

Abstract
Microbial pathogens possess an arsenal of strategies to invade their hosts, evade immune defences and promote infection. In particular, bacteria use virulence factors, such as secreted toxins and effector proteins, to manipulate host cellular processes and establish a replicative niche. Survival of eukaryotic organisms in the face of such challenge requires host mechanisms to detect and counteract these pathogen-specific virulence strategies. In this Review, we focus on effector-triggered immunity (ETI) in metazoan organisms as a mechanism for pathogen sensing and distinguishing pathogenic from non-pathogenic microorganisms. For the purposes of this Review, we adopt the concept of ETI formulated originally in the context of plant pathogens and their hosts, wherein specific host proteins ‘guard’ central cellular processes and trigger inflammatory responses following pathogen-driven disruption of these processes. While molecular mechanisms of ETI are well-described in plants, our understanding of functionally analogous mechanisms in metazoans is still emerging. In this Review, we present an overview of ETI in metazoans and discuss recently described cellular processes that are guarded by the host. Although all pathogens manipulate host pathways, we focus primarily on bacterial pathogens and highlight pathways of effector-triggered immune defence that sense disruption of core cellular processes by pathogens. Finally, we discuss recent developments in our understanding of how pathogens can evade ETI to overcome these host adaptations.
Metazoan organisms use multiple strategies to protect themselves against invading pathogens. As originally proposed by the late Charles Janeway Jr., germline-encoded pattern recognition receptors (PRRs) sense microbial structures such as cell wall components, termed pathogen-associated molecular patterns (PAMPs)1,2 (Fig. 1a). PRRs activate signalling pathways that alter the transcription of thousands of immune defence genes, including cytokines, chemokines and interferons (IFNs)3. While PRRs are necessary for innate immune responses to infection and innate instruction of adaptive immunity4–7, both invasive pathogens and commensal bacteria possess many of the same PAMPs. Added layers of immune recognition must therefore be engaged to distinguish beneficial commensals from harmful pathogens. The host’s ability to gauge the level of threat through multiple layers of sensing allows the immune system to act as a tuneable dial, rather than an on/off switch, that can fine-tune the response according to the threat. This is important because inflammatory responses are not only harmful to the pathogen, but can also be associated with tissue pathology and compromised organ function. Being able to tune the response to appropriate threat levels enables the immune system to limit collateral damage to host tissues and respond to bona fide pathogens.
Fig. 1 |. Host cells possess multiple mechanisms of pathogen detection and immune defence.
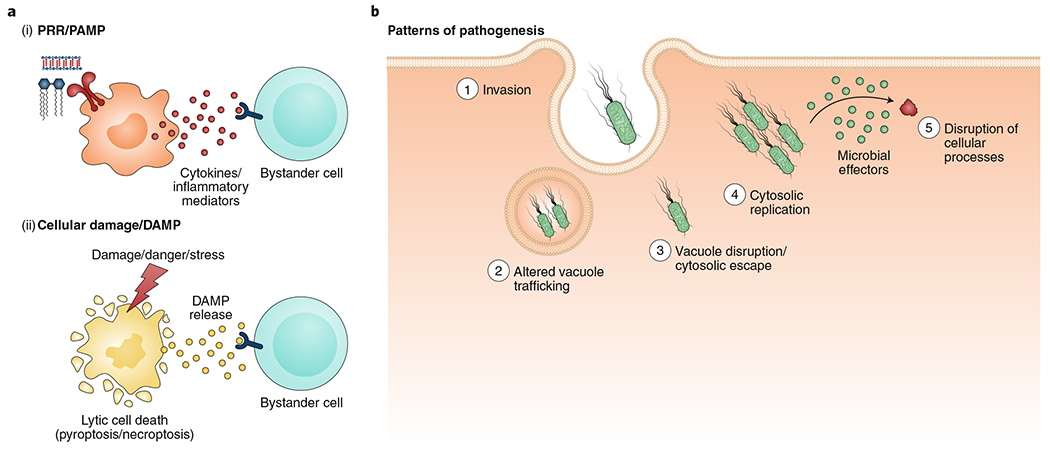
a, PRRs represent an evolutionarily conserved mechanism to directly detect microbial products (termed PAMPs) that serve as general structural features of specific categories or classes of microorganisms (i). PAMP sensing by host PRRs triggers release of inflammatory mediators from the host cell. These mediators can, in turn, activate other cells to amplify the immune response. Certain intracellular molecules associated with damaged tissues or cells that are released as a consequence of microbial infection or other kinds of acute stress stimuli also contribute to innate immune activation (ii). These molecules, termed DAMPs, are also sensed by receptors on neighbouring cells and contribute to immune activation. b, ‘Patterns of pathogenesis’ provides an additional framework for understanding immune sensing of microbial pathogens, which manipulate cellular physiology to colonize the host but provide specific signals that enable the host to detect the pathogen. Such patterns include: (1) invasion of pathogens into the host cell, (2) altered vacuole trafficking, (3) vacuole disruption or cytosolic escape of the pathogen, (4) cytosolic replication of the pathogen and (5) disruption of host cellular processes by microbial effectors.
One mechanism of pathogen sensing is to detect host molecules that are present or altered due to pathogen presence. This includes detection of damage-associated molecular patterns (DAMPs) that are normally located intracellularly but are released into the extracellular milieu as a result of microbial infection8,9. DAMPs include the nuclear protein high-mobility growth Box 1 (HMGB1), ATP and surface exposure of the endoplasmic reticulum (ER) protein calreticulin10,11 (Fig. 1a). DAMPs can also be released following physical trauma or other forms of tissue damage, independent of overt infection12. Thus, neither DAMPs nor PAMPs alone signify the presence of a pathogen. Rather, simultaneous detection of altered cellular processes and engagement of a toll-like receptor (TLR) or other PRRs provide the critical combination of signals that could only occur in the presence of an infectious microorganism.
Box 1 |. Effector-triggered immunity in plants.
Nucleotide-binding domain, LRR-containing proteins are present in both plants and animals. NLRs play an important role in innate immunity and have been extensively described in connection with ETI18,23,24,173. Several NLR proteins serve as receptors that detect pathogenic activity and trigger a downstream immune response. In the plant field, two main strategies for host defence have emerged based on studies on NLR proteins: the ‘gene-for-gene’ model and the ‘guard’ model.
‘Gene-for-gene’ model.
This model proposes that for every ‘avirulence’ (Avr) pathogen gene, there exists a ‘resistance’ (R) gene that confers protection to the host174. In this model, the R protein directly interacts with the pathogen effector and triggers an immune response. One notable example of plants that use this strategy of recognition is flax plants of the Linum genus. The rust fungus Melampsora lini infects plants such as flax, linseed and linola, causing rust disease175. Flax plants have several different R proteins that confer protection against the fungal Avr genes. Resistance proteins at the L locus of flax plants directly interact with AvrL567 genes from the rust fungus, thereby triggering a hypersensitive response, resulting in cell death176. This method of detection relies on direct recognition of the pathogen effector by the host receptor. However, in many instances, the host protein can detect the activity of many effectors, suggesting that there is an indirect mode of recognition, as described below.
‘Guard’ model.
This model proposes that resistance proteins survey host cellular processes, thereby acting as ‘guards’ of host homeostasis23,177. When pathogen effectors disturb these pathways, host guard proteins detect their activity and initiate an ETI response. Several examples of ETI that support this model have been described in plants. For example, the Arabidopsis surface receptors disease resistance protein RPM1 and disease resistance protein RPS2 both serve as guards of a third protein, RPM1-interacting protein 4 (RIN4). Modifications of RIN4 by any of the Pseudomonas syringae effectors AvrB, AvrRpm1 and AvrRpT2 are detected by RPM1 or RPS2, leading to downstream immune signalling178–180. Another example of the guard model involves the decoy strategy, whereby a host receptor that resembles the pathogen effector target is itself modified and triggers an immune response. The Arabidopsis serine/threonine protein kinase PBS1 inhibits the plasma membrane NLR RPS5 by interacting with its N-terminal domain. When the P. syringae effector AvrPphB cleaves PBS1, RPS5 is derepressed and becomes activated, leading to downstream cell death181. This example is analogous to the NLRP1B activation strategy seen in murine macrophages in response to anthrax lethal toxin30,34.
Effector-triggered immunity models in plants.
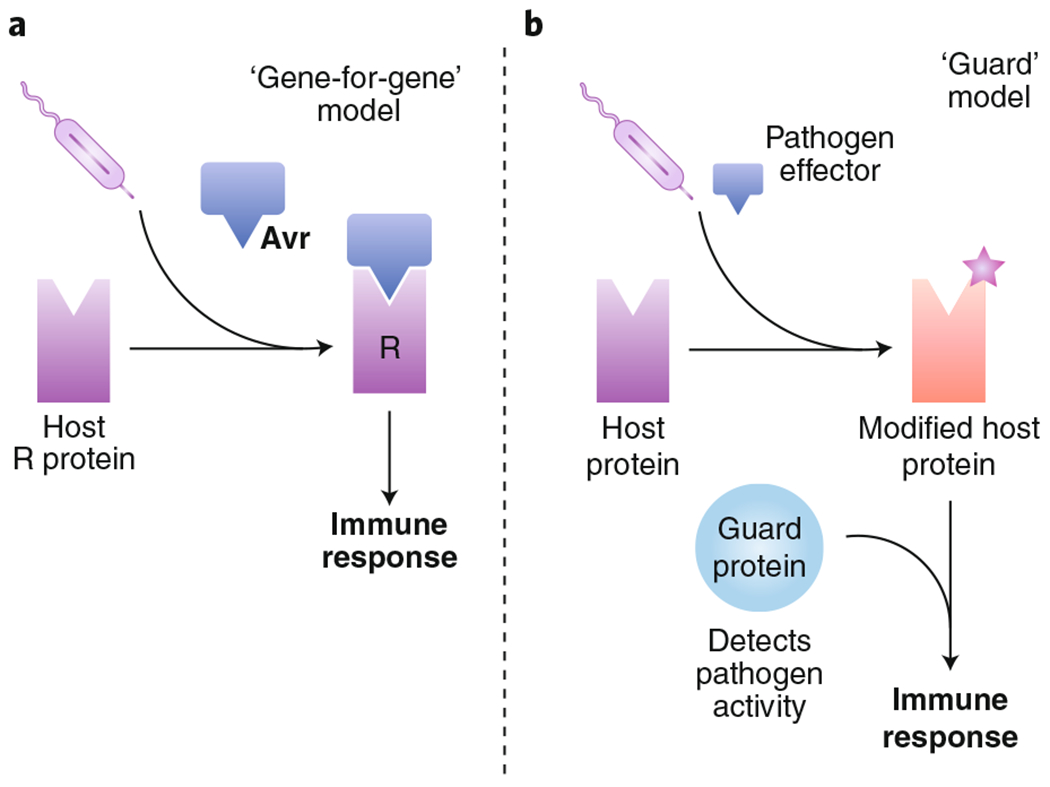
Several mechanisms of effector-triggered immune responses have been proposed in plants, including two well-studied models. a, In the ‘gene-for-gene’ model, a pathogen avirulence (Avr) protein directly interacts with a host resistance (R) protein. This interaction activates a subsequent immune response from the host. b, In a second model, the guard model proposes that host proteins ‘guard’ cellular processes, and the disruption of these processes by virulence factors can be sensed by the host. For example, a pathogen effector that modifies a host protein can trigger recognition of this modification by the host cell, which then initiates an immune response. Since the activity of the effector, rather than the effector itself, is detected, this model of ETI does not rely on direct effector recognition.
