

Abstract
As the outermost barrier tissue of the body, the skin harbors a large number of innate lymphoid cells (ILCs) that help maintain local homeostasis in the face of changing environments. How skin-resident ILCs are regulated and function in local homeostatic maintenance is poorly understood. We herein report discovery of a cold-sensing neuron-initiated pathway that activates skin group 2 ILCs (ILC2s) to help maintain thermal homeostasis. In stearoyl-CoA desaturase 1 (SCD1) knockout mice whose skin is defective in heat maintenance, chronic cold stress induced excessive activation of CCR10−CD81+ST2+ skin ILC2s and associated inflammation. Mechanistically, stimulation of the cold-sensing receptor TRPM8 expressed in sensory neurons of the skin led to increased production of IL-18, which in turn activated skin ILC2s to promote thermogenesis. Our findings reveal a neuroimmune link that regulates activation of skin ILC2s to support thermal homeostasis and promotes skin inflammation following hyperactivation.
One Sentence Summary:
Skin-resident ILC2s are activated by signals from cold-sensing neurons to aid in regulation of thermal homeostasis in skin.
INTRODUCTION
Innate lymphoid cells (ILCs) are a family of innate lymphocytes (1–4). Some members of this family, such as natural killer (NK) cells, have been known for a long time, while the others were discovered in recent years. All ILCs originate from lymphoid-primed multipotent progenitors (LMPPs) or common lymphoid progenitors (CLP) (1, 2, 5). Under direction of transcription factors and extrinsic environmental signals, the progenitor cells give rise to various subsets of ILCs (6, 7). Based on their developmental pathways and functional potentials, ILCs are commonly divided into three groups of helper ILCs (ILC1–3) in addition to NK cells and lymphoid tissue inducer (LTi) cells (1, 2). ILC1s depend on transcription factors such as T-bet for their development and function and produce type 1 cytokines such as interferon γ (IFNγ) when activated (5, 8, 9). ILC2s express the transcription factors GATA3 and RORα for their development and are able to produce type 2 cytokines such as IL-4, IL-5 and IL-13 (10–17). ILC3s depend on the transcription factor RORγt for their development and produce IL-17 and IL-22 (18–21).
ILCs preferentially localize into barrier tissues such as the skin, intestine and lung where they help protect local tissue homeostasis while their dysregulation can lead to inflammatory diseases, defective tissue development and function, and other disorders (1–4, 22–27). Depending on their functional potentials, different groups of ILCs have different functions in tissue protection and homeostatic regulation. In general, ILC1s and NK cells function in immunity against viral and bacterial infection and tumor surveillance. ILC2s are primarily involved in immunity against parasite infection as well as in metabolic homeostatic regulation and wound healing. ILC3s mainly function in early immune responses against local infection and are also implicated in tissue repair following inflammation or damage. The composition and number of ILCs in different tissues vary, likely as an adaptation to tissue-specific requirements for their optimal protection and homeostatic maintenance.
Skin is the outermost barrier tissue of the body. The integrity and proper function of the skin is crucial in maintaining thermal homeostasis and the body’s water and electrolyte balance in the face of changing environments. Among different barrier tissues, skin is enriched with one of the highest percentages of ILCs, which constitute more than 10% of resident lymphocytes in mice (27–33). Under homeostatic conditions, skin-homing ILCs are continuously generated in skin-draining lymph nodes (sLNs) and migrate into the skin where they contribute to the pool of skin ILCs (29). The chemokine receptor CCR10, through interaction with its skin-specific ligand CCL27, plays an important role in migration and homeostatic establishment of skin ILCs (29). While various groups of ILCs, including NK1.1+ NK cells and ILC1s, can be found in the healthy skin, ILC2s account for the majority of skin ILCs in adult wild-type (WT) mice (28, 29, 34–37). Skin-specific CCR10+ ILCs under homeostatic conditions express the ILC2-associated transcription factor GATA3 but not the ILC1 and ILC3-associated transcription factors T-bet and RORγt (29). However, when stimulated with phorbol myristate acetate (PMA) and ionomycin, significant percentages of skin CCR10+ ILCs can produce both IL-5 and IL-17A although few produce IFNγ (29), suggesting that they are not fully committed ILC2s. On the other hand, a significant fraction of skin CCR10+ ILCs express high levels of MHCII and play an important role in maintenance of skin T cell homeostasis in the skin (29). Skin ILC2s can also interact with mast cells to suppress their lgE-dependent cytokine release (28). Therefore, skin ILC2s likely represent a unique population of ILCs with regulatory functions under homeostatic conditions and plasticity for further differentiation into active effector cells in response to local environmental stimulation, which would allow them to optimally help maintain the skin homeostasis and function.
Various pathophysiological changes are associated with skin ILC2 activation. In wounded skin, the number of IL-5 and IL-13-producing ILC2s increases, suggesting that they are activated to help wound healing (38). On the other hand, over-activation of skin ILC2s contributes to cutaneous inflammation. In mouse models of atopic dermatitis, skin ILC2s are activated by thymic stromal lymphopoietin (TSLP) and IL-33 produced by skin keratinocytes and produce IL-5 and IL-13 that promote disease development (39–41). Prostaglandin D2, acting through its receptor CRTH2 expressed on skin ILC2s, induces migration and cytokine production of skin ILC2s in human atopic dermatitis (42). Consistent with the notion that local environments provide cues to regulate ILCs for their tissue-specific functions, ILC2s in the healthy skin respond differently to molecular regulators than ILC2s of mucosal tissues. For example, few ILC2s in the healthy skin express the IL-33 receptor ST2, which is upregulated only after their activation (39–41). On the other hand, skin ILC2s constitutively express IL-18 receptor and IL-18 was reported as an important cytokine for skin ILC2 activation (30). We reported that under various inflammatory or dysregulated conditions, CCR10+MHCII+ skin ILCs could differentiate into CCR10−MHCII− ILCs, suggesting their activation (29). However, how skin ILC2s are activated in response to changes in the ambient environment to help maintain homeostatic regulation is unknown.
Herein, we report that skin ILC2s are highly activated in mice with a defective skin lipid barrier, suggesting their involvement in thermal homeostatic regulation. We also found that a neuron-derived TRPM8 cold-sensing pathway activates skin ILC2s in an IL-18-dependent manner to help thermal homeostatic regulation. Our findings reveal that skin ILC2s are an integral component of the skin thermoregulation machinery that responds to ambient temperature changes to help maintain its physiological functions, while their dysregulation could lead to disorders of the skin and body.
RESULTS
Preferential upregulation of CD81 on activated CCR10− skin ILC2s
We previously found that while CCR10+ ILCs are dominant in healthy skin, they could differentiate into activated CCR10− ILCs in the skin of T and B cell-deficient Rag1−/− mice (29). To gain clues about the functional mechanism and regulation of ILC activation in the skin, we compared the gene expression profiles of CCR10+ trunk skin ILCs from WT mice versus CCR10− or CCR10low skin ILCs from WT and Rag1−/− mice by microarray analysis (Fig. S1A). Principal component analysis (PCA) revealed that the gene expression pattern of CCR10+ skin ILCs of WT mice was significantly different from that of CCR10low/- skin ILCs of WT or Rag1−/− mice (Fig. S1B). Relative to CCR10+ ILCs of WT mice, CCR10− or CCR10low ILCs, particularly of Rag1−/− mice, had significantly reduced expression of multiple MHCII genes (Fig. S1C), consistent with our previous report (29). Notably, relative to CCR10+ ILCs of WT mice, CCR10−/low ILCs of WT and Rag1−/− mice had increased expression of multiple cytokine receptors known to be involved in activation of ILC1s, ILC2s and ILC3s, including receptors for IL-2, IL-12, IL-15, IL-33, PGD2, TLSP, and IL-1β, IL-6, IL-23 (Fig. 1A). In addition, expression of several chemokines (CXCL2, XCL1, CCL1 and CX3CL1) involved in recruitment of inflammatory immune cells was enhanced in CCR10−/low ILCs relative to CCR10+ ILCs (Fig. 1B). Expression of cytokine/effector molecules typically associated with different ILC groups was not significantly increased except for granzyme C (Gzmc), an effector molecule specifically expressed in activated ILC1s (Fig. 1C) (9, 35, 43, 44). CD81, a tetraspanin molecule involved in signal transduction of various immune activation receptors (45, 46), was one of the most upregulated genes in CCR10−/low ILCs relative to CCR10+ ILCs while Clec12a, an immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing immune inhibitory receptor (47), was one of the most downregulated genes (Fig. 1D).
Figure 1.
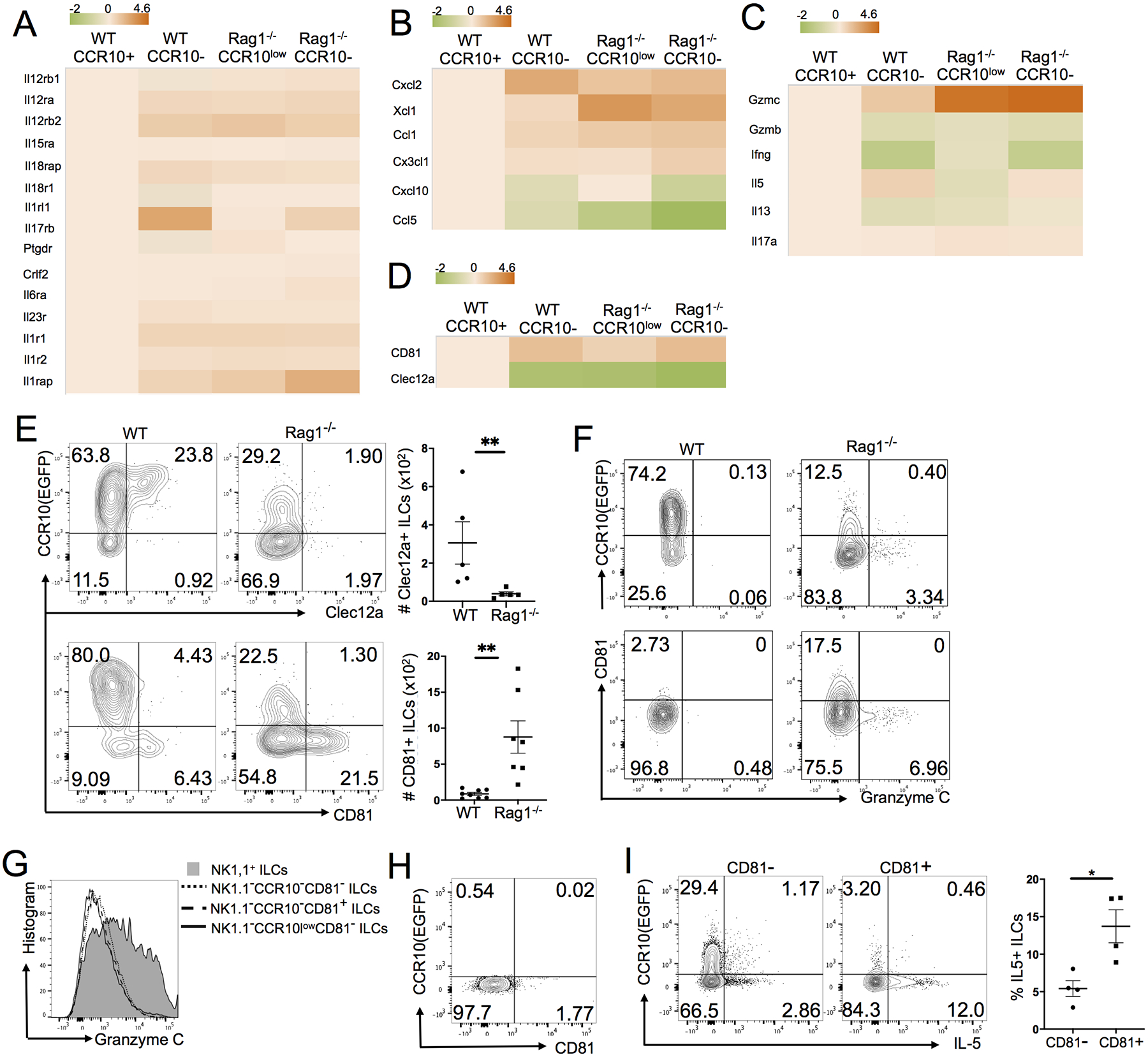
Preferential upregulation of CD81 on activated CCR10− skin ILC2s. Both WT and Rag1−/− mice carry a CCR10-knockout/EGFP-knockin (CCR10+/EGFP) allele for purpose of identifying CCR10-expressing cells with EGFP reporter (29, 93). (A-D) Heat map of gene expression of selected cytokine receptors (A), chemokines (B), cytokines/effector molecules (C) and Clec12A and CD81 (D) by different subsets of CCR10− or CCR10low skin ILCs of WT and Rag1−/− mice relative to CCR10+ skin ILCs of WT mice. (E) Flow cytometric (FC) analysis of expression of CCR10 versus Clec12a or CD81 in skin ILCs of WT and Rag1−/− mice. The dot plot shows numbers (#) of Clec12a+ and CD81+ skin ILCs. One dot represents one mouse sample. (F) FC analysis of CCR10, CD81 and granzyme C expression in skin ILCs of WT or Rag1−/− mice. (G) FC analysis of granzyme C expression on indicated subsets of skin ILCs of Rag1−/− mice. (H) FC analysis of the CCR10 versus CD81 expression in NK1.1+ skin ILC1s of Rag1−/− mice. (I) Comparison of IL-5 expression of CD81+ and CD81− skin ILCs of Rag1−/− mice. The dot plot shows average percentages of CD81+ and CD81− skin ILCs that express IL-5. One dot represents one mouse. *P<0.05. **P<0.01. Statistical significance was determined by paired T test for panel E and unpaired T test for panel I.
