

Abstract
Emerging evidence suggests that elevated concentrations of triglyceride-rich lipoprotein remnants (TRLs) derived from hepatic and intestinal sources contribute to the risk of atherosclerotic cardiovascular events. Natural selection studies support a causal role for elevated concentrations of remnant cholesterol and the pathways contributing to perturbations in metabolic pathways regulating TRLs with an increased risk of atherosclerotic cardiovascular disease events. New therapies targeting select catalytic pathways in TRL metabolism reduce atherosclerosis in experimental models, and concentrations of TRLs in patients with a vast range of triglyceride levels. Clinical trials with inhibitors of angiopoietin-like 3 protein and apolipoprotein C-III will be required to provide further guidance on the potential contribution of these emerging therapies in the paradigm of cardiovascular risk management in patients with elevated remnant cholesterol.
Keywords: angiopoietin-like 3 protein inhibitors, apolipoprotein C-III inhibitors, eicosapentaenoic acid, omega-3 fatty acids, triglyceride-rich lipoproteins, triglycerides
Evidence from epidemiological studies, biological mechanisms, genomic studies, and clinical trial data suggest that triglyceride-rich lipoprotein remnants (TRLs) contribute to atherosclerotic cardiovascular disease (ASCVD) risk. An abundance of remnant lipoproteins emanating from intravascular remodeling of triglyceride-rich lipoproteins (TGLs), chylomicrons and very low-density lipoproteins (VLDL), creates a pro-atherogenic environment that augments cardiovascular risk (1).
In this review, we provide a critical appraisal of published studies focusing on randomized clinical trials with new and emerging therapies. Accordingly, this review paper emphasizes studies with inhibitors of apolipoprotein C-III (Apo-III) and angiopoietin-like 3 protein (ANGPTL3). The essential contributions of lifestyle changes and pharmacotherapy, including peroxisome proliferator-activated receptor alpha (PPAR-α) agonists, SPPAR (selective peroxisome proliferator-activated receptor alpha modulator) agonists, and fibroblast growth factor agonists, for lowering TGLs has been recently reviewed (1,2). We provide an update on emerging developments in omega 3 fatty acid therapy for hypertriglyceridemia.
MEASUREMENT OF TRIGLYCERIDE-RICH REMNANTS
Accurate measurement of TRLs is technically challenging due to the dynamic catabolism that alters the lipid and apolipoprotein composition of chylomicrons and VLDL. With ongoing lipolysis, TRLs become smaller in size, depleted in triglycerides (TGs), and enriched in cholesterol while maintaining one apolipoprotein B (apoB) per particle (Figures 1 and 2). Although chylomicrons carry apoB48, a truncated form of apoB100, the chylomicron precursor and remnants often overlap in size and density, limiting this measure. VLDL and VLDL remnants contain apoB100, but apoB100 is also present on low-density lipoprotein (LDL) and lipoprotein (a), and thus cannot be used to measure VLDL remnants. Challenges in analytical approaches for measuring TRLs has been discussed in recent reviews (3,4).
FIGURE 1. Exogenous and Endogenous Pathways for Triglyceride Metabolism.
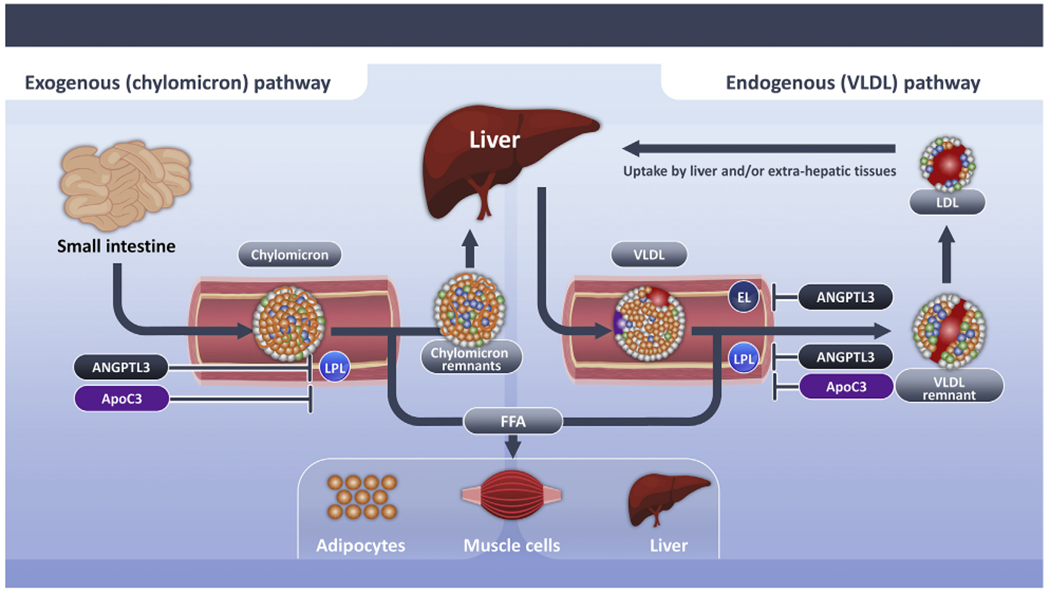
Schematic depicting exogenous and endogenous pathways for triglyceride metabolism. Triglyceride-rich lipoproteins derive from lipoproteins synthesized from exogenous sources (chylomicrons) and endogenous sources (VLDL). These triglyceride-rich lipoproteins are hydrolyzed by lipoprotein lipase, which elaborates free fatty acids that are used as an energy source by skeletal muscle or stored for future use in the liver or adipocytes. Lipoprotein lipase activity is inhibited by angiopoietin like 3 protein and apolipoprotein C-III. Delayed clearance of triglyceride-rich lipoproteins fosters formation of a more cholesterol enriched remnant particle. ANGPTL3 = angiopoietin-like 3; apoB100 = apolipoprotein B-100; apoB48 = apolipoprotein B48; apoC3 = apolipoprotein C-III; apo E = apolipoprotein E; CM = chylomicrons; EL = endothelial lipase; FFA = free fatty acid; IDL = intermediate-density lipoprotein; LDL = low-density lipoprotein; LPL = lipoprotein lipase; VLDL = very low density lipoproteins.
FIGURE 2. Regulation of VLDL Production and Clearance by ANGPTL3 and ApoC-III.