Additional mechanisms of pathogen sensing detect PAMPs or pathogen-specific activities within sites that normally exclude microorganisms. Cytosolic invasion by microorganisms, or cytosolic delivery of their products via pathogen-specific secretion systems, constitutes a ‘violation of cytosolic sanctity’13. Such activities, which include disruption of vacuolar trafficking, pathogen replication within the cytosol, disruption of the actin cytoskeleton and cell-intrinsic immune signalling, are ‘patterns of pathogenesis’ (Fig. 1b) that can be detected by host factors14. Pathogen detection may also occur via sensing of homeostasis-altering molecular processes (HAMPs), including manipulation of the actin cytoskeleton or inappropriate reactive oxygen species (ROS) production, either in the context of microbial infection or sterile inflammation15. Appropriate clearance of pathogens therefore requires not only sensing of microbial structures but also sensing pathogen-specific activity, allowing the host to assess the threat level and generate appropriate responses16,17 (Fig. 2).
Fig. 2 |. Microbial threat checkpoints gauge the level of threat posed by a pathogen and fine-tune the host immune response.
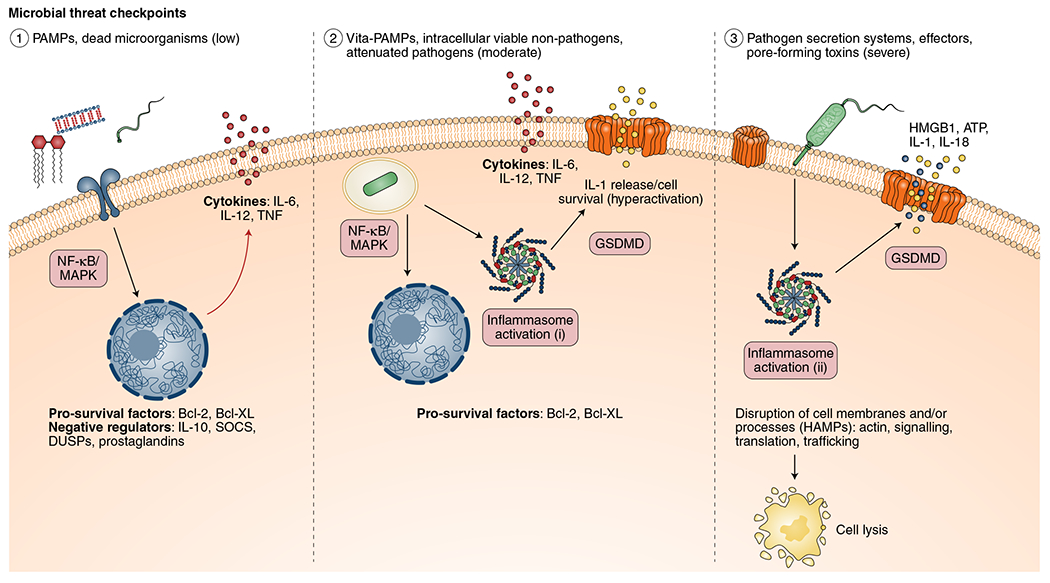
Host immune responses are tuned according to the microbial threat level. (1) Low-level threats, such as PAMPs and dead microorganisms, lead to upregulation of cytokines and pro-survival factors. Negative regulators—such as IL-10, suppressor of cytokine signalling (SOCS) proteins and dual specificity phosphatases (DUSPs)—control and prevent excessive responses under such conditions. (2) Intracellular, viable non-pathogens and vita-PAMPs (for example, bacterial mRNA) pose a moderate level threat to the host and therefore lead to limited inflammasome activation (i) and release of proinflammatory cytokines and IL-1 signalling with limited levels of cell death or, in the absence of cell death, hyperactivation. The release of IL-1 cytokines is mediated by GSDMD, which forms pores in the cell membrane. (3) Pathogens that possess secretion systems and toxins that disrupt barrier tissues and membranes, or perturb host cellular processes, lead to robust inflammasome activation (ii), resulting in cell lysis and release of intracellular DAMPs (for example, HMGB1, calreticulin and ATP, together with IL-1). Notably, both pathogenic and sterile events that alter cellular homeostasis can trigger these immune responses. An alternative framework for thinking about these types of responses is as factors that indicate disruption of cellular homeostasis or HAMPs. These checkpoints allow the host to modulate the immune response based on the level of threat posed by a microorganism or other cellular stresses.
Here, we focus on effector-triggered immunity (ETI)18,19, which senses activities of pathogen virulence factors (effectors) that are secreted during infection by bona fide microbial pathogens20. As these effectors specifically modulate host cellular processes to promote infection and bacterial replication, they can be viewed as a sine qua non of pathogenicity. The term ‘ETI’ refers to sensing of pathogen-specific virulence activities through direct protein–protein interactions between a host target protein and a bacterial effector, or through sensing of pathogen-mediated disruption of cellular activities21,22. ETI was originally hypothesized to function in plant defence via host resistance proteins that would ‘guard’ cellular processes and detect their disruption by pathogen effectors18,23,24. Notably, nucleotide-binding domain, leucine-rich repeat (NLR) proteins are evolutionarily conserved resistance proteins that detect such disturbances18,19,23 (Box 1).
ETI plays an important role in the evolutionary race between host and pathogen, as mechanisms that sense obligate features of pathogenicity, including activities that disrupt host cellular processes, are essential for metazoan survival against host-adapted pathogens. Pathogens, in their turn, are selected by these host mechanisms to evolve countermeasures to evade host detection. In this Review, we seek to integrate the previous concepts of pattern recognition and ETI with recently described mechanisms of pathogen sensing by detection of virulence activities and disruption of cellular homeostasis. We also discuss strategies used by pathogens to evade ETI. A common feature of mammalian ETI responses against pathogens, as distinct from PRR responses to commensal bacteria, is activation of inflammatory cell death, which both eliminates replicative niches and releases DAMPs. Here, we focus on common cellular processes disrupted by pathogens and the host mechanisms that sense this disruption.
Activation of NLRP1B and NLRP3 inflammasomes
Bacterial pathogens produce diverse effectors that have detrimental effects on the host. The host has evolved mechanisms to specifically detect enzymatic activities of these bacterial products, as they are clear signatures of pathogen presence when they occur in the context of TLR signalling. As in plants, nucleotide-binding leucine-rich repeat (NLR) family proteins in the mammalian cell cytosol also serve as sentinels of pathogenic activity. Certain mammalian NLRs form inflammasome complexes that mediate the release of interleukin-1 (IL-1) family cytokines and a type of cell death called pyroptosis25. NLRs serve as guardians of ‘cytosolic sanctity’, as they detect pathogenic insults in the otherwise sterile environment of the host cell cytosol13, and inflammatory cell death is a common outcome of activating ETI responses through the NLRs. These responses eliminate infected or damaged cells and release inflammatory mediators that amplify antimicrobial responses in the tissues, analogous to hypersensitive responses triggered by plants in response to the virulence factors of phytopathogens18,26.
NLRP1B and NLRP3 mediate ETI in response to a number of bacterial toxins. Although their activation mechanisms are different, both NLRs oligomerize to form inflammasomes that recruit and activate the cysteine protease caspase-1 (ref. 27). Caspase-1 cleaves specific substrates, including pro-IL-1 family cytokines and the pore-forming protein gasdermin D (GSDMD), allowing for the subsequent release of IL-1 cytokines and pyroptosis27 (Fig. 3). Other inflammasomes, including the pyrin inflammasome, also mount effector-triggered responses to pathogenic activity and are discussed in separate sections of this Review.
Fig. 3 |. Effector-triggered immunity engages inflammasomes, but can also be targeted by other pathogen virulence factors.
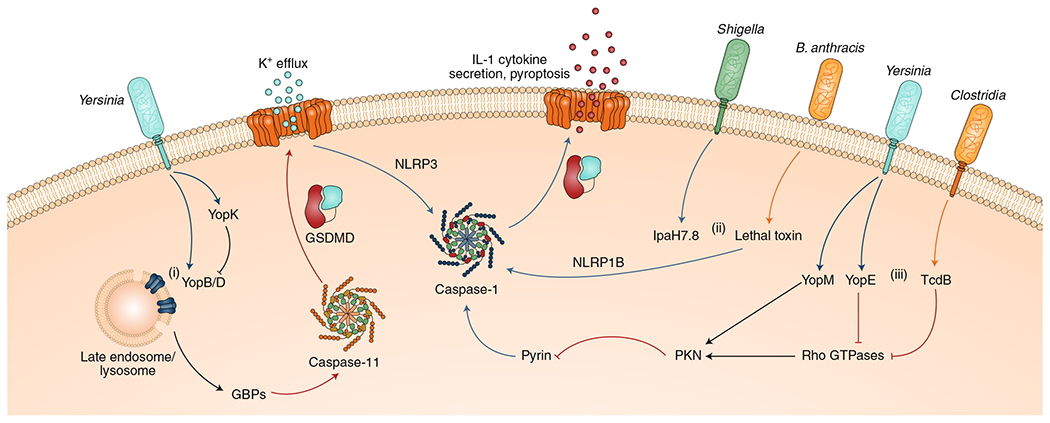
(i) Excessive injection of the Yersinia translocon proteins YopB and YopD damages intracellular endosomal membranes, leading to recruitment of GBPs and downstream activation of the caspase-11 inflammasome. Caspase-11-induced GSDMD pores also trigger K+ efflux, thereby secondarily activating the NLRP3 inflammasome. Yersinia suppresses this defence mechanism by preventing hypertranslocation of YopB and YopD via another effector, YopK. (ii) The NLRP1B inflammasome is activated by bacterial effectors such as B. anthracis LeTx or Shigella IpaH7.8, which cleave or degrade its N terminus, respectively. (iii) Effectors, such as YopE and TcdB, modulate the actin cytoskeleton by suppressing Rho GTPases and, consequently, trigger the pyrin inflammasome by inactivating the PKNs, which are sensitive to GTPase activity. Notably, Yersinia YopM overrides this sensing pathway by directly activating PKNs, thereby maintaining pyrin suppression.