We then used flow cytometry to assess expression of Clec12a, CD81 and other molecules in association with specific groups of CCR10+ and CCR10− ILCs. Consistent with the microarray result, Clec12a was expressed predominantly on a fraction of CCR10+ ILCs of WT mice, while CD81 was expressed preferentially on a fraction of CCR10− ILCs, which were expanded in the skin of Rag1−/− mice (Fig. 1E). CD81 was also more preferentially expressed on MHCII− ILCs than on MHCII+ ILCs (Fig. S1D), consistent with the notion that MHCII was mostly expressed by CCR10+ skin ILCs (29). These results suggest that upregulation of CD81 is associated with activated CCR10−MHCII− skin ILC2s. There was also increased expression of Gzmc in a small population of CCR10− ILCs of Rag1−/− mice (Fig. 1F). However, Gzmc was not expressed by CD81+ skin ILCs (Fig. 1F). Instead, it was highly expressed by activated NK1.1+ CCR10− skin ILC1s of Rag1−/− mice (Fig. 1G)(35). In contrast, CD81 was not expressed by NK1.1+CCR10− skin ILC1s (Fig. 1H), consistent with the notion that CD81 was preferentially associated with activated skin ILC2s. Further supporting this notion, CCR10−CD81+ skin ILCs expressed more IL-5 than CD81− skin ILCs (Fig. 1I). In addition, skin ILCs (excluding NK1.1+ ILC1s) of Rag1−/− and WT mice expressed GATA3 and CD127 but no Rorγt (Fig. S1E) (29, 48). A small fraction of WT skin ILCs expressed low level of T-bet (Fig. S1E–F). However, the T-bet expression was not associated with CD81 or Clec12a (Fig. S1F). Together, these results indicate that the CD81 upregulation, in combination with CCR10 downregulation, preferentially identifies differentiated skin ILC2s. However, very few skin ILCs of Rag1−/− mice expressed ST2 (Fig. S1G), suggesting that they might not be terminally differentiated ILC2s.
Predominance of CD81+ST2+ activated ILC2s in the skin of SCD1 knockout mice with defective skin barrier for heat maintenance
Considering a major function of ILC2s in regulation of metabolic and thermal homeostasis (49–52), we tested whether a skin defect in prevention of heat loss would result in increased activation of ILC2s using mice deficient of stearoyl-CoA desaturase-1 (SCD1). SCD1 is an enzyme that catalyzes generation of monounsaturated fatty acids (MUFAs) such as oleate and palmitoleate, which are major components for formation of lipid layers of the skin (53, 54). SCD1 knockout (SCD1 KO) mice have defective skin integrity, impaired maintenance of thermal homeostasis and severe skin inflammation (54–56). Skin-specific SCD1 KO mice exhibit the same phenotypes in the skin integrity, maintenance of thermal homeostasis and skin inflammation as global SCD1 KO mice, indicating that impaired skin integrity is primarily responsible for the dysregulation of thermal homeostasis and skin inflammation (57). We tested whether skin ILC2s were activated in SCD1 KO mice based on the expression of CD81 and related activation markers. Compared to WT littermates, there were significantly higher percentages of ILCs in the skin of SCD1 KO (SCD1−/−) mice (Fig. 2A). Furthermore, in contrast to skin ILCs of WT mice, nearly all skin ILCs of SCD1−/− mice had highly upregulated CD81 and downregulated MHCII (Fig. 2B). Notably, a significant fraction of skin ILCs of SCD1−/− mice expressed ST2 (Fig. 2C). In addition, compared to WT mice, the number of IL-5-expressing skin ILCs in SCD1−/− mice was increased (Fig. 2D).
Figure 2.
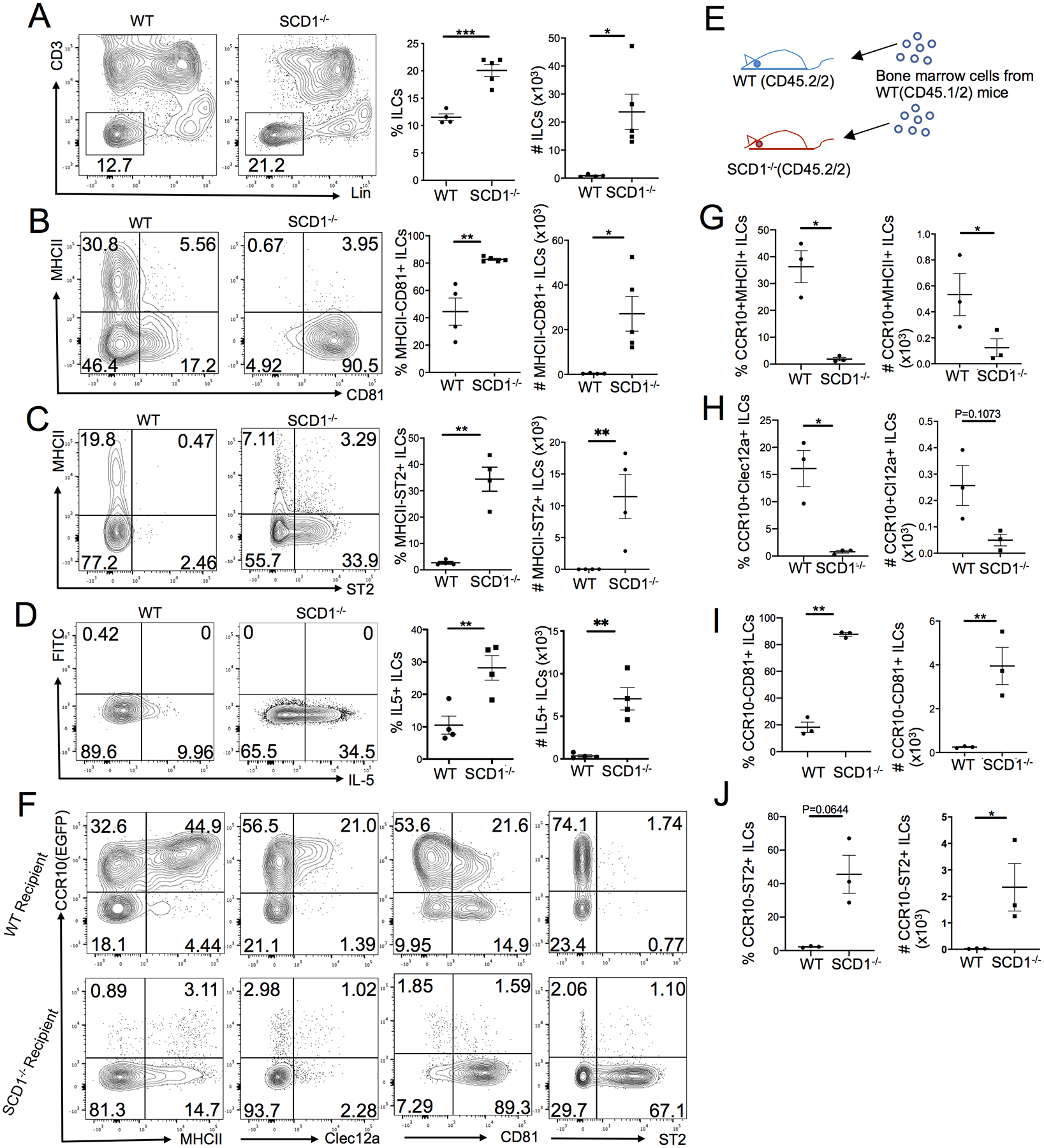
Predominance of CCR10−CD81+ST2+ activated ILC2s in the skin of SCD1 knockout mice. (A) Comparison of the percentage and number of Lin− ILCs in the skin of WT and SCD1−/− mice. (B-D) Comparison of the percentage and number of MHCII−CD81+ (B), MHCII−ST2+ (C) and IL-5+ (D) ILCs in the skin of WT and SCD1−/− mice. (E) Scheme of reconstitution of WT and SCD1−/− mice with BM cells of WT mice. (F) FC analysis of expression of CCR10 versus indicated markers in donor-derived ILCs in the skin of WT (top row) or SCD1−/− recipient mice (bottom) 6–8 weeks after the BM transfer. (G-J) Comparison of the percentage and number of donor-derived CCR10+MHCII+ (G), CCR10+Clec12a+ (H), CCR10−CD81+ (I) and CCR10−ST2+ (J) ILCs in the skin of WT versus SCD1−/− recipient mice, based on analysis of the panel F. Donor BM cells in panels F through J carry a CCR10-knockout/EGFP-knockin (CCR10+/EGFP) allele for purpose of identifying CCR10+ cells with EGFP reporter. One dot represents one mouse. *P<0.05. **P<0.01. ***P<0.001. Statistical significance was determined by unpaired T test for panels A-B and paired T test for panels C-D and G-J.
We also assessed other ILC groups and T cells in the skin and sLNs of SCD1−/− mice. Compared to WT controls, there was a higher percentage of CD3−NK1.1+ cells and T-bet+NK1.1+ ILC1s in the skin of SCD1−/− mice (Fig. S2A–B), although they accounted for a minor population of skin ILCs (2–4%). The percentage of IFNγ+ skin ILCs was lower while the percentage of IL-17A+ skin ILCs was higher in SCD1−/− mice than WT controls (Fig. S2C). These results indicate that in contrast to the high activation of ILC2s, there was a relatively limited or no increase in ILC1, NK and ILC3 cells in the skin of SCD1−/− mice. Within the T cell compartment, there were increased percentages of CD4+ T and Treg cells in the skin of SCD1−/− mice compared to WT controls (Fig. S2D). Unlike in the skin, there were similar percentages of CD81+ and ST2+ ILCs in the sLNs of WT and SCD1−/− mice (Fig. S2E), suggesting that the altered skin environment causes the ILC2 activation in the skin of SCD1−/− mice.
To further confirm that the activation of skin ILC2s in SCD1−/− mice was due to the altered skin environment but not intrinsic SCD1 deficiency in ILCs, we transferred bone marrow (BM) cells of WT mice (CD45.1+CD45.2+) to SCD1−/− or WT recipient mice (CD45.2+CD45.1−) (Fig. 2E). Six to eight weeks after the transfer, ILCs of the WT donor origin in the recipients were analyzed. In striking contrast to high expression of CCR10 by donor-derived ILCs in the skin of WT recipients, few donor-derived ILCs in the skin of SCD1−/− recipients expressed CCR10 (Fig. 2F). There were significantly fewer CCR10+MHCII+ and CCR10+Clec12a+ donor-derived ILCs in the skin of SCD1−/− compared to WT recipients (Fig. 2F, 2G, 2H). In contrast, there were substantially more CCR10−CD81+ donor-derived ILCs in the skin of SCD1−/− recipients compared to WT recipients (Fig. 2F, 2I). About half of donor-derived ILCs in the skin of SCD1−/− recipients expressed ST2 while very few of WT recipients expressed ST2 (Fig. 2F, 2J). These results revealed that there were excessively activated CCR10−CD81+ST2+ ILC2s in the skin of SCD1−/− mice, likely due to the defective skin barrier resulting from impaired lipid synthesis.
