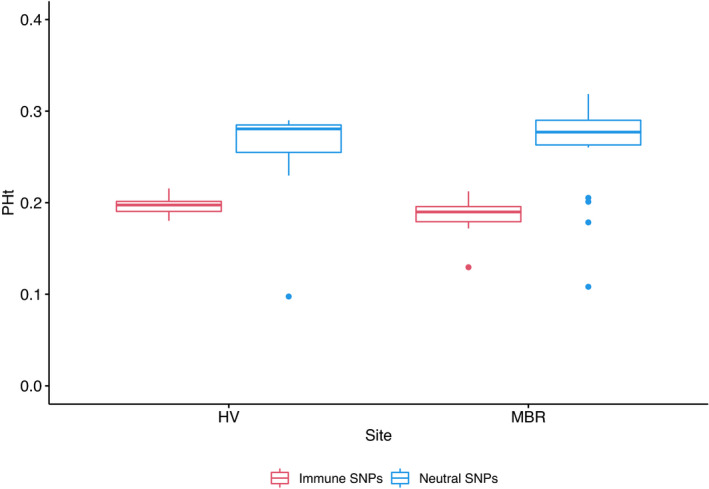
Boxplot displaying the proportion of heterozygotes (PHt) at Hidden Vale and Moreton Bay determined from immune gene SNPs (red) and genome‐wide neutral SNPs (blue). Whiskers mark the “minimum” (1Q − 1.5 × IQR) and “maximum” (3Q + 1.5 × IQR), with outliers shown as dots
3.5. Identification of candidate SNPs
Using immune gene SNPs, no significant difference in inbreeding was detected between resolvers and nonresolvers (resolvers =0.051 ± 0.10; nonresolvers 0.024 ± 0.059, p = .3977) suggesting that inbreeding was not a factor in the ability to resolve a Chlamydia infection. Our GWAS aimed to identify loci associated with the ability to resolve a Chlamydia infection and we used both a chi‐squared test and Fisher's exact test to reduce the chance of false positives whilst maintaining the greatest number of candidate SNPs possible. Using our immune gene variants, the chi‐square test in plink identified 13 SNPs had significance levels ≤.001, while Fisher's exact test identified two SNPs as significant (Figure S2). The F ST outlier test identified loci with allele frequency differences between our two groups (resolvers and nonresolvers) than would be expected under drift alone (Lotterhos & Whitlock, 2014); we identified 42 SNPs as significant using this method. A total of 13 SNPs showed signatures of selection from two or more methods and were designated as candidate SNPs (Figure S2). These 13 SNPs are located across eight genes (HSD3B7, PATZ1, RAB35, SERPINE1, STAT2, STX4, TLR5 and TOB2). Four SNPs are located within exons and result in synonymous substitutions, two SNPs were within introns and seven SNPs were within untranslated regions (Table 3).
TABLE 3.
Results of a GWAS and Weir and Cockerham's F ST (Weir & Cockerham, 1984) investigation into the association of SNPs with the ability to resolve Chlamydia infection
| Gene | GO terms | No. of SNPs in gene (No. of NS SNPs in gene) |
Scaffold Position |
A1 | A2 | F ST | Fisher's p‐value | χ2 | p‐value |
|---|---|---|---|---|---|---|---|---|---|
| TLR5 | Activation of innate immune response, innate immune response‐activating signal transduction, immune response, regulation of immune response, defence response, positive regulation of immune system process, signal transduction | 25 (3) |
14,802,487 |
C | A | 0.28 | 1.21 × 10−3 | 12.46 | 4.15 × 10−4 |
| TOB2 | Negative regulation of immune system process, regulation of haemopoiesis | 9 (4) |
15,243,211 |
G | A | 0.29 | 1.10 × 10−3 | 12.29 | 4.56 × 10−4 |
|
15,244,189 |
A | G | 0.30 | 6.51 × 10−4 | 13.57 | 2.30 × 10−4 | |||
| LAAO‐like | NA | 1 (0) |
11,680,781 |
G | C | 0.32 | 1.25 × 10−3 | 13.60 | 2.26 × 10−4 |
| SEC31A | Protein‐containing complex assembly, protein‐containing complex assembly, membrane organization, vesicle‐mediated transport, cellular response to stress, antigen processing and presentation, signal transduction, transport | 1 (0) |
13,163,640 |
C | T | 0.31 | 2.64 × 10−4 | 13.59 | 2.27 × 10−4 |
| NA | NA |
3,701,342 |
A | G | 0.57 | 9.65 × 10−6 | 22.12 | 2.57 × 10−6 | |
| STAT2 | Immune response, immune effector process, defence response, signal transduction | 14 (2) |
1,206,796 |
A | G | 0.33 | 2.63 × 10−4 | 15.74 | 7.25 × 10−5 |
| Upstream IFNγ | Signal transduction, defence response, immune response, regulation of programmed cell death, regulation of haemopoiesis, immune effector process, cell cycle, leukocyte activation, cell death, positive regulation of immune system process, regulation of immune response, regulation of leukocyte activation, regulation of immune effector process | 5 (0) |
14,472,749 |
T | C | 0.31 | 5.26 × 10−4 | 13.87 | 1.96 × 10−4 |
| OCA2 | Secondary metabolic process, transmembrane transport, transport, biosynthetic process | 2 (1) |
7,212,126 |
G | T a | 0.27 | 9.36 × 10−4 | 12.02 | 5.26 × 10−4 |
| lncRNA | NA |
3,493,071 |
C | T | 0.30 | 3.95 × 10−4 | 13.00 | 3.11 × 10−4 | |
| PATZ1 | Lymphocyte differentiation, immune system development, leukocyte activation | 23 (8) |
7,839,581 |
T | C | 0.23 | 5.00 × 10−3 | 10.84 | 9.95 × 10−4 |
|
7,864,118 |
A | C | 0.29 | 1.22 × 10−3 | 13.71 | 2.13 × 10−4 | |||
| EIF4ENIF1 | Transport, nucleocytoplasmic transport | 3 (0) |
7,972,423 |
G | A | 0.44 | 1.90 × 10−4 | 13.00 | 3.11 × 10−4 |
| C12orf4 | Positive regulation of immune system process, regulation of immune response, regulation of leukocyte activation, regulation of immune effector process | 1 (0) |
7,018,265 |
A | G | 0.27 | 7.95 × 10−4 | 11.11 | 8.58 × 10−4 |
| RAD51AP1 | Cellular response to stress, cellular nitrogen compound metabolic process, DNA metabolic process | 1 (0) |
7,018,265 |
A | G | 0.27 | 7.95 × 10−4 | 11.11 | 8.58 × 10−4 |
| lncRNA | NA |
286,263 |
C | G | 0.39 | 2.73 × 10−4 | 15.02 | 1.07 × 10−4 | |
| RAB35 | Vesicle‐mediated transport, antigen processing and presentation, cell division, mitotic cell cycle, signal transduction, cell cycle, transport | 5 (0) |
6,437,123 |
G | A | 0.25 | 2.13 × 10−3 | 11.00 | 9.11 × 10−4 |
|
6,437,706 |
T | C | 0.25 | 2.13 × 10−3 | 11.00 | 9.11 × 10−4 | |||
| NA | NA |
5,151,686 |
C | T | 0.30 | 4.47 × 10−4 | 12.99 | 3.13 × 10−4 | |
| HSD3B7 | Biosynthetic process, lipid metabolic process, catabolic process, small molecule metabolic process, cell motility, leukocyte migration | 7 (0) |
4,423,003 |
C | T | 0.25 | 2.60 × 10−3 | 10.90 | 9.61 × 10−4 |
|
4,423,041 |
T | C | 0.25 | 2.60 × 10−3 | 10.90 | 9.61 × 10−4 | |||
| STX4 | Cell–cell signalling, protein‐containing complex assembly, protein‐containing complex assembly, membrane organization, immune response, regulation of immune response, regulation of programmed cell death, regulation of leukocyte activation, regulation of immune effector process, defence response, positive regulation of immune system process, vesicle‐mediated transport, signal transduction, transport | 20 (2) |
4,444,745 |
T | C | 0.25 | 2.60 × 10−3 | 10.90 | 9.61 × 10−4 |
|
4,477,817 |
T | C | 0.25 | 2.60 × 10−3 | 10.90 | 9.61 × 10−4 | |||
| SERPINE1 | Extracellular matrix organization, ageing, regulation of leukocyte migration, defence response, regulation of programmed cell death, positive regulation of immune system process, vesicle‐mediated transport, transport | 19 (3) |
6,941,273 |
G | A | 0.23 | 5.00 × 10−3 | 10.84 | 9.95 × 10−4 |
| RBFOX1 | mRNA processing, cellular nitrogen compound metabolic process, transport | 5 (0) |
1,520,037 |
A | C | 0.31 | 1.56 × 10−3 | 12.25 | 4.65 × 10−4 |
| UCN3 | Cellular response to stress, signal transduction | 1(0) |
224,660 |
G | A | 0.50 | 4.43 × 10−6 | 23.17 | 1.48 × 10−6 |
SNPs were identified by two methods: target capture to identify immune gene SNPs and DArT to identify genome‐wide SNPs.
