

Abstract
Objectives
Despite remarkable advances in the treatment of non‐small cell lung cancer (NSCLC) with anti‐programmed death (PD)‐1 therapy; only a fraction of patients derives durable clinical benefit. In this study, we investigated whether the differentiation status of systemic CD8+ T cells predicts the outcome of PD‐1 blockade in NSCLC.
Methods
We carried out a prospective study on a total of 77 NSCLC patients receiving anti‐PD‐1 blockers, among which 47 patients were assigned as a discovery cohort and 30 patients as a validation cohort. Peripheral blood samples were obtained at baseline and upon multiple therapy cycles and analyzed by multi‐parameter flow cytometry.
Results
We found that a higher baseline ratio of PD‐1+ early effector memory CD8+ T cells (CD28+CD27−CD45RO+, TEEM) to PD‐1+ effector CD8+ T cells (CD28−CD27−CD45RO−, TE) delineated responders to PD‐1 blockade from progressors and was associated with prolonged progression‐free survival (PFS) and durable clinical benefit. Moreover, PD‐1+CD8 TEEM cells exhibited early responses after anti‐PD‐1 therapy and was the major fraction of cycling PD‐1+Ki67+CD8+ T cells to expand specifically with positive impact on PFS.
Conclusion
These findings provide insights into how the baseline differentiation status of the peripheral immune system determines responses to PD‐1‐targeted therapies.
Keywords: biomarkers, early‐like differentiation, non‐small‐cell lung cancer, PD‐1 blockade, T cells
In this study, we examined the association between PD‐1 blockade and the differentiation status of systemic CD8 T‐cell immunity in non‐small cell lung cancer. We found that early‐like differentiation status of PD‐1+CD8 T cells is associated with favorable outcome to PD‐1‐targeted immunotherapy. We further identified a baseline indicator of response to immune checkpoint blockade for implementation in clinical practice.

Introduction
Non‐small cell lung cancer (NSCLC) is among the most common malignancies accounting for 17.6% of cancer deaths worldwide, 1 with a 5‐year survival rate of < 15% following diagnosis. 2 The advent of immune checkpoint blockade (ICB) agents has remarkably prolonged overall survival rates in NSCLC patients. 3 To date, a plethora of agents targeting PD‐1 or its ligand PD‐L1 has been approved in multiple clinical settings. 4 , 5 However, such therapies are only beneficial to a limited fraction of patients; the majority of advanced NSCLC patients on progression do not respond to PD‐1 blockade. 6 Factors underlying such distinct clinical outcomes are only partially understood, and there is a need for further investigations as to how PD‐1 blockade modulates the immune system in cancer. 3 , 7 , 8
CD8+ T‐cell density in tumor biopsies has been associated with favorable clinical outcomes in response to PD‐1‐targeted therapies in multiple studies, 9 which is thought to occur mainly because of the reinvigoration of exhausted T cells. 10 , 11 However, recent evidence suggests that pre‐existing tumor‐specific T cells might have a limited re‐invigoration capacity. PD‐1 blockade might act rather by attracting novel T‐cell clones that derive from the tumor periphery, lymphoid organs or the bloodstream to the tumor regions. 12 Indeed, systemic PD‐1+CD8+ T cells were shown to expand upon PD‐1 blockade in NSCLC patients and that such activation correlated well with clinical responses. 13 Considering the diversity of the peripheral CD8+ T‐cell pool in terms of differentiation states and functionality, exploring the features of distinct systemic CD8+ T‐cell subsets responding to PD‐1 blockade might broaden our understanding on the mechanism of PD‐1 blockade and provide indicators of response to the ICB immunotherapy.
The peripheral CD8+ T‐cell repertoire is highly heterogeneous, consisting of diverse cellular populations with distinct maturation states. Upon antigenic stimuli, CD8+ T cells are activated and acquire memory and effector functions while progressively losing the expression of co‐stimulatory receptors CD28 and CD27. Based on the expression of these two molecules, CD8+ T cells transit through four differentiation states, from an early differentiated state (CD28+CD27+CD8+ T cells, TED), to an early‐like differentiated state (CD28+ CD27−CD8+ T cells, TELD), to an intermediate differentiated state (CD28−CD27+CD8+ T cells, TID) to finally reach a highly differentiated state (CD28−CD27−CD8+ T cells, THD). 14 , 15 , 16 , 17 , 18 , 19 Each of these states comprises of different cellular subsets. Relying on the linear progression model of T‐cell diversification often referred to as the decreasing potential model, where repetitive stimulation of T cells drives the proliferation and acquisition of effector functions. 20 Naïve CD8+ T cells (CD28+ CD27+CD45RO−) after first encounter with an antigenic stimuli undergo activation and acquisition of the CD45RO isoform to form central memory CD8+ T cells ‘TCM’ (CD28+CD27+CD45RO+). Following further antigenic stimulation, TCM acquire more cytolytic capacity while retaining their memory properties to form effector memory T cells ‘TEM’. CD28 and CD27 are down‐regulated stepwise as TCM become specialised TEM cells defining intermediate memory subsets; early effector memory T cells (CD28+CD27−CD45RO+, TEEM) 15 , 21 and transitional memory T cells (CD28−CD27+ CD45RO+, TTM). 19 The final stage of T‐cell differentiation in the linear progressive model is characterised by the loss of the memory marker CD45RO and formation of fully differentiated Effector T cells (CD28−CD27−CD45RO−, TE) that are endowed with higher cytolytic activity but lack proliferative potential and memory features. Although conventionally TE cells are CD28−CD27−, there is evidence of the presence of small subpopulations expressing either CD27: TpE2 (CD28−CD27+CD45RO−) or CD28: TpE3 (CD28+CD27−CD45RO−) within the TE pool. 22
Considering the heterogeneity of the peripheral CD8+ T‐cell pool in terms of states of maturation and differentiation, whether the differentiation status of CD8+ T subsets with unique functionality correlates with PD‐1 blockade in NSCLC has not yet been addressed and is worthy of investigation. Therefore, in this study, we have examined the differentiation states of circulating CD8+ T cells at baseline and analysed their prognostic values with regard to their association with progression‐free survival (PFS) and their predictive potential as a surrogate indicator of response to ICB therapy. We have also examined the kinetics of peripheral CD8+ T‐cell differentiation subsets upon the course of the treatment, which intended to further establish potential correlations between PD‐1 blockade and systemic CD8+ T‐cell differentiation states.