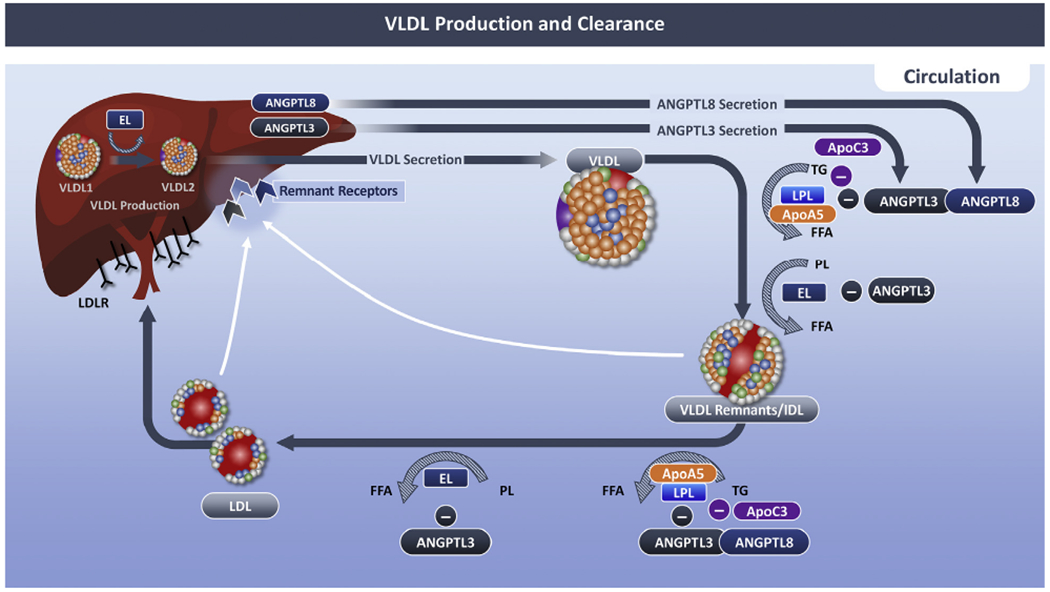
Schematic depicting the mechanism whereby angiopoietin-Like 3 (ANGPTL3) and apolipoprotein C-3 (apoC3) regulates VLDL production and clearance. During homeostasis, ANGPLT3 diminishes the activity of vascular Lipase Lipoprotein Lipase (LPL) and endothelial Lipase (EL). ANGPTL3 is a weak inhibitor of LPL, but when complexed with ANGPTL8, the ability of ANGPTL3 to bind to and inhibit LPL is enhanced. EL converts VLDL1 (nascent VLDL) to VLDL2 (mature VLDL). VLDL is secreted from the Liver where it is hydrolyzed by Lipoprotein Lipase (LPL) resulting VLDL remnant/intermediate density Lipoprotein (IDL), and subsequently Low density Lipoprotein (LDL). ApoC3 impacts lipolysis of triglyceride-rich Lipoproteins via LPL-dependent and independent manners. ApoC3 inhibits LPL activity, and it displaces LPL from TGLs, after which, ANGPTL4 inactivates LPL. PL = phospholipid; other abbreviations as in Figure 1.
Conventionally, remnant cholesterol (RC) is estimated mathematically as the sum of chylomicron, VLDL and intermediate-density lipoprotein (IDL) cholesterol, using the formula RC = total cholesterol - high-density lipoprotein cholesterol (HDL-C) - LDL cholesterol (LDL-C). Consensus statements recommend direct LDL-C measures rather than estimates from the Friedewald equation or other mathematical approximations (4,5). However, this calculation method for RC is not a specific measure of TRLs, and does not reflect the dynamics of TGL metabolism.
ATHEROGENICITY OF TRIGLYCERIDE-RICH REMNANTS
Multitudinous sources of evidence suggest TRLs contribute to the development of atherothrombosis (reviewed in Shaik and Rosenson [1], Chait et al [3], Duran and Pradhan [4]). Although chylomicrons are too large to enter the arterial wall until they are hydrolyzed by lipoprotein lipase, many cell types that are ultimately involved in atherosclerosis and thrombosis interact with chylomicron remnants in the circulation. Unlike LDL, which requires chemical modification to enter the arterial wall, chylomicron remnants are directly endocytosed by macrophages. TRL particles are theoretically more atherogenic than LDL particles by way of larger size, 20- to 40-fold higher content of cholesterol molecules per particle, and are longer retained in the intima. Interestingly, VLDL obtained from subjects with hypertriglyceridemia, regardless of size, bind with high affinity to LDL receptors and to a distinct apoB48 receptor expressed specifically by monocytes, macrophages, and endothelial cells. Receptor binding and uptake of remnant lipoproteins by macrophages result in the formation of lipid-filled, macrophage-derived “foam cells” of atherosclerotic lesions.
TRLs also regulate atherogenesis by activating inflammation (6,7). Lipoprotein lipase (LPL)-mediated hydrolysis of chylomicrons and VLDL increases production of oxidized free fatty acids and remnant lipoproteins, which then induce endothelial inflammation and increased production of intracellular adhesion molecule-1 (8). Moreover, these lipoproteins increase production of reactive oxygen species and secretion of tumor necrosis factor-α and interleukin 1β, resulting in impaired endothelium-dependent vasodilation and increased endothelial apoptosis and oxidative stress. TRLs also can induce apoptosis in endothelial cells by nicotinamide adenine dinucleotide phosphate (NAD[P]H). NAD(P)H oxidase mediates production of superoxide and cytokines via lectin-like oxidized LRP-1 activation (9).
TRLs facilitate thrombosis through multiple pathways (reviewed in Shaik and Rosenson [1] and Chait et al [3]). Mechanisms of VLDL remnant-like particles mediated platelet activation include changes in platelet lipid composition and receptor-dependent interaction between enhanced shear-induced platelet activation and increased expression of P-selectin and platelet-derived microparticles. VLDL can activate the intrinsic arm of the coagulation system, and upregulate the gene expression and activity of plasminogen activator inhibitor-1 antigen.
EVIDENCE FROM EPIDEMIOLOGICAL OBSERVATIONS
REMNANT CHOLESTEROL.