Detection of proteolytic activity by NLRP1B
Bacillus anthracis produces a pore-forming toxin called lethal toxin (LeTx). LeTx is a bipartite toxin composed of a protective antigen, which binds to cellular receptors and forms an oligomeric pore within endosomal membranes, and a lethal factor (LF) that is translocated through the pore28. Once inside the host cell, LF cleaves and inactivates mitogen-activated protein kinase (MAPK) kinases, thereby disrupting innate immune signalling pathways and triggering apoptosis28. Additionally, LeTx induces inflammatory cell death in macrophages from certain strains of inbred mice, while other mouse strains are resistant to this effect, suggesting a host-mediated mechanism of cell death29 which was mapped to polymorphisms in the Nlrp1b locus30.
NLRP1B was the first NLR described to assemble an inflammasome complex31. NLRP1B is activated in response to cleavage by B. anthracis LeTx, indicating that it acts as a guard to sense this pathogen-specific activity. However, precisely how cleavage by LF enabled NLRP1B activation was enigmatic. NLRP1B contains a function-to-find (FIIND) domain that undergoes autoproteolysis following initial biosynthesis32–34. This autoproteolysis occurs independently of any stimulus, resulting in a small polypeptide that inhibits NLRP1B activation by remaining non-covalently associated with the remaining C-terminal portion of the NLRP1B protein35,36. LF-dependent proteolytic cleavage of this auto-inhibitory fragment leads to its recognition by cellular ubiquitin ligases, followed by its degradation via the N-end rule pathway. This releases the C-terminal portion of NLRP1B, which contains the caspase activation and recruitment domain (CARD) domain that recruits and activates caspase-1 (refs. 35,36) (Fig. 3). Mouse strains containing NLRP1B that is sensitive to LeTx are thus able to induce an inflammasome response to the proteolytic activity of LF.
Interestingly, pathogen effectors that are not directly proteolytic can also activate NLRP1B. The Shigella type III-secreted effector IpaH7.8 is a ubiquitin ligase whose activity triggers assembly of the NLRP1B inflammasome36 (Fig. 3). Given that LF activates the NLRP1B inflammasome by inducing ubiquitin-dependent degradation of an auto-inhibitory fragment, NLRP1B may have evolved to sense at least two activities common to many pathogen virulence factors—proteolysis or ubiquitin ligase activity—either of which can induce degradation of the NLRP1B auto-inhibitory fragment. The protozoan parasite Toxoplasma gondii also activates the NLRP1B inflammasome37–39, but the factor responsible is unknown. NLRP1B may, therefore, sense a yet-to-be discovered T. gondii ubiquitin ligase or protease.
Mice have two additional NLRP1 homologues, a functional NLRP1A and a putative pseudogene NLRP1C, whereas humans have a single NLRP1 (ref. 31). The signals that engage murine NLRP1A and human NLRP1 are unknown. While human NLRP1 is not cleaved or activated by LeTx40, in vitro proteolytic cleavage of the human NLRP1 N terminus induces inflammasome activation, suggesting that human NLRP1 is activated via a similar proteolytic mechanism41. The inability of human NLRP1 to be cleaved by LeTx could, in part, account for the susceptibility of humans to anthrax. As with other NLRs, gain-of-function mutations in human NLRP1 are associated with a variety of autoimmune diseases, suggesting that such mutations may abrogate association of the auto-inhibitory FIIND fragment with the remainder of the protein or enable spontaneous NLRP1 activation in the presence of the FIIND fragment42,43.
Detection of pore-forming toxins by NLRP3
NLRP3 is activated in response to a diverse array of stimuli, many of which induce changes in cytosolic ion concentrations. A common theme for NLRP3 activation is a drop in intracellular potassium concentration below a critical threshold44 and/or mitochondrial dysfunction associated with excessive mitochondrial ROS (mitoROS) production45. Whether a specific link exists between mitoROS and ion flux is unclear, although NLRP3-activating stimuli that only induce mitoROS or potassium efflux have been reported44,46.
Pore formation at the plasma membrane induces potassium efflux, which is a general mechanism of NLRP3 activation44. Bacterial pore-forming toxins (PFTs) that permeabilize the plasma membrane, such as aerolysin toxin from Aeromonas spp.47 and α-hemolysin from Staphylococcus aureus48, cause potassium efflux that triggers the NLRP3 inflammasome (Fig. 3). NLRP3 may thus serve as a guard of cytosolic potassium levels, whereby a drop in potassium levels below a key threshold serves as a HAMP for NLRP3 activation, which mediates ETI by engaging caspase-1 and pyroptotic cell death27.
Detection of intracellular membrane disruption
In addition to PFTs, bacteria employ other virulence factors that have the capacity to activate the NLRP3 inflammasome through disruption of intracellular membranes. This involves guanylate binding proteins (GBPs), although the precise mechanisms remain unclear. GBPs are IFN-inducible GTPases involved in cell-intrinsic immunity against intracellular pathogens, including bacteria, viruses and protozoa49–53. GBPs are recruited to pathogen-containing vacuoles, where they are thought to deliver a variety of cargo important for host defence50. Precisely how GBPs target pathogen-containing vacuoles is unclear, but GBPs are recruited to vacuolar membranes containing secretion system components54,55 and are thought to mediate inflammasome activation as a result of damage to these membranes.
Gram-negative bacterial secretion system effectors include factors that modulate cellular signalling pathways to evade antibacterial immune defences56. However, in doing so, they also activate cellular cytosolic proteins that initiate ETI responses to counteract this activity. Yersinia species possess a type III secretion system that is formed by the Yersinia outer proteins B and D57. While these proteins form the translocation complex that mediates the secretion of other effector proteins, YopB and D themselves can also be translocated into the host cell57–62. Following injection into host cells, YopD colocalizes with markers of damaged membranes and GBP2 (ref. 55). Disruption of intracellular membranes by YopB and D leads to activation of the NLRP3 inflammasome, as well as the non-canonical caspase-11 inflammasome that detects bacterial lipopolysaccharide (LPS) in the cytosol (Fig. 3)55,62,63. GBPs are necessary for this inflammasome response55, and they are proposed to activate caspase-11 via a number of mechanisms involving disrupting pathogen-containing vacuoles or directly lysing cytosolic bacteria and releasing LPS into the cytosol49,64–68. GBPs may, therefore, act both as sensors of intracellular membrane integrity and direct effectors of membrane damage in response to pathogen-induced alteration of vacuolar membranes. Interestingly, Yersinia has evolved to evade this ETI response through the injected effector YopK, which prevents host sensing of pathogen activity by limiting translocation of YopB and D62,63,69.
Viral proteins that disrupt intracellular membranes also activate NLRP3, again linking NLRP3 activation to membrane disruption. The influenza virus M2 protein forms a proton-specific ion channel that pumps protons out of the Golgi lumen, thereby neutralizing the pH of the trans-Golgi network (TGN)70. This activity is sufficient to activate the NLRP3 inflammasome in macrophages and dendritic cells71, suggesting that NLRP3 may serve as a general guard of ion homeostasis within the cell. Intriguingly, a number of other NLRP3 inflammasome activators induce dispersal of TGN, resulting in recruitment of NLRP3 to dispersed TGN membranes via ionic interactions between a poly-basic region of NLRP3 and negatively-charged phosphatidylinositol 4-phosphotate on TGN membranes72. Interestingly, NLRP3 has also been found to bind cardiolipin on the mitochondrial membrane via interactions between cardiolipin and the leucine-rich repeat (LRR) domain46. Whether this is the mechanism by which influenza M2 protein triggers NLRP3 activation, and whether GBPs might also be involved in this response to influenza virus, is unknown.
Detection of pathogen interference with immune signalling
The seemingly paradoxical ability to generate a protective immune response against pathogens that suppress innate immune defence is critical to surviving such attacks. Pathogen-driven immune subversion thus also triggers ETI, but how such responses might be engaged and how they function to protect the host has only recently become appreciated. Many of these responses involve the host protein receptor-interacting protein kinase 1 (RIPK1), which is a central component of cell fate decisions involving pro-survival, pro-death and inflammatory gene expression pathways during infection73 (Fig. 4). RIPK1 ubiquitination following activation of TLR or tumour necrosis factor (TNF) receptors (TNFRs) promotes assembly of scaffold complexes that activate nuclear factor-κB (NF-κB) and MAPK signalling as well as cell survival programs73–77. Conversely, activation of TLR or TNFR signalling while NF-κB signalling is blocked triggers RIPK1 kinase activity, resulting in caspase-8-dependent apoptosis78–80. Alternatively, if caspase-8 is inhibited or deleted, RIPK1 kinase activity activates RIPK3-dependent necroptosis81–83. Thus, multiple checkpoints exist to release the brake on cell death if intracellular signalling is disrupted by pathogens. Interestingly, in contrast to the classic view of apoptosis as an immunosuppressive form of cell death, caspase-8 can mediate inflammatory cell death in the setting of NF-κB blockade by cleaving the pore-forming protein GSDMD independently of caspases-1 or −11 (refs. 84,85). Whether caspase-8-mediated GSDMD cleavage occurs only in the context of NF-κB inhibition is unclear; however, this may be a mechanism to couple apoptotic caspases with inflammation during pathogen inhibition of cell signalling.