Results
Characterisation of systemic CD8 + T‐cell differentiation subsets in NSCLC
To explore the distinct peripheral CD8+ T‐cell composition in NSCLC and its potential association with responses to PD‐1 blockade, we conducted a prospective analysis on PBMC samples isolated from the fresh blood of a discovery cohort of 47 patients with NSCLC before the initiation of PD‐1 blockade therapy (baseline) and every 3 weeks upon nivolumab (anti‐PD‐1 antibody therapy) allocation. Efficacy evaluation was performed every 8 weeks until disease progression. Durable clinical benefit (DCB) was evaluated at day 180, and progression‐free survival (PFS) was determined beyond 800 days (Figure 1 and Table 1). We then characterised the heterogeneity of peripheral CD8+ T cells according to the decreasing potential model leading to the definition of four general stages of differentiation as early differentiation (ED, CD28+CD27+), early‐like differentiation (ELD, CD28+CD27−), intermediate differentiation (ID, CD28−CD27+) and high differentiation (HD, CD28−CD27−). Each stage was composed of two distinct cellular subsets based on CD45RO expression leading to the identification of eight T‐cell subpopulations (Figure 2a). Furthermore, by applying FlowSom algorithm, CD8+ T cells from 10 NSCLC patients were clustered based on the expression of CD27, CD28 and CD45RO into eight distinct clusters that displayed similar phenotypes as the predefined subpopulations and exhibited a developmental trajectory aligning with the decreasing potential model (Figure 2b). It is noteworthy that the circulating CD8+ T‐cell pool from NSCLC patients is mostly composed of effector subsets including TEEM, TEM, and TE cells that were more frequent when compared with other subsets (P < 0.0001) (Figure 2c).
Figure 1.
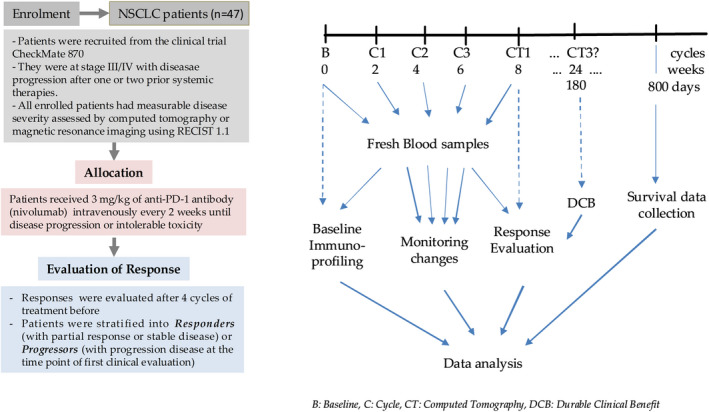
Flowchart of study design of the discovery cohort.
Table 1.
Patient characteristics and demographics
| Patients characteristics | Discovery cohort (Nivolumab, Checkmate 870) (n = 47) |
Validation cohort Anti‐PD1 immunotherapy (n = 30) |
||||
|---|---|---|---|---|---|---|
| Responders (n = 24) | Progressors (n = 23) | P | Responders (n = 18) | Progressors (n = 12) | P | |
| Age (years), median (range) | 62 (42–74) | 62 (44–77) | ns | 65.5 (45–81) | 62.5 (37–78) | ns |
| Sex, n (%) | ||||||
| Male | 21 (87.5) | 18 (78.26) | 16 (88.89) | 6 (50) | ||
| Female | 3 (12.5) | 5 (21.73) | 2 (11.12) | 6 (50) | ||
| Histological type, n (%) | ||||||
| ADC | 16 (66.67) | 15 (65.21) | 11 (61.11) | 10 (83.33) | ||
| SCC | 8 (33.34) | 8 (34.78) | 7 (38.88) | 2 (16.66) | ||
| Stage, n (%) | ||||||
| III | 6 (25) | 0 | 6 (33.33) | 2 (16.66) | ||
| IV | 18 (75) | 23 (100) | 12 (66.66) | 10 (83.33) | ||
| Anti‐PD1 agent, n (%) | ||||||
| Nivolumab | 24 (100) | 23 (100) | 7 (38.88) | 5 (41.66) | ||
| Sintilimab | 4 (22.22) | 3 (25) | ||||
| Pembrolizumab | 6 (33.33) | 1 (8.33) | ||||
| Tislelizumab | 1 (5.55) | 0 | ||||
| Toripalimab | 0 | 3 (25) | ||||
| Response, n (%) | ||||||
| CR | 0 | 0 | 0 | 0 | ||
| PR | 7 (29.16) | 0 | 3 (16.66) | 0 | ||
| SD | 17 (70.83) | 0 | 15 (83.33) | 0 | ||
| PD | 0 | 23 (100) | 0 | 12 (100) | ||
| Median PFS | 271 | 52 | < 0.0001 | 247.5 | 40.5 | < 0.0001 |
ADC, adenocarcinoma; CR, complete response; PD, progressive disease; PFS, progression‐free survival; PR, partial response; SCC, squamous cell carcinoma; SD, stable disease.
Figure 2.