Multiple epidemiological studies report statistically significant associations between TRLs and incident ASCVD (reviewed in Duran and Pradhan [4]) (Table 1) (6,10–16). Analyses conducted on nonfasting samples demonstrate stronger associations with cardiovascular risk than studies relying on fasting samples (4). A recent analysis of RC measured from fasting samples and cardiovascular events was investigated in 6,901 older individuals at high cardiovascular risk based on a diagnosis of type 2 diabetes or the presence of 3 or more risk factors (12). In a multivariate adjusted Cox model, every 10 mg/dL increment increase in RC was associated with a higher risk (hazard ratio [HR]: 1.21; 95% CI: 1.10-1.33) of a major adverse cardiac event (MACE) when compared with TG (HR: 1.04; 95% CI: 1.02-1.06) or non-HDL-C levels (HR: 1.05; 95% CI: 1.01-1.10). Baseline RC ≥ 30 mg/dL (75th percentile of the cohort) was associated with an adjusted HR of 1.83 (95% CI: 1.30-2.58). Surprisingly, LDL-C was unrelated to risk in this study. An earlier study from ARIC (Atherosclerosis Risk In Communities) measured RC enzymatically on fasting specimens from 9,334 participants (15). After adjustment for traditional risk factors including lipids, the risk of a cardiovascular event was nonsignificant. In contrast, a pooled analysis of 17,532 ASCVD-free participants from ARIC, MESA (Multi-Ethnic Study of Atherosclerosis), and CARDIA (Coronary Artery Risk Development in Young Adults) calculated RC was associated with a 1.65 (95% CI: 1.45-1.89) higher risk of ASCVD after multivariable adjustment for LDL-C and apoB (14).
TABLE 1.
Triglyceride-Rich Remnants Lipoprotein Cholesterol and Incident Cardiovascular Disease
| Study Association (Ref. #) | Sample Size | Fasting Status | Measurement | Outcome | HR (95% CI) Adjusted Risk |
|---|---|---|---|---|---|
| CGS, CCHS, CIHS (13) | 75,513 | Nonfasting | TC-LDL-C-HDL-C | Ischemic heart disease | 2.3 (1.7-3.1) for highest vs lowest quintile |
| CGS (6) | 106,213 | Nonfasting | TC-LDL-C-HDL-C | MI | Normal weight: 2.0 (1.3-3.2) Overweight: 1.9 (1.4-2.6) Obese: 2.3 (1.4-3.5); for highest vs lowest quartile |
| CGS (11) | 102,964 | Nonfasting | TC-LDL-C-HDL-C | Ischemic stroke | 1.99 (1.49-2.67) for highest vs lowest quartile |
| PREDIMED (12) | 6,901 | Overnight fast | TC-LDL-C-HDL-C | Cardiovascular event (MI, ischemic stroke, CVD death) | 1.83 (1.30-2.58) highest (>30.95 mg/dL) vs lowest quartile (<17.5 mg/dL) |
| Women’s Health Study (13,16) | 480 cases, 496 controls | Nonfasting | TRL cholesterol | Cardiovascular events (MI, ischemic stroke, PAD, CVD death) | 1.87 (1.14-3.06) |
CCHS = Copenhagen Community Health Study; CGS = Copenhagen General Study; CI = confidence interval; CIHS = Copenhagen Ischemic Heart Study; CVD = cardiovascular disease; HDL-C = high-density lipoprotein cholesterol; HR = hazard ratio; LDL-C = low-density lipoprotein cholesterol; MI = myocardial infarction; PAD = peripheral artery disease; PREDIMED = Prevencion con Dieta Mediteranea; TC = total cholesterol; TRLC = triglyceride-rich lipoprotein cholesterol.
The WHS (Women’s Health Study) evaluated the risk of incident cardiovascular disease associated with TRL cholesterol and small-dense LDL (sdLDL) (16). The risk of MACE increased across quartiles of TRL cholesterol in fully adjusted models, including cardiovascular risk factors, baseline total LDL-C and high-sensitivity C-reactive protein (multivariable adjusted HR: 1.87; 95% CI: 1.14-3.06), and separately for myocardial infarction (MI) (3.05; 95% CI: 1.46-6.39) and peripheral artery disease (2.58; 95% CI: 1.33-5.01).
VLDL CHOLESTEROL.
The risk of MI associated with TRLs was investigated in 25,480 individuals enrolled from CGPS (Copenhagen General Population Study) (17). The lipid content of TRLs was derived from NMR spectroscopy-measured cholesterol and TG content of VLDL, IDL, and LDL (13). For every 39 mg/dL increase in cholesterol content, respective multivariable adjusted HRs for VLDL, IDL, and LDL were 2.07 (95% CI: 1.81-2.36), 5.38 (95% CI: 3.73-7.75), and 1.86 (95% CI: 1.62-2.14). Overall, VLDL cholesterol (VLDL-C) explained 46% of the risk from apoB-containing lipoproteins, and the sum of IDL and LDL explained 25% of the risk. In a separate analysis, RC explained only 10% (95% CI: 0%-24%) of MI risk.
LDL-TRIGLYCERIDE AND sdLDL.
Enzymatic methods have been used to measure the TG content in LDL. Among older participants in ARIC, LDL-TG concentrations were associated with a higher risk of coronary heart disease after adjustment for traditional risk factors, including lipids; HR: 1.72; 95% CI: 1.25-2.37 per log unit increase in LDL-TG (18).
The association of sdLDL cholesterol with coronary heart disease events was examined in a meta-analysis of 21 studies, including a total of 30,628 participants who had 5,693 incident coronary heart disease events (19). The pooled estimate for high versus low category of sdLDL was 1.36 (95% CI: 1.21-1.52) and 1.07 (95% CI: 1.02-1.12) when analyzed by the top versus lowest quartile. In WHS, sdLDL was associated with a multivariable adjusted HR for MI of 3.71 (95% CI: 1.59-8.63), but not with total cardiovascular events, ischemic stroke, or peripheral arterial disease (16). In an analysis of the Framingham Offspring Study, sdLDL was the atherogenic lipoprotein parameter most strongly associated with incident atherosclerotic cardiovascular disease events (MI, angina, stroke, transient ischemic attacks, cardiovascular death, revascularization procedures). The HR for sdLDL and ASCVD was 1.42 (P < 0.0001) in models that adjusted for the pooled cohort risk equation (20).