Fig. 4 |. Pathogen manipulation of core cellular processes and signalling pathways induces host immune responses.
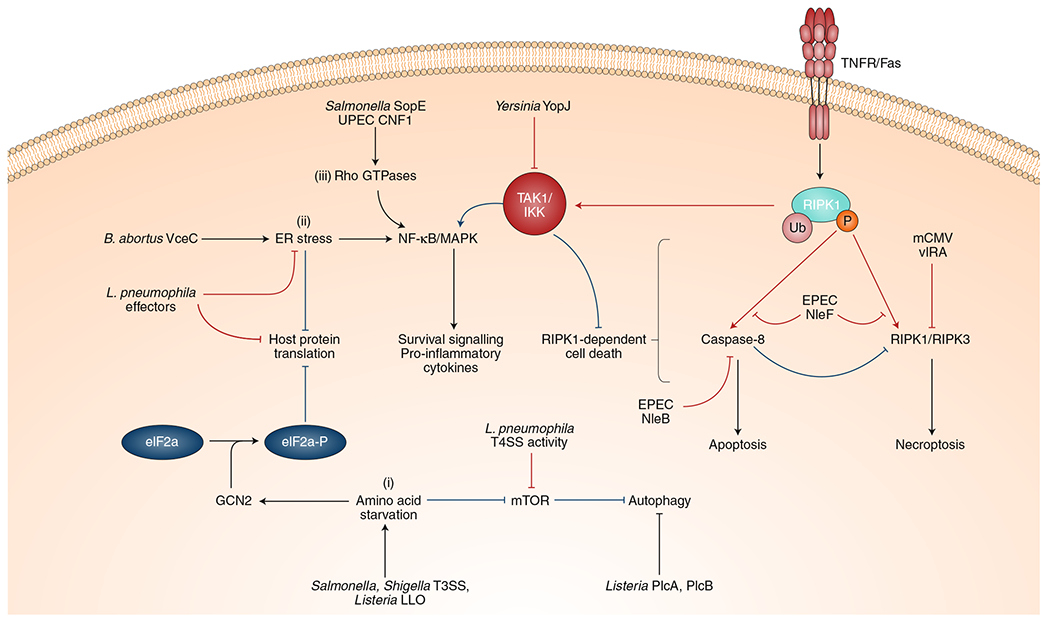
In addition to inflammasomes, the host has a myriad of other cellular defence pathways activated in response to virulent activity. Many of these pathways trigger signalling cascades that upregulate a subset of innate immune genes that promote host cell defence. Furthermore, other mechanisms, such as autophagy and cell death, serve to eliminate invading pathogens and infected cells. The following are examples of effector-triggered responses in these categories and pathogenic adaptations to evade these host responses. (i) Pathogen-induced amino acid starvation suppresses mTOR, activates autophagy and activates GCN2, leading to a block in host protein synthesis. Inhibiting protein translation may be useful against pathogens that co-opt host cellular processes for their own protein production, such as viral pathogens. Some pathogen effectors, such as Listeria PlcA and PlcB, can block mTOR signalling and autophagy, respectively, in order to evade detection and destruction. (ii) Similarly, pathogens that co-opt the host ER, such as Legionella and Brucella, induce ER stress and consequently trigger a block in protein translation. This activity triggers NF-κB and MAPK signalling and induces expression of subset of proinflammatory cytokines. To counteract this immune response, Legionella also possesses effectors that suppress ER stress and host protein translation, thereby partially masking itself from effector-triggered immunity. (iii) Activation of Rho GTPases by SopE and CNF1 similarly activates the NF-κB pathway to induce inflammatory cytokines and promote cell survival. Several pathogens have evolved to suppress immune signalling pathways in order to limit inflammation. However, in doing so, they activate an ETI pathway mediated by RIPK1 kinase activity, which induces host cell death. Effectors, such as YopJ, that suppress NF-κB and MAPK pathways induce RIPK1-dependent apoptosis. Pathogens that inhibit apoptosis may induce a back-up cell death mechanism, namely RIPK1-dependent necroptosis. Despite these fail-safes, some pathogens have evolved to suppress both of these responses: EPEC inhibits both apoptosis and necroptosis with the effectors NleB, NleF and EspL; the virus murine cytomegalovirus (mCMV) uses its effector viral inhibitor of RIP activation (vIRA) to suppress necroptosis.
Several bacterial virulence factors—including the YopJ/P proteins in Yersinia, the NleE/OspZ proteins from enteropathogenic Escherichia coli and Shigella, and the Shigella effector IpaH9.8—block innate signalling by interfering with key upstream kinases TGF-β-activated kinase 1 (TAK1) and inhibitor of κB kinase (IKK)78,86,87. YopJ belongs to a family of evolutionarily conserved virulence factors that interfere with activation of MKK family proteins by acetylating a conserved serine residue in the MKK activation domain88–90. In macrophages, this blockade triggers caspase-8-dependent apoptosis91,92 (Fig. 4), while in dendritic cells, it disrupts TIR domain-containing adaptor inducing interferon-β (TRIF)-dependent gene expression93. RIPK1 kinase activity is required for apoptosis in response to YopJ activity, and RIPK1-mediated apoptosis protects against Yersinia infection in vivo78,79,91. Thus, despite YopJ-mediated inhibition of innate immune signalling, RIPK1- and caspase-8-dependent apoptosis triggered by this activity mediates a host-protective immune response. RIPK1 kinase-induced apoptosis is thus intimately tied to the integrity of signalling pathways that RIPK1 itself regulates.
Mechanistically, the switch between RIPK1 pro-death and pro-survival functions is regulated by post-translational modifications that control RIPK1 activity, including phospho-sites on RIPK1 that are targets of the same kinases activated by TLRs or TNFR. Specifically, IKK phosphorylates RIPK1 on Ser25 (refs. 94,95), and the p38 MAPK-activated kinase, MK2, also phosphorylates RIPK1 at Ser321 and Ser336 (refs. 96,97). Phosphorylation at these sites prevents RIPK1 from autophosphorylating on Ser166, thus preventing RIPKl-mediated apoptosis and necroptosis95–97. The lack of phosphorylation at these negative regulatory sites when IKK and MKK signalling are blocked by virulence factors, such as YopJ, thus appears to release the brake on RIPK1-dependent death. RIPK1 therefore functions both as a central regulator and guardian of innate immune signalling.
Evasion of RIPK1-mediated cell death
Given the importance of maintaining cellular integrity for pathogen replication, it is not surprising that pathogens block both apoptosis and necroptosis. For example, human cytomegalovirus (hCMV) protein viral inhibitor of caspase activation (vICA) inhibits apoptosis by preventing caspase-8 activation98 (Table 1) and also suppresses necroptosis via the immediate-early 1 (IE1) gene99. The murine CMV effector vIRA performs an analogous function and suppresses RIPK3 activity during the caspase-8 blockade to inhibit necroptosis and promote viral replication100. Similarly, enteropathogenic E. coli (EPEC) possess multiple effectors that block both apoptosis and necroptosis, including NleB, NleF and EspL101,102 (Fig. 4 and Table 1). The interplay of microbial evasion and host sensing strategies is such that layers of selective pressure on both the host and pathogen have produced a system in which necroptosis is initiated in response to pathogen inhibition of caspase-8 and the pathogen, in turn, blocks necroptosis to maintain its replicative niche and limit release of intracellular DAMPs. Whether an additional layer of host recognition and pathogen evasion exists beyond programmed necrosis remains to be determined.
Table 1 |.
Examples of ETI and pathogen subversion strategies
| Pathogenic activity | Host guardee | Host guard | Host ETI response | Pathogen subversion mechanism | References |
|---|---|---|---|---|---|
| Inactivation of RhoA by C. difficile TcdB and Yersinia YopE | RhoA | Pyrin | Pyrin-dependent pyroptosis and IL-1 cytokine secretion | Yersinia YopM inhibits pyrin activation by activating PKNs | 104,108 |
| Modulation of Rho GTPases by UPEC CNF1, Salmonella SopE and SopB, and Shigella IpgB2 and OspB | Rho GTPase functions | Drosophila IMD, Rho GTPases (guard themselves) and NOD1/NOD2 | Drosophila IMD signalling and NF-κB signalling-mediated cytokine secretion | 114–121 | |
| Activation of NLRP1B by B. anthracis LeTx and Shigella flexneri IpaH7.8 | NLRP1B (decoy) | NLRP1B (guards itself) | NLRP1B-dependent pyroptosis and IL-1 cytokine secretion | 30,35,162 | |
| Activation of NLRP3 inflammasome by Aeromonas aerolysin, S. aureus α-hemolysin, and influenza A virus M2 and PB1-F2 proteins | Cytoplasmic homeostasis (intracellular K+ levels and pH) | NLRP3 | NLRP3-dependent pyroptosis and IL-1 cytokine secretion | Measles V protein suppresses NLRP3-mediated IL-1β secretion and influenza A virus NS1 protein suppresses NLRP3 and NF-κB signalling | 71,163–166 |
| Activation of caspase-11 and NLRP3 inflammasomes by Yersinia YopB and YopD | Integrity of intracellular membranes | GBPs | Caspase-11 and NLRP3-dependent pyroptosis and IL-1 cytokine secretion | Yersinia YopK prevents hypertranslocation of YopB and YopD, thereby suppressing inflammasome activation | 55,62,63,69,167 |
| Amino acid starvation induced by Shigella, Salmonella T3SS and Listeria LLO | Amino acid levels in the host cell | mTOR GCN2 | Autophagy Inhibition of host protein translation | Listeria PlcA and PlcB suppress autophagic flux, and Leishmania GP63 and vesicular stomatitis virus M protein suppress mTOR | 125–127,168,169 |
| ER stress induced by B. abortus VceC, L. pneumophila and P. aeruginosa exotoxin A | Host protein translation | IRE1α, IκB, NOD1/2 and C. elegans ZIP2 | NOD1/2-induced cytokine secretion via NF-κB signalling, NF-κB/MAPK-mediated cytokine secretion and C. elegans infection response gene 1 (irg1)-mediated defence | Legionella Lgt1 and Lgt2 block IRE1-mediated Xbp1 splicing | 138,146–148,155,170 |
| Activation of apoptosis by Yersinia YopJ, EPEC NleE and Shigella OspZ | NF-κB/MAPK pathway | RIPK1 | RIPK1/caspase-8-dependent apoptosis | EPEC NleB and NleF block caspase-8-mediated apoptosis | 78,86,101 |
| Activation of necroptosis by CMV vICA and EPEC NleB | Caspase-8 | RIPK3 | RIPK3-dependent necroptosis | EPEC NleF blocks necroptosis | 98,101 |
| Activation of apoptosis by Aspergillus fumigatus gliotoxin | Cellular attachment to extracellular matrix | RhoA | Activation of anoikis (a form of apoptosis induced by cell detachment) | 171 | |
| Activation of Toll pathway by Beauveria bassiana PR1 | Drosophila persephone protease (decoy) | Drosophila persephone protease (guards itself) | Upregulation of Drosophila Toll pathway-dependent gene expression | 172 |
Detection of Rho GTPase modification by pathogens
Rho GTPases—such as Rac1, RhoA and Cdc42—serve numerous core cellular functions, ranging from actin cytoskeletal dynamics and epithelial barrier integrity to production of ROS and antimicrobial peptides, and are thus targeted by numerous pathogens103. Similar to RIPK1, Rho GTPases are centrally positioned to regulate cellular processes and are therefore poised to be both targets and sensors of pathogen virulence activity.