High-fat diet feeding reduced activation of CD81+ST2+ ILC2s in the skin of SCD1 knockout mice
To demonstrate that the impaired fatty acid synthesis and associated defective skin barrier were responsible for increased skin ILC2 activation in SCD1−/− mice, we tested whether the skin ILC2 activation could be reduced by feeding them on a high-fat diet (HFD) containing high levels of unsaturated fatty acids. SCD1−/− mice fed with HFD for 3 weeks had a more normal fur appearance and reduced skin inflammation compared to those fed with a standard chow diet (Fig. S3A), consistent with the notion that that HFD rescues the defective skin barrier of SCD1−/− mice (54, 57). Associated with this, HFD-fed SCD1−/− mice had a significantly reduced ILCs in the skin compared to chow-fed SCD1−/− littermates (Fig. 3A). Although the majority of skin ILCs of HFD-fed SCD1−/− mice were still CD81+ and did not express MHCII (Fig. 3B), they had significantly reduced expression of ST2 compared to chow-fed SCD1−/− controls (Fig. 3C). HFD-fed SCD1−/− mice also had reduced IL-5+ skin ILCs compared to chow-fed SCD1−/− mice (Fig. 3D). These results demonstrate that HFD-feeding reduced activation of CD81+ST2+ ILC2s in the skin of SCD1−/− mice.
Figure 3.
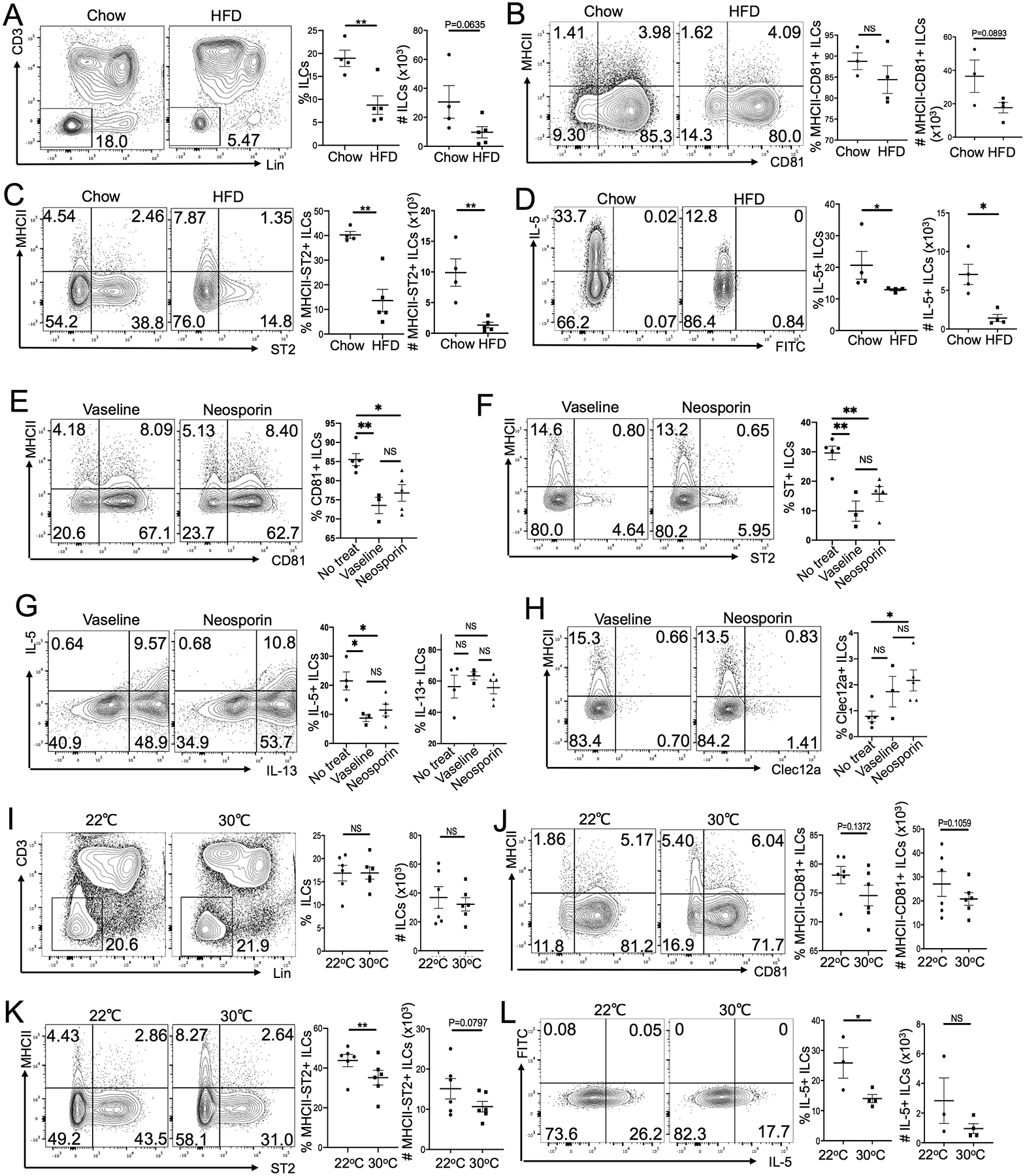
High-fat diet feeding or housing at the thermo-neutral ambient temperature reduces activation of skin ILC2s in SCD1−/− mice. (A) Comparison of the percentage and number of Lin− ILCs in the skin of SCD1−/− mice fed with chow diet versus HFD for 3 weeks. (B-D) Comparison of the percentage and number of MHCII−CD81+ (B), MHCII−ST2+ (C) and IL-5+ (D) ILCs in the skin of SCD1−/− mice fed with chow diet versus HFD for 3 weeks. (E-H) Comparison of the percentage of CD81+ (E), ST2+(F), IL-5+/IL-13+ (G) and Clec12a+ (H) skin ILCs in SCD1−/− mice treated with/without Vaseline or Neosporin for 10 days. (I) Comparison of the percentage and number of Lin− ILCs in the skin of SCD1−/− mice raised at 22°C versus 30°C for 3 weeks. (J-L) Comparison of the percentage and number of MHCII−CD81+ (J), MHCII−ST2+ (K) and IL-5+ (L) ILCs in the skin of SCD1−/− mice raised at 22°C versus 30°C for 3 weeks. One dot represents one sample. *P<0.05, **P<0.01, NS: no significant difference. Statistical significance was determined by unpaired T test for panels A-H and L, paired T test for panels I-K.
As controls, we also analyzed skin ILCs of HFD-fed WT mice. Compared to chow-fed WT mice, HFD-fed WT mice had reduced CCR10+Clec12a+ and MHCII+ and increased CCR10−CD81+ ILCs in the skin (Fig. S3B–D). These results suggest that in contrast to those of SCD1−/− mice, skin ILCs of WT mice had increased activation phenotypes in response to the HFD feeding, consistent with the previous reports that the HFD feeding induced the skin inflammation (58). However, the HFD feeding did not increase differentiation of ST2+ ILCs in the skin of WT mice (Fig. S3C). The opposite effects of HFD on the skin ILC2 activation and inflammation of WT and SCD1−/− mice are consistent with the notion that skin homeostasis is impaired in SCD1−/− mice and the HFD feeding improves their skin barrier function, while WT mice have balanced metabolic homeostasis and the HFD feeding causes metabolic dysregulation (54, 57, 58).
Housing SCD1 knockout mice at a thermoneutral temperature reduced activation of CD81+ST2+ ILC2s in the skin
SCD1−/− mice had impaired maintenance of thermal homeostasis, increased water loss, and potentially dysregulated microbiota resulting from defective monounsaturated fatty acid synthesis (54, 56, 57, 59). To dissect cause(s) of the preferential skin ILC2 activation, we treated skin ILC2s of SCD1−/− mice with oleic acid, a monounsaturated fatty acid reduced in the skin of SCD1−/− mice. However, oleic acid treatment had a little effect on the CD81 and ST2 expression on skin ILC2s (Fig. S3E). To test whether microbiota were involved, we treated SCD1−/− mice topically with Neosporin antibiotic cream. Since the topical application of Neosporin cream could also improve the skin lipid barrier, treatment with inert Vaseline cream was included as a control. There was no difference in skin ILCs of Neosporin- and Vaseline-treated SCD1−/− mice (Fig. 3E–H). However, both Neosporin- and Vaseline-treated SCD1−/− mice had lower CD81+, ST2+ and IL-5+ and higher Clec12a+ skin ILCs compared to untreated SCD1−/− mice (Fig. 3E–H). These results suggest that microbiota are not involved in skin ILC2 activation. Instead, the impaired skin lipid barrier itself is a major cause for the increased skin ILC2 activation.
SCD1−/− mice have impaired maintenance of thermal homeostasis due to the inability of their defective skin barrier to prevent heat loss at standard mouse room temperatures in the range of 22–26°C (54, 56, 57). To assess whether activation of CD81+ST2+ skin ILC2s in SCD1−/− mice might be affected by room temperatures, we housed SCD1−/− mice at the thermoneutral 30°C room temperature for 3 weeks after weaning while some of their SCD1−/− littermates were kept at 22°C. SCD1−/− mice housed at 30°C and 22°C had similar numbers of ILCs in the skin (Fig. 3I). However, compared to SCD1−/− mice housed at 22°C, SCD1−/− mice housed at 30°C had reduced percentages of ST2+ skin ILC2s (Fig. 3J–K). The percentage of IL-5+ skin ILCs was also significantly lower in SCD1−/− mice housed at 30°C than those at 22°C (Fig. 3L). These results indicate that the impaired maintenance of thermal homeostasis in SCD1−/− mice resulting from the defective skin lipid barrier causes skin ILC2 activation.
Stimulation of the cold-sensing receptor TRPM8 activates skin ILC2s in wild-type mice
Unlike SCD1−/− mice, WT mice raised at the 24°C room temperature did not have significant skin ILC2 activation, correlating with their intact skin barrier in maintenance of thermal homeostasis. However, after being placed at a colder room temperature (4°C) for 2 weeks, WT mice had increased CD81+ but decreased Clec12a+ ILCs in the skin compared to WT mice kept at 24°C, indicating their increased activation (Fig. S4A). The extent of the skin ILC activation in WT mice was not as strong as that of SCD1−/− mice, consistent with the notion that SCD1−/− mice have a severe defect in maintenance of thermal homeostasis while WT mice can better adapt to environmental changes.
The activation of skin ILC2s by cold stress suggests a role of the cold-sensing machinery in this process. To test this, we topically applied WT mice with menthol, a stimulating ligand (agonist) for transient receptor potential cation channel subfamily M member 8 (TRPM8), a major cold-sensing receptor expressed at nerve terminals of sensory neurons and by keratinocytes in the skin (60–64). After daily treatment for two weeks, the percentage and number of total ILCs in the skin treated with menthol (10% in ethanol) were highly increased compared to those in the vehicle control (ethanol)-treated skin (Fig. 4A). Percentages and/or numbers of CCR10+ ILCs, particularly MHCII+CCR10+ and Clec12a+CCR10+ ILCs, were lower in the menthol-treated skin than the control-treated skin (Fig. 4B–C). Reciprocally, there were significantly increased CCR10−CD81+ and CCR10−ST2+ ILCs in the menthol-treated skin compared to the control-treated skin (Fig. 4D–E). The percentage and number of IL-5+ ILCs were also significantly higher in the menthol-treated skin than in the control-treated skin (Fig. 4F).
Figure 4.