EVIDENCE FROM GENETIC STUDIES
Mild to moderate hypertriglyceridemia is often polygenic in etiology (21). Among patients with severe hypertriglyceridemia (≥885 mg/dL), 46.3% had polygenic hypertriglyceridemia and 1.1% had biallelic (homozygous or compound heterozygous) variants in the same gene or homozygous monogenic hypertriglyceridemia. Loss-of-function variants (LOFV) in LPL or LPL pathway genes result in moderate (fasting TGs ≥150 mg/dL) to severe (fasting TGs ≥885 mg/dL) elevations in TGs and an increased risk of ASCVD (1,4).
EVIDENCE FROM OMEGA-3 FATTY ACID CLINICAL TRIALS
Epidemiologic and observational studies have shown that consumption of marine n-3 polyunsaturated fatty acids (PUFAs) is associated with lower cardiovascular risk (22). It has been mostly speculated that the reduced cardiovascular risk associated with n-3 PUFA consumption is mediated by their TG-lowering effect (23). However, interventional clinical trials aimed at reducing cardiovascular incidents by supplementation with n-3 PUFAs have yielded inconsistent results, likely due to the heterogenicity of outcome trials with different formulations, dosages, and patient populations studied (Table 2) (24–29).
TABLE 2.
Lipid-Lowering Efficacy and Clinical Trial Evidence With Omega-3 Fatty Acids
| Study (Ref. #) | Patient Population | Sample Size | Omega-3 PUFAs | Dose (g) | Omega-3 FFA Equivalence (g) | Placebo | Duration (y) | Baseline TG (mg/dL) | Intergroup Change of TG (%) | Outcome HR (95% CI) |
|---|---|---|---|---|---|---|---|---|---|---|
| JELIS (25) | Hypercholesterolemia patients with baseline LDL-C 182 mg/dL | 18,645 | EPA EE | 1.8 | 1.6 | No placebo | 4.6 | 151 | 5.0 | 0.81 (0.69-0.95) |
| VITAL (26) | Healthy volunteers (men>50 y or women>55 y) | 25,871 | EPA EE DHA EE |
0.46 0.38 |
0.42 0.35 |
“Fish oil placebo” | 5.3 | Unknown | Unknown | 0.92 (0.8-1.06) |
| ASCEND (27) | Diabetic patients without ASCVD | 15,480 | EPA EE DHA EE |
0.46 0.38 |
0.42 0.35 |
Olive oil | 7.4 | Unknown | Unknown | 0.97 (0.87-1.08) |
| REDUCE-IT (28) | Diabetic patients and patients with ASCVD | 8,179 | EPA EE | 4 | 3.5 | Mineral oil | 4.9 | 217 | 20.5 | 0.75 (0.68-0.83) |
| STRENGTH (29) | Diabetic patients, patients with ASCVD and high-risk patients | 13,078 | EPA DHA |
0.55 0.2 |
2.2 0.8 |
Corn oil | 3.2 | 240 | 18.1 | 0.99 (0.9-1.09) |
ASCEND = A Study of Cardiovascular events in diabetes; ASCVD = atherosclerotic cardiovascular disease; DHA = docosahexaenoic acid; EE = ethyl ester; EPA = eicosapentaenoic acid; FFA = free fatty acid; HR = hazard ratio; JELIS (The Japan EPA Lipid Intervention Study); LDL-C = low-density lipoprotein cholesterol; PUFAs = polyunsaturated fatty acids; TG = triglyceride; VITAL = The VITamin D and OmegA-3 Trial; REDUCE-IT = the Reduction of Cardiovascular Events with Icosopent Ethyl-Intervention Trial; STRENGTH = Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia.
Results of n-3 PUFA cardiovascular outcome trials are inconsistent, with the exception of REDUCE-IT (the Reduction of Cardiovascular Events with Icosopent Ethyl-Intervention Trial) (Table 2) (25–29). Among patients with ASCVD and moderately elevated TGs (>135 mg/dL to 500 mg/dL) on the background of moderate to high-intensity statin (>93%), treatment with icosopent ethyl 4 g daily versus placebo reduced cardiovascular events (relative risk reduction 25%, absolute reduction 4.8%). Furthermore, the relatively modest TG-lowering effect (33 mg/dL placebo-corrected difference) is unlikely to explain the large risk reduction. Although there is debate about whether mineral oil in the placebo might have affected statin absorption because there was a modest increase of LDL-C level in the placebo group, differences in LDL-C between groups were small (5 mg/dL net difference). A recent systematic meta-analysis showed that the relative risk reduction of MACE was 0.79 per 40 mg/dL reduction in LDL-C but only 0.92 per 40 mg/dL reduction in TGs (30). Based on these data, the net difference of TGs (−33 mg/dL) and LDL-C (+5 mg/dL) should translate into only approximately 9% (6.6% due to TG + 2.8% due to LDL-C) relative risk reduction in MACE (28). JELIS (Japan EPA Lipid Intervention Study) was another EPA trial that showed a reduction in cardiovascular events (25). However, the STRENGTH (Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia) trial evaluated the efficacy of 4 g omega-3 carboxylic acid (EPA ethyl ester + docosahexaenoic acid ethyl ester) in a population similar to REDUCE-IT, but was halted due to futility (29) despite a similar level of TG reduction (34 mg/dL). Ample evidence has suggested that compared to DHA, EPA plays more favorable roles in the cardiovascular system (31–33). It is notable that the absolute EPA dose used in REDUCE-IT is much higher than that used in STRENGTH. The duration of REDUCE-IT is also significantly longer than STRENGTH. The total exposure of dose·duration of EPA is about 2.5 times higher (17.4 grams·year vs 6.6 grams·year) in REDUCE-IT compared to that in STRENGTH. Furthermore, a recent study suggests higher levels of EPA in the blood lower the risk of major cardiac events and death in patients, whereas DHA decreased the cardiovascular benefits of EPA (34). Therefore, the triglyceride independent beneficial effect of EPA but not DHA in the cardiovascular system possibly explain the controversies in the omega-3 trials. On the other hand, the contrasting effects of the comparative oils (mineral vs corn) as an explanation for the disparate outcomes in REDUCE-IT and STRENGTH was explored in a cohort study that mimicked trial designs. In the REDUCE-IT mineral oil arm, LDL cholesterol, non-HDL cholesterol, apolipoprotein B and C-reactive protein were all increased at the end of the study, whereas the STRENGTH corn oil arm was not accompanied by changes in these atherogenic lipids. Using data from the Copenhagen General Population Study of 106,088 individuals, 5,684 met inclusion criteria for REDUCE-IT and 6,832 met criteria for STRENGTH. The hazard ratio for the mineral oil vs corn oil comparators were 1.07 (1.04-1.10) vs 0.99 (0.98-0.99). When compared to the active oils (EPA in REDUCE-IT and EPA/DHA), part of the risk reduction was explained by the effects of mineral oil on lipids and CRP. Further evaluation of this hypothesis will require a clinical trial comparing all treatments in these trials (EPA, EPA plus DHA, mineral oil and corn oil) (35). However, what is urgently needed, before the initiation of a new clinical trial with an inert placebo, are well designed mechanism studies to understand differential impacts of EPA and DHA in the cardiovascular system.