Detection of RhoA inactivation by the pyrin inflammasome
Bacterial inactivation of RhoA can trigger inflammasome responses through the host protein pyrin104. Pyrin forms an inflammasome that recruits caspase-1, leading to release of IL-1 cytokines and pyroptosis (Fig. 3). Pyrin activation is normally restrained by RhoA through the activity of protein kinase C-related kinases (PKNs, also known as PRKs). When RhoA is active, one of its targets, the PKNs, phosphorylate pyrin, creating binding sites for suppressive 14-3-3 regulatory proteins105,106. Thus, under basal conditions, the pyrin inflammasome is maintained in an inactive state. Similar to RIPK1 apoptotic activity being kept in check by IKK-dependent phosphorylation, pathogen inactivation of RhoA triggers the pyrin inflammasome by preventing phosphorylation of pyrin. Importantly, a number of pathogens are sensed via the linking of pyrin to RhoA activity.
Burkholderia cenocepacia inactivates RhoA via direct deamidation, thereby disrupting macrophage effector functions104,107. However, the resulting derepression of pyrin leads to an inflammatory response that controls B. cenocepacia infection104. Similarly, other RhoA-inactivating toxins, such as Clostridium botulinum C3, Clostridium difficile TcdB, Vibrio parahaemolyticus VopS and Histophilus somni IbpA, also activate the pyrin inflammasome104. Only catalytically active bacterial effectors trigger this inflammasome response, indicating that pyrin senses the activity of effectors and their disruption of RhoA function104.
In an elegant interplay of host–pathogen co-evolution strategies, pathogenic Yersinia have evolved to evade the pyrin inflammasome in response to manipulation of RhoA (Fig. 3 and Table 1). Yersinia effectors YopE and YopT inactivate the Rho GTPases RhoA, Rac1 and Cdc42 to prevent phagocytosis of the bacteria56, which would trigger pyrin by disrupting its interactions with 14-3-3 proteins108–110. However, the Yersinia effector, YopM, specifically activates PKN kinases to phosphorylate pyrin, thereby maintaining pyrin’s association with 14-3-3 proteins and masking YopE/T interference with Rho GTPases108.
Gain-of-function mutations in Mediterranean Fever (MEFV), the gene encoding human pyrin, are associated with Familial Mediterranean fever, an illness characterized by recurrent inflammation in the absence of infection111. The high prevalence of mutant MEFV alleles in Mediterranean populations may be the result of plague epidemics within Europe and the possibility that MEFV carriers have increased protection from Y. pestis infection108,112,113. YopM is among the most polymorphic of Yersinia virulence factors, suggesting that YopM itself may be a target of host selective pressure. There may exist other pathogen virulence factors that similarly overcome host immune defences by targeting other guard proteins.
PRR signalling in response to Rho GTPase disruption
In addition to inflammasome activation, pathogen modulation of Rho GTPases can induce conserved transcriptional responses via NF-κB. The toxin cytotoxic necrotizing factor 1 (CNF1) from uropathogenic E. coli (UPEC) constitutively activates the Drosophila Rho GTPase Rac2 by preventing GTP hydrolysis114. Modified Rac2 interacts with the Drosophila homologue of RIPK1, immune deficiency (IMD), and induces transcription of Drosophila antimicrobial peptides that control bacterial infection114. A constitutively active Rac2 mutant also induces IMD-dependent NF-κB activation and Rac2-deficient flies succumb to UPEC infection114, indicating that one function of Rac2 is to detect the activity of pathogen virulence factors such as CNF1 (ref. 114). Importantly, CNF1-modified human Rac2 interacts with the human homologues of IMD and also induces expression of antimicrobial peptide genes114. These findings imply that detection of bacterial toxins that target Rac2 represents an evolutionarily conserved ETI pathway.
Similarly to CNF1, the Salmonella effectors SopB, SopE and SopE2 activate Rho GTPases, thus triggering innate immune activation115–117. While SopB/E/E2 are necessary for Salmonella intracellular invasion, their injection activates NF-κB and MAPK signalling in epithelial cells, indicating that inappropriate activation of Rho GTPases engages a conserved immune defence circuit. Ectopic expression of SopE induces NF-κB activation via a complex containing Rho GTPases, NOD1, RIPK2 and heat shock protein 90 (HSP90)115. However, inflammatory responses to these effectors can also occur independently of RIPK2 (refs. 116,117). Indeed, SopE/E2/B activity can activate p21-activated kinase (PAK1) together with recruitment of TNF receptor-associated factor 6 (TRAF6) and TAK1, resulting in NF-κB and MAPK activation independently of RIPK2 (ref. 117). Distinct signalling platforms may have the capacity to engage NF-κB in response to pathogen manipulation of Rho GTPase activity, perhaps depending on cell type, route of delivery or expression level of pathogen effectors.
Like Salmonella, Shigella interacts intimately with the actin cytoskeleton during invasion of intestinal epithelial cells, but also intracellularly when it employs actin-based motility. Shigella’s manipulation of the actin cytoskeleton also induces NF-κB signalling via NOD1/2 (refs. 118–120). In unpolarized epithelial cells or macrophages, the phosphatase slingshot 1 (SSH1), which regulates actin depolymerization through cofilin, activates NOD1-dependent NF-κB signalling during Shigella infection, suggesting that disruption of actin filaments triggers the NOD1 pathway118. Consistently, chemical inhibitors of actin polymerization, such as latrunculin B and cytochalasin D, also activate NOD1 (ref. 118). In polarized epithelial cells, cytoskeleton disruption by Shigella effectors IpgB2 and OspB activate NOD1 in a manner dependent on the Rho-associated protein kinase (ROCK)119. Collectively, these and other studies that link modulation of Rho GTPase activity to NOD1/2 signalling120,121 demonstrate that actin-regulating Rho GTPases are central regulators of ETI responses and serve as important guardians of actin cytoskeleton dynamics.
Detection of pathogen-induced stress responses
Adaptation to environmental stresses is essential for cell survival and growth. The integrated stress response (ISR) is activated by a variety of stimuli, including amino acid starvation, oxidative stress and ER stress, and is required for cell survival122. The ISR blocks general mRNA translation while selectively allowing expression of genes that enable specific adaptations that promote cellular recovery122. The mammalian target of rapamycin (mTOR) is also an important monitor of nutrient starvation and intersects with the ISR123. Notably, both the ISR and mTOR pathways are also coupled to inflammation and cell death in response to pathogen manipulation of amino acid pools and ER stress122, highlighting the link between disruption of core cellular physiologic functions to immune responses during infection.
Detection of pathogen-induced amino acid starvation
Metabolic stress in the form of amino acid starvation is sensed by the cell via two signalling cascades: the mammalian target of rapamycin (mTOR) and eIF2α123. While both pathways sense amino acid availability independently, they intersect with and regulate many other cellular responses, including cell division, gene expression, cell survival and cell death123. Similar to other core processes discussed above, these pathways are both targeted by intracellular pathogens and also play a key role in sensing pathogen-specific disruption of the cell.
The mTOR complex 1 (mTORC1) is normally found on the membrane of late endosomes and lysosomes where it regulates gene transcription via eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-P1) and S6 kinase 1 (S6K1), and also negatively regulates autophagy under nutrient-replete conditions123. mTORC1 senses amino acid availability inside lysosomes via the transmembrane protein SLC38A9 and in the cytosol via Sestrins and CASTOR proteins124. In response to a drop in amino acid levels, mTORC1 dissociates from the membrane, which initiates autophagy to break down cellular components in order to replenish nutrients123. Therefore, mTORC1 acts as a guard of intracellular amino acid levels and induces an autophagic response when these levels drop.
Intracellular pathogens compete with the host cell for essential building blocks, including amino acids123. Thus, similarly to T3SSs above, an activity necessary for pathogen survival or virulence simultaneously enables host detection and immune defence. Intriguingly, disruption of mTOR signalling during bacterial infection can co-opt the autophagic machinery to engulf the pathogen itself, leading to degradation of the pathogen via xenophagy123. Damage to phagosomal membranes induced by bacteria that invade the cytosol is thought to induce amino acid starvation and activate xenophagy123. In this manner, both the Listeria pore-forming toxin listeriolysin O (LLO) that breaks down the pathogen-vacuole membrane125 and the T3SSs from Shigella and Salmonella induce host membrane damage that triggers xenophagy126,127. Since the amino acid sensor SLC38A9 is necessary for transporting leucine out of lysosomes to interact with and activate mTORC1 (ref. 128), pathogen-induced membrane disruption may impede the ability of SLC38A9 to activate mTORC1.
Given the antibacterial nature of this pathway, successful intracellular pathogens must possess mechanisms to either subvert or escape downstream consequences of mTOR signalling. An example of such an activity are the Listeria phospholipases, PlcA and PlcB, which stall maturation of autophagy vesicles125. Other pathogens, such as Toxoplasma gondii129 and certain viruses130–132, promote mTOR signalling, likely to maintain this pathway’s function and avoid autophagy.
