

Abstract
Pulmonary arterial hypertension (PAH) is a fatal vasculopathy that ultimately leads to elevated pulmonary pressure and death by right ventricular (RV) failure, which occurs in part due to decreased fatty acid oxidation and cytotoxic lipid accumulation. In this study, we tested the hypothesis that decreased fatty acid oxidation and increased lipid accumulation in the failing RV is driven, in part, by a relative carnitine deficiency. We then tested whether supplementation of l‐carnitine can reverse lipotoxic RV failure through augmentation of fatty acid oxidation. In vivo in transgenic mice harboring a human BMPR2 mutation, l‐carnitine supplementation reversed RV failure by increasing RV cardiac output, improving RV ejection fraction, and decreasing RV lipid accumulation through increased PPARγ expression and augmented fatty acid oxidation of long chain fatty acids. These findings were confirmed in a second model of pulmonary artery banding‐induced RV dysfunction. In vitro, l‐carnitine supplementation selectively increased fatty acid oxidation in mitochondria and decreased lipid accumulation through a Cpt1‐dependent pathway. l‐Carnitine supplementation improves right ventricular contractility in the stressed RV through augmentation of fatty acid oxidation and decreases lipid accumulation. Correction of carnitine deficiency through l‐carnitine supplementation in PAH may reverse RV failure.
Keywords: metabolism, pulmonary arterial hypertension, right heart failure
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a progressive, fatal vasculopathy of the pulmonary arteries. 1 , 2 Despite current therapies, 3 more than 50% of patients die within 5 years of diagnosis of PAH. 4 The most common cause of death in PAH is right ventricular (RV) failure which has no effective treatment, 5 highlighting the need for therapies that directly improve RV function in PAH. PAH is associated with systemic abnormalities in glucose and lipid metabolism that are thought to result from abnormalities in fatty acid processing, 6 , 7 and a consequence of these alterations in metabolism in the PAH RV is a decrease in fatty acid oxidation and concomitant accumulation of lipotoxic metabolites in the failing RV. 8 , 9 , 10 , 11 , 12
To date, there are no effective strategies for reducing lipid accumulation and improving fatty acid oxidation in the diseased RV. While incompletely understood, one mechanism that is thought to contribute to decreased β‐oxidation of fatty acids is a deficiency of carnitine in the failing RV. 11 , 13 Carnitines are a family of compounds considered indispensable for β‐oxidation of fatty acids that are involved in the conjugation of and transport of long chain fatty acids across the mitochondrial membranes, thereby facilitating β‐oxidation and adenosine triphosphate (ATP) generation. 14 Multiple studies have now demonstrated an accumulation of long chain fatty acids in the plasma and RV of patients with PAH, 8 , 11 , 13 , 15 perhaps a consequence of decreased fatty acid oxidation. One strategy for improving RV fatty acid oxidation may be through supplementation of l‐carnitine to improve conjugation and transport of long chain fatty acids into mitochondria. Carnitine supplementation has not yet been studied in the context of RV failure in PAH, and additionally the mechanisms by which l‐carnitine may affect cardiac function are also not well understood.
In this study, we sought to test the hypothesis that l‐carnitine supplementation reverses RV dysfunction and increases fatty acid β‐oxidation in the diseased RV. We studied the effects of l‐carnitine supplementation on the hemodynamic and structural remodeling of the RV as well as fatty acid oxidation capacity in two models of RV failure in a mouse model harboring a clinically relevant mutation in the BMPR2 gene 16 and a model of load‐stress on the RV imposed by pulmonary artery banding. We further tested the mechanism of carnitine's effect on cardiomyocytes using cell culture techniques.
METHODS
Patient samples and plasma collection
Patients with idiopathic PAH or heritable PAH followed at the Vanderbilt University Medical Center over a 2‐year period, aged ≥18 years and without a known diagnosis of polycystic ovarian syndrome, diabetes mellitus, therapy with an oral hypoglycemic agent or other diabetic therapy were eligible to participate. Plasma was collected from patients who underwent clinically indicated right heart catheterization. Plasma was also collected from age‐, sex‐, and body mass index‐matched healthy volunteers free from cardiovascular disease or diabetes mellitus. After sample collection, plasma was immediately isolated by centrifugation and stored at −80°C for metabolomic analysis as previously described. 8
Animal studies
Wild type C57BL6/J mice and transgenic mice harboring a disease‐causing human mutation in BMPR2 were used for this study. 16 Each group contained roughly equal numbers of female and male mice, and all mice were 10–12 weeks old at the start of the study. For BMPR2 mutant mouse studies, mice were fed a high‐fat as previously described for 6 weeks to cause RV lipid accumulation as previously described. 17 After 6 weeks, mice were given either control water or water supplemented with 200 mg/kg/day of l‐carnitine for 10 weeks. As a second model of right heart strain induced by load stress, wild type C57BL6/J mice underwent sham surgery or pulmonary artery banding with a high‐fat diet as previously described. 15 Mice were then treated immediately after surgery with l‐carnitine (200 mg/kg/day) or vehicle (water) control in their water for 2 weeks. At the end of the study period, all mice underwent either echocardiography or cardiac magnetic resonance imaging (MRI) (see Supporting Information for detailed methods of cardiac MRI), cardiac catheterization as previously described, 18 isolation of tissue for mitochondrial respiration studies (detailed methods in Supporting Information), and tissue harvest for analysis of RNA, protein, histology, and mass spectrometry for lipid metabolites as previously described. 11 , 18 Primer sequences for RT‐PCR and antibodies with concentrations used for immunostaining are detailed in the Supporting Information.
Cell studies
The H9C2 rat myoblast cell line was transfected with either a plasmid containing a cytoplasmic mutation in the BMPR2 gene (2579‐2580 delT) 19 or empty vector control. To measure lipid accumulation in cells, cells were stained with BODIPY 493/502 stain (Invitrogen) at a concentration of 0.1 μg/ml and counterstained with DAPI stain after fixation in 4% paraformaldehyde. A subset of cells was also assayed for mitochondrial respiration using a Seahorse XFe96 Analyzer (see Supporting Information for detailed methods).
Statistical analysis
Differences in measurements in animal studies were assessed using one‐way analysis of variance with the Tukey post hoc test. A p < 0.05 was considered significant. Differences in plasma metabolite levels of free carnitine and long chain acylcarnitines in human samples were assessed using the Mann–Whitney test comparing samples from patients with pulmonary hypertension versus control. A p < 0.05 was considered significant.
RESULTS
Long‐chain fatty acid to carnitine ratios are increased in patients with pulmonary arterial hypertension
We first measured free carnitine (C0), medium‐chain (C10), and long chain (C18) acylcarnitine levels in the plasma of patients with pulmonary arterial hypertension (n = 27) and control patients without pulmonary hypertension (n = 37). Compared to controls, the ratio of long chain (C18) to free carnitine levels was increased in PAH patients. Medium‐chain (C10) to free carnitine levels were unchanged between groups (Figure 1, Supporting Information: Figure S1). Relative to the circulating levels of long chain fatty acids in patients with PAH, free carnitine levels were not correspondingly elevated (Supporting Information: Figure S1), suggestive of a relative free carnitine deficiency in patients with PAH.