The change in nucleotide results in a nonsynonymous mutation.
Using genome‐wide SNPs, the chi‐squared association test in plink identified 12 SNPs with significance levels ≤.001, while Fisher's exact test identified 11 SNPs and the F ST outlier test identified 32 significant SNPs. A total of 12 SNPs showed signatures of selection from two or more methods and were also designated as candidate SNPs (Figure S2). Seven SNPs are located within gene regions (LAAO‐like, C12orf4 and RAD51AP1, EIF4ENIF1, OCA2, RBFOX1, SEC31A and UCN3), two are within noncoding RNA and one is 7000 bp downstream from IFNγ (Table 3). Of the seven SNPs located within genes, six are within introns and one SNP, located in OCA2, is located within an exon and results from the change of a cysteine amino acid to a tryptophan in the peptide sequence. Of the 17 candidate genes we identified, additional SNPs were identified within 12 of these genes and seven of these genes contained nonsynonymous substitutions (OCA2, PATZ1, SERPINE1, STAT2, STX4, TLR5 and TOB2) that may influence biological function (Table 3). STX4 and IFNγ had the most associated GO terms (13), highlighting the importance of these two genes in the overall immune response to pathogen infection (Figure 4), while SERPINE1 was associated with eight GO terms (Figure S3). The most implicated GO terms were signal transduction, transport, defence response, positive regulation of immune system process and immune response.
FIGURE 4.
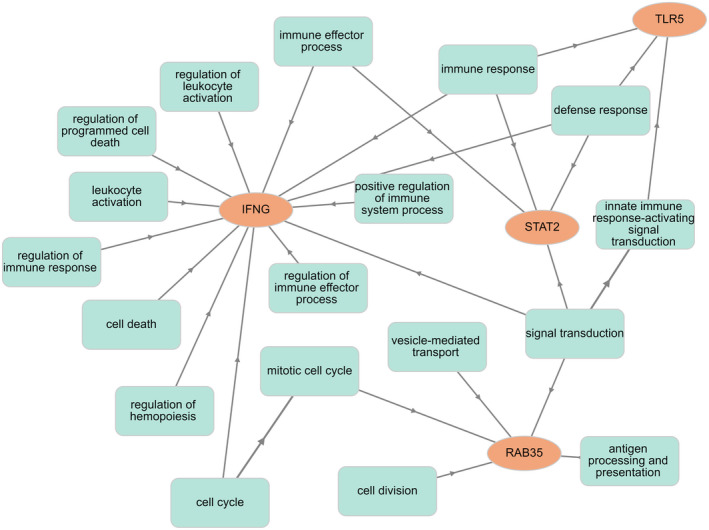
Interaction of four genes (IFNγ, RAB35, STAT2 andTLR5) with GO terms, determined using GOnet (Pomaznoy et al., 2018), with genes in orange circles and GO terms in blue rectangles. For the full interaction plot, see Figure S3
3.6. MHC analysis
Our target probe captured sequence from 24 of the 35 genes we annotated, and within these 24 genes we identified 846 biallelic SNPs within MHC genes with 69 haplotypes in 11 MHC Class I genes and 71 haplotypes across 12 MHC Class II genes and no variation in one MHC Class II gene, DBα2 (Table 1). Two MHC Class I genes, MHCI‐4 (UA), MHCI‐8 (UC), had significant differences in allele frequency between individuals that resolved a Chlamydia infection and individuals that did not (Table 1, Figure 5). Similar levels of variation within MHC Class I (and II) genes were observed in our study, with 17 and four alleles in UA and UC genes compared to the seven and nine alleles identified by Cheng et al. (2018). In Class II genes we identified 29, 11 and 13 alleles in the genes DAβ, DBα and DBβ, respectively, compared with eight, three and five alleles identified by Lau et al. (2013). We conducted AICC model selection on all alleles for UA (N = 17) and UC (N = 4) to determine alleles influencing the ability to resolve a Chlamydia infection. For the UA gene we identified one model ≥2 AICC from the base model with the presence of allele UA*6 more prevalent in koalas that resolved Chlamydia infection (25%, 3/12) than those that did not resolve an infection (0%, 0/31) (Table 4). For the UC gene we failed to identify any models ≥2 AICC and interpret this as heterozygosity at UC having the greatest impact on being able to resolve a Chlamydia infection. Koalas that were heterozygous at UC were more likely to resolve a Chlamydia infection (50%, 6/12) than those that did not resolve an infection (9.68%, 3/31) (Table 4).
FIGURE 5.
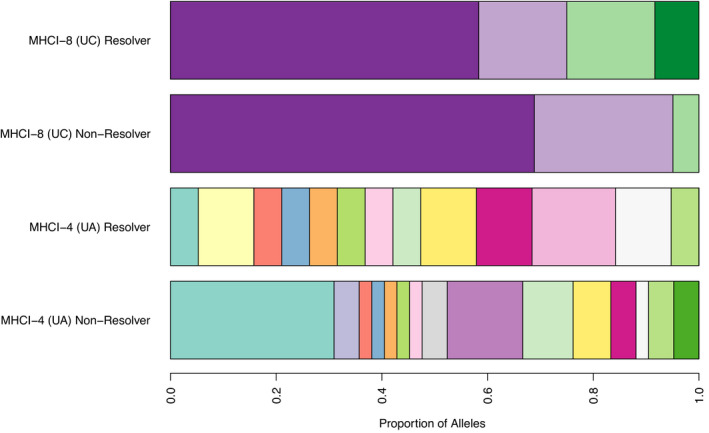
Bar plot representing the differences in proportion of each allele present between koalas that resolved a Chlamydia infection and koalas that did not resolve an infection in the genes UA and UC
TABLE 4.