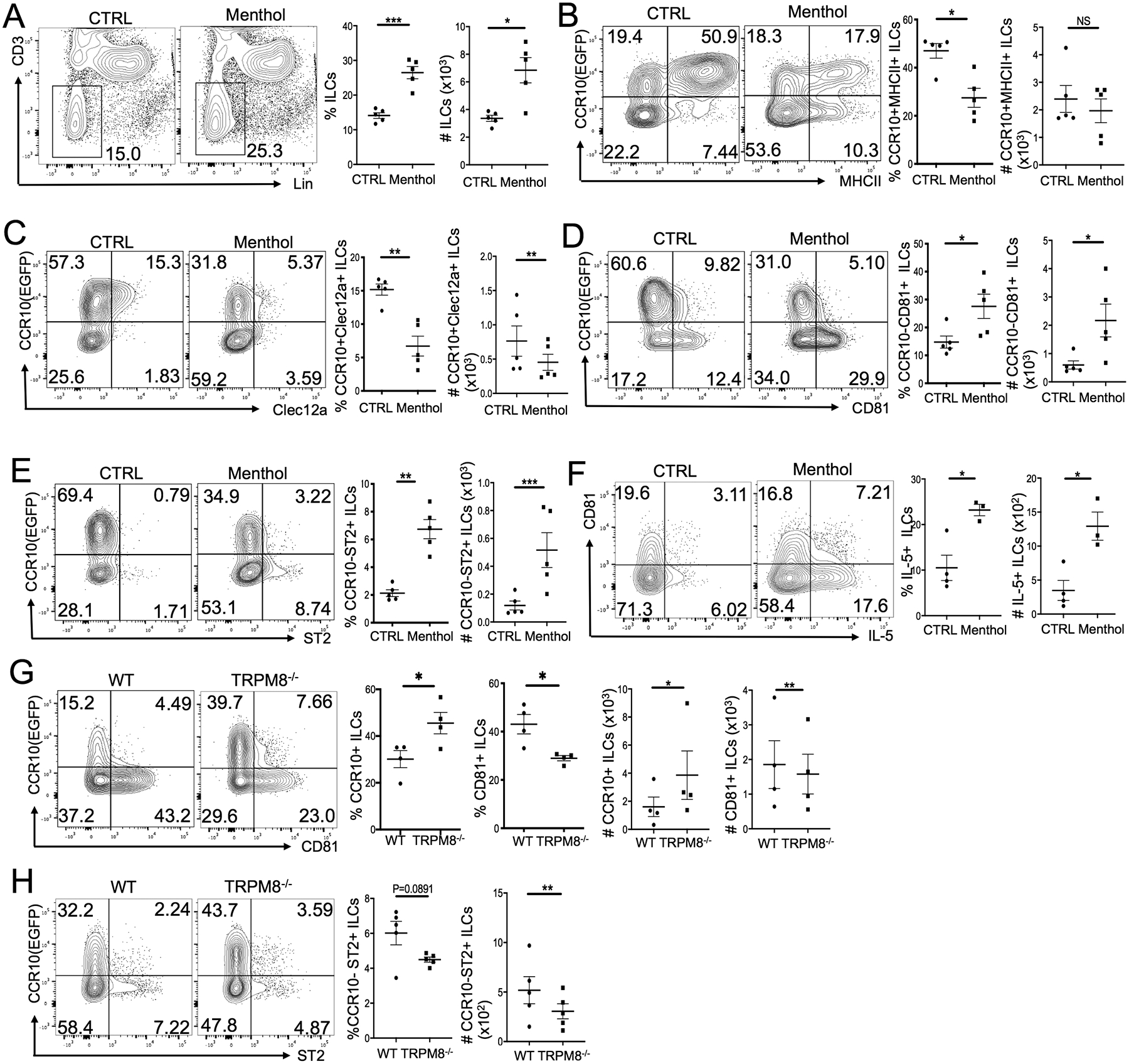
Stimulation of the cold-sensing receptor TRPM8 activates skin ILC2s. (A) Percentages and numbers of Lin− ILCs in the menthol versus control (ethanol)-treated skin of WT mice. (B-E) Comparison of the percentage and number of CCR10+MHCII+ ILCs (B), CCR10+Clec12a+ ILCs (C), CCR10−CD81+ (D) and CCR10−ST2+ (E) ILCs in the menthol versus control (ethanol)-treated skin of WT mice. (F) Analysis of IL-5 expression by ILCs of the menthol versus control (ethanol)-treated skin of WT mice. (G) Analysis of CCR10 and CD81 expression on skin ILCs in WT mice versus TRPM8−/− mice treated with menthol daily for 2 weeks. (H) Analysis of CCR10 and ST2 expression on ILCs of the menthol-treated skin of WT versus TRPM8−/− mice. Both WT and TRPM8−/− mice carry a CCR10-knockout/EGFP-knockin (CCR10+/EGFP) allele for purpose of identifying CCR10+ cells with EGFP. One dot is one sample. *P<0.05, **P<0.01. ***P<0.001, ***P<0.0001, NS: no significant difference. Statistical significance was determined by unpaired T test for the panel F, paired T test for panels A-E, G-H.
In the skin, CCR10+ resident ILCs were located close to sensory neuron fibers under the epidermis or around hair follicle areas (Fig. S4B), supporting their possible interaction. To confirm that stimulation of TRPM8 led to activation of skin ILC2s, we topically applied menthol to a strain of TRPM8 KO (TRPM8−/−) mice in which TRPM8 was deleted from sensory neurons while an alternatively spliced form of TRPM8 was still functional in keratinocytes (62, 65). There was no significant difference in the percentage and number of CCR10+ and CCR10− ILCs or MHCII+ and MHCII− ILCs and their expression of Clec12a, CD81 and ST2 in the untreated skin of TRPM8−/− and WT mice (Fig. S4C–E). There were also similar percentages of IL-5+ and IL-17A+ ILCs in the untreated skin of TRPM8−/− and WT mice (Fig. S4F). However, menthol-treated TRPM8−/− mice had more CCR10+ but fewer CD81+ ILCs in the skin than menthol-treated WT mice (Fig. 4G). In addition, menthol-treated TRPM8−/− mice also had lower percentages and numbers of ST2+ skin ILCs than menthol-treated WT mice (Fig. 4H). These results indicate that stimulation of TRPM8 expressed on sensory neurons leads to the skin ILC2 activation.
Stimulation of TRPM8 induces production of IL-18 for activation of skin ILC2s
We searched for candidate molecules that potentially mediated the function of TRPM8-activated sensory neurons in the ILC2 activation. Based on the microarray analysis, CCR10+ skin ILCs expressed receptors for several neurotransmitters and cytokines involved in immune regulation, including calcitonin gene-related peptide (CGRP) and IL-18 (30, 66). However, treatment of skin ILCs with CGRP in vitro did not significantly increase their expression of the type 2 cytokine IL-13 compared to the untreated control (Fig. S5A). Neuromedin U (NMU), another neurotransmitter capable of activating ILC2s of the lung and intestine (67, 68), also did not induce expression of IL-13 by skin ILCs (Fig. S5A). On the other hand, IL-18 stimulated skin ILCs to produce IL-13 (Fig. S5A), suggesting that it could be involved in the cold stress-induced skin ILC2 activation. Consistent with this notion, it was reported that the skin of SCD1−/− mice had significantly higher IL-18 expression than WT mice (69). The skin of WT mice raised at 4°C also had higher IL-18 expression than mice maintained at 22°C (Fig. S5B–C). Menthol-treated skin of WT mice had increased expression of IL-18 transcripts and proteins compared to control (ethanol)-treated skin, indicating that IL-18 was induced by stimulation of TRPM8 and could be important in mediating ILC2 activation (Fig. 5A–C). Correlating with this, nearly all skin ILCs of WT and SCD1−/− mice expressed IL-18 receptor (Fig. S5D).
Figure 5.
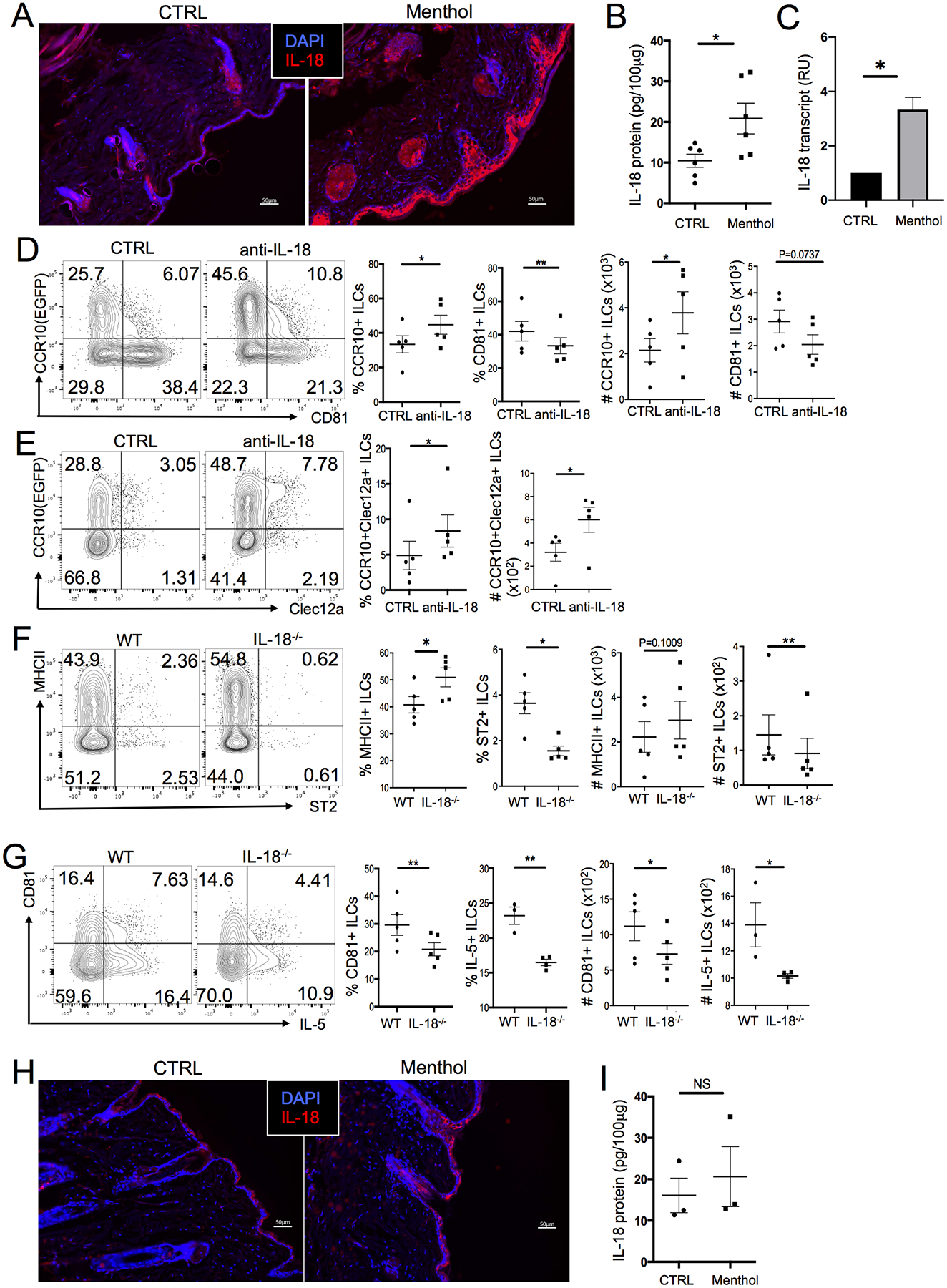
Stimulation of TRPM8 induces production of IL-18 that promotes activation of skin ILC2s. (A) Immunofluorescent microscopic imaging of anti-IL-18 antibody staining of menthol and control(ethanol)-treated skin sections of WT mice. (B) ELISA analysis of IL-18 in the menthol and control-treated skin of WT mice. (C) qRT-PCR analysis of IL-18 expression in the menthol and control-treated skin of WT mice. N =3 each. (D-E) CCR10 vs. CD81 (D) and Clec12a (E) expression in ILCs of the menthol-treated skin of WT mice injected with anti-IL-18 or isotype control antibodies. Mice carry a CCR10-knockout/EGFP-knockin (CCR10+/EGFP) allele. (F-G) MHCII and ST2 (F) and CD81 and IL-5 (G) expression in ILCs of the menthol and control-treated skin of WT or IL-18−/− mice. (H) Immunofluorescent microscopic images of anti-IL-18 antibody staining of menthol and control-treated skin sections of TRPM8−/− mice. (I) ELISA analysis of IL-18 in the menthol and control-treated skin of TRPM8−/− mice. One dot is of one sample. *P<0.05, **P<0.01, NS: no significant difference. Statistical significance was determined by unpaired T test for panels B, G and I (IL-5), paired T test for panels D-G (CD81).
To directly assess whether IL-18 was important in mediating skin ILC2 activation induced by TRPM8 stimulation, we injected IL-18-neutralizing antibodies into menthol-treated WT mice. Menthol-treated WT mice injected with anti-IL-18 antibodies had higher percentages of CCR10+ and CCR10+Clec12a+ ILCs and lower percentages of CD81+ ILCs in the skin than menthol-treated mice injected with isotype-matched control antibodies, indicating that neutralization of IL-18 reduced the skin ILC2 activation (Fig. 5D–E). Consistent with the neutralizing anti-IL-18 antibody results, menthol-treated skin of IL-18 KO (IL-18−/−) mice also had higher percentages of MHCII+ ILCs but lower percentages of CD81+ and ST2+ ILCs than menthol-treated skin of WT mice (Fig. 5F–G). The percentage of IL-5-producing skin ILC2s was also lower in menthol-treated IL-18−/− mice compared to menthol-treated WT mice (Fig. 5G). These results suggest that IL-18 is induced by menthol stimulation of TRPM8 for activation of skin ILC2s. Further supporting this notion, menthol treatment did not affect or only slightly increased levels of IL-18 in the skin of TRPM8−/− mice (Fig. 5H–I) compared to WT mice. Together, these results demonstrate that stimulation of TRPM8 leads to increased expression of IL-18, which in turn promotes activation of skin ILC2s.