Figure 1.
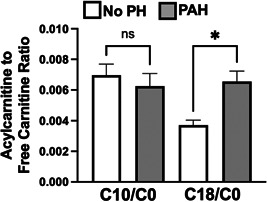
Medium chain (C10) and long chain (C18) fatty acid to free carnitine (C0) ratios in patients without pulmonary hypertension (No PH—white) and pulmonary arterial hypertension (PAH—gray). *p < 0.05 by Mann–Whitney test. N = 38 for the no PH controls. N = 27 for the PAH patients. PAH, pulmonary arterial hypertension.
l‐Carnitine supplementation improves systolic of the RV in R899X BMPR2 mutant mice
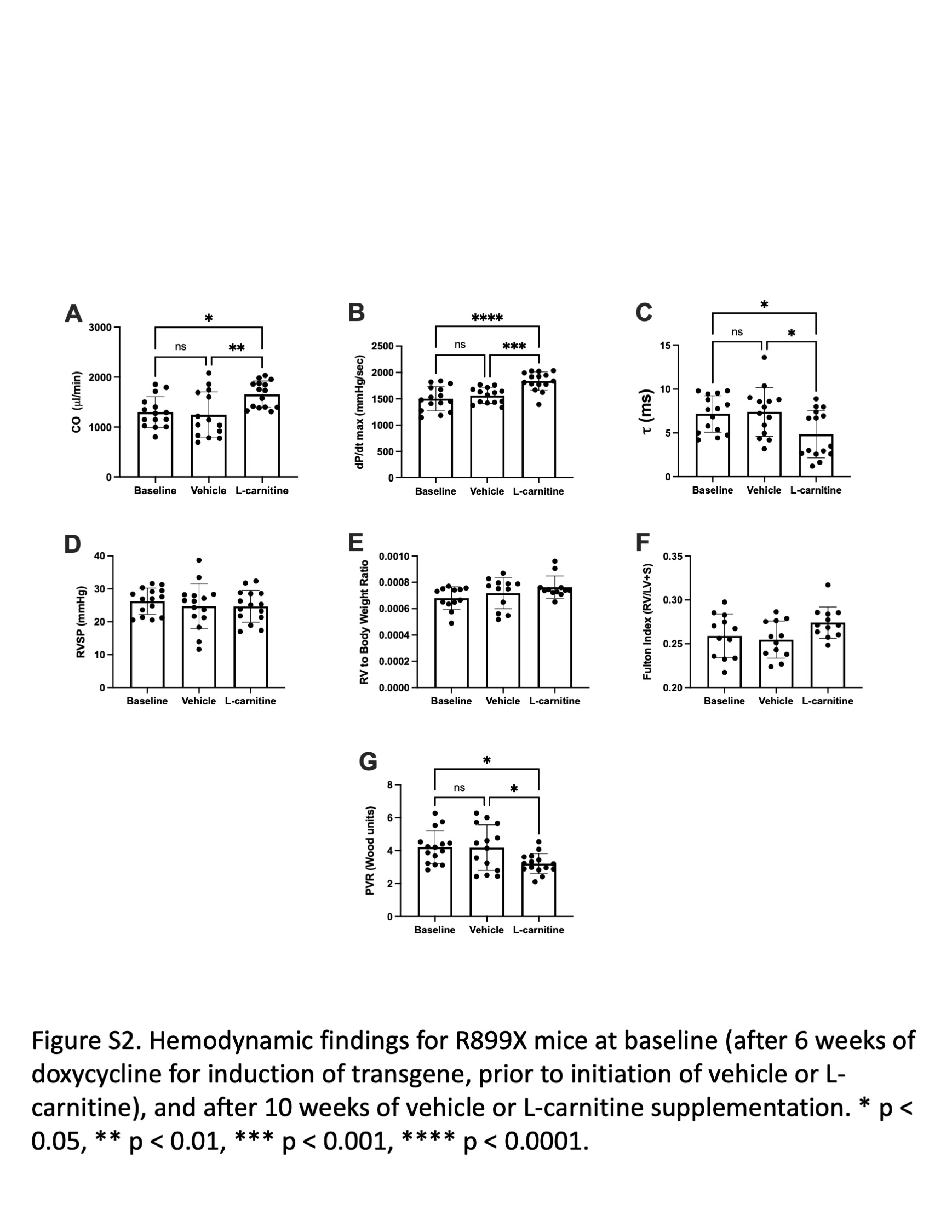
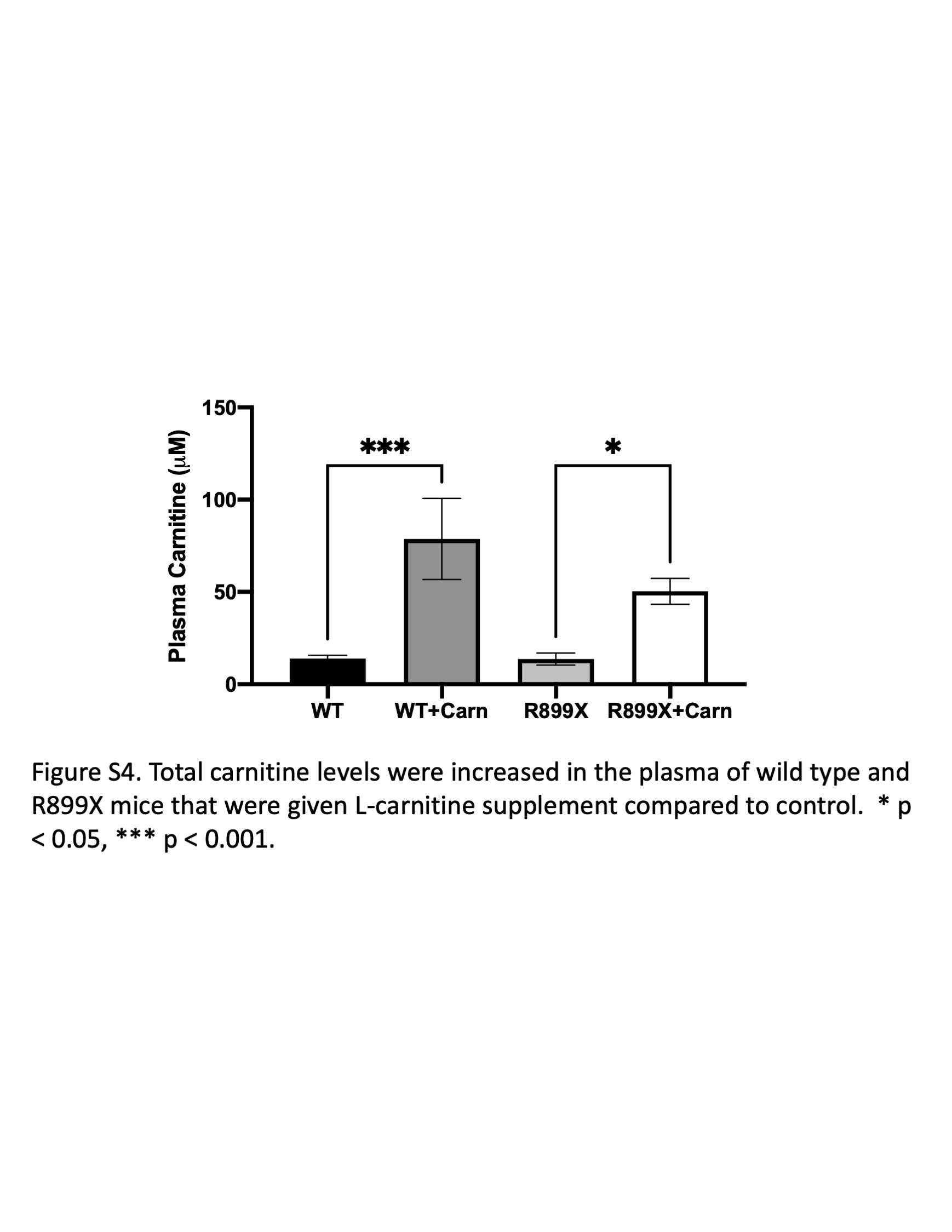
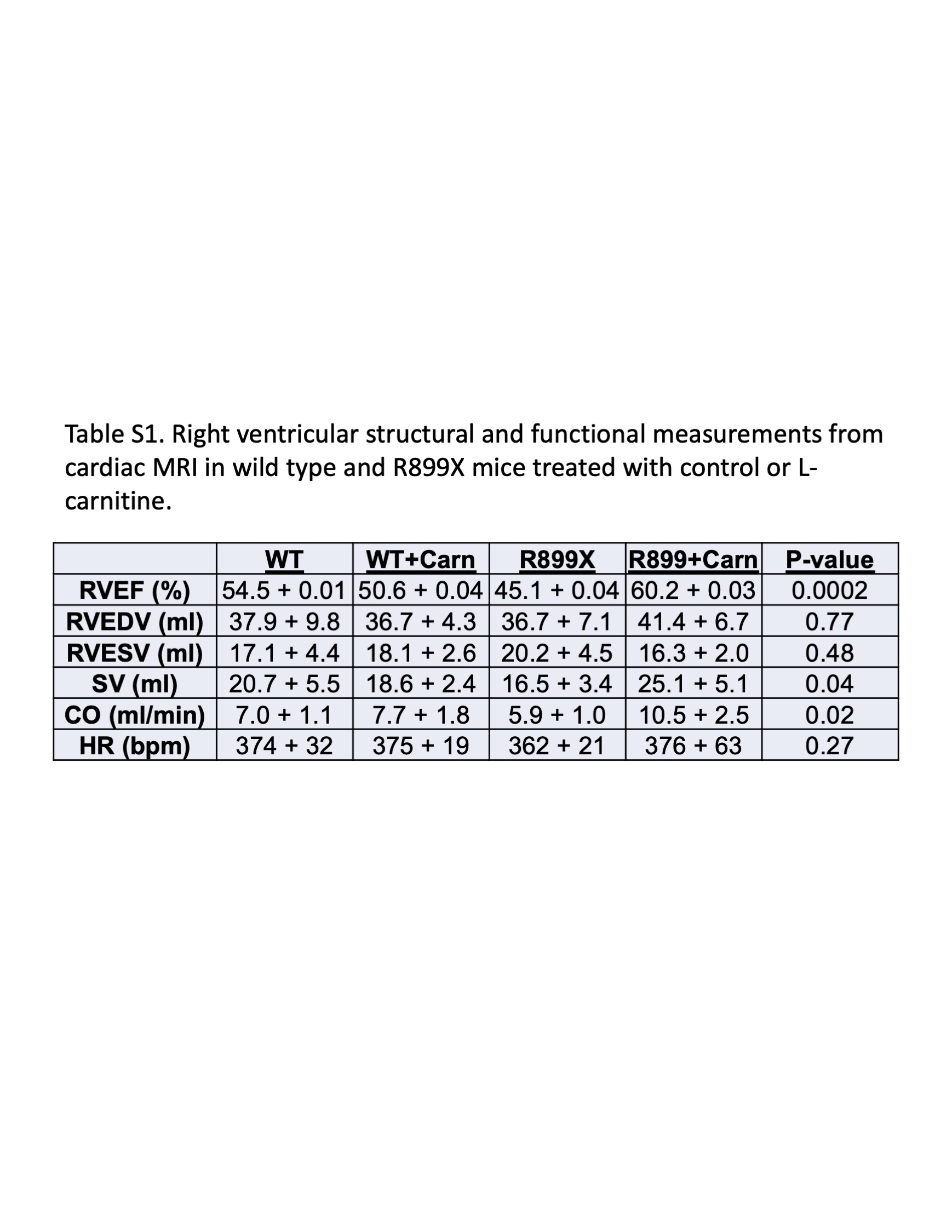
We next sought to determine whether supplementation of l‐carnitine could improve RV function in a mouse model of PAH harboring a clinically relevant mutation in BMPR2 (R899X). Whereas there were no significant differences in wild type given control or l‐carnitine supplementation, l‐carnitine in BMPR2 mutant mice led to an increase in measured cardiac output, maximum contractility (dP/dt max), decrease in diastolic relaxation constant τ, and decrease in pulmonary vascular resistance (Figure 2, Supporting Information: Figure S2). When the diastolic relaxation constant, τ, was normalized to resting heart rate, no significant changes were noted (Supporting Information: Figure S3). There were no changes in RV systolic pressure, RV to body weight ratio, or Fulton Index. Magnetic resonance imaging of the hearts of mice also confirmed that R899X mutant mice, but not wild type mice, given l‐carnitine supplementation showed an increase in RV ejection fraction, RV cardiac output, and RV stroke volume (Figure 3a, Supporting Information: Table S1). Plasma analysis confirmed that l‐carnitine supplementation led to an increase in plasma concentrations of l‐carnitine in both wild type and R899X mice (Supporting Information: Figure S4).
Figure 2.