Effect of MHC Class I genotypes on the ability to resolve a Chlamydia infection for the UA (a) and UC (b) genes
| Model | k | Deviance | AICC | ΔAICC | AICCWt |
|---|---|---|---|---|---|
| (a) UA | |||||
| Base + UA*6 | 3 | 42.6 | 49.22 | 0 | 0.4 |
| Base + UA*10 | 3 | 44.3 | 50.92 | 1.7 | 0.17 |
| Base + UA*1 | 3 | 45.14 | 51.75 | 2.54 | 0.11 |
| Base | 3 | 46.01 | 52.63 | 3.41 | 0.07 |
| Base + UA*2 | 3 | 46.03 | 52.65 | 3.43 | 0.07 |
| Base + UA*7 | 3 | 48.19 | 54.81 | 5.59 | 0.02 |
| Base + UA*9 | 3 | 48.99 | 55.61 | 6.39 | 0.02 |
| Base + UA*17 | 3 | 48.99 | 55.61 | 6.39 | 0.02 |
| Base + UA*5 | 3 | 49.13 | 55.75 | 6.53 | 0.02 |
| Base + UA*11 | 3 | 49.48 | 56.1 | 6.88 | 0.01 |
| Base + UA*4 | 3 | 49.72 | 56.33 | 7.11 | 0.01 |
| Base + UA*15 | 3 | 49.87 | 56.48 | 7.26 | 0.01 |
| Base + UA*16 | 3 | 49.87 | 56.48 | 7.26 | 0.01 |
| Base + UA*14 | 3 | 49.87 | 56.48 | 7.26 | 0.01 |
| Base + UA*12 | 3 | 50.13 | 56.75 | 7.53 | 0.01 |
| Base + UA*13 | 3 | 50.13 | 56.75 | 7.53 | 0.01 |
| Base + UA*3 | 3 | 50.37 | 56.99 | 7.77 | 0.01 |
| Base + UA*8 | 3 | 50.43 | 57.04 | 7.82 | 0.01 |
| (b) UC | |||||
| Base | 2 | 43.15 | 47.45 | 0 | 0.31 |
| Base + UC*3 | 3 | 41.25 | 47.86 | 0.42 | 0.25 |
| Base + UC*4 | 3 | 41.25 | 47.86 | 0.42 | 0.25 |
| Base + UC*2 | 3 | 43.12 | 49.74 | 2.29 | 0.1 |
| Base + UC*1 | 3 | 43.14 | 49.76 | 2.31 | 0.1 |
The model in bold is the best model supported by the data.
Abbreviations: AICCWt, model weight; k, number of parameters; ΔAICC, increase in AICC compared to the top model.
4. DISCUSSION
In this study, we used a targeted sequencing approach, complemented with reduced representation sequencing, to investigate the role of immune genes in chlamydial infection resolution in koalas. Despite our two study sites being only 80 km from each other, genome‐wide SNP data show significant population structuring (Figure 1) consistent with previous identification of the Brisbane Valley as a biogeographical barrier to gene flow (Johnson et al., 2018). We identified higher heterozygosity in the RRS data set than in the immune gene data set as expected, as neutral regions have a higher differentiation between populations, which is why they are used for population genetics (Holderegger et al., 2006). Variation within functional regions is expected to be lower as these regions are under selection with variants only maintained if they are beneficial to the population (Horscroft et al., 2019). We could also have potentially biased our heterozygosity measures of functional regions by targeting genes, such as TLR genes, that are highly conserved and therefore unlikely to contain high levels of variation.
Across the two methods, we identified 25 SNPs located in 17 genes. Of these 17 genes, seven are predicted to be involved in transport (EIF4AENIF1, OCA2, RAB35, RBFOX1, SEC31A, SERPINE1 and STX4) and signalling (IFNγ, RAB35, SEC31A, STAT2, STX4, TLR5 and UCN4), and six in regulation of the immune response (C12orf4, IFNγ, SERPINE1, STX4, TLR5 and TOB2). Five genes are involved in defence (IFNγ, SERPINE1, STAT2, STX4 and TLR5). Others played a role in lymphocyte differentiation (PATZ1), the cellular response to stress (RAD51AP1) and leukocyte migration (HSD3B7), while LAAO‐like had no GO term associations. Of the 17 genes, only three have previously been associated with disease caused by Chlamydia infection in other species (TLR5, STAT2 and IFNγ) (Beckett et al., 2012; Derbigny et al., 2005; Hosey et al., 2015; Perfettini et al., 2003; Sixt, 2021). We also identified two MHC Class I genes associated with disease progression.
Our study identified TLR5, STAT2 and IFNγ as genes significantly associated with the ability of a koala to resolve a Chlamydia infection. Toll‐like receptor (TLR) genes have previously been shown to play a critical role in response to urogenital infection of mice with Chlamydia muridarum (Beckett et al., 2012; Derbigny et al., 2005). TLR genes are membrane‐bound receptors which bind to pathogen‐associated molecule patterns (PAMPs) (Akira et al., 2006; Kawai & Akira, 2010) and are involved in the innate immune system response and signalling (Figure 4). Binding of TLRs to PAMPs initiates the host immune response and synthesis of cytokines and chemokines, such as IFNs (Akira et al., 2006; Derbigny et al., 2005). It is therefore plausible that in the koala, variation in TLR genes could result in variable levels of PAMPs binding to Chlamydia causing variable immune responses to infection. In addition, STAT genes have been hypothesized to be involved in IFN‐β production through the JAX/STAT pathway during infection with C. muridarum (Hosey et al., 2015). Therefore, it is significant that we also identified that variation in both STAT2 and IFNγ is associated with infection resolution in the koala. IFNγ has a role as an immunomodulator produced by natural killer (NK) and T lymphocytes (Farrar & Schreiber, 1993). It has been shown that the presence of IFNγ in cells infected by Chlamydia can halt the developmental cycle of Chlamydia, resulting in a persistent infection (Perfettini et al., 2003; Sixt, 2021). IFNγ is required for activation of the antibody‐mediated response to Chlamydia infection (Hafner & Timms, 2018; Naglak et al., 2016). The role of IFNγ in the adaptive immune system is through its expression in CD4 TH1 and CD8 cytotoxic T lymphocyte effector T cells (Schoenborn & Wilson, 2007).
Our study found RAB35 to be significantly associated with the ability to resolve a Chlamydia infection. RAB proteins are regulators of membrane trafficking and the docking of vesicles and vesicle budding (Zerial & McBride, 2001). RAB35 is required for the recycling of endocytosed MHC‐I and MHC‐II complexes (Klinkert & Echard, 2016), and as such, variation in RAB35 in koalas may also impact MHC function. SERPINE1, STX4 and SEC31A, along with RAB35, are also involved in membrane trafficking and the innate immune response, which is an important initial host response to infection of cells with Chlamydia bacteria (Dockterman & Coers, 2021).
We identified additional genes with no previously published associations with Chlamydia infection, but these were all involved in similar immune pathways (Figure S3). For example, STX4 plays a vital role in exocytosis (Kennedy et al., 2010) and has been shown to participate in increasing the Salmonella‐containing vacuole (Stevenin et al., 2019). Also involved in transport and membrane organization is SEC31A, which has been implicated in the spread of Listeria between cells (Gianfelice et al., 2015). Natural variation in RBFOX1 has been shown, in Drosophila, to alter the phagocytotic ability in response to infection with Staphylococcus aureus (Nazario‐Toole et al., 2018). C12orf4 is a relatively undescribed gene found on human chromosome 12, is conserved across the animal kingdom and appears to be involved in mast cell degranulation (Dudkiewicz & Pawlowski, 2019; Mazuc et al., 2014). Despite other identified genes having no published examples of involvement in bacterial infections, GO term analysis shows the interaction between each identified gene and numerous aspects of the immune response from cell signalling, transport and the innate immune response to lymphocyte differentiation and aspects of humoral immunity. The complex interaction of genes and immune processes emphasizes the importance of both innate and humoral immunity to clear infection (Beckett et al., 2012; Khan et al., 2016; Lad et al., 2005; Robbins et al., 2020). Also vital is genetic variation in immune‐related genes allowing populations to respond to a range of pathogens (Flanagan et al., 2018; Nandakumar & Ishtiaq, 2020; Savage et al., 2016).