The eIF2α pathway similarly senses amino acid starvation via the cytosolic kinase general control nonderepressible 2 (GCN2). GCN2 is activated in response to a drop in amino acid concentrations and phosphorylates eIF2α, inducing a block in host translation and the formation of mRNA-containing stress granules123. Like the mTOR pathway, GCN2 is activated during bacterial infection as a result of membrane damage126, and both pathways can be activated simultaneously during bacterial infection125,126. Notably, despite global translational repression induced by inactivation of mTOR and activation of GCN2, expression of key inflammatory genes was selectively upregulated in response to Shigella infection126. Thus, both GCN2 and mTOR act as guards of amino acid levels in the host cell and their activation, in response to pathogen virulence activity, promotes anti-microbial responses.
Sensing disruption of protein synthesis and ER homeostasis
ER perturbance also engages the integrated stress response pathway and can be coupled to ETI. Legionella, Chlamydia trachomatis and Brucella spp. all replicate within an ER-derived compartment133. However, these pathogens can be detected via their interactions with the ER stress and unfolded protein response (UPR) pathways133,134. The UPR engages three different transmembrane receptors, inositol requiring enzyme 1 (IRE1α), double-stranded RNA-activated protein kinase (PKR)-like ER kinase (PERK) and activating transcription factor 6 (ATF6), which sense ER stress and induce downstream responses to alleviate the stress and restore cellular homeostasis134. Like GCN2, PERK phosphorylates eIF2α, leading to attenuated protein synthesis123. However, a prolonged UPR response that fails to be resolved, or an acute response that is extreme in amplitude, triggers autophagy or apoptosis of infected cells134. Activation of UPR in response to bacterial virulence activity serves as an ETI mechanism whereby receptors of the UPR pathway act as guards of ER homeostasis in the context of infection.
The Legionella pneumophila type IV secretion system (T4SS) injects a large panel of effectors to establish an ER-derived vacuole and replicate within macrophages135. Notably, L. pneumophila infection induces UPR in a manner dependent on T4SS activity, suggesting the presence of an ETI response to pathogen disruption of the ER136,137. However, L. pneumophila also encodes at least 11 effectors that inhibit host translation, perhaps to alleviate ER stress138–145. Indeed, several of these effectors, particularly Lgt1 and Lgt2, block the ATF6 and IRE1 branches of the UPR136,137. Nevertheless, in another example of the intimate interactions between host and pathogen, the translational block induced by these Legionella effectors triggers another layer of immune response, as it prevents resynthesis of the NF-κB inhibitor IκB, leading to prolonged NF-κB signalling138. The effector-driven translational block also activates p38 and c-Jun N-terminal kinase (JNK) MAPKs146, and this enhanced NF-κB and MAPK signalling amplifies expression of immune genes, including Il1a and Il1b138,143,146–149. Although infected macrophages produce IL-1α and IL-1β proteins, they are unable to synthesize other key cytokines, including TNF and IL-12, due to the translational block149. Instead, secretion of IL-1α and IL-1β by infected macrophages is critical for production of TNF and IL-12 by uninfected bystander monocytes and other recruited myeloid cells within the lung149, indicating that collaboration between infected and uninfected cells is critical for appropriate host defence.
Intriguingly, TLR stimulation coupled with pharmacological inhibition of host translation induces an overlapping set of immune response genes to Legionella infection138, indicating that this program represents an immune response disrupting host protein synthesis. How the infected host cell maintains its ability to synthesize a subset of proteins despite the translational block is unclear, but superinduction of a subset of mRNAs is involved144,148. Collectively, these findings reveal that although Legionella effectors inhibit host translation to alleviate ER stress, this activity triggers an ETI response that promotes NF-κB and MAPK signalling as well as superinduction of immune genes, which allows infected macrophages to synthesize IL-1α and IL-1β. Subsequent release of these DAMPs elicits inflammatory cytokine production from uninfected bystander cells, thus providing a backup mechanism to ensure inflammatory responses. Such a strategy may be a general mechanism employed by the immune system to overcome immunoevasive pathogens that inhibit host translation.
In C. elegans, sensing pathogen disruption of core cellular processes is the primary pathogen detection mechanism, as direct receptors for PAMPs have yet to be identified. Infection with pathogenic bacteria, or pharmacological disruption of translation, cytoskeletal components, mitochondrial function or proteasome activity, triggers a concerted set of cellular protective responses known as ‘surveillance immunity’150,151. This response includes upregulation of an antimicrobial transcriptional effector program as well as organismal responses, such as avoidance behaviour, reduced ingestion and xenobiotic detoxification152. The Pseudomonas aeruginosa effector exotoxin A (ToxA) blocks host translation by ADP-ribosylating the translation elongation factor-2 (EF-2)153, which paradoxically triggers induction of the transcription factor BZIP domain-containing protein (ZIP-2) and subsequent expression of a suite of infection response genes that mediate anti-bacterial defence154–156. The ToxA-mediated translational block promoted ZIP-2 protein expression through a mechanism involving an upstream open reading frame (uORF) that normally suppresses ZIP-2 translation in the absence of infection155. Interestingly, uORF-regulated translation is an evolutionarily conserved mechanism in yeast and mammals that is also employed during non-infectious cellular stress to enable selective translation of genes important for stress adaptation157–159. Thus, uORF-regulated protein expression may be similarly used in mammalian cells to enable immune defence against pathogens that block host translation.
These studies highlight the role of the ER stress response pathway as a sensor of pathogens that co-opt and replicate in ER-derived compartments. Interestingly, pathogens have evolved to block host protein synthesis as a strategy to prevent the host from mounting an immune response. To counteract this, the host detects this disruption by overcoming the translation block and triggering defence mechanisms.
Concluding remarks and future perspectives
The concept that innate immune systems of eukaryotes can detect the activity of pathogen virulence factors, termed ‘ETI’, was proposed in the early 1980s, with the discovery that plant pathogen virulence (Avr) proteins are sensed by plant innate immune pathways18,19,23,160. The Pattern Recognition Theory, proposed by Janeway in 1989 and confirmed experimentally in the subsequent decade, provided important insight into innate recognition of microbial pathogens in animal systems1,3,161. However, metazoans require additional mechanisms to detect pathogens, since commensal microorganisms engage in intimate interactions with their eukaryotic hosts and possess PAMPs that are sensed by PRRs2. Thus, pathogen manipulation of key cellular processes must be detected by cellular sensors to enable host survival in the face of such a pathogenic insult. The existence of ETI responses as an additional layer of immune defence that functions in concert with PRR-induced responses is necessitated by the presence of microorganisms that have developed mechanisms to evade, subvert or take advantage of PRR-driven immune defence. In some instances, PRR signalling ‘primes’ host responses to engage ETI in response to a ‘second signal’ that involves manipulation of cell physiology. In this sense, ETI functions as a threat assessment system that detects particularly problematic pathogens, because engagement of ETI only occurs if a pathogen has penetrated other cellular defences. Engagement of ETI, therefore, usually leads to some form of immunologic or inflammatory cell death. Given the potential pathological inflammation that can be elicited by ETI, it makes sense that engaging this pathway would generally be limited to situations in which multiple signals denote pathogen presence.
Combinatorial engagement of PRRs and ETI engages responses that are distinct from those triggered by PAMPs alone, and are therefore particularly important for immune defence against pathogens. Interestingly, as might be expected, given the selective pressure put on pathogens by ETI systems, pathogen mechanisms for evasion of ETI have also emerged. Thus, metazoan-adapted pathogens likely have an array of effector mechanisms that limit or blunt the generation of effective immune responses. This is likely to be the case, particularly in the setting of chronic infections—a class of infection for which we still lack effective antimicrobial treatments or vaccines. Further understanding of ETI pathways and the way in which pathogens specifically subvert these responses may facilitate development of antimicrobials and vaccine strategies.