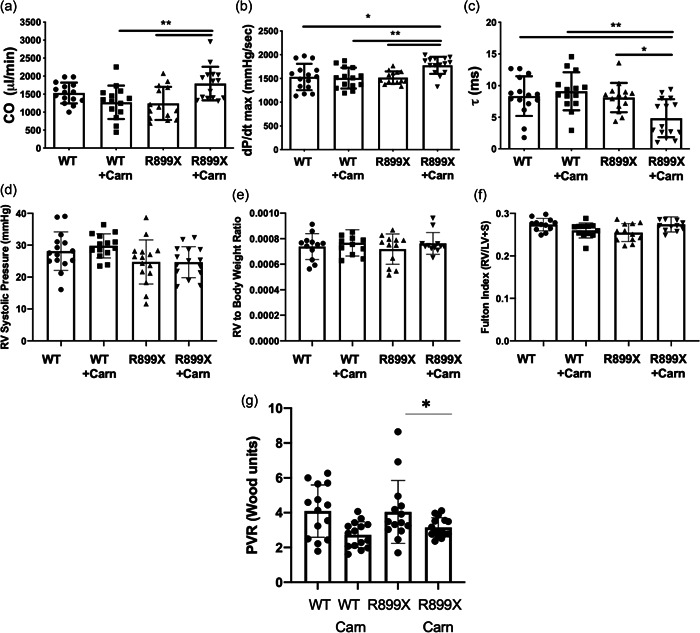
Invasively measured (a) cardiac output, (b) systolic contractility (dP/dt max), (c) diastolic relaxation (τ), (d) RV systolic pressure, (e) RV to body weight ratio, (f) Fulton Index (RV to LV+ septal weight ratio), and (g) pulmonary vascular resistance (PVR) in wild type and R899X mutant mice after 10 weeks of supplementation with l‐carnitine. *p < 0.05. **p < 0.01. Error bars represent standard deviation. N = 13–15 for each group. Carn, l‐carnitine; LV, left ventricular;RV, right ventricular.
Figure 3.
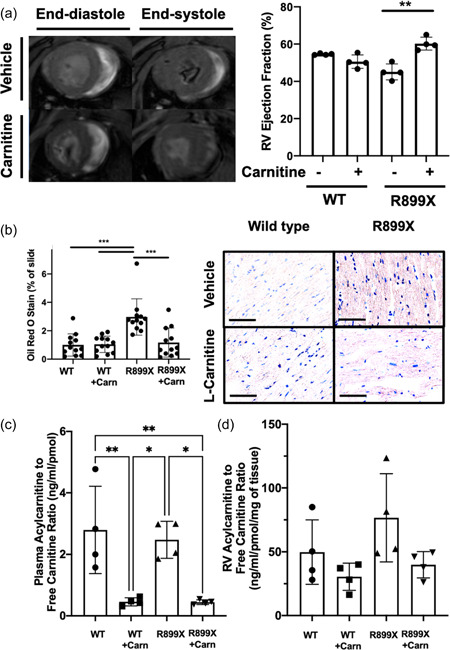
(a) l‐Carnitine treatment improves right ventricular ejection fraction by cardiac MRI in mutant R899X, but not wild type, mice (N = 4 per group). (b) Quantified Oil Red O stain in wild type and R899X mice given either vehicle control or l‐carnitine supplement in water for 10 weeks (N = 12 per group). (c) Measured plasma acylcarnitine to free carnitine ratio in each group. (d) Measured right ventricular acylcarnitine to free carnitine ratio in each group (N= 4 per group). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (Bar = 50 µm). MRI, magnetic resonance imaging; RV, right ventricle.
l‐Carnitine decreases fatty acid accumulation in the RV of R899X BMPR2 mutant mice and increases acylcarnitines
We next used Oil Red O in histologic sections to determine whether there was a change in measured fat within the RV of mice R899X mice given l‐carnitine. Compared to vehicle control, l‐carnitine supplementation led to a 61% decrease in Oil Red O staining on sections (Figure 3b, Supporting Information: Figure S5). Metabolomic analysis by mass spectrometry of lipid contents of the plasma and RV demonstrated that l‐carnitine treatment resulted in a 82% decrease in the acylcarnitine to free carnitine ratio in the plasma of R899X mice (Figure 3c). There was a nonsignificant trend towards a decrease in acylcarnitine to free carnitine ratio in the RV of R899X mice that were treated with l‐carnitine (Figure 3d).
l‐Carnitine increases fatty acid mediated mitochondrial respiration and β‐oxidation of fatty acids
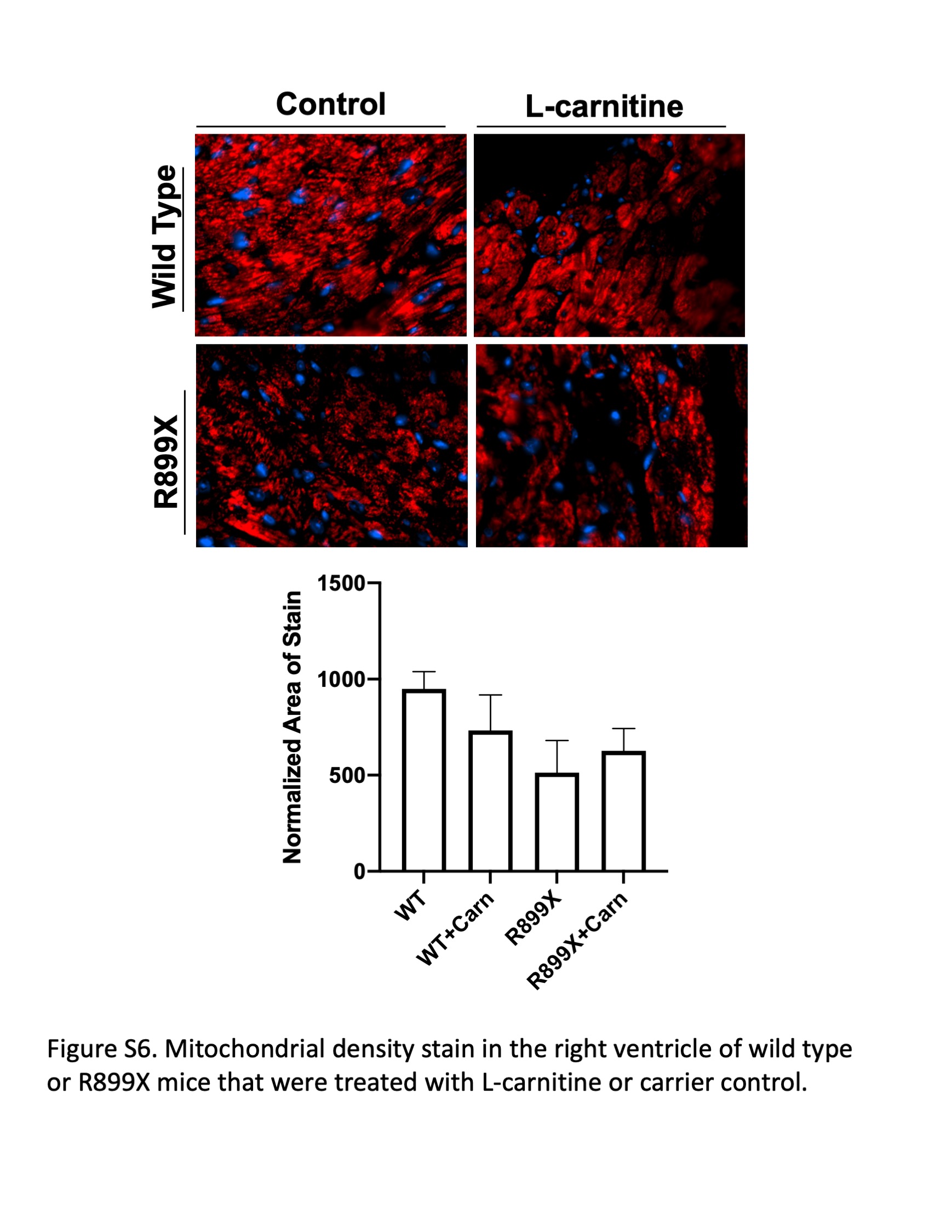
To assess whether l‐carnitine supplementation improved fatty acid‐mediated mitochondrial respiration, a subset of mice were killed and the RV tissue was immediately permeabilized for measurement of oxygen respiration in response to various substrates and inhibitors of mitochondrial respiration. All data were normalized to mitochondrial density in the RV by Tom20 stain (Supporting Information: Figure S6). The RVs in R899X mice treated with l‐carnitine showed increased respiratory capacity via complex I‐mediated respiration (glutamate/malate) (Figure 4a), long chain fatty acid‐mediated oxidation (palmitoyl‐carnitine) (Figure 4b), and complex II‐mediated respiration (succinate) (Figure 4d) compared to controls. There were no differences between R899X and control RVs in response to medium‐chain fatty acids (octanoyl‐carnitine) as a result of l‐carnitine treatment (Figure 4c).