Our findings provide additional evidence of the importance of the MHC in response to Chlamydia infection in koalas. We found associations between two MHC Class I genes (UA and UC) and disease progression. UA and UC are classical MHC Class I genes (Cheng et al., 2018). We found 17 UA alleles and four UC alleles in our data set. Class I MHC genes are responsible for presenting peptides derived from intracellular pathogens to cytotoxic T cells (Cresswell et al., 2005), and Chlamydia is an intracellular bacterium (Horn, 2008; Zuck et al., 2017). We identified that presence of the UA*6 allele results in a koala being more likely to resolve a Chlamydia infection, as does being heterozygous at UC. Investigating koalas from additional populations as well as identifying the role of specific MHC alleles in clearing a Chlamydia infection will further contribute to our understanding of the factors involved in the progression of Chlamydia infection.
We did not find any associations with MHC Class II genes, despite previously published associations. Lau et al. (2014) identified a single MHC Class II variant (DAβ*10) that was more prevalent in koalas with Chlamydia infection and a second MHC Class II variant (DBβ*04) associated with high Chlamydia‐hsp60 antibody levels. These genes were targeted in our probe set but these alleles were not present in our sample set, possibly due to the two studies using koalas from two different locations (Port Macquarie and southeast Queensland). Further research should investigate diversity within MHC genes across multiple koala populations. Similarly to Lau et al. (2014), we found Class II diversity to be high and the majority of koalas shared only a few DAβ alleles. We identified 29 alleles occurring across three DAβ genes. Robbins et al. (2020) showed that four MHC alleles from the MHC Class II (DCβ*3, DBβ*4, DAβ*10) and Class I (UC*01:01) genes were associated with disease progression. We also identified alleles DCβ*3 and UC*01:01 in our study animals and an association between UC alleles and the ability to resolve a Chlamydia infection.
Despite our probe including 88 genes associated with the GO term “phagocytosis” (GO:0006909) as Chlamydia is phagocytosed by macrophages (Mitchell et al., 2009; Quigley & Timms, 2020; Zuck et al., 2017), none were associated with Chlamydia disease traits. Our probe also included additional genes previously associated with Chlamydia, including IL‐4, IL‐6, IL‐17A, IFNγ and TNF‐α with association only seen in this study with IFNγ.
In conclusion, through targeted sequencing and RRS, we identified 25 SNPs in 17 genes associated with progression of Chlamydia infection to clinical disease, with 14 of these genes not previously associated with Chlamydia infection. Most of these genes are involved in the innate immune response, indicating that the initial host immune response to infection with C. pecorum is vital to resolving the infection. We build on the work of others to show association between MHC genes and chlamydial infection. Future work will include confirming candidate SNPs in other populations and carrying out GWAS using whole genome resequencing now that we have a strong proof of concept that genomic associations exist. Also of interest will be investigating MHC diversity across numerous populations, as currently information is only available on koalas from three populations (Port Macquarie, Hidden Vale and Moreton Bay) (Cheng et al., 2018; Lau et al., 2013, 2014; Quigley et al., 2018; Robbins et al., 2020). It is interesting to note that we found two scaffolds (MSTS01000038.1 and MSTS01000065.1) with significant SNPs from both target capture and RRS methods, suggesting that a functionally important SNP is present within these two regions but not sequenced using our assay (Johnson et al., 2018).
The results generated here will assist conservation biologists to effectively manage wild koala populations. In general, maintenance of genome‐wide variation to allow for adaptive potential and population persistence (Ralls et al., 2018) is more important than managing a population to increase the proportion of individuals with specific disease resistance variants (Hohenlohe et al., 2021). However, as koala populations exist in a fragmented landscape, which limits their gene flow (Johnson et al., 2018), conservation biologists should consider our findings if population viability is being negatively impacted by chlamydial disease. These populations may benefit from the input of certain individuals with genetic variants associated with the ability to resolve a Chlamydia infection.
AUTHOR CONTRIBUTIONS
This study was designed by K.B., C.J.H. and Y.C. Tissue samples and clinical disease data were provided by P.T., B.L.Q. and A.R. Laboratory work and bioinformatics was primarily conducted by L.S. with assistance and guidance from Y.C. Data analysis was conducted by L.S. with assistance from Y.C. L.S. wrote the paper with feedback and revisions provided by all authors.
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
OPEN RESEARCH BADGES
This article has earned an Open Data, for making publicly available the digitally‐shareable data necessary to reproduce the reported results. The data is available at https://registry.opendata.aws/australasian‐genomics/.
Supporting information
Supplementary Material
Fig S1‐S3
Table S1‐S4
ACKNOWLEDGEMENTS
We thank the reviewers for constructive comments that significantly improved the manuscript. We thank the many people who participated in sample collection and veterinary examinations of koalas, as well as the Queensland Department of Transport and Main Roads and the Turner Family Foundation for their support of this research. L.S. is supported by a scholarship funded by the Australian Research Council Linkage Project: LP180100244. This work was funded by the University of Sydney. Open access publishing facilitated by The University of Sydney, as part of the Wiley ‐ The University of Sydney agreement via the Council of Australian University Librarians.
Silver, L. W. , Cheng, Y. , Quigley, B. L. , Robbins, A. , Timms, P. , Hogg, C. J. , & Belov, K. (2022). A targeted approach to investigating immune genes of an iconic Australian marsupial. Molecular Ecology, 31, 3286–3303. 10.1111/mec.16493
DATA AVAILABILITY STATEMENT
Locations of probes in the genome are provided as a Supporting Information file to this article. The raw and aligned sequence data for target enrichment and DArTSeq are available through the Amazon Web Services Open Datasets Program: https://registry.opendata.aws/australasian‐genomics/. Fasta sequences for alleles identified in genes UA and UC are provided as Supporting Information (UA_UC_alleles.docx).