Acknowledgements
We thank members of the Brodsky and Shin laboratories for scientific discussion. We thank J. Kagan for scientific discussion and critical reading. Work in the Brodsky laboratory is supported by NIH/NIAID grants AI128530, AI139102 and AI135421. Work in the Shin laboratory is supported by NIH/NIAID grants AI118861 and AI123243, and the Linda Pechenik Montague Investigator Award from the University of Pennsylvania Perelman School of Medicine. Both I.E.B. and S.S. are recipients of the Burroughs-Wellcome Fund Investigators in the Pathogenesis of Infectious Disease Award. N.N. is a recipient of the American Heart Association Predoctoral Fellowship. N.L.F. is a recipient of the Institutional Ruth L. Kirschstein National Research Service Award T32GM07229 and the HHMI James H. Gilliam, Jr. Fellowship for Advanced Study.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Janeway CA Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol 54, 1–13 (1989). [DOI] [PubMed] [Google Scholar]
- 2.Ausubel FM Are innate immune signaling pathways in plants and animals conserved? Nat. Immunol 6, 973 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Janeway CA & Medzhitov R Innate immune recognition. Annu. Rev. Immunol 20, 197–216 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Iwasaki A & Medzhitov R Control of adaptive immunity by the innate immune system. Nat. Immunol 16, 343–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasare C & Medzhitov R Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Pasare C & Medzhitov R Toll-dependent control mechanisms of CD4 T cell activation. Immunity 21, 733–741 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Pasare C & Medzhitov R Control of B-cell responses by Toll-like receptors. Nature 438, 364–368 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Matzinger P Tolerance, danger, and the extended family. Annu. Rev. Immunol 12, 991–1045 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Seong SY & Matzinger P Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol 4, 469–478 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Kono H & Rock KL How dying cells alert the immune system to danger. Nat. Rev. Immunol 8, 279–289 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vénéreau E, Ceriotti C & Bianchi ME DAMPs from cell death to new life. Front. Immunol 6, 422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen GY & Nuñez G Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol 10, 826–837 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamkanfi M & Dixit VM Inflammasomes: guardians of cytosolic sanctity. Immunol. Rev 227, 95–105 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Vance RE, Isberg RR & Portnoy DA Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6, 10–21 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liston A & Masters SL Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol 17, 208 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Blander JM & Sander LE Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat. Rev. Immunol 12, 215–225 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Evavold CL & Kagan JC Inflammasomes: threat-assessment organelles of the innate immune system. Immunity 51, 609–624 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones JD & Dangl JL The plant immune system. Nature 444, 323–329 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Chisholm ST, Coaker G, Day B & Staskawicz BJ Host–microbe interactions: shaping the evolution of the plant immune response. Cell 124, 803–814 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Galan JE & Waksman G Protein-injection machines in bacteria. Cell 172, 1306–1318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stuart LM, Paquette N & Boyer L Effector-triggered versus pattern-triggered immunity: how animals sense pathogens. Nat. Rev. Immunol 13, 199–206 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajamuthiah R & Mylonakis E Effector triggered immunity: activation of innate immunity in metazoans by bacterial effectors. Virulence 5, 697–702 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dangl JL & Jones JD Plant pathogens and integrated defence responses to infection. Nature 411, 826–833 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Jones JD, Vance RE & Dangl JL Intracellular innate immune surveillance devices in plants and animals. Science 354, aaf6395 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Cookson BT & Brennan MA Pro-inflammatory programmed cell death. Trends Microbiol. 9, 113–114 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Scheel D Resistance response physiology and signal transduction. Curr. Opin. Plant Biol 1, 305–310 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Lamkanfi M & Dixit VM Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Friebe S, van der Goot FG & Bürgi J The ins and outs of anthrax toxin. Toxins 8, 69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedlander AM, Bhatnagar R, Leppla SH, Johnson L & Singh Y Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect. Immun 61, 245–252 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyden ED & Dietrich WF Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet 38, 240–244 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Martinon F, Burns K & Tschopp J The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Frew BC, Joag VR & Mogridge J Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 8, e1002659 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levinsohn JL et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8, e1002638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chavarría-Smith J & Vance RE Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 9, e1003452 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okondo MC et al. Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem. Biol 25, 262–267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sandstrom A et al. Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 5, eaau1330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ewald SE, Chavarria-Smith J & Boothroyd JC NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun 82, 460–468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cirelli KM et al. Inflammasome sensor NLRP1 controls rat macrophage susceptibility to Toxoplasma gondii. PLoS Pathog. 10, e1003927 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorfu G et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio 5, e01117–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moayeri M, Sastalla I & Leppla SH Anthrax and the inflammasome. Microbes Infect. 14, 392–400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chavarría-Smith J, Mitchell PS, Ho AM, Daugherty MD & Vance RE Functional and evolutionary analyses identify proteolysis as a general mechanism for NLRP1 inflammasome activation. PLoS Pathog. 12, e1006052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong FL et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 167, 187–202 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Jin Y et al. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med 356, 1216–1225 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Muñoz-Planillo R et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leemans JC, Cassel SL & Sutterwala FS Sensing damage by the NLRP3 inflammasome. Immunol. Rev 243, 152–162 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iyer SS et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abrami L, Fivaz M & van der Goot FG Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol. 8, 168–172 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Bhakdi S & Tranum-Jensen J Alpha-toxin of Staphylococcus aureus. Microbiol. Rev 55, 733–751 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pilla DM et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc. Natl Acad. Sci. USA 111, 6046–6051 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim B-H et al. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat. Immunol 17, 481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Man SM, Place DE, Kuriakose T & Kanneganti TD Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. J. Leukoc. Biol 101, 143–150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meunier E & Broz P Interferon-inducible GTPases in cell autonomous and innate immunity. Cell. Microbiol 18, 168–180 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Pilla-Moffett D, Barber MF, Taylor GA & Coers J Interferon-inducible GTPases in host resistance, inflammation and disease. J. Mol. Biol 428, 3495–3513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feeley EM et al. Galectin-3 directs antimicrobial guanylate binding proteins to vacuoles furnished with bacterial secretion systems. Proc. Natl Acad. Sci. USA 114, E1698–E1706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zwack EE et al. Guanylate binding proteins regulate inflammasome activation in response to hyperinjected yersinia translocon components. Infect. Immun 85, e00778–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Viboud GI & Bliska JB Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol 59, 69–89 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Neyt C & Cornelis GR Insertion of a Yop translocation pore into the macrophage plasma membrane by Yersinia enterocolitica: requirement for translocators YopB and YopD, but not LcrG. Mol. Microbiol 33, 971–981 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Nordfelth R & Wolf-Watz H YopB of Yersinia enterocolitica is essential for YopE translocation. Infect. Immun 69, 3516–3518 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosqvist R, Persson C, Håkansson S, Nordfeldt R & Wolf-Watz H Translocation of the Yersinia YopE and YopH virulence proteins into target cells is mediated by YopB and YopD. Contrib. Microbiol 13, 230–234 (1995). [PubMed] [Google Scholar]
- 60.Dewoody R, Merritt PM & Marketon MM YopK controls both rate and fidelity of Yop translocation. Mol. Microbiol 87, 301–317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Francis MS & Wolf-Watz H YopD of Yersinia pseudotuberculosis is translocated into the cytosol of HeLa epithelial cells: evidence of a structural domain necessary for translocation. Mol. Microbiol 29, 799–813 (1998). [DOI] [PubMed] [Google Scholar]
- 62.Zwack EE et al. Inflammasome activation in response to the Yersinia type III secretion system requires hyperinjection of translocon proteins YopB and YopD. mBio 6, e02095–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brodsky IE et al. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coers J Sweet host revenge: Galectins and GBPs join forces at broken membranes. Cell Microbiol. 19, e12793 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rühl S & Broz P Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol 45, 2927–2936 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Meunier E et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509, 366–370 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Man SM et al. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell 167, 382–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Finethy R et al. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in Chlamydia-infected macrophages. Infect. Immun 83, 4740–4749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holmström A et al. YopK of Yersinia pseudotuberculosis controls translocation of Yop effectors across the eukaryotic cell membrane. Mol. Microbiol 24, 73–91 (1997). [DOI] [PubMed] [Google Scholar]
- 70.Sakaguchi T, Leser GP & Lamb RA The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J. Cell. Biol 133, 733–747 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ichinohe T, Pang IK & Iwasaki A Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol 11, 404–410 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen J & Chen ZJ PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ofengeim D & Yuan J Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell. Biol 14, 727–736 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Hsu H, Huang J, Shu HB, Baichwal V & Goeddel DV TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4, 387–396 (1996). [DOI] [PubMed] [Google Scholar]
- 75.Kelliher MA et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8, 297–303 (1998). [DOI] [PubMed] [Google Scholar]
- 76.Meylan E & Tschopp J The RIP kinases: crucial integrators of cellular stress. Trends Biochem. Sci 30, 151–159 (2005). [DOI] [PubMed] [Google Scholar]
- 77.Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA & Kelliher MA Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-{kappa}B activation but does not contribute to interferon regulatory factor 3 activation. J. Biol. Chem 280, 36560–36566 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Peterson LW et al. RIPK1-dependent apoptosis bypasses pathogen blockade of innate signaling to promote immune defense. J. Exp. Med 214, 3171–3182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peterson LW et al. Cell-extrinsic TNF collaborates with TRIF signaling to promote Yersinia-induced apoptosis. J. Immunol 197, 4110–4117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yuan J, Najafov A & Py BF Roles of caspases in necrotic cell death. Cell 167, 1693–1704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tenev T et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448 (2011). [DOI] [PubMed] [Google Scholar]
- 82.Feoktistova M et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moriwaki K & Chan FK RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 27, 1640–1649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sarhan J et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl Acad. Sci. USA 115, E10888–E10897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Orning P et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Newton HJ et al. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 6, e1000898 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ashida H et al. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat. Cell Biol 12, 66–73 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Orth K et al. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science 290, 1594–1597 (2000). [DOI] [PubMed] [Google Scholar]
- 89.Mukherjee S, Hao Y-H & Orth K A newly discovered post-translational modification–the acetylation of serine and threonine residues. Trends Biochem. Sci 32, 210–216 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Cheong MS et al. AvrBsT acetylates Arabidopsis ACIP1, a protein that associates with microtubules and is required for immunity. PLoS Pathog. 10, e1003952 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Philip NH et al. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc. Natl Acad. Sci. USA 111, 7385–7390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weng D et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl Acad. Sci. USA 111, 7391–7396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosadini CV et al. A single bacterial immune evasion strategy dismantles both MyD88 and TRIF signaling pathways downstream of TLR4. Cell Host Microbe 18, 682–693 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dondelinger Y et al. NF-kappaB-independent role of IKKalpha/IKKbeta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Dondelinger Y et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun 10, 1729 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jaco I et al. MK2 Phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 66, 698–710 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Menon MB et al. p38 MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol 19, 1248–1259 (2017). [DOI] [PubMed] [Google Scholar]