Figure 4.
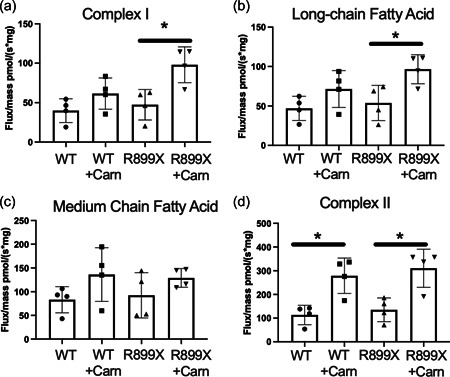
Mitochondrial respiration and oxygen flux response to (a) glutamate/malate treatment (Complex I), (b) palmitoyl‐carnitine (long chain fatty acid), (c) octanoyl‐carnitine (medium chain fatty acid), and (d) succinate (Complex II). *p < 0.05. N = 4 per group.
Figure 5.
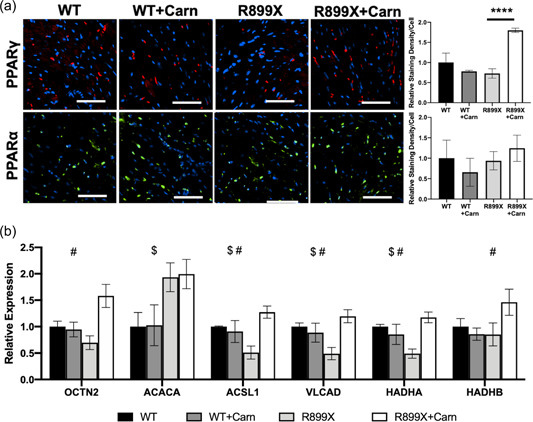
(a) Immunostain of the RV for PPARγ and PPARα in wild type and R899X mice with and without l‐carnitine supplementation (****p < 0.0001, n = 3 samples for each group). (b) RT‐PCR for mRNA transcript expression in wild type and R899X mice with and without l‐carnitine supplementation for regulators of fatty acid oxidation ($ p < 0.05 between wild type and R899X mice, # p < 0.05 between R899X mice treated with l‐carnitine vs. control) (bar = 50 µm). N = 6 animals per group. mRNA, messenger ribonucleic acid; PPAR, peroxisome proliferator‐activated receptors; RT‐PCR, real time reverse transcription–polymerase chain reaction; RV, right ventricle.
Oxidized lipids are also a natural ligand for peroxisome proliferator‐activated receptors (PPAR), a family of nuclear receptors that regulate mitochondrial respiration and metabolism that have been shown to be diminished in the PAH RV. 20 , 21 , 22 Thus, we next investigated whether metabolic changes in response to l‐carnitine treatment may be, in part, mediated by changes in PPAR expression. Immunostain for PPARα and PPARγ demonstrated an increase in expression of PPARγ in the RV of R899X mice treated with l‐carnitine (Figure 5a). As a result, downstream transcription of regulators of fatty acid oxidation was also increased in R899X mice that received l‐carnitine as opposed to control (Figure 5b).
l‐Carnitine decreases lipid accumulation and increases Cpt‐1‐dependent fatty acid oxidation in vitro
We finally sought to confirm whether the effects of l‐carnitine on fatty acid oxidation were directly attributable to cardiomyocytes through the use of an in vitro model of cardiomyocyte‐like cells harboring a BMPR2 mutation or controls. H9C2 cells transfected with either empty vector control or a cytoplasmic domain mutation in the BMPR2 gene 19 were treated with l‐carnitine or loading control in the setting of bovine serum albumin (BSA):Palmitate and subsequently fixed and stained for lipid accumulation. l‐Carnitine improved lipid accumulation in BMPR2‐mutant H9C2 cells but had no effect on wild type H9C2 cells (Figure 6a,b). Additionally, BMPR2 mutant and wild type H9C2 cells were analyzed for the ability of their mitochondria to undergo mitochondrial respiration in the presence of palmitate conjugated BSA in vitro. Cells were sequentially treated with Oligomycin, FCCP, and Rotenone/Antimycin A to determine basal respiration, ATP‐linked respiration, maximal respiratory capacity, nonmitochondrial respiration, and reserve capacity. In BMPR2 mutant H9C2 cells, there was a decrease in the overall maximal respiratory capacity of fatty acids compared to wild type H9C2 cells, consistent with a decreased ability to undergo fatty acid β‐oxidation. With supplementation of 0.5 mM l‐carnitine, the overall mitochondrial respiratory capacity of fatty acids was increased by 65% in H9C2 cells harboring the BMPR2 mutation but not wild type H9C2 cells (Figure 6c,d). Treatment with a Cpt1 inhibitor, etoximir, abolished all oxygen consumption in both cell types, confirming that the assay was indeed measuring mitochondrial oxidation of fatty acid oxidation and that l‐carnitine's effect is Cpt1‐dependent. Taken together, these findings confirm that l‐carnitine directly regulates fatty acid oxidation in RV tissue, specifically cardiomyocytes, in a CPT1 dependent manner to improve mitochondrial function.
Figure 6.
