

SUMMARY
Replacement of mitochondria through nuclear transfer between oocytes of two different women has emerged recently as a strategy for preventing inheritance of mtDNA diseases. Although experiments in human oocytes have shown effective replacement, the consequences of small amounts of mtDNA carryover have not been studied sufficiently. Using human mitochondrial replacement stem cell lines, we show that, even though the low levels of heteroplasmy introduced into human oocytes by mitochondrial carryover during nuclear transfer often vanish, they can sometimes instead result in mtDNA genotypic drift and reversion to the original genotype. Comparison of cells with identical oocyte-derived nuclear DNA but different mtDNA shows that either mtDNA genotype is compatible with the nucleus and that drift is independent of mitochondrial function. Thus, although functional replacement of the mitochondrial genome is possible, even low levels of heteroplasmy can affect the stability of the mtDNA genotype and compromise the efficacy of mitochondrial replacement.
In Brief
Yamada et al. show, using human cells, that even small amounts of mtDNA carried over during nuclear transfer for mitochondrial replacement can lead to mtDNA genotype reversion. This situation would need to be avoided for clinical application and stable prevention of mtDNA diseases.
Graphical Abstract
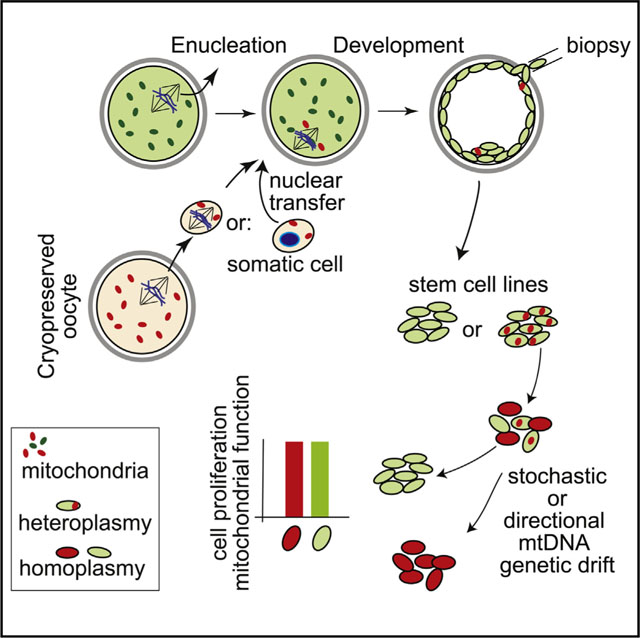
mtDNA is transmitted maternally via the cytoplasm of the oocyte in humans. Thereby, the inheritance of maternal nuclear and mitochondrial genomes is linked, whereas paternal nuclear genomes combine with a new mitochondrial genotype at fertilization. The reasons for this asymmetry are incompletely understood beyond the fact that differences in the size of the egg and sperm are not compatible with symmetric contribution to the developing embryo. In mice, artificially induced symmetric inheritance of two normal mtDNA genotypes results in mtDNA genotype instability (Sharpley et al., 2012); uniparental inheritance assures the stability of the mitochondrial genotype. It has also been suggested that co-inheritance of maternal nuclear and mtDNA ensures functional interactions of nuclear and mitochondrial gene products (Dunham-Snary and Ballinger, 2015; Poulton et al., 2010; Reinhardt et al., 2013). According to this hypothesis, oocyte mitochondrial replacement might affect the function of respiratory chain complexes that are composed of both nuclear and mitochondrial proteins.
To address the consequences of small amounts of heteroplasmy introduced into human oocytes, we transferred the nuclear genome between oocytes of women with different mitochondrial haplotypes, followed by parthenogenesis and stem cell derivation. Parthenogenesis—the development of oocytes without fertilization—allows us to examine mitochondrial function after combination of the maternal nuclear genome with an exogenous mitochondrial genome. The lack of a sperm genome excludes potential functional complementation by paternal nuclear alleles. The research was reviewed and approved by the Columbia University Medical Center Institutional Review Board.