- 98.Skaletskaya A et al. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl Acad. Sci. USA 98, 7829–7834 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Omoto S et al. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J. Biol. Chem 290, 11635–11648 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Upton JW, Kaiser WJ & Mocarski ES DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pollock GL et al. Distinct roles of the antiapoptotic effectors NleB and NleF from enteropathogenic Escherichia coli. Infect. Immun 85, e01071–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pearson JS et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat. Microbiol 2, 16258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lemichez E & Aktories K Hijacking of Rho GTPases during bacterial infection. Exp. Cell Res 319, 2329–2336 (2013). [DOI] [PubMed] [Google Scholar]
- 104.Xu H et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 (2014). [DOI] [PubMed] [Google Scholar]
- 105.Park YH, Wood G, Kastner DL & Chae JJ Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol 17, 914–921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gao W, Yang J, Liu W, Wang Y & Shao F Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl Acad. Sci. USA 113, E4857–E4866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aubert DF et al. A Burkholderia type VI effector deamidates Rho GTPases to activate the Pyrin inflammasome and trigger inflammation. Cell Host Microbe 19, 664–674 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Chung LK et al. The Yersinia virulence factor YopM hijacks host kinases to inhibit Type III effector-triggered activation of the Pyrin inflammasome. Cell Host Microbe 20, 296–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ratner D et al. The Yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathog. 12, e1006035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Medici NP, Rashid M & Bliska JB Characterization of pyrin dephosphorylation and inflammasome activation in macrophages as triggered by the Yersinia effectors YopE and YopT. Infect. Immun 87, e00822–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heilig R & Broz P Function and mechanism of the pyrin inflammasome. Eur. J. Immunol 48, 230–238 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Schaner P et al. Episodic evolution of pyrin in primates: human mutations recapitulate ancestral amino acid states. Nat. Genet 27, 318–321 (2001). [DOI] [PubMed] [Google Scholar]
- 113.Jamilloux Y, Magnotti F, Belot A & Henry T The pyrin inflammasome: from sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes. Pathog. Dis 76, fty020 (2018). [DOI] [PubMed] [Google Scholar]
- 114.Boyer L et al. Pathogen-derived effectors trigger protective immunity via activation of the Rac2 enzyme and the IMD or Rip kinase signaling pathway. Immunity 35, 536–549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Keestra AM et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 496, 233–237 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bruno VM et al. Salmonella Typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog. 5, e1000538 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sun H, Kamanova J, Lara-Tejero M & Galán JE Salmonella stimulates pro-inflammatory signalling through p21-activated kinases bypassing innate immune receptors. Nat. Microbiol 3, 1122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bielig H et al. The cofilin phosphatase slingshot homolog 1 (SSH1) links NOD1 signaling to actin remodeling. PLoS Pathog. 10, e1004351 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fukazawa A et al. GEF-H1 mediated control of NOD1 dependent NF-κB activation by Shigella effectors. PLoS Pathog. 4, e1000228 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Legrand-Poels S et al. Modulation of Nod2-dependent NF-κB signaling by the actin cytoskeleton. J. Cell Sci 120, 1299–1310 (2007). [DOI] [PubMed] [Google Scholar]
- 121.Eitel J et al. β-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J. Immunol 181, 2664–2671 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Pakos-Zebrucka K et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tsalikis J, Croitoru DO, Philpott DJ & Girardin SE Nutrient sensing and metabolic stress pathways in innate immunity. Cell. Microbiol 15, 1632–1641 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Wolfson RL & Sabatini DM The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metabol. 26, 301–309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tattoli I et al. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 32, 3066–3078 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tattoli I et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575 (2012). [DOI] [PubMed] [Google Scholar]
- 127.Tattoli I, Philpott DJ & Girardin SE The bacterial and cellular determinants controlling the recruitment of mTOR to the Salmonella-containing vacuole. Biol. Open 1, 1215–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wyant GA et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Y, Weiss LM & Orlofsky A Intracellular parasitism with Toxoplasma gondii stimulates mTOR-dependent host cell growth despite impaired signaling to S6K1 and 4E-BP1. Cell. Microbiol 11, 983 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Soares JA et al. Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J. Virol 83, 6883–6899 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Clippinger AJ, Maguire TG & Alwine JC Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J. Virol 85, 9369–9376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Surviladze Z, Sterk RT, DeHaro SA & Ozbun MA Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol 87, 2508–2517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Celli J & Tsolis RM Bacteria, the endoplasmic reticulum and the unfolded protein response: friends or foes? Nat. Rev. Microbiol 13, 71–82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Walter P & Ron D The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011). [DOI] [PubMed] [Google Scholar]
- 135.Isberg RR, O’Connor TJ & Heidtman M The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol 7, 13–24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hempstead AD & Isberg RR Inhibition of host cell translation elongation by Legionella pneumophila blocks the host cell unfolded protein response. Proc. Natl Acad. Sci. USA 112, E6790–E6797 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Treacy-Abarca S & Mukherjee S Legionella suppresses the host unfolded protein response via multiple mechanisms. Nat. Commun 6, 7887 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fontana MF et al. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog 7, e1001289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Belyi Y et al. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl Acad. Sci. USA 103, 16953–16958 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Belyi Y, Tabakova I, Stahl M & Aktories K Lgt: a family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J. Bacteriol 190, 3026–3035 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.De Leon JA et al. Positive and negative regulation of the master metabolic regulator mTORC1 by two families of Legionella pneumophila effectors. Cell Rep. 21, 2031–2038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shen X et al. Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell. Microbiol 11, 911–926 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Barry KC, Fontana MF, Portman JL, Dugan AS & Vance RE IL-1α signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J. Immunol 190, 6329–6639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Barry KC, Ingolia NT & Vance RE Global analysis of gene expression reveals mRNA superinduction is required for the inducible immune response to a bacterial pathogen. eLife 6, e22707 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ivanov SS & Roy CR Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR. Nat. Immunol 14, 1219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fontana MF, Shin S & Vance RE Activation of host mitogen-activated protein kinases by secreted Legionella pneumophila effectors that inhibit host protein translation. Infect. Immun 80, 3570–3575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shin S et al. Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog. 4, e1000220 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Asrat S, Dugan AS & Isberg RR The frustrated host response to Legionella pneumophila is bypassed by MyD88-dependent translation of pro-inflammatory cytokines. PLoS Pathog. 10, e1004229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Copenhaver AM, Casson CN, Nguyen HT, Duda MM & Shin S IL-1R signaling enables bystander cells to overcome bacterial blockade of host protein synthesis. Proc. Natl Acad. Sci. USA 112, 7557–7562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Reddy KC, Dunbar TL, Nargund AM, Haynes CM & Troemel ER The C. elegans CCAAT-enhancer-binding protein gamma is required for surveillance immunity. Cell Rep. 14, 1581–1589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pukkila-Worley R Surveillance immunity: an emerging paradigm of innate defense activation in Caenorhabditis elegans. PLoS Pathog. 12, e1005795 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Melo JA & Ruvkun G Inactivation of conserved C. elegans genes engages pathogen-and xenobiotic-associated defenses. Cell 149, 452–466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ogata M, Chaudhary VK, Pastan I & FitzGerald DJ Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J. Biol. Chem 265, 20678–20685 (1990). [PubMed] [Google Scholar]
- 154.Estes KA, Dunbar TL, Powell JR, Ausubel FM & Troemel ER bZIP transcription factor zip-2 mediates an early response to Pseudomonas aeruginosa infection in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 107, 2153–2158 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dunbar TL, Yan Z, Balla KM, Smelkinson MG & Troemel ER C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 11, 375–386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.McEwan DL, Kirienko NV & Ausubel FM Host translational inhibition by Pseudomonas aeruginosa Exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 11, 364–374 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Abastado J, Miller PF, Jackson BM & Hinnebusch AG Suppression of ribosomal reinitiation at upstream open reading frames in amino acid-starved cells forms the basis for GCN4 translational control. Mol. Cell. Biol 11, 486–496 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fernandez J et al. Ribosome stalling regulates IRES-mediated translation in eukaryotes, a parallel to prokaryotic attenuation. Mol. Cell 17, 405–416 (2005). [DOI] [PubMed] [Google Scholar]
- 159.Barbosa C, Peixeiro I & Romão L Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 9, e1003529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Staskawicz BJ, Dahlbeck D & Keen NT Cloned avirulence gene of Pseudomonas syringae pv. glycinea determines race-specific incompatibility on Glycine max (L.) Merr. Proc. Natl Acad. Sci. USA 81, 6024–6028 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Medzhitov R Approaching the asymptote: 20 years later. Immunity 30, 766–775 (2009). [DOI] [PubMed] [Google Scholar]
- 162.Sandstrom A et al. Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364, eaau1330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gurcel L, Abrami L, Girardin S, Tschopp J & van der Goot FG Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126, 1135–1145 (2006). [DOI] [PubMed] [Google Scholar]
- 164.Craven RR et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 4, e7446 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Komune N, Ichinohe T, Ito M & Yanagi Y Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1β secretion. J. Virol 85, 13019–13026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cheong WC et al. Influenza A virus NS1 protein inhibits the NLRP3 inflammasome. PLoS ONE 10, e0126456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Thorslund SE et al. The RACK1 signaling scaffold protein selectively interacts with Yersinia pseudotuberculosis virulence function. PloS ONE 6, e16784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jaramillo M et al. Leishmania repression of host translation through mTOR cleavage is required for parasite survival and infection. Cell Host Microbe 9, 331–341 (2011). [DOI] [PubMed] [Google Scholar]
- 169.Dunn EF & Connor JH Dominant inhibition of Akt/protein kinase B signaling by the matrix protein of a negative-strand RNA virus. J. Virol 85, 422–431 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Keestra-Gounder AM et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 532, 394–397 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Haun F et al. Identification of a novel anoikis signalling pathway using the fungal virulence factor gliotoxin. Nat. Commun 9, 3524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gottar M et al. Dual detection of fungal infections in Drosophila via recognition of glucans and sensing of virulence factors. Cell 127, 1425–1437 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cui H, Tsuda K & Parker JE Effector-triggered immunity: from pathogen perception to robust defense. Annu. Rev. Plant Biol 66, 487–511 (2015). [DOI] [PubMed] [Google Scholar]
- 174.Flor HH Current status of the gene-for-gene concept. Annu. Rev. Phytopathol 9, 275–296 (1971). [Google Scholar]
- 175.Lawrence GJ, Dodds PN & Ellis JG Rust of flax and linseed caused by Melampsora lini. Mol. Plant Pathol 8, 349–364 (2007). [DOI] [PubMed] [Google Scholar]
- 176.Ellis JG, Dodds PN & Lawrence GJ Flax rust resistance gene specificity is based on direct resistance-avirulence protein interactions. Annu. Rev. Phytopathol 45, 289–306 (2007). [DOI] [PubMed] [Google Scholar]
- 177.Van Der Biezen EA & Jones JD Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem. Sci 23, 454–456 (1998). [DOI] [PubMed] [Google Scholar]
- 178.Mackey D, Holt BF III, Wiig A & Dangl JL RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell 108, 743–754 (2002). [DOI] [PubMed] [Google Scholar]
- 179.Mackey D, Belkhadir Y, Alonso JM, Ecker JR & Dangl JL Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell 112, 379–389 (2003). [DOI] [PubMed] [Google Scholar]
- 180.Axtell MJ & Staskawicz BJ Initiation of RPS2-specified disease resistance in Arabidopsis is coupled to the AvrRpt2-directed elimination of RIN4. Cell 112, 369–377 (2003). [DOI] [PubMed] [Google Scholar]
- 181.Ade J, DeYoung BJ, Golstein C & Innes RW Indirect activation of a plant nucleotide binding site-leucine-rich repeat protein by a bacterial protease. Proc. Natl Acad. Sci. USA 104, 2531–2536 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]