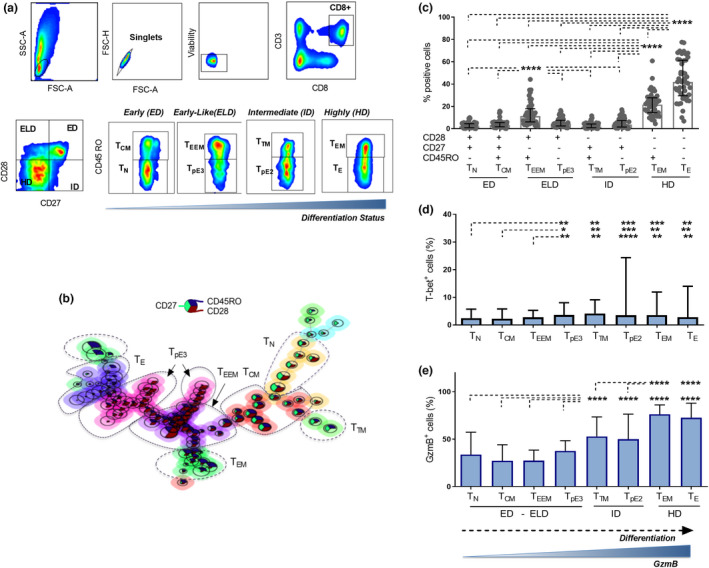
Characterisation of peripheral CD8+ T‐cell differentiation subsets. (a) Gating strategy for CD8+ T‐cell differentiation subsets in flow cytometry. Four differentiation states were defined based on the expression of CD28 and CD27, including an early differentiated state (CD28+CD27+, ED), an early‐like differentiated state (CD28+CD27−, ELD), an intermediate differentiated state (CD28−CD27+, ID) and a highly differentiated state (CD28−CD27−, HD). Peripheral CD8+ T cells were further subgrouped into eight subsets based on the additional expression of CD45RO including naïve CD8+ T cells (CD28+CD27+CD45RO−, TN) and central memory CD8+ T cells (CD28+CD27+CD45RO+, TCM) in ED state (second plot), early effector memory T cells (CD28+CD27−CD45RO+, TEEM) and effector T cells (CD28+CD27−CD45RO−, TpE3) in ELD state (third plot), transitional memory T cells (CD28−CD27+CD45RO+, TTM) and effector T cells (CD28−CD27+CD45RO−, TpE2) in ID state (forth plot), and effector memory T cells (CD28−CD27−CD45RO+, TEM) and fully differentiated effector T cells (CD28−CD27−CD45RO−, TE) in HD state (fifth plot). (b) FlowSOM generated CD8+ T cell clusters from 10 NSCLC samples showing the different differentiation profiles of CD8+ T cell subpopulations. (c) Baseline frequencies of CD8+ T cell differentiation subsets (TN, TCM, TEEM, TpE3, TTM, TpE2, TEM and TE) from 47 NSCLC patients. (d) Baseline frequencies of T‐bet‐positive cells in each differentiation subset. (e) Baseline frequencies of GzmB expressing cells in each differentiation subset. Data presented as median with interquartile range. Statistical analyses were performed using the Wilcoxon matched‐pairs signed‐rank test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Cytotoxic responses and effector function of CD8+ T cells depend on transcription factors T‐bet and Bcl6, 20 whereas Foxp3 and RORγt were reported to define distinct subsets within the CD8+ T‐cell population with regulatory or proinflammatory properties, respectively. 23 , 24 In our data set, the transcription factors Foxp3 and RORγt were barely expressed by CD8+ T‐cell subsets and Bcl6 did not show a specific trend between the subsets (Supplementary figure 1). Meanwhile, T‐bet expression was slightly increased within subsets exhibiting higher differentiation states (ID and HD) including effector subpopulations CD8+ TpE3, TpE2, TEM and TE as well as TTM cells when compared with CD8+ TN, TCM and TEEM cells (Figure 2d). More notably, granzyme B expression showed significantly increased expression in subsets with an intermediate differentiation state (TTM and TpE2) as compared to those with an early and early‐like differentiation status (TN, TCM,TEEM and TpE3) (P < 0.0001), and an even more enhanced profile was observed with cells displaying a highly differentiated profile (TEM and TE) when compared with all other subsets either with an early, early‐like or intermediate differentiation state (P < 0.0001) (Figure 2e).
These results indicate that in the periphery of NSCLC patients, the baseline composition of systemic CD8+ T cells is heterogeneous and includes subpopulations with distinct cytolytic capacity which is proportional to their differentiation state.
The cytolytic capacity of CD8 + T‐cell differentiation subsets at baseline does not distinguish responders to nivolumab from progressors
We investigated the potential link between the baseline differentiation of CD8+ T‐cell subsets and responses to PD‐1‐targeted immunotherapy. We first compared the composition of the CD8+ T‐cell population between responders to nivolumab in the Checkmate 870 cohort who had stable disease (SD) or partial response (PR) at the first evaluation point and progressors who had progressive disease (PD). We found that the frequencies of each defined subset were not significantly different between the two groups at baseline (Supplementary figure 2a). We then examined the frequencies of GzmB and T‐bet‐positive cells in each CD8+ T‐cell subset and found no significant differences (Supplementary figure 2b and c). These results imply that PD‐1 blockade‐induced clinical responses are not linked to the cytolytic ability of circulating CD8+ T cells.
The baseline ratio of PD‐1+ early effector memory T cells to PD‐1+ fully differentiated effector CD8 + T cell predicts responses to PD‐1 blockade in NSCLC
PD‐1 expression might constitute a potential indicator of response to anti‐PD‐1 therapy. Therefore, we next examined the expression of PD‐1 on each CD8+ T‐cell differentiation subset (Figure 3a). We found that PD‐1 expression differed between subsets and was mainly observed on cells displaying a memory phenotype, including TCM, TTM, TEEM and TEM cells (Figure 3b). To assess the predictive value of PD‐1 expression on CD8+ T cell subsets, we first compared the frequencies of PD‐1+ cells in the total CD8+ T‐cell population between patients who responded to nivolumab therapy and those who did not. We found no significant differences between both groups (Supplementary figure 3a), and no prognostic value was observed as well (Supplementary figure 3b). We then compared the frequencies of PD‐1+ cells from each individual subset between both groups. We found that responders to nivolumab treatment exhibited slightly higher frequencies of PD‐1+cells in the TEEM subset (P = 0.05) and TN (P = 0.03), whereas lower levels were observed in TE (P = 0.03) than in progressors (Figure 3c).
Figure 3.