TRPM8-induced activation of skin ILC2s is critical in promoting UCP1 expression in the skin
Activation of TRPM8 was reported to promote thermogenesis (70, 71). The activation of skin ILC2s by stimulation of TRPM8 suggests its involvement in regulation of thermal homeostasis. To address this, we assayed the menthol-treated skin for expression of uncoupling protein 1 (UCP1), a molecule whose major function is promoting thermogenesis (70, 72). Compared to control (ethanol)-treated skin, menthol-treated skin of WT mice had increased levels of UCP1 transcripts and protein (Fig. 6A–B). The menthol-treated skin also had increased thickness of the epidermal and dermal layers compared to the control-treated skin (Fig. 6C). Consistent with this, SCD1-KO mice raised at 30°C had reduced UCP1 expression and skin thickness compared to SCD1-KO mice raised at 22°C (Fig. S6A–B). In contrast, menthol treatment did not significantly upregulate UCP1 in the skin of TRPM8−/− and IL-18−/− mice (Fig. 6D–E, Fig. S6C–D). Interestingly, menthol-treated skin of TRPM8−/− but not IL-18−/− mice still had highly increased epidermal thickness compared to vehicle control-treated skin (Fig. 6F–G, Fig. S6E–F), correlating with the fact that the epidermis of TRPM8−/− mice still expressed functional TRPM8 (62, 65).
Figure 6.
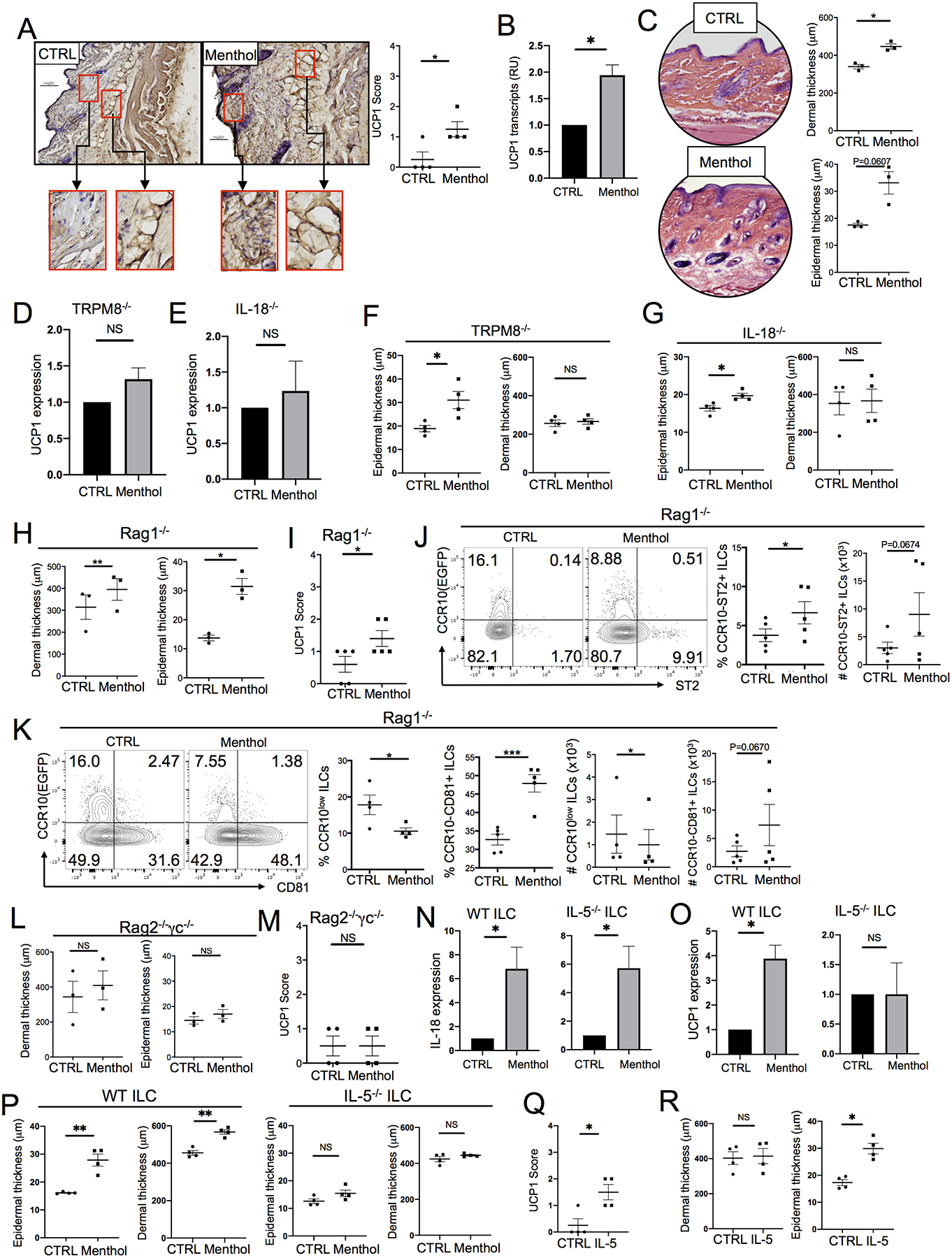
TRPM8-induced activation of skin ILC2s is critical in promoting thermogenesis in the skin. (A) IHC staining for the UCP1 expression in the menthol and control(ethanol)-treated skin of WT mice. (B) qRT-PCR analysis of UCP1 expression in the menthol and control-treated skin of WT mice. Normalized on β-actin. N=3. (C) H&E staining of the menthol and control-treated skin of WT mice and their epidermal and dermal thickness. (D-E) qRT-PCR analysis of UCP1 expression in the menthol and control-treated skin of TRPM8−/− (D) and IL-18−/− (E) mice. N=3 each. (F-G) The epidermal and dermal thickness of the menthol and control-treated skin of TRPM8−/− (F) and IL-18−/− (G) mice. (H) The epidermal and dermal thickness of the menthol and control-treated skin of Rag1−/− mice. (I) Levels of UCP1 staining intensities in the menthol and control (ethanol)-treated skin of Rag1−/− mice. (J-K) FC analysis of ST2 (J) or CD81 (K) vs. CCR10 expression in ILCs of the menthol and control-treated skin of Rag1−/− mice. (L) Epidermal and dermal thickness of the menthol and control-treated skin of Rag2−/−γc−/− mice. (M) Levels of the UCP1 staining intensities in the menthol and control-treated skin of Rag2−/−γc−/− mice. (N-O) qRT-PCR analysis of IL-18 (N) and UCP1 (O) expression in the menthol and control-treated skin of Rag2−/−γc−/− mice transferred with WT or IL-5−/− donor ILCs. N=4 for WT and 3 for IL-5−/− each. (P) The epidermal and dermal thickness of the menthol and control-treated skin of Rag2−/−γc−/− mice transferred with WT or IL-5−/− donor ILCs. (Q) Levels of UCP1 staining intensities (Q) and epidermal and dermal thickness (R) of rIL-5 (1μg for 4 days) and control (PBS,0.1% BSA)-treated skin of IL-18−/− mice. One dot is of one mouse. *P<0.05, **P<0.01. ***P<0.001, NS: no significant difference. Statistical significance was determined by unpaired T test for the panels A, I, M and Q, paired T test for the panels B-H,J-L, N-P, and R.
To further determine whether activation of ILC2s was involved in promoting the UCP1 upregulation and skin thickness increase, we analyzed the menthol vs. vehicle control skin of Rag1−/− and Rag2−/−γc−/−mice. Rag1−/− mice lack T and B cells while Rag2−/−γc−/− mice lack T and B cells as well as ILCs. As in WT mice, menthol treatment increased the thickness of the epidermal and dermal layers of Rag1−/− mice (Fig. 6H, Fig. S6G). In addition, there were higher anti-UCP1 staining intensities in the menthol-treated skin compared to the vehicle control-treated skin of Rag1−/− mice (Fig. 6I, Fig. S6H). Menthol-treated skin of Rag1−/− mice had more CCR10−ST2+ and CCR10−CD81+ ILCs than control-treated skin of Rag1−/− mice (Fig. 6J–K), suggesting that stimulation of TRPM8 led to the increased skin ILC2 activation in Rag1−/− mice. In contrast to WT or Rag1−/− mice, the menthol-treated skin of Rag2−/−γc−/− mice did not have increased thickness of epidermal and dermal layers or an increase in UCP1 expression (Fig. 6L–M, Fig. S6I–J). Together, these results suggest that activation of ILC2s by stimulation of TRPM8 is important in regulation of thermal homeostasis in the skin.
To confirm that lack of ILCs was responsible for the reduced inflammatory responses and UCP1 expression in the skin of Rag2−/−γc−/− mice in response to TRPM8 stimulation, we transferred WT or IL-5−/− ILCs into Rag2−/−γc−/− mice and then treated them with menthol. Both WT and IL-5−/− donor ILCs were activated in menthol-treated skin of Rag2−/−γc−/− recipients, as indicated by their increased CD81 and reduced CCR10 expression in comparison to those of control-treated skin of the Rag2−/−γc−/− recipients (Fig. S7A–B). Menthol-treated skin of Rag2−/−γc−/− mice receiving WT or IL-5−/− donor ILCs had increased IL-18 expression compared to their vehicle control-treated skin (Fig. 6N). However, menthol treatment significantly increased the UCP1 expression in the skin of Rag2−/−γc−/− mice receiving WT but not IL-5−/− donor ILCs (Fig. 6O, Fig. S7C). Associated with this, menthol treatment significantly increased the epidermal and dermal thickness of the skin of Rag2−/−γc−/− mice receiving WT but not IL-5−/− donor ILCs (Fig. 6P, Fig. S7D). These results indicate that activated ILC2s are important in induction of UCP1 expression in response to the TRPM8 stimulation by producing IL-5. Further supporting this, injection of rIL-5 into the skin of IL-18−/− mice could also increase UCP1 expression and epidermal thickness of the skin (Fig. 6Q–R, Fig. S7E–F).
DISCUSSION
As the external barrier tissue, the skin functions in maintaining thermal homeostasis and the body’s water and electrolyte balance. A high number of ILCs reside in the skin. How skin ILCs are regulated to help functions of the skin is not well understood. In this report, we found that stimulation of the TRPM8 cold-sensing pathway promotes activation of skin ILC2s for thermo-homeostatic regulation, revealing that skin-resident ILC2s are an integral component of thermo-homeostatic regulatory machinery (illustrated in Fig. 7).
Figure 7.
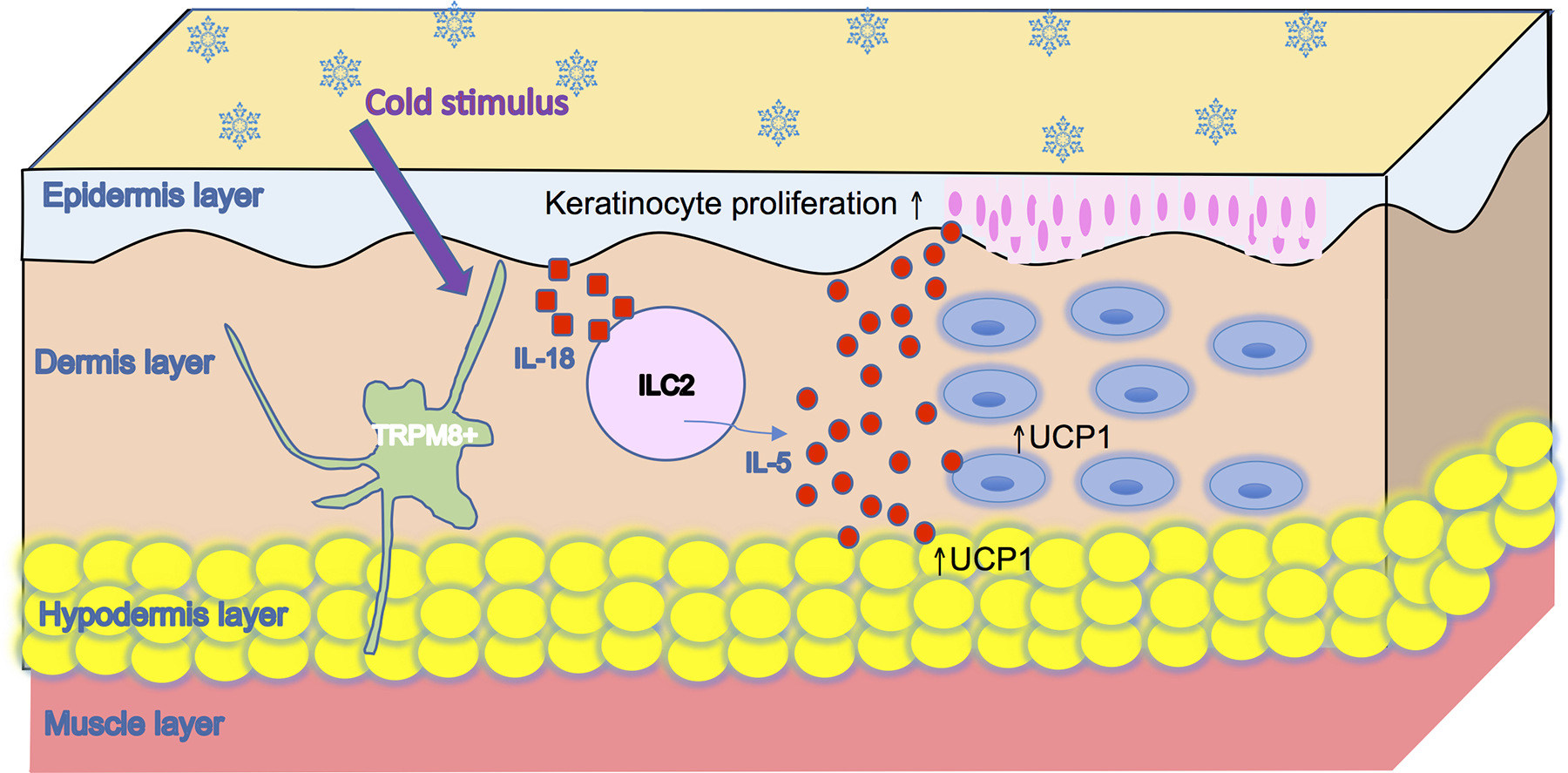
Schematic illustration of a neuroimmune axis that links TRPM8 sensing of environmental cold stress and activation of skin-resident ILC2s to the regulation of cutaneous thermal homeostasis. In response to an environmental cold stimulus, signals from activated TRPM8+ neurons induce increased production of IL-18, which in turn activates skin ILC2s. Effector cytokines produced by activated ILC2s (such as IL-5) promote UCP1 expression in adipocytes of the hypodermis and dermal cells (potentially fibroblasts).