APOLIPOPROTEIN C-III.
The APOC3 gene encodes apolipoprotein C-III (apoC-III) and is expressed primarily in hepatocytes and, to a lesser extent, in enterocytes. The distribution of apoC-III among TG-rich lipoproteins (TGRLs) varies in fasting and postprandial states and based on plasma TG levels. Although glucose increases hepatic expression of APOC3, insulin and PPAR-α reduce its transcription rate. As a result, those with type 2 diabetes have higher secretion rates of apoC-III. When a glucagonlike peptide-1 analog is added to improve glycemic control, apoC-III secretion rate and thus apoC-III levels are significantly reduced (36).
GENETIC EVIDENCE.
Numerous genetic and epidemiologic studies highlight apoC-III as a key regulator of TG metabolism. Increased APOC3 expression has been shown to result in hypertriglyceridemia, whereas LOFVs associate with lower TG levels. In 75,725 participants in CCHS (Copenhagen Community Health Study) and CGPS, heterozygosity for APOC3 loss-of-function mutations (LOFVs) was associated with a 44% reduction in nonfasting TGs levels when compared with noncarriers (37). This further corresponded to a 36% reduction in ischemic heart disease (38). The Exome Sequencing Project demonstrated that heterozygous carriers for any of 4 APOC3 LOFVs were associated with lower apoC-III levels of 46% and TG levels of 39% and cardiovascular risk of 40% (38). In another genome-wide association study (GWAS), 5% of Lancaster Amish, carriers of the apoC-III-R19X null mutation, had 50% lower apoC-III levels, 35% lower TG levels, and significantly lower coronary artery calcification scores when compared with noncarriers (39).
MECHANISM OF ACTION.
The mechanisms whereby apoC-III impacts TGL lipolysis are still not fully elucidated; however, apoC-III appears to work in both LPL-dependent and -independent manners (Figure 3). ApoC-III inhibits LPL activity, and increasing apoC-III levels correlate with reduced TGRL lipolysis. It was thought that apoC-III inhibits LPL by directly competing with apoC-II; however, this has been demonstrated only when the ratio of apoC-III to apoC-II molecules is greater than physiological levels (40). Instead, apoC-III appears to displace LPL from TGLs, after which, ANGPTL4 inactivates LPL.
FIGURE 3. Triglyceride Remnant Lipoproteins and Mechanisms of Atherogenicity.
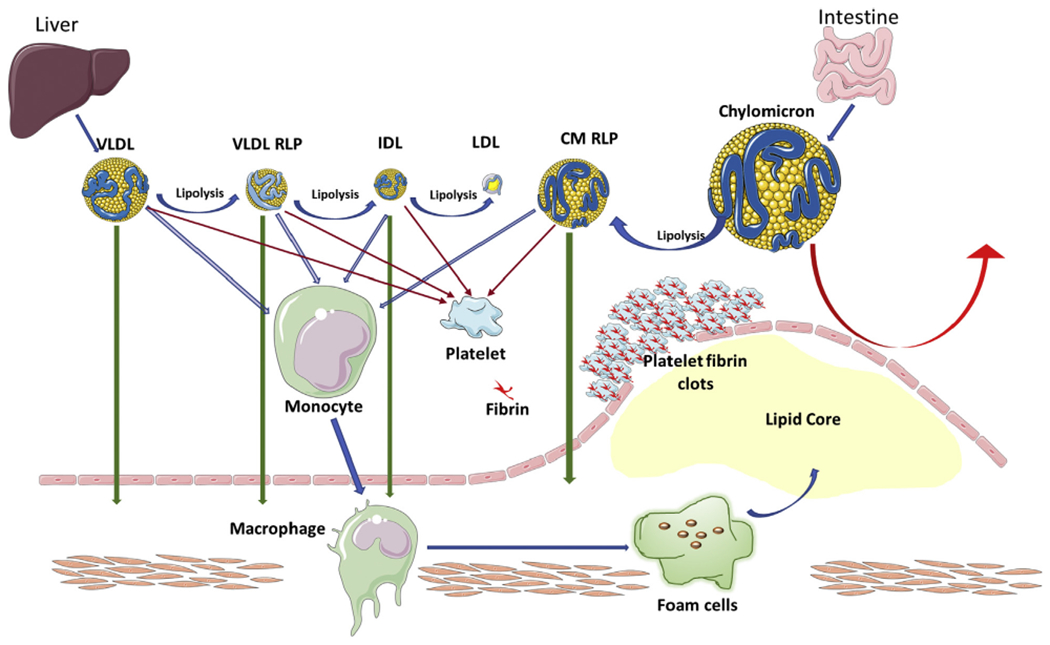
Schematic depicting the mechanism whereby triglyceride remnant lipoproteins increase atherosclerosis. Triglyceride remnants have multifarious contributions to atherosclerosis. Chylomicrons are too large to enter the arterial wall. Lipoprotein lipase hydrolyzes chylomicrons and CM RLP can then infiltrate into the subendothelium, uptake by macrophages can contribute the lipid core. Monocytes can also directly uptake all triglyceride remnants in circulation before migrating into the subendothelium. Triglyceride remnants can also activate platelets and the coagulation pathway, and support the assembly of the prothrombinase complex and formation of microthrombosis. CM = chylomicron; RLP = remnant like protein; other abbreviations as in Figure 1.