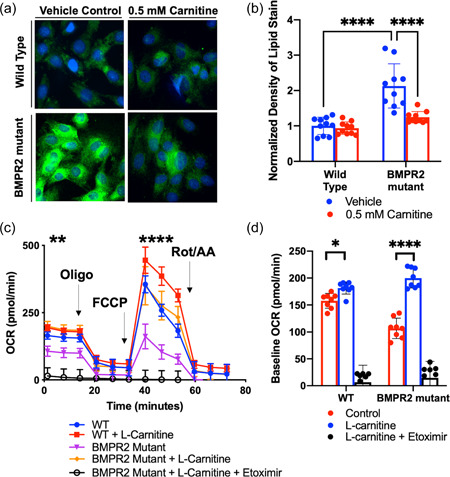
(a, b) Lipid stain in H9C2 cells transfected with either empty vector (wild type) or a human cytoplasmic domain mutation in BMPR2 and treated with l‐carnitine or vehicle control. (c) Mitochondrial oxygen consumption rate in H9C2 cells transfected with either empty vector control (WT) or a human cytoplasmic domain mutation in BMPR2. (d) Maximal mitochondrial respiratory capacity in H9C2 cells with and without BMPR2 mutation after l‐carnitine supplementation and Etoximir (Cpt1 inhibitor). *p < 0.05, **p < 0.01, ****p < 0.0001. Technical replicates N = 10 per group. Biologic replicates N = 3 per group.
l‐Carnitine improves right ventricular function and fatty acid oxidation after pulmonary artery banding
We utilized a second model of RV stress induced by pulmonary banding and high‐fat diet in wild type mice to determine if the effects of l‐carnitine on RV function were independent of any potential effect on pulmonary vasculature, and to determine if its effect was selective to mice harboring a BMPR2 mutation or more generalizable to RV load stress (Supporting Information: Figure S8). Two weeks after wild type C57/BL6 mice underwent pulmonary artery banding and high‐fat diet initiation as metabolic stress, mice were given either l‐carnitine supplementation in the water or control for 2 weeks. Mice given l‐carnitine supplementation demonstrated increased cardiac output, stroke work, stroke volume, decreased end‐arterial elastance (Ea), increased end‐systolic elastance (Ees), and overall decreased Ea/Ees ratio compared to control mice that did not receive l‐carnitine (Figure 7).
Figure 7.
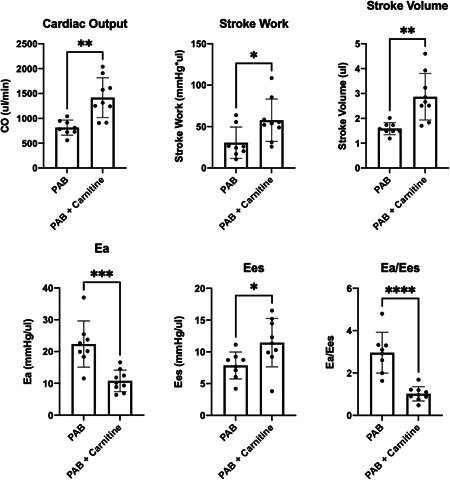
Hemodynamic measurements from invasive catheterization of mice who underwent pulmonary artery banding with and without l‐carnitine treatment. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. N = 8–9 per group.
DISCUSSION
The present study investigated whether l‐carnitine supplementation could reverse RV lipid accumulation and RV dysfunction in two separate models. We further demonstrated that this occurs through increased fatty acid oxidation, decreased lipid accumulation, and enhancement of PPARγ‐mediated transcription of regulators of fatty acid oxidation (Figure 8). The diseased RV in PAH displays diminished PPARγ activity and diminished fatty acid oxidation capacity, 22 , 23 , 24 both of which contribute to the accumulation of lipotoxic intermediates in the myocardium. 12 , 17 , 25 Thus, our findings suggest that l‐carnitine treatment may improve lipotoxicity in the diseased RV in part through the restoration of normal levels of fatty acid oxidation. Furthermore, our findings confirming a similar benefit in pulmonary artery‐banded wild type mice also suggest that the effect of l‐carnitine may be applicable more generally to RV dysfunction that occurs as a result of load stress.
Figure 8.
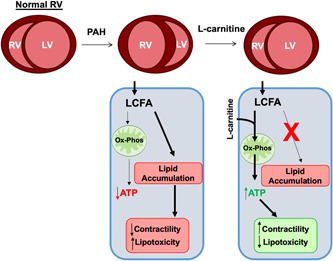
Overall schematic of l‐carnitine supplementation's effect upon fatty acid oxidation, oxidative phosphorylation, lipid accumulation, and myocardial contractility/lipotoxicity in the stressed right ventricle.
Most studies to date have attempted to target metabolic remodeling in the RV by preferentially decreasing fatty acid oxidation further and increasing glucose oxidation through pyruvate dehydrogenase kinase inhibition. 26 While successful in preclinical models, this approach has yielded mixed results in a clinical trial. 27 A logical approach toward targeting metabolic remodeling in the RV would be to attempt to restore the normal balance of fatty acid and glucose oxidation for energy generation. Studies of the pressure‐loaded left ventricle have indeed demonstrated that preservation of fatty acid oxidation can prevent the pathologic decrease in contractility associated with cardiac hypertrophy. 28 , 29 , 30 Therapies such as metformin have been shown to reduce lipid accumulation in preclinical models and triglyceride content in human RVs with PAH, 17 , 31 but whether these changes are driven by fatty acid oxidation‐related changes or not is not known.
Although previous studies have suggested that augmentation of fatty acid oxidation through genetic means may prevent cardiomyopathy, 22 , 28 , 29 our study utilized a readily available pharmacologic therapy that is readily translatable to humans and demonstrated that augmentation of fatty acid oxidation can reverse RV failure. This is particularly relevant for two reasons. The first is that features of metabolic dysfunction are disproportionately prevalent in patients with PAH, occurring in patient demographics that would otherwise not be considered “at risk” for development of complications such as insulin resistance, hyperglycemia, and hyperlidemia. 6 , 12 The second reason is that our therapeutic approach via l‐carnitine supplementation is known to be a safe intervention, even at high doses. 32 The findings of our study support previous work by our group and others that have demonstrated that PAH is characterized by an acquired state of deficient levels of carnitine, 11 , 13 , 33 , 34 which is important for shuttling of fatty acids to the mitochondrial membrane for β‐oxidation. The findings of the present study demonstrate that exogenous supplementation of l‐carnitine can, at least partially, increase acylcarnitine levels in the RV and increase fatty acid oxidation.