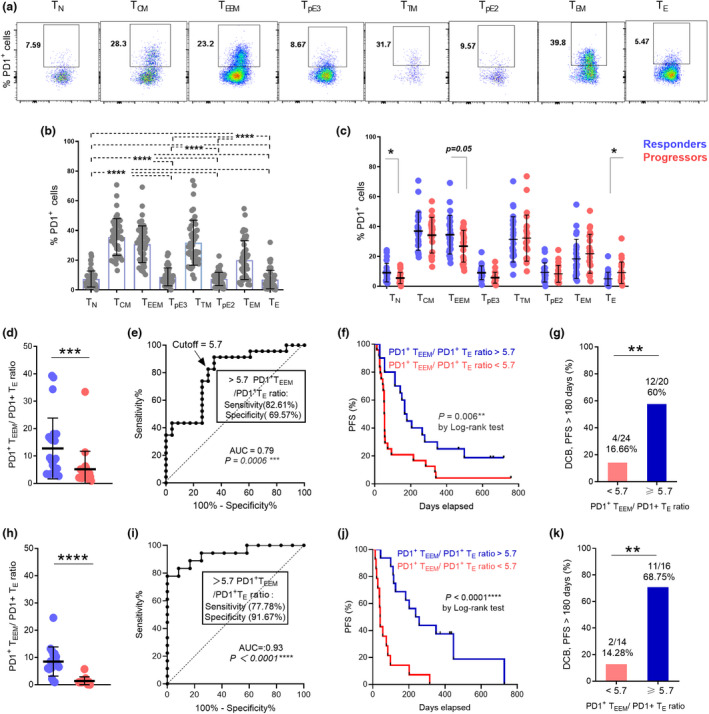
Baseline ratio of PD‐1+CD8+ TEEM to TE cells predicts favorable responses to nivolumab treatment in advanced NSCLC. (a) Representative flow cytometry data plot of PD‐1 expressions in peripheral CD8+ T differentiation subsets from one NSCLC patient. (b) Baseline frequencies of PD‐1+ cells in CD8+ T subsets. (c) Comparison of PD‐1 expressions at baseline in peripheral CD8+ T subsets between responders (n = 24, blue dots) and progressors (n = 23, red dots) to nivolumab therapy. (d) Comparison of PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio at baseline between responders and progressors (discovery cohort). (e) ROC analysis of baseline PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio (f) Kaplan–Meier PFS curves of PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio at baseline (discovery cohort). (g) Proportions of NSCLC patients (discovery cohort) with DCB responses at 6 months with PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio > 5.7 or < 5.7. (h) Comparison of PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio at baseline between responders and progressors from the validation cohort (n = 30). (i) ROC analysis of baseline PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio (validation dataset). (j) Kaplan–Meier PFS curves of PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio at baseline (validation data set). (k) Proportions of NSCLC patients (validation cohort) with DCB responses at 6 months with PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio > 5.7 or < 5.7. Data were analysed using the Wilcoxon matched‐pairs signed‐rank test (b) and the Mann–Whitney test (c, d, g, h, k). *P < 0.05, ***P < 0.001, ****P < 0.0001.
When we explored the predictive value of PD‐1 expression on a given CD8+ T‐cell subset, we found that frequencies of PD‐1+CD8+ TE correlated slightly with PFS (P = 0.05), whereas no clinical significance was detected among other subsets (Supplementary figure 4a–e). However, by integrating the baseline frequencies of PD‐1+ TEEM and PD‐1+ TE into the baseline ratio ‘PD‐1+CD8+ TEEM to PD‐1+CD8+ TE’, we observed striking differences between responders and progressors (P < 0.0005) (Figure 3d). Furthermore, the ratio had a high predictive value for response to nivolumab therapy with the area under curve (AUC) reaching 0.79 [95% confidence interval (CI) 0.66–0.92, P = 0.0006, Figure 3e]. An optimal cut‐off value of 5.7 for the PD‐1+ TEEM to PD‐1+ TE ratio was determined from the receiver operator characteristic (ROC) curve with a sensitivity and specificity reaching up to 82.61% and 69.57%, respectively. Interestingly, PD‐1+ TEEM to PD‐1+ TE ratio exhibited a significant positive association with PFS, NSCLC patients with a ratio higher than 5.7 had a notable prolonged PFS with a median of 173 days, whereas those with values < 5.7 had a PFS median of only 54 days (P = 0.006) (Figure 3f). Moreover, the probability of DCB (with response lasting up to 180 days) was significantly elevated when the ratio was higher than 5.7 (P = 0.004) (Figure 3g).
To validate these findings, we included an independent cohort of NSCLC patients receiving different anti‐PD‐1 agents (validation cohort) (Table 1 and Supplementary table 1). Consistently, responders from the validation cohort also displayed a significant increase in PD‐1+ TEEM to PD‐1+ TE ratio at baseline when compared with progressors (P < 0.0001) (Figure 3h). In addition, PD‐1+ TEEM to PD‐1+ TE ratio significantly predicted response to PD‐1 blockade [AUC 0.93, 95% confidence interval (CI) 0.85–1, P < 0.0001, Figure 3i] and the predefined cut‐off value of 5.7 had 77.78% and 91.67% of sensitivity and specificity, respectively (Figure 3i). The median PFS was 247.5 days for patients with a ratio higher than 5.7 and 40.5 days for those with a ratio < 5.7 (Figure 3j). Moreover, the probability of DCB was significantly higher when the ratio was more than 5.7 in the validation cohort as well (P = 0.003) (Figure 3k).
Taken together, we have identified the baseline PD‐1+ TEEM to PD‐1+ TE ratio as an indicator of response to PD‐1 blockade in NSCLC. These findings also establish a clear association between PD‐1 expression, the differentiation status of systemic CD8+ T cells and responses to PD‐1 blockade in NSCLC.
PD‐1+ CD8 + TEEM cells are the early responders to PD‐1 blockade
The association between the baseline differentiation status of PD‐1+CD8+ T cells and the response to PD‐1 blockade also suggests that this blockade might have selective effects on CD8+ T‐cell differentiation subsets. We therefore monitored responses of PD‐1+CD8+ T‐cell subsets at baseline and 2 weeks after the first cycle of nivolumab therapy (Checkmate 870) through measuring Ki67 expression, a cell cycle marker expressed in cycling or recently divided cells 25 (Figure 4a). Interestingly, we found that the increase in Ki67 expression occurred mainly in PD‐1+ cells displaying an early effector memory phenotype (P = 0.004 in responders and P = 0.06 in progressors), yet no significant differences were observed among other differentiation subsets (Figure 4b). Nonetheless, further monitoring of subset kinetics upon several time points (C2–C10) (Supplementary figure 5) reveals that frequencies of cycling PD‐1+CD8+ TEEM were increasing from baseline to C4 specifically in responders to nivolumab but not in progressors. Furthermore, delayed activation (after 4 weeks) was observed in other PD‐1+CD8+ subsets mainly in those expressing CD28 (TCM and TpE3). However, increased proliferation of PD‐1+CD8+ TCM cells was also observed in progressor patients after 4 weeks of nivolumab initiation (Supplementary figure 5).
Figure 4.
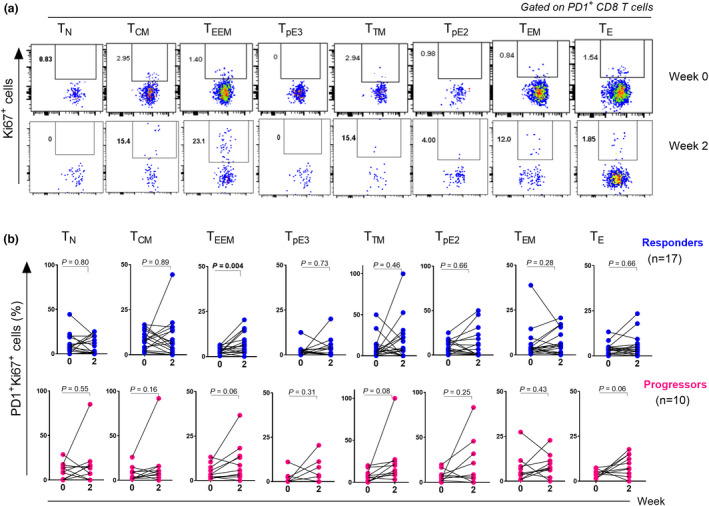
PD‐1+CD8+ TEEM cells are the early responders to PD‐1 blockade. (a) Representative flow cytometry dot plots showing the expression of Ki67 in PD‐1+CD8+ T cell subsets at baseline and 2 weeks after nivolumab therapy (Discovery Cohort). (b) Graphs showing the kinetics of cycling Ki67+PD‐1+CD8+ T subsets upon one cycle of nivolumab therapy in responders (n = 17) (upper row) and progressors (n = 10) (lower row). Data were analysed using the Wilcoxon matched‐pairs signed‐rank test.