To enable such a transfer, we used vitrification instead of synchronized retrieval of oocytes (Figure S1A). Vitrification of either the nuclear genome or of the cytoplasm allowed development to the blastocyst stage and stem cell derivation. Development was most efficient when the nuclear genome was cryopreserved and the recipient cytoplasm was fresh, suggesting differential effects of cryopreservation on nuclear and cytoplasmic components (Figures S1B and S1C). The percentage of mtDNA “carryover” transferred along with the nuclear genome was between 0.0% and 2.2%, with an average of 0.2% (Figure 1A), and remained constant throughout preimplantation development (Figure 1A). Blastocysts of high quality with expanding trophectoderm and a distinct inner cell mass (Figure S1C) were used to derive eight mitochondrial replacement pluripotent stem cell lines designated MR-PS5–12 (Figure S2). These cell lines were used to determine the replicative stability of the mtDNA genotype. No carryover (heteroplasmy) was detected in four of eight cell lines, whereas four cell lines contained 0.2%1.7% heteroplasmy at derivation (0.42% on average). For all but one cell line, mitochondrial heteroplasmy decreased below the limit of detection by passage 6 and remained stable for more than 30 passages or more than 6 months of culture (Figure 1B; Table S1).
Figure 1. Mitochondrial Genotype Drift after MR.

(A) mtDNA carryover during preimplantation development. Parthenotes without MR were used as a control for background noise of the assay (blue line). The red line indicates the level of detection. TE, trophectoderm; PB, polar body.
(B) Heteroplasmy in MR-PS cell lines during extended culture.
(C) Heteroplasmy in MR-PS12 cells made with a mitochondrial donor of the L3 haplotype and a nuclear donor of the H1 haplotype. Black line, bulk cultures; blue dots, colonies grown from single cells at specific passages.
(D) Heteroplasmy in MR-PS12 cultures expanded from single cells. F, T, P, N, R, and O indicate individual clonal cell lines.
(E) Heteroplasmy in nuclear transfer ESCs. NT1018 is diploid, whereas soPS cells contain both a somatic and oocyte nuclear genome (Noggle et al., 2011).
(F) Sanger sequencing of diploid nuclear transfer ESCs generated with a somatic cell of the K1 haplotype and an oocyte of the L0 haplotype.
(G) Allele refractory mutation system (ARMS)-qPCR for heteroplasmy in three diploid nuclear transfer ESC lines generated with a somatic cell of the K1 haplotype
and an oocyte of the L0 haplotype. Black symbols, bulk cultures; colored squares of dots, genotype dynamics in colonies grown from single cells at the indicated passages.
(H) Heteroplasmy in differentiated fibroblasts, cardiomyocytes, and teratomas from stem cells.
(I) Cell competition assay. Shown is the percentage of cells with the K1 haplotype (NT6 and NT8) mixed at the indicated ratio with cells of haplotype L0 (NT5).
However, among the eight MR-PS cell lines, in one cell line, MR-PS12, heteroplasmy of the mtDNA H1 haplotype carried over following genome transfer increased from 1.3% at derivation to 53.2% at passage 36 (Figure 1C) while maintaining a normal karyotype of 46XX (Figure S2). Upon further passaging, the H1 haplotype decreased to 1.0% at passage 59, after 23 weeks of culture (Figure 1C; Table S1). Clonal expansion from single cells at passages 18, 30, 36, 40, 50, and 60 resulted in colonies with diverse mtDNA heteroplasmy, ranging from 0%–90% of the H1 haplotype (Figure 1C, blue dots). Continued culture of these colonies again showed stochastic drift of mitochondrial heteroplasmy (Figure 1D). A few colonies became homoplasmic for either haplotype, resulting in stem cell lines isogenic for the nuclear DNA but with mtDNA of either haplotype H1 of the nuclear genome donor or haplotype L3 of the cytoplasm donor.