REFERENCES
- Adams‐Hosking, C. , McBride, M. F. , Baxter, G. , Burgman, M. , de Villiers, D. , Kavanagh, R. , & McAlpine, C. A. (2016). Use of expert knowledge to elicit population trends for the koala (Phascolarctos cinereus). Diversity and Distributions, 22(3), 249–262. 10.1111/ddi.12400 [DOI] [Google Scholar]
- Akira, S. , Uematsu, S. , & Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell, 124(4), 783–801. 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Ashburner, M. , Ball, C. A. , Blake, J. A. , Botstein, D. , Butler, H. , Cherry, J. M. , Davis, A. P. , Dolinski, K. , Dwight, S. S. , Eppig, J. T. , Harris, M. A. , Hill, D. P. , Issel‐Tarver, L. , Kasarskis, A. , Lewis, S. , Matese, J. C. , Richardson, J. E. , Ringwald, M. , Rubin, G. M. , & Sherlock, G. (2000). Gene ontology: tool for the unification of biology. Nature Genetics, 25(1), 25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson, E. , Ratnakumar, A. , Arendt, M.‐L. , Maqbool, K. , Webster, M. T. , Perloski, M. , Liberg, O. , Arnemo, J. M. , Hedhammar, Å. , & Lindblad‐Toh, K. (2013). The genomic signature of dog domestication reveals adaptation to a starch‐rich diet. Nature, 495(7441), 360–364. 10.1038/nature11837 [DOI] [PubMed] [Google Scholar]
- Batley, K. C. , Sandoval‐Castillo, J. , Kemper, C. M. , Attard, C. R. M. , Zanardo, N. , Tomo, I. , Beheregaray, L. B. , & Möller, L. M. (2019). Genome‐wide association study of an unusual dolphin mortality event reveals candidate genes for susceptibility and resistance to cetacean morbillivirus. Evolutionary Applications, 12(4), 718–732. 10.1111/eva.12747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batley, K. C. , Sandoval‐Castillo, J. , Kemper, C. M. , Zanardo, N. , Tomo, I. , Beheregaray, L. B. , & Moller, L. M. (2021). Whole genomes reveal multiple candidate genes and pathways involved in the immune response of dolphins to a highly infectious virus. Molecular Ecology, 15, 6434–6448. 10.1111/mec.15873 [DOI] [PubMed] [Google Scholar]
- Beckett, E. L. , Phipps, S. , Starkey, M. R. , Horvat, J. C. , Beagley, K. W. , Foster, P. S. , & Hansbro, P. M. (2012). TLR2, but not TLR4, is required for effective host defence against Chlamydia respiratory tract infection in early life. PLoS One, 7(6), e39460. 10.1371/journal.pone.0039460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender, R. , & Lange, S. (2001). Adjusting for multiple testing – when and how? Journal of Clinical Epidemiology, 54(4), 343–349. 10.1016/s0895-4356(00)00314-0 [DOI] [PubMed] [Google Scholar]
- Berger, L. , Speare, R. , Daszak, P. , Green, D. E. , Cunningham, A. A. , Goggin, C. L. , Slocombe, R. , Ragan, M. A. , Hyatt, A. D. , McDonald, K. R. , Hines, H. B. , Lips, K. R. , Marantelli, G. , & Parkes, H. (1998). Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proceedings of the National Academy of Sciences of the United States of America, 95(15), 9031–9036. 10.1073/pnas.95.15.9031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer, H. L. , de Villiers, D. , Loader, J. , Robbins, A. , Stigner, M. , Forbes, N. , & Hanger, J. (2018). Management of multiple threats achieves meaningful koala conservation outcomes. Journal of Applied Ecology, 55(4), 1966–1975. 10.1111/1365-2664.13127 [DOI] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon, S. , Douglass, E. , Good, B. M. , Unni, D. R. , Harris, N. L. , Mungall, C. J. , Basu, S. , Chisholm, R. L. , Dodson, R. J. , Hartline, E. , Fey, P. , Thomas, P. D. , Albou, L.‐P. , Ebert, D. , Kesling, M. J. , Mi, H. , Muruganujan, A. , Huang, X. , Mushayahama, T. , … Elser, J. (2021). The gene ontology resource: enriching a GOld mine. Nucleic Acids Research, 49(D1), D325–D334. 10.1093/nar/gkaa1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall, A. , & Pirofski, L. A. (1999). Host‐pathogen interactions: redefining the basic concepts of virulence and pathogenicity. Infection and Immunity, 67(8), 3703–3713. 10.1128/iai.67.8.3703-3713.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall, A. , & Pirofski, L. A. (2015). What is a host? Incorporating the microbiota into the damage‐response framework. Infection and Immunity, 83(1), 2–7. 10.1128/iai.02627-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013). Stacks: an analysis tool set for population genomics. Molecular Ecology, 22(11), 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y. , Polkinghorne, A. , Gillett, A. , Jones, E. A. , O'Meally, D. , Timms, P. , & Belov, K. (2018). Characterisation of MHC class I genes in the koala. Immunogenetics, 70(2), 125–133. 10.1007/s00251-017-1018-2 [DOI] [PubMed] [Google Scholar]
- Cockram, F. A. , & Jackson, A. R. B. (1974). Isolation of a Chlamydia from cases of keratoconjuctivitis in koalas. Australian Veterinary Journal, 50(2), 82–83. 10.1111/j.1751-0813.1974.tb05265.x [DOI] [PubMed] [Google Scholar]
- Coulon, A. (2010). genhet: an easy‐to‐use R function to estimate individual heterozygosity. Molecular Ecology Resources, 10(1), 167–169. 10.1111/j.1755-0998.2009.02731.x [DOI] [PubMed] [Google Scholar]
- Cresswell, P. , Ackerman, A. L. , Giodini, A. , Peaper, D. R. , & Wearsch, P. A. (2005). Mechanisms of MHC class I‐restricted antigen processing and cross‐presentation. Immunological Reviews, 207, 145–157. 10.1111/j.0105-2896.2005.00316.x [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , & Durbin, R. , 1000 Genomes Project Analysis Group (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbigny, W. A. , Kerr, M. S. , & Johnson, R. M. (2005). Pattern recognition molecules activated by Chlamydia muridarum infection of cloned murine oviduct epithelial cell lines. The Journal of Immunology, 175(9), 6065–6075. 10.4049/jimmunol.175.9.6065 [DOI] [PubMed] [Google Scholar]
- Dique, D. S. , Thompson, J. , Preece, H. J. , Penfold, G. C. , de Villiers, D. L. , & Leslie, R. S. (2003). Koala mortality on roads in south‐east Queensland: the koala speed‐zone trial. Wildlife Research, 30(4), 419–426. 10.1071/Wr02029 [DOI] [Google Scholar]
- Dockterman, J. , & Coers, J. (2021). Immunopathogenesis of genital Chlamydia infection: Insights from mouse models. Pathogens and Disease, 79(4), ftab012. 10.1093/femspd/ftab012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudkiewicz, M. , & Pawlowski, K. (2019). A novel conserved family of macro‐like domains‐putative new players in ADP‐ribosylation signaling. PeerJ, 7, e6863. 10.7717/peerj.6863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar, M. A. , & Schreiber, R. D. (1993). The molecular cell biology of the interferon‐gamma and its receptor. Annual Review of Immunology, 11, 571–611. 10.1146/annurev.iy.11.040193.003035 [DOI] [PubMed] [Google Scholar]