PD‐1 blockade‐induced expansion is dominated by cells displaying a PD‐1+ TEEM phenotype
To shed further light on the effects of PD‐1 blockade on CD8+ T‐cell differentiation, we aimed to characterize the contribution of each differentiation phenotype to the total cycling Ki67+PD‐1+CD8+ T‐cell population at different time points following nivolumab therapy (0, 2, 4, 6 and 8 weeks) and its association with progression‐free survival and clinical benefit (Figure 5a).
Figure 5.
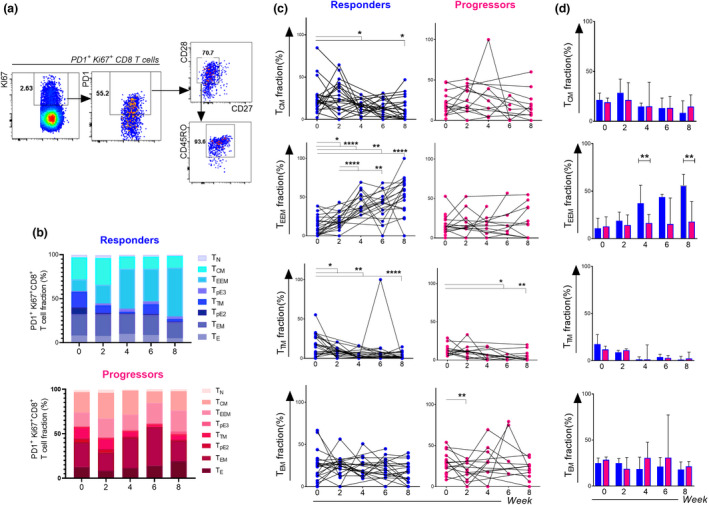
PD‐1 blockade induces the preferential expansion of PD‐1+CD8+ T cells with an early effector memory phenotype (Discovery Cohort). (a) Gating strategy to determine the phenotype of proliferating PD‐1+ Ki67+CD8+ T‐cell population based on the expression of CD27, CD28 and CD45RO with TEEM fraction. (b) Subset fractions of Ki67+PD‐1+CD8+ T cells at week 0, 2, 4, 6 and 8 after PD‐1 blockade in the discovery cohort. (c) Kinetics of the proportions of major CD8+ memory T cell subsets (TCM, TTM, TEEM and TEM) expressing PD‐1 and Ki67 cells at week 0, 2, 4, 6 and 8 in responders (Blue dots) and progressors (Red dots). (d) Comparison of PD‐1+Ki67+CD8+ T cell differentiation subsets between responders (n = 24 at week 0, n = 17 at week 2, n = 14 at week 4, n = 15 at week 6, n = 20 at week 8) and progressors (n = 20 at week 0, n = 10 at week 2, n = 7 at week 4, n = 5 at week 6 and n = 11 at week 8) at different time points, data are presented as median with interquartile ranges. Data were analysed using the Wilcoxon matched‐pairs signed‐rank test (c) and the Mann–Whitney test (d). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We found that, in responders, the major fraction of expanding Ki67+ PD‐1+CD8+ T cells in the blood upon the course of nivolumab therapy (0, 2, 4, 6 and 8 weeks) was of a TEEM phenotype (Figure 5b). Indeed, the TEEM fraction constituted 13.5% of cycling cells at week 0 and increased significantly to reach 20% at week 2, then 56% at week 8 in NSCLC patients who experienced response to nivolumab therapy. However, no changes were observed in progressors (Figure 5b). Importantly, this increase was specific to the TEEM phenotype as other differentiation phenotypes did not display similar patterns (Figure 5c and d).
In addition, the TEEM fraction of Ki67+ PD‐1+CD8+ T cells was significantly elevated in responders as compared with progressors at week 4 (P = 0.001) and week 8 (P = 0.001) (Figures 5d and 6a) and predicted response both at week 4 [AUC 0.92, 95% CI 0.78–1; P = 0.0023; Figure 6b] and week 8 [AUC 0.84, 95% CI 0.70–0.98; P = 0.0017; Figure 6b]. Optimal cut‐off values of 29.43% (week 4) and 21.86% (week 8) (Figure 6b) were associated with higher sensitivity (85.7% at week 4 and 63.6% at week 8) and specificity (91.7% at week 4 and 95% at week 8). Patients with TEEM fractions higher than 29.43% at week 4 and higher than 21.86% at week 8 had a prolonged PFS (P < 0.0001, week 4 and P = 0.0005, week 8) (Figure 6c). In addition, the TEEM fractions at weeks 4 and 8 were strongly correlated with durable benefit from nivolumab therapy, and the probability of DCB was higher when TEEM fractions exceeded 29.43% at week 4 (P = 0.03) and 21.86% at week 8 (P = 0.007) (Figure 6d).
Figure 6.