It has recently been shown that mitochondria can be replaced by somatic cell nuclear genome transfer into human oocytes (Chung et al., 2014; Ma et al., 2015; Noggle et al., 2011). With somatic nuclear genome transfer (as with oocyte nuclear genome transfer), a few thousand mtDNA copies are introduced into an egg containing several hundred thousand copies of a different mtDNA genotype (Birket et al., 2011; Paull et al., 2013). Therefore, both methods reveal the dynamics of low-level mtDNA heteroplasmy and the potential consequences of mitochondrial replacement on cell function. We analyzed mtDNA heteroplasmy in a total of 12 nuclear transfer ESC (SCNT ESC) lines, four of which were diploid and contained only the somatic cell genome (NT5, NT6, NT8, and NT1018) (Yamada et al., 2014), whereas eight also contained the maternal nuclear genome (soPS cells) (Noggle et al., 2011). Nine of twelve SCNT ES cell lines showed the mtDNA genotype of the oocyte donor with low heteroplasmy from the somatic cell between 0.0% and 0.6% (Figure 1E). However, for one specific combination of somatic cell and oocyte cytoplasm of the same donor (Table S1), two diploid cell lines showed a rapid mtDNA drift between passages 0 and 10 and reached homoplasmy of the somatic mtDNA genotype (haplotype K1c1c) between passages 15 and 25 (Figures 1F and 1G). Clonal expansion of single cells at passage 6 revealed mtDNA heteroplasmy ranging from 5%–96% (Figure 1G). The genetic changes were consistently directional, toward the K1 haplotype even when the starting cells had a majority of mtDNA of the L0 haplotype; passaging of clones showed increases in mtDNA haplotype K1 and decreases of haplotype L0 (Figure 1G). Remarkably, a third nuclear transfer ESC line from the same haplotype combination contained no detectable K1 mtDNA at the point of derivation and remained homoplasmic for the L0 haplotype for over 40 passages (Figure 1G). Therefore, we were able to isolate stable cell lines isogenic for the nuclear DNA but with either the L0 or the K1 mitochondrial genotype. Overall, among 24 cell lines with 14 different combinations of mtDNA haplotypes (Table S1), three cell lines (NT6, NT8, and MR-PS12) of two different combinations (K1 with L0 and H1 with L3) showed mtDNA genotype instability.
We next determined whether mtDNA instability also occurred during stem cell differentiation. Differentiation of NT6 and NT8 ES cells to fibroblasts or cardiomyocytes resulted in a directional shift to the K1 haplotype, while MR-PS12 showed both decrease or increase during differentiation (Figure 1H). Cells with homoplasmy remained stable as stem cells and during differentiation in vitro or in vivo as teratomas (Figure 1H). Therefore, mtDNA genotype instability was not specific to pluripotent stem cells.
We considered two potential mechanisms for mtDNA drift. First, specific mitochondrial nuclear genotype combinations might confer cellular survival and/or proliferative advantages because of differences in mitochondrial function. According to this hypothesis, colonies with a greater proportion of the K1 mtDNA genotype, as shown in Figure 1G, would outgrow cells containing predominantly the L0 genotype. Alternatively, the mtDNA genotype drift could occur at the level of mtDNA amplification, independent of mitochondrial function. To distinguish between these possibilities, we mixed cells homoplasmic for haplotype L0 or haplotype K1 at different ratios and quantified the proportion of each mtDNA genotype over 8 passages or 6 weeks. These two cell lines differ in 12 non-synonymous polymorphisms in mitochondrially encoded proteins (Table S1). Because the cells mixed are homoplasmic, quantification of mitochondrial genotypes within the culture can be used as a readout for the proportion of cells containing either haplotype L0 or haplotype K1. We confirmed that cells remained homoplasmic throughout the experiment by analysis of colonies grown from single cells. Remarkably, the composition of cultures remained stable, showing no increase in the proportion of cells with the haplotype K1 (Figure 1I). Therefore, the haplotype K1 did not confer a survival or proliferation advantage compared with cells with haplotype L0.