The presence of LPL-independent mechanisms of apoC-III-mediated lipolysis is supported by TG reduction with apoC-III ASO (antisense oligonucleotide) in patients with familial chylomicronemia syndrome (FCS) with a genetic deficiency of LPL (41). Human kinetic studies show that LDLR- and LRP1-mediated clearance of TRLs containing apoC-III is slower than those without apoC-III (42,43). In lipid-rich conditions, apoC-III also increases hepatic secretion of VLDL molecules and prevents apoE binding to hepatic LDLR, LRP1, and syndecan-1 receptors (44).
ApoC-III INHIBITION IN HYPERTRIGLYCERIDEMIA.
Antisense oligonucleotide.
Volanesorsen (ISIS-APOCIIIRX) is a second-generation ASO that selectively binds apoC-III messenger RNA (mRNA), preventing its translation and prompting its degradation, thus resulting in reduced plasma apoC-III and TG levels (Table 3) (45–50). The APPROACH (A Study of Volanesorsen [Formerly IONIS-APOCIIIRx] in Patients With Familial Chylomicronemia Syndrome) phase 3 randomized, double-blind, placebo-controlled trial examined the effect of volanesorsen in 66 patients with FCS and severe hypertriglyceridemia (fasting TG >750 mg/dL) (47). Patients who were receiving volanesorsen had an 84% decrease in apoC-III levels at 3 months and a 77% reduction in mean TG levels, compared with an increase of 6.1% and 18%, respectively, in the placebo group (47). In the COMPASS (Efficacy and Safety of Volanesorsen in Patients With Multifactorial Chylomicronaemia) trial, 113 patients with severe hypertriglyceridemia (500-1,261 mg/dL) were randomized to receive either weekly volanesorsen or placebo for 26 weeks. TG levels were reduced by 73% in the volanesorsen group compared with 2% in the placebo group (48).
TABLE 3.
Lipid-Lowering Efficacy of ApoC-III Inhibitors
| Therapy | Trial Phase (Ref. #) | Study Design | Sample Size | Study Population | Dose | ApoC-III | TG | LDL-C | HDL-C | Non-HDL-C | ApoB-48 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Volanesorsen | 1 (45) | Double-blind, placebo-controlled, ascending dose | 33 | Healthy volunteers 18-55 y | 50 mg SC QW | −20% | −20% | +18% | +19% | — | — |
| 100 mg SC QW | −17% | −25% | −4% | 0% | — | — | |||||
| 200 mg SC QW | −70.5% | −43% | −3% | +14% | — | — | |||||
| 400 mg SC QW | −78% | −44% | −4% | +8% | — | — | |||||
| 2 (46) | Randomized, double-blind, placebo-controlled, dose-ranging | 57 | Severe hypertriglyceridemia (TG 350-2000 mg/dL) | 100 mg SC QW | −40% | −38% | +48% | +27% | −7% | −20% | |
| 200 mg SC QW | −64% | −70% | +79% | +36% | −7% | −39% | |||||
| 300 mg SC QW | −80% | −72% | +118% | +46% | −11% | −61% | |||||
| 3 (47) | Randomized, double-blind, placebo-controlled | 66 | FCS (TG >750 mg/dL) | 300 mg SC QW | −84% | −77% | +136% | +46% | −46% | +20% | |
| 3 (48) | Randomized, double-blind, placebo-controlled | 113 | Severe hypertriglyceridemia (TG > 500 mg/dL) | 300 mg SC QW | — | −72% | — | — | — | — | |
|
| |||||||||||
| AKCEA-APOC-III-LRX | 1/2a (49) | Double-blind, placebo-controlled, single ascending dose | 40 | Healthy volunteers | 10 mg SC | 0% | −12% | — | — | — | — |
| 30 mg SC | −42% | −7% | — | — | — | — | |||||
| 60 mg SC | −73% | −42% | — | — | — | — | |||||
| 90 mg SC | −81% | −73% | — | — | — | — | |||||
| 120 mg SC | −92% | −77% | — | — | — | — | |||||
| Double-blind, placebo-controlled, multiple ascending dose | 17 | 15 mg SC QW | −65% | −61% | −3% | +50% | −22% | — | |||
| 30 mg SC QW | −84% | −71% | −17% | +56% | −30% | — | |||||
| 60 mg SC Q4W | −80% | −61% | −10% | +64% | −27% | — | |||||
|
| |||||||||||
| ARO-APOC3 | 1 (50) | Double-blind, placebo-controlled | 4 | Hypertriglyceridemia (TG >300 mg/dL) | 50 mg SC single dose | −96% | −78% | — | +71% | −33% | — |
| 6 | MCS (TG >880 mg/dL) | 50 mg single dose | −96% | −92% | — | +136% | −64% | — | |||
ApoB = apolipoprotein B; ApoC-III = apolipoprotein C-III; FCS = familial chylomicronemia syndrome; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; MCS = multifactorial chylomicronemia syndrome; Non-HDL-C = non-high-density lipoprotein cholesterol; SC = subcutaneously; TG = triglycerides; QW = every week; Q4W = every 4 weeks.
Ligand conjugated antisense.
AKCEA-APOCIII-LRX is an N-acetyl galactosamine-conjugated ASO that selectively inhibits hepatic apoC-III synthesis. A phase 1/2a double-blind, placebo-controlled, dose-escalation study (NCT02900027) was conducted in healthy volunteers with TGs >90 or >200 mg/dL. In the single-dose cohort, there were dose-dependent reductions in apoC-III (92% with 120 mg compared with 42% with 30 mg) and TG levels (77% and 7% with 120 mg and 30 mg, respectively) 14 days after dosing. In the multiple-dose cohorts, reductions of 66%, 84%, and 89% in apoC-III were seen in the 15-mg weekly, 30-mg weekly, and 60 mg every 4 weeks groups, respectively, 1 week after the last dose (49). There were also significant reductions in total cholesterol, apoB, non-HDL-C, and VLDL-C, as well as an increase in HDL-C. The phase 3 BALANCE randomized, double-blind, placebo-controlled study in patients with FCS is ongoing at the time of this publication (NCT04568434).
Small interfering RNA.