- Flanagan, S. P. , Forester, B. R. , Latch, E. K. , Aitken, S. N. , & Hoban, S. (2018). Guidelines for planning genomic assessment and monitoring of locally adaptive variation to inform species conservation. Evolutionary Applications, 11(7), 1035–1052. 10.1111/eva.12569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianfelice, A. , Le, P. H. , Rigano, L. A. , Saila, S. , Dowd, G. C. , McDivitt, T. , & Ireton, K. (2015). Host endoplasmic reticulum COPII proteins control cell‐to‐cell spread of the bacterial pathogen Listeria monocytogenes . Cellular Microbiology, 17(6), 876–892. 10.1111/cmi.12409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout, E. J. , Madaline, T. , Casadevall, A. , Bearman, G. , & Pirofski, L. A. (2020). The damage response framework and infection prevention: From concept to bedside. Infection Control and Hospital Epidemiology, 41(3), 337–341. 10.1017/ice.2019.354 [DOI] [PubMed] [Google Scholar]
- Goldingay, R. L. , & Dobner, B. (2014). Home range areas of koalas in an urban area of north‐east New South Wales. Australian Mammalogy, 36(1), 74–80. 10.1071/Am12049 [DOI] [Google Scholar]
- Griffith, J. E. , Dhand, N. K. , Krockenberger, M. B. , & Higgins, D. P. (2013). A retrospective study of admission trends of koalas to a rehabilitation facility over 30 years. Journal of Wildlife Diseases, 49(1), 18–28. 10.7589/2012-05-135 [DOI] [PubMed] [Google Scholar]
- Grueber, C. E. , Wallis, G. P. , & Jamieson, I. G. (2013). Genetic drift outweighs natural selection at toll‐like receptor (TLR) immunity loci in a re‐introduced population of a threatened species. Molecular Ecology, 22(17), 4470–4482. 10.1111/mec.12404 [DOI] [PubMed] [Google Scholar]
- Hafner, L. M. , & Timms, P. (2018). Development of a Chlamydia trachomatis vaccine for urogenital infections: novel tools and new strategies point to bright future prospects. Expert Review of Vaccines, 17(1), 57–69. 10.1080/14760584.2018.1417044 [DOI] [PubMed] [Google Scholar]
- Hanger, J. J. , Bromham, L. D. , McKee, J. J. , O'Brien, T. M. , & Robinson, W. F. (2000). The nucleotide sequence of koala (Phascolarctos cinereus) retrovirus: a novel type C endogenous virus related to gibbon ape leukemia virus. Journal of Virology, 74(9), 4264–4272. 10.1128/jvi.74.9.4264-4272.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger, J. J. , Loader, J. , Wan, C. , Beagley, K. W. , Timms, P. , & Polkinghorne, A. (2013). Comparison of antigen detection and quantitative PCR in the detection of chlamydial infection in koalas (Phascolarctos cinereus). Veterinary Journal, 195(3), 391–393. 10.1016/j.tvjl.2012.07.024 [DOI] [PubMed] [Google Scholar]
- Hawkins, C. E. , Baars, C. , Hesterman, H. , Hocking, G. J. , Jones, M. E. , Lazenby, B. , & Wiersma, J. (2006). Emerging disease and population decline of an island endemic, the Tasmanian devil (Sarcophilus harrisi). Biological Conservation, 131(2), 307–324. 10.1016/j.biocon.2006.04.010 [DOI] [Google Scholar]
- Hedrick, P. W. , & Garcia‐Dorado, A. (2016). Understanding inbreeding depression, purging, and genetic rescue. Trends in Ecology & Evolution, 31(12), 940–952. 10.1016/j.tree.2016.09.005 [DOI] [PubMed] [Google Scholar]
- Hohenlohe, P. A. , Funk, W. C. , & Rajora, O. P. (2021). Population genomics for wildlife conservation and management. Molecular Ecology, 30(1), 62–82. 10.1111/mec.15720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderegger, R. , Kamm, U. , & Gugerli, F. (2006). Adaptive vs. neutral genetic diversity: Implications for landscape genetics. Landscape Ecology, 21(6), 797–807. 10.1007/s10980-005-5245-9 [DOI] [Google Scholar]
- Horn, M. (2008). Chlamydiae as symbionts in eukaryotes. Annual Review of Microbiology, 62, 113–131. 10.1146/annurev.micro.62.081307.162818 [DOI] [PubMed] [Google Scholar]
- Horscroft, C. , Ennis, S. , Pengelly, R. J. , Sluckin, T. J. , & Collins, A. (2019). Sequencing era methods for identifying signatures of selection in the genome. Briefings in Bioinformatics, 20(6), 1997–2008. 10.1093/bib/bby064 [DOI] [PubMed] [Google Scholar]
- Hosey, K. L. , Hu, S. S. , & Derbigny, W. A. (2015). Role of STAT1 in Chlamydia‐induced type‐1 interferon production in oviduct epithelial cells. Journal of Interferon and Cytokine Research, 35(11), 901–916. 10.1089/jir.2015.0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelocnik, M. , Islam, M. M. , Madden, D. , Jenkins, C. , Branley, J. , Carver, S. , & Polkinghorne, A. (2017). Development and evaluation of rapid novel isothermal amplification assays for important veterinary pathogens: Chlamydia psittaci and Chlamydia pecorum . Peerj, 5, e3799. 10.7717/peerj.3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, R. N. , O’Meally, D. , Chen, Z. , Etherington, G. J. , Ho, S. Y. W. , Nash, W. J. , Grueber, C. E. , Cheng, Y. , Whittington, C. M. , Dennison, S. , Peel, E. , Haerty, W. , O’Neill, R. J. , Colgan, D. , Russell, T. L. , Alquezar‐Planas, D. E. , Attenbrow, V. , Bragg, J. G. , Brandies, P. A. , … Belov, K. (2018). Adaptation and conservation insights from the koala genome. Nature Genetics, 50(8), 1102–1111. 10.1038/s41588-018-0153-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. (2008). adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics, 24(11), 1403–1405. 10.1093/bioinformatics/btn129 [DOI] [PubMed] [Google Scholar]
- Jones, M. E. , Jarman, P. J. , Lees, C. M. , Hesterman, H. , Hamede, R. K. , Mooney, N. J. , Mann, D. , Pukk, C. E. , Bergfeld, J. , & McCallum, H. (2007). Conservation management of tasmanian devils in the context of an emerging, extinction‐threatening disease: Devil facial tumor disease. EcoHealth, 4(3), 326–337. 10.1007/s10393-007-0120-6 [DOI] [Google Scholar]
- Kawai, T. , & Akira, S. (2010). The role of pattern‐recognition receptors in innate immunity: Update on toll‐like receptors. Nature Immunology, 11(5), 373–384. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- Kennedy, M. J. , Davison, I. G. , Robinson, C. G. , & Ehlers, M. D. (2010). Syntaxin‐4 defines a domain for activity‐dependent exocytosis in dendritic spines. Cell, 141(3), 524–535. 10.1016/j.cell.2010.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. A. , Polkinghorne, A. , Waugh, C. , Hanger, J. , Loader, J. , Beagley, K. , & Timms, P. (2016). Humoral immune responses in koalas (Phascolarctos cinereus) either naturally infected with Chlamydia pecorum or following administration of a recombinant chlamydial major outer membrane protein vaccine. Vaccine, 34(6), 775–782. 10.1016/j.vaccine.2015.12.050 [DOI] [PubMed] [Google Scholar]
- Klinkert, K. , & Echard, A. (2016). Rab35 GTPase: A central regulator of phosphoinositides and F‐actin in endocytic recycling and beyond. Traffic, 17(10), 1063–1077. 10.1111/tra.12422 [DOI] [PubMed] [Google Scholar]
- Korte, A. , & Farlow, A. (2013). The advantages and limitations of trait analysis with GWAS: A review. Plant Methods, 9, 29. 10.1186/1746-4811-9-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lad, S. P. , Fukuda, E. Y. , Li, J. L. , de la Maza, L. M. , & Li, E. G. (2005). Up‐regulation of the JAK/STAT1 signal pathway during Chlamydia trachomatis infection. Journal of Immunology, 174(11), 7186–7193. 10.4049/jimmunol.174.11.7186 [DOI] [PubMed] [Google Scholar]