To further examine whether genetic drift is caused by changes in mitochondrial function, we analyzed mitochondrial activity in both pluripotent stem cells (Figures 2A–2D) and after differentiation into fibroblasts (Figures 2E–2Q). The availability of primary skin fibroblasts from the oocyte donor or the somatic cell donor provided controls for mitochondrial function without MR. MR-PS12 cells containing predominantly haplotype H1 or L3 or containing both haplotypes at an approximately equal ratio (40%–60% L3 and 60%–40% H1) showed no significant differences in mitochondrial respiratory chain enzyme (RCE) activities (Figure 2B). We also found no significant differences in RCE activities in nuclear transfer ESCs containing haplotype K1 or L0 or NT-ESCs with 40%–60% heteroplasmy (Figure 2C). Comparison of an additional eight MR-PS cells (MR-PS5–11) with parthenogenetic stem cells and iPSC lines that had not undergone mitochondrial replacement also revealed no significant differences in RCE activity (Figure 2D).
Figure 2. Mitochondrial Drift Is Not Driven by Nuclear-Mitochondrial Incompatibility.

Shown is the analysis of mitochondrial function in cells with isogenic nuclear DNA but different mitochondrial genotypes because of genotype drift.
(A) Immunostaining of MR-PS cells expressing pluripotency markers.
(B and C) RCE activities in cells with identical nuclear DNA but different indicated mitochondrial haplotypes. (B) MR-PS12. (C) NT-ESCs. CS, citrate synthase; COX, cytochrome c oxidase (also known as complex IV).
(D) RCE activity in MR-PS cells, parthenogenetic ESCs, and iPS cell lines. RCE activity values were normalized to CS, an index of mitochondrial mass.
(E) Fibroblasts differentiated from MR-PS cells. ASMA, alpha smooth muscle actin.
(F and G) Heteroplasmy in differentiated fibroblasts from MR-PS12 (F) or SCNT ESC lines (G).
(H and I) RCE activity in MR-PS12 cells of different heteroplasmy (H), and in SCNT ES cells and skin fibroblasts (I) of the oocyte donor or the nucleus donor.
(J–Q) Analysis of mitochondrial function using Seahorse XF24 Analyzer for oxygen consumption rate (OCR) per 70,000 cells
(J and N) OCR profile of NT-ESC (J) or MR-PS12 (N). From left to right: basal oxygen consumption, oxygen consumption due to proton leakage, oxygen consumption with uncoupling agent FCCP, non-mitochondrial oxygen consumption.
(K and O) Coupling efficiency of NT-ESC (K) or MR-PS12 (O).
(L and P) Cell respiratory control ratio of NT-ESC (L) or MR-PS12 (P).
(M and Q) Spare capacity of NT-ESC (M) or MR-PS12 (Q) measured by the ratio of the uncoupled OCR to basal OCR.
Error bars denote SD. Scale bars, 50 μm. Numbers indicate biological replicates.
Because oxidative phosphorylation and dependence on mitochondrial function increases with cellular differentiation, we differentiated stem cells with identical nuclear DNA but with divergent mtDNA genotypes into fibroblasts (Figure 2E). The mtDNA genotypes remained stable, reflecting the genotypes of the pluripotent stem cells prior to differentiation (Figures 2F and 2G). As in pluripotent stem cells, no consistent differences in RCE activities were detected (Figures 2H and 2I). We also examined mitochondrial function by analyzing oxygen consumption, including coupling efficiency, cell respiratory control ratio, and spare capacity (Figures 2J–2Q) and detected no significant differences in fibroblasts derived from NT-ESCs with identical nuclear DNA and either haplotype L0 or K1. Furthermore, no significant differences in mitochondrial function were found when these in vitro-differentiated cells were compared with primary BJ skin fibroblasts with the identical nuclear genome and either the original mtDNA haplotype, K1, or haplotype L0 of the oocyte donor. Likewise, fibroblasts of MR-PS12 cells with either the L3 or H1 genotype or a combination of the two showed no differences in oxidative phosphorylation. Therefore, mtDNA genotype instability is not driven by changes in mitochondrial function or by a competitive advantage of cells with a specific mitochondrial-nuclear DNA combination.