An acquired carnitine deficiency has been reported in multiple forms of cardiomyopathies, primarily ischemic and nonischemic left heart failure. In a meta‐analysis of small clinical studies investigating the efficacy of l‐carnitine supplementation in improving left heart failure, there was a 4% increase in LV ejection fraction and a mild 0.47 cm decrease in end‐diastolic diameter, but overall no change was noted in adverse events, mortality, or symptoms. 32 One study of patients with PAH showed that intravenous l‐carnitine supplementation (5 g/day) for 7 days improved functional markers such as 6‐min walk distance and WHO functional class, and simultaneously led to a decrease in measured B‐type natriuretic peptide levels (58.16 vs. 33.29 ng/L), supporting the potential benefit of l‐carnitine therapy in RV dysfunction as well. 35 Whether l‐carnitine may itself improve RV function, lipotoxicity, or longer‐term outcomes such as heart failure hospitalization, or mortality is not clear. RV failure does not respond to conventional therapies used for medical management of LV failure, and mechanistically metabolic derangement appears to be a more central mechanism that drives RV failure compared to LV failure. 36 Carnitine deficiency is less well studied in diseases of the right ventricle, but previous studies from our group and others suggest prominent defects in fatty acid oxidation underlie the development and progression of RV failure in PAH. 11 , 25 , 37 Most notably, elevated plasma acylcarnitine levels are both associated with both worse RV function and overall worse prognosis in PAH. 11 , 13 Although the mechanisms underlying elevated acylcarnitine levels in PAH are not completely understood, one plausible mechanism may be that acylcarnitines accumulate due to defective transport into mitochondria for fatty acid oxidation. 11 , 13
In our study, exogenous supplementation of l‐carnitine decreased the accumulation of cytoplasmic acylcarnitines by directly increasing fatty acid β‐oxidation in mitochondria. We saw no effect of l‐carnitine upon total mitochondrial density, but our data suggest a selective increase in mitochondrial β‐oxidation of long chain fatty acids in response to l‐carnitine supplementation in both in vivo and in vitro models through a Cpt1‐dependent mechanism. Augmentation of fatty acid oxidation also led to a feed‐forward loop through the activation of master metabolic regulator PPARγ and increased transcription of fatty acid oxidation‐related genes.
Our study has limitations. Our first model of transgenic mice harboring the BMPR2 mutant gene are incompletely penetrant and in this experiment did not develop elevated pulmonary pressures, as noted in our results. They do, however, recapitulate many cardiac and systemic features that are present in heritable PAH due to BMPR2 mutation, such as lipid accumulation, decreased ejection fraction, and insulin resistance. 10 , 11 , 12 , 17 A number of studies have suggested that the presence of a BMPR2 mutation in heritable PAH is associated with cardiomyopathic changes independent of the load stress imposed by pulmonary vascular remodeling, and thus the BMPR2 mutant mouse model in our study is still applicable to patients with heritable PAH or idiopathic PAH associated with decreases in BMPR2 expression. 11 , 12 , 38 , 39 , 40 To address the potential effects of load stress and to partially account for limitations of the BMPR2 transgenic mice used in this study, we used a second model of mechanically imposed load stress on the RV in wild type C57/BL6 by pulmonary artery banding to assess effects of l‐carnitine supplementation independent of any effect on pulmonary vascular remodeling. Future studies with other models in which pulmonary vascular remodeling occurs (e.g., Sugen‐hypoxia or monocrotaline) may further clarify the effect of l‐carnitine supplementation on RV dysfunction. A second limitation of our study was that l‐carnitine supplementation was provided through the drinking water, and thus the exact amount of supplement given to each animal cannot be entirely standardized. While our data show a significant increase in plasma carnitine levels with supplementation, our study did not control for the amount supplemented per mouse. Finally, our study was also limited to an assessment of l‐carnitine supplementation's effect on RV structure and function. We note that l‐carnitine supplementation led to a greater decrease in plasma acyl:free carnitine ratio compared to RV, and thus extra‐cardiac effects of l‐carnitine supplementation cannot be ruled out. Future studies will be needed to investigate the possibility of interorgan crosstalk as a contributing mechanism to the efficacy of l‐carnitine supplementation.
In conclusion, we demonstrate that augmentation of fatty acid oxidation in the PAH RV through supplementation of l‐carnitine improves relative carnitine deficiency, increases mitochondrial fatty acid oxidation, and improves RV function.
AUTHOR CONTRIBUTIONS
Vineet Agrawal, Anna R. Hemnes, Nicholas J. Shelburne, Niki Fortune, James D. West, and Evan L. Brittain designed research studies. Vineet Agrawal, Nicholas J. Shelburne, Niki Fortune, Julio L. Fuentes, Dan Colvin, Marion W. Calcutt, Megha Talati, and Emily Poovey conducted experiments and acquired data. Vineet Agrawal, Anna R. Hemnes, Nicholas J. Shelburne, Niki Fortune, James D. West, and Evan L. Brittain analyzed data. Vineet Agrawal, Anna R. Hemnes, and Evan L. Brittain wrote the manuscript.
CONFLICTS OF INTEREST
V. A., E. L. B., A. R. H., and J. D. W. have all received grants from the NIH/NHLBI. V. A. has received funding from the American Heart Association, Team Phenomenal Hope Foundation, and Pulmonary Hypertension Association. A. R. H. has served as a consultant to Janssen, Complexa, GossamerBio, Bayer, United Therapeutics. A. R. H. has received funding from CMREF. A. R. H. owns stock in Tenax Thera.
ETHICS STATEMENT
All human studies were approved by the Vanderbilt University Medical Center Institution Review Board (protocols 9401, 130712, and 140983). All animal studies were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee (protocol M1800073). All animal studies were approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC). The collection of human plasma samples was approved by the Institutional Review Board at Vanderbilt University Medical Center, and all subjects provided written informed consent.
Supporting information
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
{kind=link}
Supplementary information.
ACKNOWLEDGMENTS
NIH 5R01HL122417 (A. R. Hemnes), 3R01HL122417‐04S1 (A. R. Hemnes), Team Phenomenal Hope Award (V. Agrawal), Pulmonary Hypertension Association Young Investigator Award (V. Agrawal), NIH 1K08HL153956 (V. Agrawal).