- Lau, Q. , Griffith, J. E. , & Higgins, D. P. (2014). Identification of MHCII variants associated with chlamydial disease in the koala (Phascolarctos cinereus). Peerj, 2, e443. 10.7717/peerj.443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, Q. , Jobbins, S. E. , Belov, K. , & Higgins, D. P. (2013). Characterisation of four major histocompatibility complex class II genes of the koala (Phascolarctos cinereus). Immunogenetics, 65(1), 37–46. 10.1007/s00251-012-0658-5 [DOI] [PubMed] [Google Scholar]
- Legione, A. R. , Patterson, J. L. S. , Whiteley, P. L. , Amery‐Gale, J. , Lynch, M. , Haynes, L. , Gilkerson, J. R. , Polkinghorne, A. , Devlin, J. M. , & Sansom, F. M. (2016). Identification of unusual Chlamydia pecorum genotypes in Victorian koalas (Phascolarctos cinereus) and clinical variables associated with infection. Journal of Medical Microbiology, 65(5), 420–428. 10.1099/jmm.0.000241 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longcore, J. E. , Pessier, A. P. , & Nichols, D. K. (1999). Batrachochytrium dendrobatidis gen et sp nov, a chytrid pathogenic to amphibians. Mycologia, 91(2), 219–227. 10.2307/3761366 [DOI] [Google Scholar]
- Lopez‐Domingo, F. J. , Florido, J. P. , Rueda, A. , Dopazo, J. , & Santoyo‐Lopez, J. (2014). ngsCAT: A tool to assess the efficiency of targeted enrichment sequencing. Bioinformatics, 30(12), 1767–1768. 10.1093/bioinformatics/btu108 [DOI] [PubMed] [Google Scholar]
- Lotterhos, K. E. , & Whitlock, M. C. (2014). Evaluation of demographic history and neutral parameterization on the performance of F‐ST outlier tests. Molecular Ecology, 23(9), 2178–2192. 10.1111/mec.12725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher, I. E. , Griffith, J. E. , Lau, Q. , Reeves, T. , & Higgins, D. P. (2014). Expression profiles of the immune genes CD4, CD8beta, IFNgamma, IL‐4, IL‐6 and IL‐10 in mitogen‐stimulated koala lymphocytes (Phascolarctos cinereus) by qRT‐PCR. Peerj, 2, e280. 10.7717/peerj.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matchett, M. R. , Biggins, D. E. , Carlson, V. , Powell, B. , & Rocke, T. (2010). Enzootic plague reduces black‐footed ferret (Mustela nigripes) survival in Montana. Vector‐Borne and Zoonotic Diseases, 10(1), 27–35. 10.1089/vbz.2009.0053 [DOI] [PubMed] [Google Scholar]
- Mathew, M. , Beagley, K. W. , Timms, P. , & Polkinghorne, A. (2013). Preliminary characterisation of tumor necrosis factor alpha and interleukin‐10 responses to Chlamydia pecorum infection in the koala (Phascolarctos cinereus). PLoS One, 8(3), e59958. 10.1371/journal.pone.0059958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew, M. , Pavasovic, A. , Prentis, P. J. , Beagley, K. W. , Timms, P. , & Polkinghorne, A. (2013). Molecular characterisation and expression analysis of interferon gamma in response to natural Chlamydia infection in the koala, Phascolarctos cinereus. Gene, 527(2), 570–577. 10.1016/j.gene.2013.06.019 [DOI] [PubMed] [Google Scholar]
- Mathew, M. , Waugh, C. , Beagley, K. W. , Timms, P. , & Polkinghorne, A. (2014). Interleukin 17A is an immune marker for chlamydial disease severity and pathogenesis in the koala (Phascolarctos cinereus). Developmental and Comparative Immunology, 46(2), 423–429. 10.1016/j.dci.2014.05.015 [DOI] [PubMed] [Google Scholar]
- Mazuc, E. , Guglielmi, L. , Bec, N. , Parez, V. , Hahn, C. S. , Mollevi, C. , Parrinello, H. , Desvignes, J.‐P. , Larroque, C. , Jupp, R. , Dariavach, P. , & Martineau, P. (2014). In‐cell intrabody selection from a diverse human library identifies C12orf4 protein as a new player in rodent mast cell degranulation. PLoS One, 9(8), e104998. 10.1371/journal.pone.0104998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine, C. A. , Rhodes, J. R. , Callaghan, J. G. , Bowen, M. E. , Lunney, D. , Mitchell, D. L. , Pullar, D. V. , & Possingham, H. P. (2006). The importance of forest area and configuration relative to local habitat factors for conserving forest mammals: A case study of koalas in Queensland, Australia. Biological Conservation, 132(2), 153–165. 10.1016/j.biocon.2006.03.021 [DOI] [Google Scholar]
- McCallum, H. (2008). Tasmanian devil facial tumour disease: lessons for conservation biology. Trends in Ecology & Evolution, 23(11), 631–637. 10.1016/j.tree.2008.07.001 [DOI] [PubMed] [Google Scholar]
- McColl, K. A. , Martin, R. W. , Gleeson, L. J. , Handasyde, K. A. , & Lee, A. K. (1984). Chlamydia infection and infertility in the female koala (Phascolarctos Cinereus). Veterinary Record, 115(25–6), 655. 10.1136/vr.115.25-26.655 [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, C. M. , Mathews, S. A. , Theodoropoulos, C. , & Timms, P. (2009). In vitro characterisation of koala Chlamydia pneumoniae: morphology, inclusion development and doubling time. Veterinary Microbiology, 136(1–2), 91–99. 10.1016/j.vetmic.2008.10.008 [DOI] [PubMed] [Google Scholar]
- Naglak, E. K. , Morrison, S. G. , & Morrison, R. P. (2016). Gamma interferon is required for optimal antibody‐mediated immunity against genital Chlamydia infection. Infection and Immunity, 84(11), 3232–3242. 10.1128/iai.00749-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar, M. , & Ishtiaq, F. (2020). Genetic drift and bottleneck do not influence diversity in Toll‐like receptor genes at a small spatial scale in a Himalayan passerine. Ecology and Evolution, 10(21), 12246–12263. 10.1002/ece3.6855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazario‐Toole, A. E. , Robalino, J. , Okrah, K. , Corrada‐Bravo, H. , Mount, S. M. , & Wu, L. P. (2018). The splicing factor RNA‐binding fox protein 1 mediates the cellular immune response in Drosophila melanogaster . Journal of Immunology, 201(4), 1154–1164. 10.4049/jimmunol.1800496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfettini, J. L. , Hospital, V. , Stahl, L. , Jungas, T. , Verbeke, P. , & Ojcius, D. M. (2003). Cell death and inflammation during infection with the obligate intracellular Chlamydia, Biochimie . Biochimie, 85(8), 763–769. 10.1016/j.biochi.2003.08.006 [DOI] [PubMed] [Google Scholar]
- Phillips, S. , Robbins, A. , Loader, J. , Hanger, J. , Booth, R. , Jelocnik, M. , Polkinghorne, A. , & Timms, P. (2018). Chlamydia pecorum gastrointestinal tract infection associations with urogenital tract infections in the koala (Phascolarctos cinereus). PLoS One, 13(11), e0206471. 10.1371/journal.pone.0206471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polkinghorne, A. , Hanger, J. , & Timms, P. (2013). Recent advances in understanding the biology, epidemiology and control of chlamydial infections in koalas. Veterinary Microbiology, 165(3–4), 214–223. 10.1016/j.vetmic.2013.02.026 [DOI] [PubMed] [Google Scholar]