It was well established that the cold-sensing receptor TRPM8 is involved in thermal regulation (73)(60, 62–64, 74). Activation of TRPM8 induces a series of cold defenses such as brown adipose tissue thermogenesis, shivering thermogenesis and cutaneous vasoconstriction, while disruption of TRPM8, through either gene knockout or pharmaceutical inhibition, leads to impaired cold tolerance (60, 61, 75, 76). However, the effector molecules and cells mediating the TRPM8-initiated thermogenesis process are not well defined. We found that IL-18 is a key molecular mediator induced by TRPM8 activation, which in turn activates skin ILC2s for production of type 2 cytokines to promote thermogenesis. Our findings fill a critical gap in the regulation of thermal homeostasis by demonstrating how sensory neurons and immune effector cells cooperate to support local tissue thermal homeostasis, which is important in maintaining biological functions from adequate blood supply to immune responses against infection (77–79). Considering that local metabolic conditions of the skin have a significant effect on the whole-body physiology (57, 77, 80), TRPM8-regulated activation of skin ILC2s might also contribute to systemic metabolic homeostatic regulation. Previous studies have found that impairment of either the TRPM8 or IL-18 pathways could lead to obesity and other metabolic symptoms (71, 81). It will be interesting to test whether skin ILC2s are involved in these processes.
Our findings also have significant clinical implications. The excessive activation of ILC2s in SCD1 KO mice suggests that the ambient temperature plays a role in inflammatory diseases of skin. This is consistent with clinical observations that various skin diseases, such as atopic dermatitis and allergic urticaria, display cold temperature sensitivities and seasonal changes of severity (82, 83). Inflammatory skin diseases, such as atopic dermatitis, are associated with a defective skin barrier (84, 85). In light of our findings, it will be interesting to test whether skin ILC2s are activated by cold-sensing neuron-derived signals as part of the pathogenesis of these diseases. In some patients with allergic urticaria, physical pressure is a major cause of the disease onset (86). Signals from activated touch neurons could be involved in their pathogenesis (87, 88). On the other hand, since metabolic conditions of the skin have an important role in systemic metabolic homeostasis, skin ILC2 activation could potentially be manipulated to treat metabolic diseases.
The detailed molecular mechanisms underlying TRPM8-promoted skin ILC2 activation need further investigation. Our study found that TRPM8 expressed on sensory neurons is only partially responsible for the skin ILC2 activation. TRPM8 expressed in keratinocytes could also be involved in sensing environmental temperature to promote the local ILC2 activation (65). How stimulation of TRPM8 induces IL-18 expression and cellular sources of IL-18 contributing to skin ILC2 activation remain to be dissected. In this regard, it was recently reported that neuron-derived IL-18 in the gut plays an important role in immunity against Salmonella infection (89).
SCD1 is crucial in maintaining thermal homeostasis and its impairment leads to increased energy expenditure (54, 57). SCD1 KO mice have severe skin inflammation although the immune basis of the skin inflammation has not been identified (54, 57). Our findings suggest that enhanced ILC2 activation might be a major cause of skin inflammation symptoms in these mice. However, ILC2-mediated skin inflammation is likely a secondary consequence since the primary goal of the skin ILC2 activation is to increase thermogenesis to compensate for increased heat loss in these mice. Consistent with this notion, other strains of mice with a defective skin permeability barrier, such as mice deficient in the cholesterol esterification enzyme acyl-CoA:cholesterol acyltransferase-1, have similar skin inflammatory symptoms as SCD1 KO mice (90). It is likely a general paradigm that cold-sensing receptor-derived signals activate skin ILC2s to promote energy expenditure but also cause skin inflammatory symptoms. Further supporting this notion, nicotine-induced activation of TRPA1, another cold-sensing receptor, has been implicated in skin inflammation in some smokers who used nicotine replacement patches (91). Understanding how different neuron-derived sensing receptors are involved in homeostatic and inflammatory regulation of the skin is worth further study.
Consistent with the notion that local environments control functions of tissue-resident ILCs, regulation of localization and function of ILCs in the skin are significantly different from those of mucosal barrier tissues such as the lung and intestine. We have found that under homeostatic conditions, skin-homing CCR10+ ILCs are imprinted in sLNs for their preferential migration into skin (29). ILC2s of the healthy skin play an important role in homeostatic regulation of T cells and mast cells (28, 29). However, upon different stimulations, CCR10+ skin ILC2s differentiate into active effector cells to participate in specific functions (29). These findings suggest that CCR10+ ILC2s of the healthy skin are plastic, which might be important for their functions in helping to maintain local tissue homeostasis in response to changing ambient environments. Further supporting this notion, a recent report of single-cell RNA-sequencing profiles of skin ILCs of mice found that they are at a naive and effector ILC2 state under homeostatic conditions but could transit into an active ILC3-like state upon treatment with IL-23 or imiquimod to induce psoriasis-like inflammation (92).
Unlike homeostatic ILC2s in the healthy skin, activated skin ILC2s have increased CD81 and reduced Clec12a expression. CD81 is likely involved in enhancing ILC2 function since it can form a partnership with various receptors to help signal transduction and cell activation (45, 46). On the other hand, Clec12a was reported to transduce immune inhibitory signals (47), and its reduction in activated skin ILC2s might allow them to more effectively produce effector molecules. However, exact roles and importance of these molecules in skin ILC2 activation and homeostasis are yet to be determined.
MATERIALS AND METHODS
Study Design
The goal of this study was to dissect the mechanisms underlying activation of skin-resident ILCs by the cold-sensing receptor TRPM8 and the participation of ILCs in regulation of cutaneous thermal homeostasis in mice. Multiple strains of mice with defined mutations were used. Individual experiments used mice of the same sex and age with different genotypes. Both female and male mice were used.
Mice
CCR10-knockout/EGFP-knockin mice were previously described (93). Rag1−/− (002216), IL-18−/− (004130), TRPM8−/− (008198), SCD1−/− (006201) and C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Rag2−/−γc−/− mice (4111) were purchased from Taconic Biosciences. Rag1−/−, TRPM8−/− and IL-18−/− mice were crossed with CCR10-knockout/EGFP-knockin mice to introduce one CCR10-knockout/EGFP-knockin allele (CCR10+/EGFP) for purpose of reporting CCR10 expression with EGFP. All mice were on the C57BL/6 (CD45.2) background and raised in specific pathogen-free conditions unless indicated otherwise. Comparison was made between different knockout mice and their WT littermates when possible. All animal experiments were approved by Institutional Animal Care and Use Committees of the Pennsylvania State University and University of Texas Health Science Center at San Antonio.
Reagents
Anti-mouse CD90.2 (53–2.1), anti-mouse MHCII (M5–114.15.2), anti-mouse IL-17A (TC11–18H10.1), anti-mouse CD45.1 (A20), anti-mouse CD45.2 (104), anti-mouse CD3ε (145–2C11 or 17A2)), anti-mouse CD81(Eat-2), anti-mouse IL-5 (TRFK5), anti-mouse ST2 (DIH9), anti-mouse Clec12a (5D3/CLEC12A),Purified anti-NF-H(Poly28226) and anti-mouse granzyme C (SFC1D8) were purchased from BioLegend (San Diego, CA). Anti-mouse NK1.1 (PK136), anti-mouse α4β7 (DATK32) and PE-CF594-Streptavidin were purchased from BD Biosciences (San Jose, CA). Anti-mouse IL-13(eBio13A), anti-mouse IFNγ (XMG1.2), was purchased from eBioscience (San Diego, CA). A cocktail of antibodies against the lineage (Lin) markers [CD5, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), 7–4, and Ter-119 antibodies] were purchased from Miltenyi Biotec (San Diego, CA). IL-18 neutralization antibody for depletion (YIGIF74–1G7) and rat IgG2a isotype control (2A3) were purchased from Bio × Cell (West Lebanon, NH). Mouse IL-18 ELISA Kit was purchased from Invitrogen (Carlsbad, CA). Pierce BCA Protein Assay Kit and Halt Protease Inhibitor Cocktail were purchased from Thermo Fisher Scientific (Waltham, MA). Anti-mouse IL-18 antibody (12E7.1) for IF staining was purchased from EMD Millipore Corp (Burlington, MA). Mouse IgM isotype control, rat anti-mouse IgM antibody and goat anti-chicken IgY were purchased from Invitrogen (Carlsbad, CA). ImmPRESS Excel Amplified Polymer Kit was purchased from Vector Laboratories Inc (Burlingame, CA). TRIzol Reagent was purchased from Invitrogen (Carlsbad, CA). PrimeScript RT reagent kit with gDNA Eraser was purchased from TaKaRa (San Jose, CA). PowerUp SYBR Green Master Mix was purchased from Thermo Fisher Scientific (Waltham, MA). Oleic acid was purchased from Sigma-Aldrich Inc, (St. Louis, MO).
Cell isolation, staining, flow cytometric analysis
Isolation of immune cells from the skin was performed as previously described (94)(29). Briefly, mouse skin was excised, trimmed of subcutaneous fat, minced, and digested with a cocktail of collagenases type I and IV (Worthington, Lakewood, NJ) and hyaluronidase type I (Sigma-Aldrich, St. Louis, MO) to generate a single cell suspension. Mononuclear cells were enriched from the cell preparations using a Percoll gradient (40%/80%) centrifugation and used for cell analysis. For surface molecule staining, cells were incubated with fluorescently labeled antibodies in a staining buffer for 25 min at 4°C and, if necessary, with secondary antibodies for 15 min at 4°C. For intracellular molecular staining of cytokines and granzyme C, cells were stimulated with PMA (1 μg/mL) and ionomycin (2 μg/mL) (Millipore Sigma) for 3 hours in presence of brefeldin A (1 μg/mL) and then stained first for extracellular proteins. The cells were then fixed with 4% paraformaldehyde, permeabilized with a permeabilization buffer (eBioscience), and stained with antibodies to the intracellular proteins. Flow cytometric analyses were performed on FC500 (Beckman Coulter) or BD Fortessa LSRII (BD Biosciences). Data were analyzed with FlowJo software (FlowJo LLC., Ashland, OR).