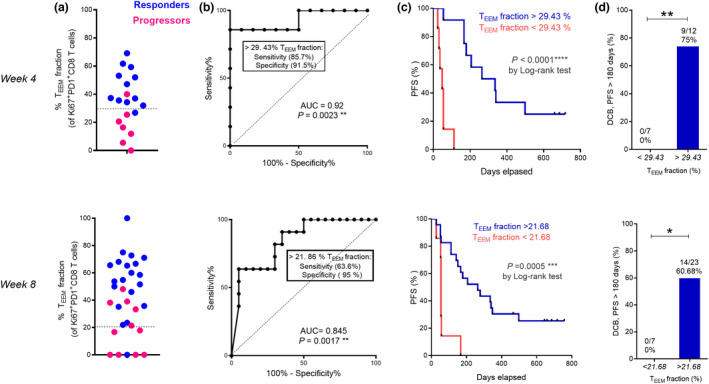
The TEEM fraction of cycling PD‐1+Ki67+CD8+ T cells is associated with better prognosis (Discovery Cohort). (a) Percentages of TEEM cells in PD‐1+Ki67+CD8+ T at week 4 (up) and week 8 (low) of responders (blue dots) (n = 14 at week 4 and n = 20 at week 8) and progressors (red dots) (n = 7 at week 4 and n = 11 at week 8). (b) ROC analysis of PD‐1+Ki67+CD8+ TEEM fraction at week 4 (up) and week 8 (low). (c) Kaplan–Meier PFS curves of PD‐1+Ki67+CD8+ TEEM fraction at week 4 (up) and week 8 (low) after nivolumab treatment. (d) Proportions of NSCLC patients (discovery cohort) showing DCB responses at 6 months stratified based on the cutoff values of the proportions of Ki67+PD‐1+CD8+ TEEM cells at week 4 or week 8. Data were analysed using the Mann–Whitney test (d). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In summary, our data demonstrates that nivolumab therapy‐induced T‐cell expansion is mostly observed with cells displaying a PD‐1+CD8+ TEEM cell phenotype which is associated with positive impact on progression‐free survival in NSCLC patients.
Discussion
Emerging evidence suggests that PD‐1 blockade might induce the remodelling of the peripheral T‐cell immunity including CD8+ T cells 13 , However, this action has not been fully elucidated. Considering the distinct functional properties of CD8+ T‐cell differentiation subsets in the periphery, we hypothesized that the differentiation spectrum of peripheral PD‐1+CD8+ T cells might determine responses to PD‐1 blockade. Hence, in this study, we analyzed the distinct differentiation subsets within circulating PD‐1+CD8+ T cells in advanced NSCLC patients before and upon PD‐1 immunotherapy. Of interest, we found that the early‐like differentiation status at baseline represented by the ratio of PD‐1+CD8+ TEEM to PD‐1+CD8 TE was associated with longer progression‐free survival and durable clinical benefit of anti‐PD‐1 therapy in NSCLC. Moreover, PD‐1+CD8+ TEEM was the subset exhibiting rapid expansion after nivolumab treatment with a significant increase in Ki67 expression, which constituted the larger fraction of cycling PD‐1+CD8+ T cells following multiple cycles of nivolumab immunotherapy.
PD‐1 expression is induced by TCR signalling upon antigenic stimulation and is mainly upregulated on antigen‐experienced T cells. 26 In our analysis of the peripheral compartment of NSCLC patients, we also noticed that PD‐1 expression was more frequent on CD8+ T cells displaying memory phenotypes including TCM, TTM, TEEM and TEM subsets than on TN, TE, TpE2 and TpE3 effector subsets in the peripheral compartment of NSCLC patients (Figure 3b). A recent study conducted in patients with epithelial cancers demonstrated that peripheral PD‐1+CD8+ T cells exhibited solely a memory phenotype. Unique and shared tumor neoantigen‐specific TCR sequences detected in those patients were restricted to the peripheral memory T‐cell pool. 27 Accordingly, our observation suggests that in the periphery, the memory repertoire might be more enriched with tumor‐specific cells as compared with effector cells.
To elucidate the clinical significance of PD‐1 expression on peripheral CD8+ T‐cell subsets, we compared the baseline frequencies of PD‐1+ cells among different subsets between responders and progressors. Interestingly, we found a statistical trend towards higher PD‐1 expression on CD8+ TEEM cells in patients who responded to the therapy as compared with those who did not. In contrast, the percentages of PD‐1+ TE were significantly lower in responders.
On the one hand, CD8+ TEEM cells are transitional memory cells with an intermediate differentiation state between CD8+ TCM and CD8+ TEM. 15 , 16 , 19 CD28 expression distinguishes them from CD8+ TEM and render them a potential target population for PD‐1 blockade‐induced activation that is contingent on CD28 expression and engagement. 28 On the contrary, effector CD8+ T cells with low PD‐1 incidence were found to be associated with clinical benefit to nivolumab therapy and prolonged PFS in a cohort of NSCLC patients, suggesting that presence of lower percentages of intra‐tumoral PD‐1+ TE cells is in favor of more proficient antitumor immunity. 29 This is consistent with our observation in the peripheral blood where PD‐1+ TE cells were found mostly in progressors to nivolumab therapy as compared with responders. Furthermore, the divergence in the differentiation status from early‐like differentiated (ELD) for TEEM to highly differentiated (HD) for TE infers more significance in considering integrating these two subsets into one novel parameter which is the baseline ratio PD‐1+CD8+ TEEM/PD‐1+CD8+ TE. By doing so, we were more able to better identify the NSCLC patients who were most likely to derive clinical benefit form PD‐1 blockade (Figure 3). Therefore, PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio, which reflects the differentiation state in the periphery, might display the status of systemic immune system at baseline for susceptibility to anti PD‐1 therapy. In this model with more PD‐1+CD8+ TEEM and less PD‐1+CD8+ TE in the peripheral blood at baseline, NSCLC patients could mount proficient anti‐tumor immunity upon inhibition of PD‐1 pathway. This model is supported by the high sensitivity and specificity of the ratio (Figure 3e and i) to distinguish responders from progressors before the start of the immunotherapeutic approach. These findings thus demonstrate the relevance of the baseline T‐cell differentiation status in determining responses to PD‐1‐targeted immunotherapies and provide a novel predictor of benefit to PD‐1 blockade in NSCLC. It is noteworthy that the identified ratio constitutes a promising indicator of response to ICB in NSCLC for implementation in clinical routine use. 30 It is associated with a high negative predictive value, with a specificity reaching up to 69.57% in the discovery cohort and 91.67% in the validation cohort for a cut‐off value of 5.7. The PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio is pre‐treatment indicator that is non‐invasive and blood accessible allowing early identification of patients who might respond PD‐1 blockade.