- Pomaznoy, M. , Ha, B. , & Peters, B. (2018). GOnet: a tool for interactive Gene Ontology analysis. BMC Bioinformatics, 19, 10.1186/s12859-018-2533-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M. A. R. , Bender, D. , Maller, J. , Sklar, P. , de Bakker, P. I. W. , Daly, M. J. , & Sham, P. C. (2007). PLINK: a tool set for whole‐genome association and population‐based linkage analyses. American Journal of Human Genetics, 81(3), 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye, R. J. , Pemberton, D. , Tovar, C. , Tubio, J. M. C. , Dun, K. A. , Fox, S. , Darby, J. , Hayes, D. , Knowles, G. W. , Kreiss, A. , Siddle, H. V. T. , Swift, K. , Lyons, A. B. , Murchison, E. P. , & Woods, G. M. (2016). A second transmissible cancer in Tasmanian devils. Proceedings of the National Academy of Sciences of the United States of America, 113(2), 374–379. 10.1073/pnas.1519691113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley, B. L. , Carver, S. , Hanger, J. , Vidgen, M. E. , & Timms, P. (2018). The relative contribution of causal factors in the transition from infection to clinical chlamydial disease. Scientific Reports, 8(1), 8893. 10.1038/s41598-018-27253-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley, B. L. , & Timms, P. (2020). Helping koalas battle disease – Recent advances in Chlamydia and koala retrovirus (KoRV) disease understanding and treatment in koalas. FEMS Microbiology Reviews, 44(5), 583–605. 10.1093/femsre/fuaa024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2021). R: a language and environment for statistical computing. R Foundation for Statistical Computing. Retrieved from https://www.r‐project.org/ [Google Scholar]
- Ralls, K. , Ballou, J. D. , Dudash, M. R. , Eldridge, M. D. B. , Fenster, C. B. , Lacy, R. C. , Sunnucks, P. , & Frankham, R. (2018). Call for a paradigm shift in the genetic management of fragmented populations. Conservation Letters, 11(2), e12412. 10.1111/conl.12412 [DOI] [Google Scholar]
- Rhodes, J. R. , Beyer, H. L. , Preece, H. J. , & McAlpine, C. A. (2015). South East Queensland koala population modelling study. UniQuest. [Google Scholar]
- Rhodes, J. R. , Ng, C. F. , de Villiers, D. L. , Preece, H. J. , McAlpine, C. A. , & Possingham, H. P. (2011). Using integrated population modelling to quantify the implications of multiple threatening processes for a rapidly declining population. Biological Conservation, 144(3), 1081–1088. 10.1016/j.biocon.2010.12.027 [DOI] [Google Scholar]
- Robbins, A. , Hanger, J. , Jelocnik, M. , Quigley, B. L. , & Timms, P. (2019). Longitudinal study of wild koalas (Phascolarctos cinereus) reveals chlamydial disease progression in two thirds of infected animals. Scientific Reports, 9(1), 13194. 10.1038/s41598-019-49382-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, A. , Hanger, J. , Jelocnik, M. , Quigley, B. L. , & Timms, P. (2020). Koala immunogenetics and chlamydial strain type are more directly involved in chlamydial disease progression in koalas from two south east Queensland koala populations than koala retrovirus subtypes. Scientific Reports, 10(1), 15013. 10.1038/s41598-020-72050-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage, A. E. , Terrell, K. A. , Gratwicke, B. , Mattheus, N. M. , Augustine, L. , & Fleischer, R. C. (2016). Reduced immune function predicts disease susceptibility in frogs infected with a deadly fungal pathogen. Conservation Physiology, 4, cow011. 10.1093/conphys/cow011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenborn, J. R. , & Wilson, C. B. (2007). Regulation of interferon‐gamma during innate and adaptive immune responses. Advances in immunology, 96, 41–101. 10.1016/S0065-2776(07)96002-2 [DOI] [PubMed] [Google Scholar]
- Sepil, I. , Lachish, S. , & Sheldon, B. C. (2013). Mhc‐linked survival and lifetime reproductive success in a wild population of great tits. Molecular Ecology, 22(2), 384–396. 10.1111/mec.12123 [DOI] [PubMed] [Google Scholar]
- Sixt, B. S. (2021). Host cell death during infection with Chlamydia: A double‐edged sword. FEMS Microbiology Reviews, 45(1), fuaa043. 10.1093/femsre/fuaa043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M. , & Scheet, P. (2005). Accounting for decay of linkage disequilibrium in haplotype inference and missing‐data imputation. American Journal of Human Genetics, 76(3), 449–462. 10.1086/428594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M. , Smith, N. J. , & Donnelly, P. (2001). A new statistical method for haplotype reconstruction from population data. American Journal of Human Genetics, 68(4), 978–989. 10.1086/319501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stévenin, V. , Chang, Y.‐Y. , Le Toquin, Y. , Duchateau, M. , Gianetto, Q. G. , Luk, C. H. , Salles, A. , Sohst, V. , Matondo, M. , Reiling, N. , & Enninga, J. (2019). Dynamic growth and shrinkage of the Salmonella‐containing vacuole determines the intracellular pathogen niche. Cell Reports, 29(12), 3958–3973. 10.1016/j.celrep.2019.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonegawa, S. (1983). Somatic generation of antibody diversity. Nature, 302(5909), 575–581. 10.1038/302575a0 [DOI] [PubMed] [Google Scholar]
- Ujvari, B. , & Belov, K. (2015). Characterization of antibody V segment diversity in the Tasmanian devil (Sarcophilus harrisii). Veterinary Immunology and Immunopathology, 167(3–4), 156–165. 10.1016/j.vetimm.2015.08.001 [DOI] [PubMed] [Google Scholar]
- Wan, C. , Loader, J. , Hanger, J. , Beagley, K. W. , Timms, P. , & Polkinghorne, A. (2011). Using quantitative polymerase chain reaction to correlate Chlamydia pecorum infectious load with ocular, urinary and reproductive tract disease in the koala (Phascolarctos cinereus). Australian Veterinary Journal, 89(10), 409–412. 10.1111/j.1751-0813.2011.00827.x [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. Y. , & Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S. , & Cockerham, C. C. (1984). Estimating F‐statistics for the analysis of population structure. Evolution, 38(6), 1358–1370. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- Williams, E. S. , Mills, K. , Kwiatkowski, D. R. , Thorne, E. T. , & Boergerfields, A. (1994). Plague in a black‐footed ferret (Mustella nigripes). Journal of Wildlife Diseases, 30(4), 581–585. 10.7589/0090-3558-30.4.581 [DOI] [PubMed] [Google Scholar]
- Wright, B. , Farquharson, K. A. , McLennan, E. A. , Belov, K. , Hogg, C. J. , & Grueber, C. E. (2019). From reference genomes to population genomics: comparing three reference‐aligned reduced‐representation sequencing pipelines in two wildlife species. Bmc Genomics, 20(1), 453. 10.1186/s12864-019-5806-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial, M. , & McBride, H. (2001). Rab proteins as membrane organizers. Nature Reviews Molecular Cell Biology, 2(2), 107–117. 10.1038/35052055 [DOI] [PubMed] [Google Scholar]
- Zuck, M. , Ellis, T. , Venida, A. , & Hybiske, K. (2017). Extrusions are phagocytosed and promote Chlamydia survival within macrophages. Cellular Microbiology, 19(4), e12683. 10.1111/cmi.12683 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Fig S1‐S3
Table S1‐S4
Data Availability Statement
Locations of probes in the genome are provided as a Supporting Information file to this article. The raw and aligned sequence data for target enrichment and DArTSeq are available through the Amazon Web Services Open Datasets Program: https://registry.opendata.aws/australasian‐genomics/. Fasta sequences for alleles identified in genes UA and UC are provided as Supporting Information (UA_UC_alleles.docx).