Our results show that the maternal genome is fundamentally compatible with different mitochondrial genotypes and that vertical inheritance is not required for normal mitochondrial function. Like the paternal genome at fertilization, the maternal genome can form a novel combination of nuclear and mitochondrial alleles that is fully functional. This is not unexpected because maternal nuclear alleles may resegregate with a new mtDNA genotype after inheritance through a son. The continuous re-combination in human reproduction therefore requires fundamental compatibility of nuclear and mitochondrial genomes. However, independent of mitochondrial function, low levels of mtDNA heteroplasmy can result in mitochondrial genotype instability. mtDNA segregation and genetic drift have been observed in both mice and monkey embryonic development, using experimental systems with very high levels of heteroplasmy (Burgstaller et al., 2014; Lee et al., 2012; Sharpley et al., 2012). Here we report both random and directional genetic changes with complete conversion from heteroplasmy levels at or below 1% to 100% in human cells. All currently available methods of MR introduce low levels of heteroplasmy, although the transfer of either a single spindle or a polar body (Figure 1A) into unfertilized oocytes carries less mitochondria than the transfer of two pronuclei after fertilization (Craven et al., 2010; Paull et al., 2013; Tachibana et al., 2013; Wang et al., 2014). Biopsies of early embryos can be used to quantify heteroplasmy and to exclude embryos with high carryover. Nevertheless, this assay may not be sufficient to reliably predict long-term outcomes. Replication of mtDNA is greatly reduced during preimplantation development (McConnell and Petrie, 2004) and, hence, biopsies taken at the cleavage or the blastocyst stage do not reveal replicative dynamics of mtDNA genotypes. Furthermore, segregation of 1,000 or fewer copies of mtDNA introduced during transfer (Paull et al., 2013) into 250 cells at the blastocyst stage may not be homogenous. This might explain why half of pluripotent stem cell lines are homoplasmic at derivation (e.g., NT5 in Figure 1H). Although mtDNA genotype stability in ESCs may differ from human development in vivo, we also observed mtDNA genotype instability during cell differentiation (Figure 1H). Furthermore, several studies in animals also show that heteroplasmy of different mitochondrial genotypes can be unstable. In mice and monkeys born after MR, changes in mitochondrial genotypes are particularly notable in the female germline (Lee et al., 2012) and in the offspring (Wang et al., 2014). Instability of mtDNA genotypes would not only compromise the durable elimination of mutant mtDNA but also result in variable composition of two normal mitochondrial genotypes in differentiated tissues. Although each mtDNA genotype can support normal mitochondrial function on its own, work in mice suggests that co-existence of two normal mtDNA genotypes at approximately equal levels can result in altered mitochondrial function (Sharpley et al., 2012). Therefore, although vertical inheritance of mtDNA is not required, it is critical to ensure inheritance of a single maternal mtDNA lineage. Unlike after fertilization, where ubiquitination of sperm mitochondria mediates the elimination of low-level heteroplasmy (Sutovsky et al., 1999), avoiding heteroplasmy in MR will require technical improvements and a mechanistic understanding of mtDNA genotype drift.
Supplementary Material
Highlights.
Cryopreservation allows coordinated nuclear transfer in human oocytes
Stem cell lines and derivatives show low-level carryover of transferred mtDNA
Recipient mtDNA is functionally compatible with donor nuclear DNA
Genetic drift can lead to restoration of the original donor mitochondrial genotype
ACKNOWLEDGMENTS
This research was supported by the New York Stem Cell Foundation (NYSCF) and the Bernard and Anne Spitzer Fund. We thank Mr. Futoshi Inoue (Kitazato Corporation) for developing and supplying new oocyte freezing and thawing media. D.E. is a NYSCF-Robertson Investigator. We thank Salvatore DiMauro for comments on the manuscript.