Our proposed model is further supported by the findings that PD‐1+CD8+ TEEM cells constitute the main subset of peripheral CD8+ T cells responding to PD‐1 blockade as early as 2 weeks of the treatment (Figure 4). More interestingly, the major fraction of Ki67+PD‐1+CD8+ T cells that undergoes active replication upon nivolumab treatment is of TEEM phenotype and shows significant associations with longer PFS at weeks 4 and 8. These data suggest that peripheral PD‐1+CD8+ TEEM cells are activated and expand upon nivolumab therapy. They can be detected cycling in the bloodstream of patients up until 8 weeks of treatment, and then most probably migrate to the tumor tissues as the percentages start to decline significantly from week 10 (Supplementary figure 7). This is in line with the concept of clonal replacement theory where T‐cell responses to checkpoint blockade are shown to derive from a distinct repertoire of T‐cell clones that may enter the tumor upon PD‐1 blockade. 12 Moreover, it is noteworthy that despite the proliferative potential of peripheral CD8+ TEEM observed upon PD‐1 blockade, these cells did not exhibit a superior cytolytic ability as compared with other effector CD8+ T subsets neither at baseline (Figure 2e and Supplementary figure 8) nor at different time points of therapy (Supplementary figure 8). These observations suggest that TEEM cells might instead serve as progenitors or precursors of effector CD8+ T cells that are essential to mount antitumor immune responses. Consequently, this peripheral CD8+ T‐cell subpopulation shares essential features with the TCF1+ stem‐like T cells that has been observed in several studies, which is reported to be associated with proliferation burst observed upon PD‐1 blockade. 31 The presence of precursor effector CD8+ T cells in the peripheral bloodstream highlights the roles of systemic immunity in the efficacy of ICBs approaches.
Taken together, our study identified the baseline PD‐1+CD8+ TEEM/PD‐1+CD8+ TE ratio as a novel non‐invasive pre‐treatment indicator of response to PD‐1 blockade in NSCLC patients and describes a model in which the differentiation state of peripheral CD8+ T cells before PD‐1 therapy in part orients the arm of antitumor immunity through regulating the expansion of TEEM fractions and therefore conditioning responses to PD‐1‐targeted immunotherapy in NSCLC.
Methods
Patients
A total of 77 advanced NSCLC patients (61 men and 16 women) with a median age of 62.25 years were recruited at Shanghai Chest Hospital affiliated to Shanghai Jiao Tong University, China. All of them had measurable disease severity assessed by computed tomography or magnetic resonance imaging using Response Evaluation Criteria in Solid Tumours version 1.1 (RECIST 1.1). Forty‐seven patients were part of the clinical trial CheckMate 870 [NCT03195491] and were designated as a discovery cohort. They received 3 mg kg−1 of anti‐PD‐1 antibody nivolumab intravenously every 2 weeks until disease progression or intolerable toxicity. Thirty patients did not enrol to any clinical trial and served as a validation cohort, also received anti‐PD‐1 therapy. They were treated with nivolumab (180 mg every 2 weeks), pembrolizumab (200 mg every 3 weeks), tislelizumab (200 mg every 3 weeks), sintilimab (200 mg every 3 weeks) and toripalimab (240 mg every 4 weeks), respectively. Responses to immunotherapy were evaluated every 4 cycles of the therapy according to RECIST 1.1 evaluation. Patients were stratified into responders (with partial response or stable disease) or progressors (with progression disease at the time point of first clinical evaluation) according to the RECIST 1.1 evaluation. Durable clinical benefit (DCB) was defined as a response (partial response or stable disease) lasting longer than 6 months from the initiation of the therapy (Figure 1, Table 1, Supplementary table 1).
Study approval
The study was approved by the Ethics Committee of Shanghai Chest Hospital (Number KS1732). All enrolled patients agreed to participate in this study by providing informed consent forms. This study was conducted in accordance with the Declaration of Helsinki.
Flow cytometric analysis
Isolation of peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMCs) were isolated from the whole blood by density gradient centrifugation using Ficoll Lymphoprep™ reagent (STEMCELL Technologies) according to the manufacturer's instructions. Two million PBMCs were resuspended in 200 μL FACS buffer [PBS containing 2% foetal bovine serum (FBS)] for subsequent multi‐colour flow cytometry tests.
Surface staining
Cells were first incubated with the mixtures of fluorochrome‐conjugated monoclonal antibodies including BV510‐conjugated anti‐human CD3, PECY7‐conjugated anti‐human CD8, FITC‐conjugated anti‐human CD45RO, APC‐H7‐conjugated anti‐human CD27, BV605‐conjugated anti‐human CD28 and BV711‐conjugated anti‐human PD‐1 antibodies from BD Biosciences (San Jose, CA, USA) for 40 min at 4°C. Cells were then washed with 1 mL FACS buffer. Anti‐human IgG4 Fc (HP6025) from Southern Biotech was used together with anti‐PD‐1 antibody staining to define PD‐1+ cells for subset monitoring study.
Intracellular staining
After surface staining, PBMCs were suspended by adding 1 mL of Fix/Perm working solution (Transcription Factor Buffer Set; BD Biosciences), and incubated at 4°C for 40 min in the dark. Subsequently, the fixed and permeabilised cells were washed with 1 mL Perm/wash buffer, and then incubated with fluorescent antibodies specific for intracellular proteins [including PE‐conjugated anti‐human Granzyme B, AF700‐conjugated anti‐human Ki67, AF674‐conjugated anti‐human T‐bet, PE‐CF594‐conjugated anti‐human FOXP3, BV650‐conjugated anti‐human RORγt and BV421‐conjugated anti‐human BCL‐6 (BD Biosciences)] at 4°C for 40 min. Cells were then washed with 1 mL Perm/wash buffer and resuspended in 200 μL FACS buffer for acquisition by Fortessa X20 Flow cytometer (BD Biosciences). Data analysis was performed with FlowJo version 10 software (Tree Star Inc., Ashland, OR, USA).
Statistical analyses
Data are presented as median with interquartile ranges. Unpaired samples were analysed with the Mann–Whitney test. A paired t‐test was used for paired samples that showed Gaussian distribution while the Wilcoxon matched‐pairs signed‐rank test was used for paired samples without Gaussian distribution. Clustering analysis was conducted using FlowSOM algorithm implemented in FlowJo software (FlowJo, LLC). Survival curves for the PFS were estimated using the Kaplan–Meier method (log‐rank test). Receiver‐operating characteristic (ROC) analysis was conducted to assess the sensitivity and specificity for signatures associated with the response.
The statistical analysis approach for this study was exploratory with no pre‐specified statistical plan, and no correction of P‐values for multiple testing included. All P‐values were two‐sided, and P < 0.05 was considered statistically significant. Statistical tests were performed with GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA, USA).
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Asma Khanniche: Conceptualization; data curation; formal analysis; investigation; methodology; project administration; writing – original draft; writing – review and editing. Yi Yang: Data curation; formal analysis; resources. Jie Zhang: Data curation; formal analysis; resources. Shiqing Liu: Data curation; resources. Liliang Xia: Data curation; formal analysis. Huangqi Duan: Data curation. Yaxian Yao: Data curation; resources. Bingrong Zhao: Data curation; resources. Guo‐Ping Zhao: Conceptualization; supervision. Chengping Hu: Resources. Ying Wang: Conceptualization; funding acquisition; resources; supervision; writing – original draft; writing – review and editing. Shun Lu: Conceptualization; funding acquisition; resources; supervision.