Microarray analysis
Microarray analysis was performed similarly as previously described (95). Skin cells enriched in mononuclear cells were stained and sorted using a gating strategy as shown in Fig. S1A with a BD Cytopeia Influx cytometer (BD Biosciences) to yield populations of CCR10(EGFP)+ and CCR10(EGFP)− ILCs (from CCR10+/EGFP mice) and CCR10(EGFP)low and CCR10(EGFP)− ILCs (from Rag1−/−CCR10+/EGFP mice). The different populations of ILCs were sorted directly into TRIzol (Invitrogen). The yield was about 0.5–2 × 104 cells per mouse with ~95% purity. Independent duplicates or triplicates were sorted. RNA processing and microarray analysis with the Affymetrix MoGene 1.0 ST array was done at the ImmGen processing center (http://www.immgen.org/Protocols/ImmGen Cell prep). Raw data were normalized by the robust multi-array average method with quantile normalization and background correction (Expression File Creator module of Gene Pattern). PCA analysis was performed by Python. Average gene expression levels of CCR10(EGFP)− or CCR10low skin ILCs from CCR10+/EGFP or Rag1−/−CCR10+/EGFP mice were normalized to the levels of CCR10(EGFP)+ skin ILCs from CCR10+/EGFP mice and presented as heat maps generated by Excel.
In vivo antibody neutralization of IL-18
The experiment was performed similarly as previously reported (96). Mice were injected retro-orbitally with anti-IL-18 or isotype-matched control antibodies (15mg/kg).
Topical menthol treatment of mice
600 μl L-menthol (10% w/v in ethanol) were applied to the dorsal skin of mice once a day. Ten days - 2 weeks after starting the treatment, mice were euthanized for analysis. L-menthol was purchased from Sigma-Aldrich (St. Louis, MO).
Immunofluorescence staining of skin sections
Skin samples were fixed with 4% paraformaldehyde overnight, embedded in O.C.T compound and frozen. Six μm-thick sections were cut of the frozen skin on a cryostat. Skin sections were blocked for 2 hours in 10% goat serum in PBT (0.3% TritonX-100 in PBS) at room temperature (RT) and then incubated in 200μl anti-IL-18 or anti-NEFH antibody solution at 4°C in a humid chamber overnight. Next day, the skin sections were washed with PBT and then incubated in 300μl solution of fluorescently labeled goat anti-mouse IgM antibody or goat anti-chicken IgY for 2 hours at RT in a dark humid chamber. The stained skin sections were washed with PBT, stained with DAPI for one minute, applied with 2–3 drops of Mount, covered with coverslips and then let dry for 1 hour. Nail polish was applied to seal edges of coverslips. Stained skin sections were imaged under a Keyence microscope.
Bone marrow transplant
Bone marrow transplant experiment was performed similarly as previously reported (29). Bone marrow cells from CD45.1/2+ CCR10+/EGFP mice were transferred into irradiated CD45.2/2+ SCD1+/+ and SCD1−/− mice. Six-eight weeks after the transfer, mice were euthanized for analysis of donor-derived immune cells in the skin.
ILC transfer experiment
Skin CD3−Lin− ILCs sorted from wild-type mice and IL-5−/− mice were transferred into Rag2−/−γc−/− mice. Two weeks after the transfer, mice were treated with 600 μl L-menthol (10% w/v in ethanol) or vesicle control (ethanol) once a day for additional 2 weeks.
Hematoxylin and eosin staining of skin sections
The skin samples were fixed in 4% paraformaldehyde and embedded in paraffin. Samples were sectioned and stained with hematoxylin and eosin (H&E) by the animal diagnostic laboratory in Penn State University and the pathology laboratory in University of Texas Health Science Center at San Antonio.
Immunohistochemistry staining and imaging analysis of skin sections for UCP1
Immunohistochemistry (IHC) was performed using a standard protocol provided by the manufacturer (ImmPRESS Excel Amplified Polymer Kit, Vector Laboratories Inc). Briefly, 5 μm skin sections were deparaffinized and rehydrated. The antigen was recovered by microwave heating in Tris-EDTA buffer (pH=9.0). Endogenous peroxidase activity was blocked, and then the tissues were incubated with rabbit anti-UCP1 (IgG, Abcam) or isotype-matched control antibody at 4°C overnight. Next day, the sections were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG polymer antibodies, followed by the DAB staining (Vector Laboratories Inc). Stained sections were imaged under a Keyence microscope equipped with BZ-X800 Viewer and BZ-X800 Analyzer. At least 3 different fields were selected from a skin section and 4–5 sections per animal were evaluated to obtain a mean value. The IHC score was calculated based on the histogram profile generated for each image using Image J with a IHC Profiler plugin to evaluate staining intensities for UCP1 in skin sections, performed as reported (97, 98). The score was presented in a semiquantitative way (high positive, positive, low positive and negative, from 3–0).
ELISA
Skin samples were homogenized in ice-cold PBS containing a protease-inhibitor cocktail. The homogenates were centrifuged for 20 minutes at 10,000g to remove debris and insoluble materials. Aliquots of the supernatants were assayed for total protein contents by the BCA method and assayed for IL-18 by an ELISA kit according to the manufacturer’s instruction (EMD Millipore).
RT-PCR
Skin samples were homogenized in ice-cold TRIzol (Invitrogen) and RNA was extracted following manufacturer’s protocol. Residual genomic DNA was removed and the reverse transcription reaction was performed using the PrimeScript RT reagent kit with gDNA Eraser (TaKaRa Bio). UCP1 primers (sense, 5’-CGACTCAGTCCAAGAGTACTTCTCTTC-3’ ; antisense, 5’-GCCGGCTGAGATCTTGTTTC-3’), IL-18 primers (sense, 5’-GCCTCAAACCTTCCAAATCA-3’; antisense, 5’-TGGATCCATTTCCTCAAAGG-3’) and β-actin primers (sense, 5’-AGGCCCAGAGCAAGAGAGG-3’; antisense, 5’-TACATGGCTGGGGTGTTGAA-3’) were used for RT-PCR. The RT-PCR was performed using Power SYBR Green Master Mix and protocol from Thermo Fisher Scientific.
Cold (4°C) and thermoneutral (30°C) treatment
Mice were housed in a Memmert constant climate chamber HPP for 2 weeks. The temperature was controlled as indicated. The humidity was 50% and daylight/dark cycle was 12h/12h.
Statistical analyses
Paired or unpaired two-tailed Student’s T tests were used for statistical analysis. For comparison of cell numbers in the skin, the ratio paired T test was used except for the experiments in which the sample numbers of the compared groups were different. P < 0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS:
We thank the staff of the flow cytometry facility and the animal diagnostic laboratory at Pennsylvania State University and of the flow cytometry facility and pathology laboratory of the University of Texas Health Science Center at San Antonio for excellent technical support.
FUNDING:
Research reported in this publication was supported by the National Institute of Arthritis, Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers R01AR070887 and AR064831 (to N.X.) and R01AR073364 (through a subaward to N.X). The microarray data was generated by the Immunological Genome Project Consortium (5R24AI072073, PI: C. Benoist). Y.S.K is supported by National Institutes of Health grants (R01DE026677 and R01DE031477), UTHSCSA startup fund and a Rising STAR Award from University of Texas system.
Footnotes
SUPPLEMENTARY MATERIALS
COMPETING INTERESTS: The authors declare no competing interests.
DATA AND MATERIALS AVAILABILITY:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The microarray data was deposited in the NCBI GEO repository under accession number GSE199703. CCR10-knockout/EGFP-knockin mice are available through an MTA upon request to the corresponding author.
REFERENCES AND NOTES
- 1.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E, Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol 13, 145–149 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H, Innate Lymphoid Cells: 10 Years On. Cell 174, 1054–1066 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Colonna M, Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 48, 1104–1117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Artis D, Spits H, The biology of innate lymphoid cells. Nature 517, 293–301 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, Huntington ND, Belz GT, Carotta S, Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med 211, 1733–1740 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, Domingues RG, Veiga-Fernandes H, Arnold SJ, Busslinger M, Dunay IR, Tanriver Y, Diefenbach A, Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A, A committed precursor to innate lymphoid cells. Nature 508, 397–401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M, Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38, 769–781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, Marvel J, Yoh K, Takahashi S, Prinz I, de Bernard S, Buffat L, Walzer T, T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med 211, 563–577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S, Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie AN, Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM, Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A 107, 11489–11494 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, te Velde AA, Fokkens WJ, van Drunen CM, Spits H, The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 37, 649–659 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, Voehringer D, Busslinger M, Diefenbach A, The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 37, 634–648 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, Barlow JL, Neill DR, Panova V, Koch U, Radtke F, Hardman CS, Hwang YY, Fallon PG, McKenzie AN, Transcription factor RORalpha is critical for nuocyte development. Nat Immunol 13, 229–236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker JA, McKenzie AN, Development and function of group 2 innate lymphoid cells. Curr Opin Immunol 25, 148–155 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F, Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity 37, 463–474 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, Eberl G, Di Santo JP, Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29, 958–970 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M, A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457, 722–725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D, Dalod M, Littman DR, Vivier E, Tomasello E, Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol 10, 75–82 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H, Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol 10, 66–74 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Moretta L, Locatelli F, Innate lymphoid cells in normal and disease: An introductory overview. Immunol Lett 179, 1 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Ebbo M, Crinier A, Vely F, Vivier E, Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol 17, 665–678 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Klose CS, Artis D, Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol 17, 765–774 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Gronke K, Kofoed-Nielsen M, Diefenbach A, Innate lymphoid cells, precursors and plasticity. Immunol Lett 179, 9–18 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Kim BS, Artis D, Group 2 innate lymphoid cells in health and disease. Cold Spring Harb Perspect Biol 7, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Zhao L, Xu M, Xiong N, Establishment and function of tissue-resident innate lymphoid cells in the skin. Protein Cell 8, 489–500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, Mitchell AJ, Tay SS, Jain R, Forbes-Blom E, Chen X, Tong PL, Bolton HA, Artis D, Paul WE, Fazekas de St Groth B, Grimbaldeston MA, Le Gros G, Weninger W, Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol 14, 564–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Hu S, Zhao L, Kaplan DH, Perdew GH, Xiong N, Selective programming of CCR10(+) innate lymphoid cells in skin-draining lymph nodes for cutaneous homeostatic regulation. Nat Immunol 17, 48–56 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, Vaka D, Eckalbar WL, Molofsky AB, Erle DJ, Locksley RM, Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol 19, 1093–1099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salimi M, Ogg G, Innate lymphoid cells and the skin. BMC Dermatol 14, 18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim BS, Innate lymphoid cells in the skin. J Invest Dermatol 135, 673–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi T, Ricardo-Gonzalez RR, Moro K, Skin-Resident Innate Lymphoid Cells - Cutaneous Innate Guardians and Regulators. Trends Immunol 41, 100–112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebert LM, Meuter S, Moser B, Homing and function of human skin gammadelta T cells and NK cells: relevance for tumor surveillance. J Immunol 176, 4331–4336 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Restori KH, Xu M, Song EH, Zhao L, Hu S, Lyu P, Wang WB, Xiong N, Preferential Perinatal Development of Skin-Homing NK1.1(+) Innate Lymphoid Cells for Regulation of Cutaneous Microbiota Colonization. iScience 23, 101014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi T, Voisin B, Kim DY, Kennedy EA, Jo JH, Shih HY, Truong A, Doebel T, Sakamoto K, Cui CY, Schlessinger D, Moro K, Nakae S, Horiuchi K, Zhu J, Leonard WJ, Kong HH, Nagao K, Homeostatic Control of Sebaceous Glands by Innate Lymphoid Cells Regulates Commensal Bacteria Equilibrium. Cell 176, 982–997 e916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, Nutman TB, Urban JF Jr., Wang J, Ramalingam TR, Bhandoola A, Wynn TA, Belkaid Y, Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343, 432–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rak GD, Osborne LC, Siracusa MC, Kim BS, Wang K, Bayat A, Artis D, Volk SW, IL-33-Dependent Group 2 Innate Lymphoid Cells Promote Cutaneous Wound Healing. J Invest Dermatol 136, 487–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, Nakanishi K, Yamanishi K, Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci U S A 110, 13921–13926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Van Voorhees AS, Comeau MR, Artis D, TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med 5, 170ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST, McKenzie AN, Fallon PG, Ogg GS, A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med 210, 2939–2950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, Klenerman P, Ogg G, Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol 133, 1184–1194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M, Immunological Genome C, Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 16, 306–317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, Levy M, Salame TM, Weiner A, David E, Shapiro H, Dori-Bachash M, Pevsner-Fischer M, Lorenzo-Vivas E, Keren-Shaul H, Paul F, Harmelin A, Eberl G, Itzkovitz S, Tanay A, Di Santo JP, Elinav E, Amit I, The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell 166, 1231–1246 e1213 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Levy S, Todd SC, Maecker HT, CD81 (TAPA-1): a molecule involved in signal transduction and cell adhesion in the immune system. Annu Rev Immunol 16, 89–109 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Levy S, Function of the tetraspanin molecule CD81 in B and T cells. Immunol Res 58, 179–185 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Neumann K, Castineiras-Vilarino M, Hockendorf U, Hannesschlager N, Lemeer S, Kupka D, Meyermann S, Lech M, Anders HJ, Kuster B, Busch DH, Gewies A, Naumann R, Gross O, Ruland J, Clec12a is an inhibitory receptor for uric acid crystals that regulates inflammation in response to cell death. Immunity 40, 389–399 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Roediger B, Weninger W, Group 2 innate lymphoid cells in the regulation of immune responses. Adv Immunol 125, 111–154 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, Chawla A, Locksley RM, Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 210, 535–549 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, Yun K, Locksley RM, Chawla A, Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 160, 74–87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashiguchi M, Kashiwakura Y, Kojima H, Kobayashi A, Kanno Y, Kobata T, IL-33 activates eosinophils of visceral adipose tissue both directly and via innate lymphoid cells. Eur J Immunol 45, 876–885 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, Thome JJ, Farber DL, Lutfy K, Seale P, Artis D, Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paton CM, Ntambi JM, Biochemical and physiological function of stearoyl-CoA desaturase. Am J Physiol Endocrinol Metab 297, E28–37 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sampath H, Ntambi JM, Role of stearoyl-CoA desaturase-1 in skin integrity and whole body energy balance. J Biol Chem 289, 2482–2488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng Y, Eilertsen KJ, Ge L, Zhang L, Sundberg JP, Prouty SM, Stenn KS, Parimoo S, Scd1 is expressed in sebaceous glands and is disrupted in the asebia mouse. Nat Genet 23, 268–270 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Lee SH, Dobrzyn A, Dobrzyn P, Rahman SM, Miyazaki M, Ntambi JM, Lack of stearoyl-CoA desaturase 1 upregulates basal thermogenesis but causes hypothermia in a cold environment. J Lipid Res 45, 1674–1682 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Sampath H, Flowers MT, Liu X, Paton CM, Sullivan R, Chu K, Zhao M, Ntambi JM, Skin-specific deletion of stearoyl-CoA desaturase-1 alters skin lipid composition and protects mice from high fat diet-induced obesity. J Biol Chem 284, 19961–19973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakamizo S, Honda T, Adachi A, Nagatake T, Kunisawa J, Kitoh A, Otsuka A, Dainichi T, Nomura T, Ginhoux F, Ikuta K, Egawa G, Kabashima K, High fat diet exacerbates murine psoriatic dermatitis by increasing the number of IL-17-producing gammadelta T cells. Sci Rep 7, 14076 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Binczek E, Jenke B, Holz B, Gunter RH, Thevis M, Stoffel W, Obesity resistance of the stearoyl-CoA desaturase-deficient (scd1−/−) mouse results from disruption of the epidermal lipid barrier and adaptive thermoregulation. Biol Chem 388, 405–418 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A, A TRP channel that senses cold stimuli and menthol. Cell 108, 705–715 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Tajino K, Matsumura K, Kosada K, Shibakusa T, Inoue K, Fushiki T, Hosokawa H, Kobayashi S, Application of menthol to the skin of whole trunk in mice induces autonomic and behavioral heat-gain responses. Am J Physiol Regul Integr Comp Physiol 293, R2128–2135 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, Jordt SE, Julius D, The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 448, 204–208 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Colburn RW, Lubin ML, Stone DJ Jr., Wang Y, Lawrence D, D’Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N, Attenuated cold sensitivity in TRPM8 null mice. Neuron 54, 379–386 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Dhaka A, Murray AN, Mathur J, Earley TJ, Petrus MJ, Patapoutian A, TRPM8 is required for cold sensation in mice. Neuron 54, 371–378 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Bidaux G, Borowiec AS, Gordienko D, Beck B, Shapovalov GG, Lemonnier L, Flourakis M, Vandenberghe M, Slomianny C, Dewailly E, Delcourt P, Desruelles E, Ritaine A, Polakowska R, Lesage J, Chami M, Skryma R, Prevarskaya N, Epidermal TRPM8 channel isoform controls the balance between keratinocyte proliferation and differentiation in a cold-dependent manner. Proc Natl Acad Sci U S A 112, E3345–3354 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, Li Y, Zhang S, Ho J, Davis BM, Albers KM, Kaplan DH, Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 178, 919–932 e914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, Hofree M, Cuoco MS, Rodman C, Farouq D, Haas BJ, Tickle TL, Trombetta JJ, Baral P, Klose CSN, Mahlakoiv T, Artis D, Rozenblatt-Rosen O, Chiu IM, Levy BD, Kowalczyk MS, Regev A, Kuchroo VK, The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, Monticelli LA, Moriyama S, Putzel GG, Rakhilin N, Shen X, Kostenis E, Konig GM, Senda T, Carpenter D, Farber DL, Artis D, The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Flowers MT, Paton CM, O’Byrne SM, Schiesser K, Dawson JA, Blaner WS, Kendziorski C, Ntambi JM, Metabolic changes in skin caused by Scd1 deficiency: a focus on retinol metabolism. PLoS One 6, e19734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma S, Yu H, Zhao Z, Luo Z, Chen J, Ni Y, Jin R, Ma L, Wang P, Zhu Z, Li L, Zhong J, Liu D, Nilius B, Zhu Z, Activation of the cold-sensing TRPM8 channel triggers UCP1-dependent thermogenesis and prevents obesity. J Mol Cell Biol 4, 88–96 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Reimundez A, Fernandez-Pena C, Garcia G, Fernandez R, Ordas P, Gallego R, Pardo-Vazquez JL, Arce V, Viana F, Senaris R, Deletion of the Cold Thermoreceptor TRPM8 Increases Heat Loss and Food Intake Leading to Reduced Body Temperature and Obesity in Mice. J Neurosci 38, 3643–3656 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Enerback S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, Kozak LP, Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387, 90–94 (1997). [DOI] [PubMed] [Google Scholar]
- 73.Tan CL, Knight ZA, Regulation of Body Temperature by the Nervous System. Neuron 98, 31–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dhaka A, Earley TJ, Watson J, Patapoutian A, Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J Neurosci 28, 566–575 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Almeida MC, Hew-Butler T, Soriano RN, Rao S, Wang W, Wang J, Tamayo N, Oliveira DL, Nucci TB, Aryal P, Garami A, Bautista D, Gavva NR, Romanovsky AA, Pharmacological blockade of the cold receptor TRPM8 attenuates autonomic and behavioral cold defenses and decreases deep body temperature. J Neurosci 32, 2086–2099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gavva NR, Davis C, Lehto SG, Rao S, Wang W, Zhu DX, Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol Pain 8, 36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Romanovsky AA, Skin temperature: its role in thermoregulation. Acta Physiol (Oxf) 210, 498–507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moriyama M, Ichinohe T, High ambient temperature dampens adaptive immune responses to influenza A virus infection. Proc Natl Acad Sci U S A 116, 3118–3125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Foxman EF, Storer JA, Vanaja K, Levchenko A, Iwasaki A, Two interferon-independent double-stranded RNA-induced host defense strategies suppress the common cold virus at warm temperature. Proc Natl Acad Sci U S A 113, 8496–8501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kruse V, Neess D, Faergeman NJ, The Significance of Epidermal Lipid Metabolism in Whole-Body Physiology. Trends Endocrinol Metab 28, 669–683 (2017). [DOI] [PubMed] [Google Scholar]
- 81.Netea MG, Joosten LA, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, Tack CJ, van Krieken H, Kim SH, Stalenhoef AF, van de Loo FA, Verschueren I, Pulawa L, Akira S, Eckel RH, Dinarello CA, van den Berg W, van der Meer JW, Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med 12, 650–656 (2006). [DOI] [PubMed] [Google Scholar]
- 82.Engebretsen KA, Johansen JD, Kezic S, Linneberg A, Thyssen JP, The effect of environmental humidity and temperature on skin barrier function and dermatitis. J Eur Acad Dermatol Venereol 30, 223–249 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Maltseva N, Borzova E, Fomina D, Bizjak M, Terhorst-Molawi D, Kosnik M, Kulthanan K, Meshkova R, Thomsen SF, Maurer M, Committee C-CS, Cold urticaria - What we know and what we do not know. Allergy 76, 1077–1094 (2021). [DOI] [PubMed] [Google Scholar]
- 84.Kim BE, Leung DYM, Significance of Skin Barrier Dysfunction in Atopic Dermatitis. Allergy Asthma Immunol Res 10, 207–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Segre JA, Epidermal barrier formation and recovery in skin disorders. J Clin Invest 116, 1150–1158 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abajian M, Schoepke N, Altrichter S, Zuberbier T, Maurer M, Physical urticarias and cholinergic urticaria. Immunol Allergy Clin North Am 34, 73–88 (2014). [DOI] [PubMed] [Google Scholar]
- 87.Abraira VE, Ginty DD, The sensory neurons of touch. Neuron 79, 618–639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ranade SS, Syeda R, Patapoutian A, Mechanically Activated Ion Channels. Neuron 87, 1162–1179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jarret A, Jackson R, Duizer C, Healy ME, Zhao J, Rone JM, Bielecki P, Sefik E, Roulis M, Rice T, Sivanathan KN, Zhou T, Solis AG, Honcharova-Biletska H, Velez K, Hartner S, Low JS, Qu R, de Zoete MR, Palm NW, Ring AM, Weber A, Moor AE, Kluger Y, Nowarski R, Flavell RA, Enteric Nervous System-Derived IL-18 Orchestrates Mucosal Barrier Immunity. Cell 180, 50–63 e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yagyu H, Kitamine T, Osuga J, Tozawa R, Chen Z, Kaji Y, Oka T, Perrey S, Tamura Y, Ohashi K, Okazaki H, Yahagi N, Shionoiri F, Iizuka Y, Harada K, Shimano H, Yamashita H, Gotoda T, Yamada N, Ishibashi S, Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J Biol Chem 275, 21324–21330 (2000). [DOI] [PubMed] [Google Scholar]
- 91.Talavera K, Gees M, Karashima Y, Meseguer VM, Vanoirbeek JA, Damann N, Everaerts W, Benoit M, Janssens A, Vennekens R, Viana F, Nemery B, Nilius B, Voets T, Nicotine activates the chemosensory cation channel TRPA1. Nat Neurosci 12, 1293–1299 (2009). [DOI] [PubMed] [Google Scholar]
- 92.Bielecki P, Riesenfeld SJ, Hutter JC, Torlai Triglia E, Kowalczyk MS, Ricardo-Gonzalez RR, Lian M, Amezcua Vesely MC, Kroehling L, Xu H, Slyper M, Muus C, Ludwig LS, Christian E, Tao L, Kedaigle AJ, Steach HR, York AG, Skadow MH, Yaghoubi P, Dionne D, Jarret A, McGee HM, Porter CBM, Licona-Limon P, Bailis W, Jackson R, Gagliani N, Gasteiger G, Locksley RM, Regev A, Flavell RA, Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature 592, 128–132 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jin Y, Xia M, Sun A, Saylor CM, Xiong N, CCR10 is important for the development of skin-specific gammadeltaT cells by regulating their migration and location. J Immunol 185, 5723–5731 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xia M, Hu S, Fu Y, Jin W, Yi Q, Matsui Y, Yang J, McDowell MA, Sarkar S, Kalia V, Xiong N, CCR10 regulates balanced maintenance and function of resident regulatory and effector T cells to promote immune homeostasis in the skin. J Allergy Clin Immunol 134, 634–644 e610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Narayan K, Sylvia KE, Malhotra N, Yin CC, Martens G, Vallerskog T, Kornfeld H, Xiong N, Cohen NR, Brenner MB, Berg LJ, Kang J, Immunological Genome Project C, Intrathymic programming of effector fates in three molecularly distinct gammadelta T cell subtypes. Nat Immunol 13, 511–518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cohen TS, Boland ML, Boland BB, Takahashi V, Tovchigrechko A, Lee Y, Wilde AD, Mazaitis MJ, Jones-Nelson O, Tkaczyk C, Raja R, Stover CK, Sellman BR, aureus S Evades Macrophage Killing through NLRP3-Dependent Effects on Mitochondrial Trafficking. Cell Rep 22, 2431–2441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Potts SJ, Krueger JS, Landis ND, Eberhard DA, Young GD, Schmechel SC, Lange H, Evaluating tumor heterogeneity in immunohistochemistry-stained breast cancer tissue. Lab Invest 92, 1342–1357 (2012). [DOI] [PubMed] [Google Scholar]
- 98.Varghese F, Bukhari AB, Malhotra R, De A, IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One 9, e96801 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The microarray data was deposited in the NCBI GEO repository under accession number GSE199703. CCR10-knockout/EGFP-knockin mice are available through an MTA upon request to the corresponding author.