Agrawal V, Hemnes AR, Shelburne NJ, Fortune N, Fuentes JL, Colvin D, Calcutt MW, Talati M, Poovey E, West JD, Brittain EL. l‐Carnitine therapy improves right heart dysfunction through Cpt1‐dependent fatty acid oxidation. Pulm Circ. 2022;12:e12107. 10.1002/pul2.12107
Vineet Agrawal and Anna R. Hemnes contributed equally to this study.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Prins KW, Thenappan T. World Health Organization group I pulmonary hypertension. Cardiol Clin. 2016;34:363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Humbert M, Lau EMT, Montani D, Jaïs X, Sitbon O, Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014;130:2189–208. [DOI] [PubMed] [Google Scholar]
- 3. Lau EMT, Giannoulatou E, Celermajer DS, Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol. 2017;14:603–14. [DOI] [PubMed] [Google Scholar]
- 4. Benza RL, Miller DP, Gomberg‐Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long‐term pulmonary arterial hypertension disease management (REVEAL). Circulation. 2010;122:164–72. [DOI] [PubMed] [Google Scholar]
- 5. Inampudi C, Hemnes AR, Briasoulis A. Approach to a patient with pulmonary hypertension. J Geriatr Cardiol . 2019;16:478–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pugh ME, Robbins IM, Rice TW, West J, Newman JH, Hemnes AR. Unrecognized glucose intolerance is common in pulmonary arterial hypertension. J Hear Lung Transplant. 2011;30:904–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trammell AW, Talati M, Blackwell TR, Fortune NL, Niswender KD, Fessel JP, Newman JH, West JD, Hemnes AR. Pulmonary vascular effect of insulin in a rodent model of pulmonary arterial hypertension. Pulm Circ. 2017;7:624–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hemnes AR, Luther JM, Rhodes CJ, Burgess JP, Carlson J, Fan R, Feesel JP, Fortune N, Gerszten RE, Halliday SJ, Hekmat R, Howard L, Newman JH, Niswender KD, Pugh ME, Robbins IM, Sheng Q, Shibao CA, Shyr Y, Sumner S, Talati M, Wharton J, Wilkins MR, Ye F, Yu C, West J, Brittain EL. Human PAH is characterized by a pattern of lipid‐related insulin resistance. JCI Insight. 2019;4:e123611. 10.1172/jci.insight.123611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Archer SL, Fang YH, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ. 2013;3:144–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Talati MH, Brittain EL, Fessel JP, Penner N, Atkinson J, Funke M, et al. Mechanisms of lipid accumulation in the bone morphogenetic protein receptor type 2 mutant right ventricle. Am J Respir Crit Care Med. 2016;194:719–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brittain EL, Talati M, Fessel JP, Zhu H, Penner N, Calcutt MW, et al. Fatty acid metabolic defects and right ventricular lipotoxicity in human pulmonary arterial hypertension. Circulation. 2016;133:1936–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189:325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luo N, Craig D, Ilkayeva O, Muehlbauer M, Kraus WE, Newgard CB, et al. Plasma acylcarnitines are associated with pulmonary hypertension. Pulm Circ. 2017;7:211–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Longo N, Amat Di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet‐Semin Med Genet. 2006;142C:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189:325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. West J, Niswender KD, Johnson JA, Pugh ME, Gleaves L, Fessel JP, et al. A potential role for insulin resistance in experimental pulmonary hypertension. Eur Respir J. 2013;41:861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brittain EL, Talati M, Fortune N, Agrawal V, Meoli DF, West J, et al. Adverse physiologic effects of Western diet on right ventricular structure and function: role of lipid accumulation and metabolic therapy. Pulm Circ. 2019;9:204589401881774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Agrawal V, Fortune N, Yu S, Fuentes J, Shi F, Nichols D, et al. Natriuretic peptide receptor C contributes to disproportionate right ventricular hypertrophy in a rodent model of obesity‐induced heart failure with preserved ejection fraction with pulmonary hypertension. Pulm Circ. 2019;9:2045894019878599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson JA, Hemnes AR, Perrien DS, Schuster M, Robinson LJ, Gladson S, et al. Cytoskeletal defects in Bmpr2‐associated pulmonary arterial hypertension. Am J Physiol‐Lung Cell Mol Physiol. 2012;302:474–84. 10.1152/ajplung.00202.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grygiel‐Górniak B. Peroxisome proliferator‐activated receptors and their ligands: nutritional and clinical implications—a review. Nutr J. 2014;13:17. 10.1186/1475-2891-13-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hansmann G, Calvier L, Risbano MG, Chan SY. Activation of the metabolic master regulator PPARγ: a potential PIOneering therapy for pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2020;62:143–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Legchenko E, Chouvarine P, Borchert P, Fernandez‐Gonzalez A, Snay E, Meier M, et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. 2018;10:eaao0303. 10.1126/scitranslmed.aao0303 [DOI] [PubMed] [Google Scholar]
- 23. Chouvarine P, Giera M, Kastenmüller G, Artati A, Adamski J, Bertram H, et al. Trans‐right ventricle and transpulmonary metabolite gradients in human pulmonary arterial hypertension. Heart. 2020;106:1332–41. 10.1136/heartjnl-2019-315900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meyers JL, Zhang J, Chorlian DB, Pandey AK, Kamarajan C, Wang JC, et al. Molecular mechanisms of right ventricular dysfunction in pulmonary arterial hypertension: focus on the coronary vasculature, sex hormones, and glucose/lipid metabolism. Cardiovasc Diagnosis Ther. 2020;26:5040–52. http://cdt.amegroups.com/article/view/44903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Talati M, Hemnes A. Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ. 2015;5:269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Piao L, Fang YH, Cadete VJJ, Wietholt C, Urboniene D, Toth PT, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med. 2010;88:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. 2017;9:eaao4583. 10.1126/scitranslmed.aao4583 [DOI] [PubMed] [Google Scholar]
- 28. Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, et al. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res. 2020;126:182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kolwicz SC, Olson DP, Marney LC, Garcia‐Menendez L, Synovec RE, Tian R. Cardiac‐specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure‐overload hypertrophy. Circ Res. 2012;111:728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu A, Schreier D, Tian L, Eickhoff JC, Wang Z, Hacker TA, et al. Direct and indirect protection of right ventricular function by estrogen in an experimental model of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2014;307:H273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brittain EL, Niswender K, Agrawal V, Chen X, Fan R, Pugh ME, et al. Mechanistic phase II clinical trial of metformin in pulmonary arterial hypertension. J Am Heart Assoc. 2020;9:e018349. 10.1161/JAHA.120.018349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Song X, Qu H, Yang Z, Rong J, Cai W, Zhou H. Efficacy and safety of l‐Carnitine treatment for chronic heart failure: a meta‐analysis of randomized controlled trials. BioMed Res Int. 2017;2017:1–11. 10.1155/2017/6274854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharma S, Aramburo A, Rafikov R, Sun X, Kumar S, Oishi PE, et al. l‐Carnitine preserves endothelial function in a lamb model of increased pulmonary blood flow. Pediatr Res. 2013;74:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, et al. A metabolic basis for endothelial‐to‐mesenchymal transition. Mol Cell. 2018;69:689–98.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu X‐Q, Jing Z‐C, Jiang X, Zhao QH, He J, Dai LZ, et al. Clinical efficacy of intravenous l‐carnitine in patients with right‐sided heart failure induced by pulmonary arterial hypertension. Zhongua Xin Xue Guan Bing Za Zhi. 2010;38:152–55. [PubMed] [Google Scholar]
- 36. Koop AC, Bossers GPL, Ploegstra MJ, Hagdorn Q, Berger R, Silljé H, et al. Metabolic remodeling in the pressure‐loaded right ventricle: shifts in glucose and fatty acid Metabolism—a systematic review and meta‐analysis. J Am Heart Assoc. 2019;8:012086. 10.1161/JAHA.119.012086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rabinovitch M. PPARγ and the pathobiology of pulmonary arterial hypertension. Adv Exp Med Biol, 661:447–58. 10.1007/978-1-60761-500-2_29 [DOI] [PubMed] [Google Scholar]
- 38. Evans JD, Girerd B, Montani D, Wang XJ, Galiè N, Austin ED, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta‐analysis. Lancet Respir Med. 2016;4:129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van der Bruggen CE, Happé CM, Dorfmüller P, Trip P, Spruijt OA, Rol N, et al. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial Hypertension CLINICAL PERSPECTIVE. Circulation. 2016;133:1747–60. [DOI] [PubMed] [Google Scholar]
- 40. Girerd B, Montani D, Eyries M, Yaici A, Sztrymf B, Coulet F, et al. Absence of influence of gender and BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res. 2010;11:73. 10.1186/1465-9921-11-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.