ARO-APOC3 is a small interfering RNA that preferentially targets hepatocyte APOC3 mRNA. A phase 1 study (NCT03783377) with single subcutaneous doses of ARO-APOC3 demonstrated reductions in apoC-III and TG levels in healthy volunteers when compared with placebo (50). Initial results of a subsequent study of patients with hypertriglyceridemia (TGs >300 mg/dL) or multifactorial chylomicronemia syndrome (MCS) showed a 96% reduction of apoC-III levels in both groups 4 weeks after the first dose of ARO-APOC3. In the hypertriglyceridemia group, mean TG levels decreased by 78% and by 92% in the MCS group (50). A phase 2b dose-finding study in patients with severe hypertriglyceridemia and a phase 3 study in patients with FCS is ongoing (NCT04863014). This medication can potentially be dosed quarterly or semi-annually, which has important implications for treatment adherence.
ANGPTL3.
ANGPTL3 inhibits LPL and endothelial lipase (EL), and is thus a key regulator of lipid metabolism (Figure 4). LOFVs in ANGPTL3 are associated with lower levels of LDL-C, TGs, and HDL-C. Large GWAS have confirmed these observations. In a pooled analysis of 58,335 participants in the Discover Electronic Health Record human genetics study, the 212 carriers versus the 49,017 noncarriers of LOFVs in ANGPTL3 had lower serum levels of TGs (94 [interquartile range (IQR): 75-125] vs 130 mg/dL [IQR: 94-179], P = 2.5 × 10−21), LDL-C levels (112 [IQR: 90-136] vs 121 [IQR: 100-146] mg/dL, P = 2.8 × 10−5), HDL-C (46 [IQR: 38-56] vs 49 [IQR: 40-59] mg/dL, P = 0.02) (51). This study reported an odds ratio of 0.60 (0.41-0.86) for coronary artery disease (CAD) in carriers versus noncarriers of ANGPTL3 LOFV. An analysis of 18,231 participants in the Myocardial Infarction Genetics Consortium reported that LOFVs in ANGPTL3 were associated with lower levels of LDL-C of −11.8% (95% CI: −21.5 to −2.1) and TGs of −17.3% (95% CI: −31 to −3.4), but no difference in HDL-C (−5.2 [95% CI: −12.8 to 2.3]) (52). In this meta-analysis of Mendelian randomization studies, carriers had an odds ratio for CAD of 0.66 (95% CI: 0.44-0.98) compared with noncarriers.
FIGURE 4. ANGPTL-3 Protein Inhibition Enhanced Remnant Lipoprotein Clearance.
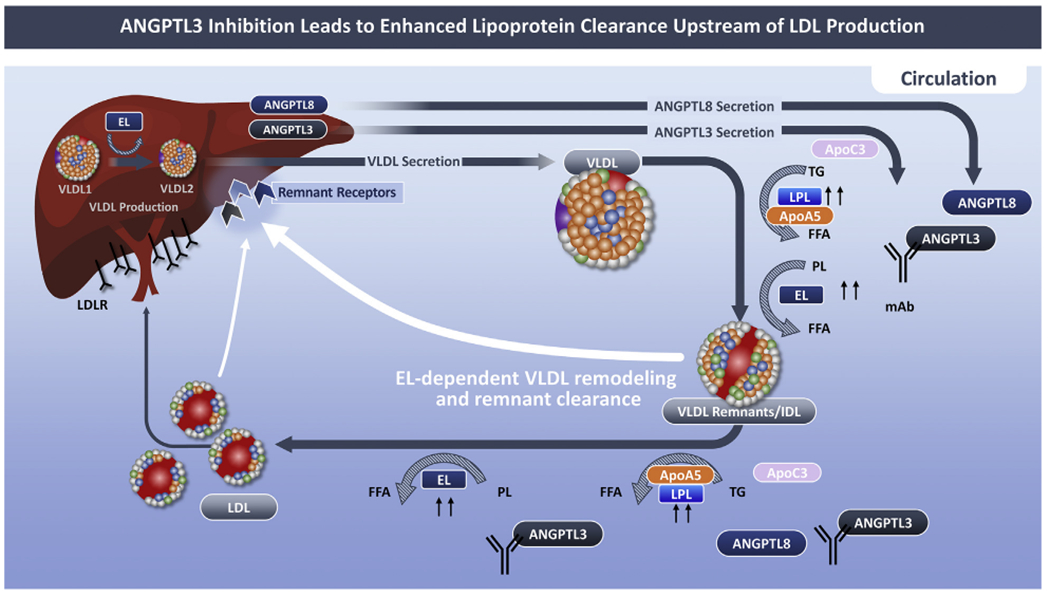
ANGPTL3 inhibition unblocks the inhibitory effect of ANGPTL3 on both lipases, promoting VLDL remodeling and removal of VLDL remnants from the circulation by hepatic remnant receptors. The effects of a fully human monoclonal antibody, evinacumab, are depicted. By enhancing clearance of VLDL remnants, the pool for LDL is reduced resulting in a reduction in LDL cholesterol. ApoA5 lowers triglycerides by suppressing ANGPTL3/8-mediated LPL inhibition. Lowering of apo3 concentrations is another potential mechanism for the increased LPL activity with ANGPTL3 inhibition. FCS = familial chylomicronemia syndrome; MCS = multifactorial chylomicronemia syndrome; other abbreviations as in Figure 1.
These studies of genetic epidemiology are supported by classical epidemiology. Among older adults participating in ARIC, ANGPTL3 levels were independent predictors of ASCVD events after adjustment for traditional risk factors, including lipids (HR: 1.63; 95% CI: 1.17-2.28 per log unit increase inANGPTL3) (18).
MECHANISM OF ACTION.
Studies in experimental models have shown that ANGPTL3 represses LPL and EL (Figure 4). ANGPTL3 is a weak inhibitor of LPL, but when complexed with ANGPTL8, the ability of ANGPTL3 to bind to and inhibit LPL is enhanced (53,54). In contrast, apoA5 lowers TGs by suppressing ANGPTL3/8-mediated LPL inhibition (55). Inhibition of ANGPTL3 or ANGPTL8 increases LPL and decreases TG levels (56). Lowering of apoCIII concentrations is another potential mechanism for the increased LPL activity with ANGPTL3 inhibition.
ANGPTL3 inhibition with evinacumab mediates an EL-dependent pathway that reduces VLDL lipid content and particle size, generating remnant particles that are efficiently cleared from the circulation (57). Consequently, evinacumab reduces LDL-C (58).
ANGPTL3 PROTEIN INHIBITION IN HYPERTRIGLYCERIDEMIA.
Anti-ANGPTL3 human monoclonal antibodies.