Supporting information
Supplementary figures 1–8
Supplementary table 1
Acknowledgments
This work was supported by the National Key R&D Program of China (2016YFC1303303), National Natural Science Foundation of China (82073152, 81802264, 82030045), National Multidisciplinary Cooperative Diagnosis and Treatment Capacity Building Project for Major Diseases (Lung Cancer), and Technology Innovation Program of Shanghai (19411950500). The authors acknowledge Tomasz Maj (Institute of Molecular Medicine, Shanghai Jiao Tong University School of Medicine, China) for kind help with revising and editing the manuscript.
Contributor Information
Asma Khanniche, Email: asmakhanniche@hotmail.com.
Ying Wang, Email: ywang@sibs.ac.cn.
Shun Lu, Email: shun.lu@me.com.
References
- 1. Parkin DM, Bray F, Ferlay J et al. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol 2006; 24: 2137–2150. [DOI] [PubMed] [Google Scholar]
- 3. Hui R, Garon EB, Goldman JW et al. Pembrolizumab as first‐line therapy for patients with PD‐L1‐positive advanced non‐small cell lung cancer: a phase 1 trial. Ann Oncol 2017; 28: 874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farhat FS, Houhou W. Targeted therapies in non‐small cell lung carcinoma: what have we achieved so far? Ther Adv Med Oncol 2013; 5: 249–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Medina PJ, Adams VR. PD‐1 pathway inhibitors: immuno‐oncology agents for restoring antitumor immune responses. Pharmacotherapy 2016; 36: 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maleki Vareki S, Garrigos C, Duran I. Biomarkers of response to PD‐1/PD‐L1 inhibition. Crit Rev Oncol Hematol 2017; 116: 116–124. [DOI] [PubMed] [Google Scholar]
- 7. Yokosuka T, Takamatsu M, Kobayashi‐Imanishi W et al. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012; 209: 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol 2015; 36: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geng Y, Shao Y, He W et al. Prognostic role of tumor‐infiltrating lymphocytes in lung cancer: a meta‐analysis. Cell Physiol Biochem 2015; 37: 1560–1571. [DOI] [PubMed] [Google Scholar]
- 10. Sakuishi K, Apetoh L, Sullivan JM et al. Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J Exp Med 2010; 207: 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yost KE, Satpathy AT, Wells DK et al. Clonal replacement of tumor‐specific T cells following PD‐1 blockade. Nat Med 2019; 25: 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamphorst AO, Pillai RN, Yang S et al. Proliferation of PD‐1+ CD8 T cells in peripheral blood after PD‐1‐targeted therapy in lung cancer patients. Proc Natl Acad Sci USA 2017; 114: 4993–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Plunkett FJ, Franzese O, Finney HM et al. The loss of telomerase activity in highly differentiated CD8+CD28−CD27− T cells is associated with decreased Akt (Ser473) phosphorylation. J Immunol 2007; 178: 7710–7719. [DOI] [PubMed] [Google Scholar]
- 15. Appay V, Dunbar PR, Callan M et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med 2002; 8: 379–385. [DOI] [PubMed] [Google Scholar]
- 16. Appay V, van Lier RA, Sallusto F et al. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A 2008; 73: 975–983. [DOI] [PubMed] [Google Scholar]
- 17. Khan N, Shariff N, Cobbold M et al. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol 2002; 169: 1984–1992. [DOI] [PubMed] [Google Scholar]
- 18. van Baarle D, Tsegaye A, Miedema F et al. Significance of senescence for virus‐specific memory T cell responses: rapid ageing during chronic stimulation of the immune system. Immunol Lett 2005; 97: 19–29. [DOI] [PubMed] [Google Scholar]
- 19. van Aalderen MC, Remmerswaal EB, Verstegen NJ et al. Infection history determines the differentiation state of human CD8+ T cells. J Virol 2015; 89: 5110–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 2012; 12: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takata H, Takiguchi M. Three memory subsets of human CD8+ T cells differently expressing three cytolytic effector molecules. J Immunol 2006; 177: 4330–4340. [DOI] [PubMed] [Google Scholar]
- 22. Romero P, Zippelius A, Kurth I et al. Four functionally distinct populations of human effector‐memory CD8+ T lymphocytes. J Immunol 2007; 178: 4112–4119. [DOI] [PubMed] [Google Scholar]
- 23. Chellappa S, Hugenschmidt H, Hagness M et al. CD8+ T cells that coexpress RORγt and T‐bet are functionally impaired and expand in patients with distal bile duct cancer. J Immunol 2017; 198: 1729–1739. [DOI] [PubMed] [Google Scholar]
- 24. Yu Y, Ma X, Gong R et al. Recent advances in CD8+ regulatory T cell research. Oncol Lett 2018; 15: 8187–8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gerdes J, Lemke H, Baisch H et al. Cell cycle analysis of a cell proliferation‐associated human nuclear antigen defined by the monoclonal antibody Ki‐67. J Immunol 1984; 133: 1710–1715. [PubMed] [Google Scholar]
- 26. Duraiswamy J, Ibegbu CC, Masopust D et al. Phenotype, function, and gene expression profiles of programmed death‐1hi CD8 T cells in healthy human adults. J Immunol 2011; 186: 4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cafri G, Yossef R, Pasetto A et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun 2019; 10: 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kamphorst AO, Wieland A, Nasti T et al. Rescue of exhausted CD8 T cells by PD‐1‐targeted therapies is CD28‐dependent. Science 2017; 355: 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mazzaschi G, Madeddu D, Falco A et al. Low PD‐1 expression in cytotoxic CD8+ tumor‐infiltrating lymphocytes confers an immune‐privileged tissue microenvironment in NSCLC with a prognostic and predictive value. Clin Cancer Res 2018; 24: 407–419. [DOI] [PubMed] [Google Scholar]
- 30. Lesterhuis WJ, Bosco A, Millward MJ et al. Dynamic versus static biomarkers in cancer immune checkpoint blockade: unravelling complexity. Nat Rev Drug Discov 2017; 16: 264–272. [DOI] [PubMed] [Google Scholar]
- 31. Im SJ, Hashimoto M, Gerner MY et al. Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature 2016; 537: 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–8
Supplementary table 1