ACCESSION NUMBERS
The accession numbers for the _mtDNA sequences reported in this paper are GenBank: KX079702, KX079703, KX079704, and KX079705.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, two figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2016.04.001.
REFERENCES
- Birket MJ, Orr AL, Gerencser AA, Madden DT, Vitelli C, Swistowski A, Brand MD, and Zeng X (2011). A reduction in ATP demand and mitochondrial activity with neural differentiation of human embryonic stem cells. J. Cell Sci. 124, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller JP, Johnston IG, Jones NS, Albrechtova J, Kolbe T, Vogl C, Futschik A, Mayrhofer C, Klein D, Sabitzer S, et al. (2014). MtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 7, 2031–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YG, Eum JH, Lee JE, Shim SH, Sepilian V, Hong SW, Lee Y, Treff NR, Choi YH, Kimbrel EA, et al. (2014). Human somatic cell nuclear transfer using adult cells. Cell Stem Cell 14, 777–780. [DOI] [PubMed] [Google Scholar]
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, et al. (2010). Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 465, 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham-Snary KJ, and Ballinger SW (2015). GENETICS. Mitochondrialnuclear DNA mismatch matters. Science 349, 1449–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Ma H, Juanes RC, Tachibana M, Sparman M, Woodward J, Ramsey C, Xu J, Kang EJ, Amato P, et al. (2012). Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell Rep. 1, 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Folmes CD, Wu J, Morey R, Mora-Castilla S, Ocampo A, Ma L, Poulton J, Wang X, Ahmed R, et al. (2015). Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 524, 234–238. [DOI] [PubMed] [Google Scholar]
- McConnell JM, and Petrie L (2004). Mitochondrial DNA turnover occurs during preimplantation development and can be modulated by environmental factors. Reprod. Biomed. Online 9, 418–424. [DOI] [PubMed] [Google Scholar]
- Noggle S, Fung HL, Gore A, Martinez H, Satriani KC, Prosser R, Oum K, Paull D, Druckenmiller S, Freeby M, et al. (2011). Human oocytes reprogram somatic cells to a pluripotent state. Nature 478, 70–75. [DOI] [PubMed] [Google Scholar]
- Paull D, Emmanuele V, Weiss KA, Treff N, Stewart L, Hua H, Zimmer M, Kahler DJ, Goland RS, Noggle SA, et al. (2013). Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493, 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton J, Chiaratti MR, Meirelles FV, Kennedy S, Wells D, and Holt IJ (2010). Transmission of mitochondrial DNA diseases and ways to prevent them. PLoS Genet. 6, e1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt K, Dowling DK, and Morrow EH (2013). Medicine. Mitochondrial replacement, evolution, and the clinic. Science 341, 1345–1346. [DOI] [PubMed] [Google Scholar]
- Sharpley MS, Marciniak C, Eckel-Mahan K, McManus M, Crimi M, Waymire K, Lin CS, Masubuchi S, Friend N, Koike M, et al. (2012). Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, and Schatten G (1999). Ubiquitin tag for sperm mitochondria. Nature 402, 371–372. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner-Hedges R, Kang E, Lee HS, et al. (2013). Towards germline gene therapy of inherited mitochondrial diseases. Nature 493, 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Sha H, Ji D, Zhang HL, Chen D, Cao Y, and Zhu J (2014). Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell 157, 1591–1604. [DOI] [PubMed] [Google Scholar]
- Yamada M, Johannesson B, Sagi I, Burnett LC, Kort DH, Prosser RW, Paull D, Nestor MW, Freeby M, Greenberg E, et al. (2014). Human oocytes reprogram adult somatic nuclei of a type 1 diabetic to diploid pluripotent stem cells. Nature 510, 533–536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.