Evinacumab was initially investigated in several phase 1 studies (Table 4) (51,59,60). Among patients with TG levels ≥450 mg/dL to <1,500 mg/dL, intravenous evinacumab 10 mg/kg lowered TGs by a median of 81.5% at day 4 versus 20.6% with placebo. TG lowering with evinacumab 20 mg/kg intravenously was highly variable among patients with severe hypertriglyceridemia (>1,000 mg/dL with LPL pathway mutations) ranging from 0.9% to 93.2% on day 3 with sustained efficacy through day 22 in most subjects. The efficacy and safety of evinacumab 15 mg/kg intravenously in patients with severe hypertriglyceridemia (≥500 mg/dL on 2 occasions and ≥1,000 mg/dL on 1 occasion) with 1 or more hospitalizations for acute pancreatitis has been evaluated in a double-blind placebo-controlled phase 2 trial of 52 participants (NCT03452228) (61). Eligible participants were randomized into 3 cohorts based on genotyping for LOFVs in LPL or LPL pathway genes. Cohort 1 comprised patients with FCS with homozygous or compound heterozygous LOFVs in LPL pathway (LPL) mutations; cohort 2 included patients with heterozygous LOFVs in LPL pathway mutations MCS, and cohort 3 included patients with no LPL pathway mutations. Study participants were randomized 2:1 to evinacumab or placebo. The doubleblind treatment period was followed by a single-blind period of 12 weeks in which all participants received treatment with evinacumab 15 mg per kg intravenously every 4 weeks. The primary efficacy endpoint was mean percent change in TGs from baseline to week 24. Secondary endpoints included changes in non-HDL-C, ApoB48, ApoB100, and apoC-III.
TABLE 4.
Lipid-Lowering Efficacy of ANGPLT3 Inhibitors
| Therapy | Trial Phase (Ref. #) | Sample Size | Study Design | Dose | TG | Non-HDL-C | Remnant Cholesterol | LDL-C | Total ApoB | ApoB100 | ApoB48 | Apo C-III | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Evinacumab | HTG | 1 (59) | 83 | Single dose Cohort A: (TGs ≥150 and >450 mg/dL) |
5 mg/kg IV | −80.3% (Day 4) | −30.6% (Day 4) | — | −18.2% (Day 8) | −24.6% (Day 4) | — | — | — |
| 10 mg/kg IV | −88.0% (Day 4) | −31.2% (Day 11) | — | −19.0% (Day 4) −22.8% (Day 8) −14.5% (Day 22) |
−21.4% (Day 11) | — | — | — | |||||
| 20 mg/kg IV | −83.9% (Day 4) | −35.4% (Day 11) | — | −16.3% (Day 4) −14.8% (Day 43) |
−26.8% (Day 11) | — | — | — | |||||
| 75 mg SC | −20.8% (Day 4) | −6.9% (Day 43) | — | −12.4% (Day 11) | −3.4% (Day 43) | — | — | — | |||||
| 150 mg SC | −40.8 (Day 4) | −12.9% (Day 4) | — | −12.9% (Day 3) −15.9% (Day 8) |
−8.3% (Day 8) | — | — | — | |||||
| 250 mg SC | −55.5% (Day 4) | −23.7% (Day 11) | — | −20.6% (Day 8) −20.3% (Day 11) |
−13.6% (Day 43) | — | — | — | |||||
|
| |||||||||||||
| Evinacumab | HTG | 1 (60) | 16 | Single dose Cohort B: High HTG (≥450 and <1,500 mg/dL) Cohort C: Severe HTG (>1,000 mg/dL) |
10 mg/kg IV [Cohort B] | -81.8% (Day 4) | −32.0 (Day 8) | — | +54.4% (Day 4) | — | — | — | — |
| 20 mg/kg IV [Cohort C] | −0.9 to −93.2% (Day 3) | −57.8 (Day 22) | — | +74.4% (Day 29) | — | — | — | — | |||||
| 250 mg SC [Cohort C] | −37.8% (Day 8) | −27.4 (Day 8) | — | +79.6% (Day 64) | — | — | — | — | |||||
| HTG | 1 (61) | 56 | Multiple ascending dose | 20 mg/kg IV Q4W | −88.2% (Day 2) | −45.8% (Day 36) | — | −25.1% (Day 57) | −30.7% (Day 57) | — | — | — | |
| −39.8% (Day 57) | |||||||||||||
| 150 mg SC QW | −42.0% (Day 57) | −24.5% (Day 57) | — | −12.4% (Day 57) | −17.9% (Day 36) | — | — | — | |||||
| 300 mg SC Q2W | −45.5% (Day 36) | −14.7% (Day 57) | — | −8.6% (Day 8) | −18.5% (Day 36) | — | — | — | |||||
| 300 mg SC QW | −51.9% (Day 15) | −37.5% (Day 57) | — | −22.0% (Day 57) | −23.7% (Day 57) | — | — | — | |||||
| 450 mg SC Q2W | −52.8% (Day 57) | −22.6% (Day 57) | — | −13.1% (Day 8) | −17.8% (Day 57) | — | — | — | |||||
| 450 mg SC QW | −50.3% (Day 15) | −26.2% (Day 57) | — | −12.0% (Day 57) | −17.4% (Day 36) | — | — | — | |||||
| HeFH | 2 (65) | 252 | Double-blind, placebo-controlled. Evinacumab 15 mg/kg IV Q4W. 2:1 randomization (Exclusion criteria: TG >400 mg/dL and >300 mg/dL for patients without and with history of diabetes mellitus, respectively) | 5 mg/kg Q4W | −25.2% | −23.7% | — | −24.2% | −16.6% | — | — | — | |
| 15 mg/kg Q4W | −45.9% | −50.9% | — | −50.5% | −39.4% | — | — | — | |||||
| 300 mg SC Q2W | −46.l% | −39.3% | — | −38.5% | −26.6% | — | — | — | |||||
| 300 mg SC QW | |||||||||||||
| 450 mg SC QW | −55.8% | −53.8% | — | −52.9% | −42.0% | — | — | — | |||||
| −61.5% | −58.5% | — | −56.0% | −45.5% | — | — | — | ||||||
| HoFH | 3(66) | 64 | Double-blind, placebo-controlled Evinacumab 15 mg/kg IV QW. 2:1 randomization (Inclusionc criteria: untreated TG <300 mg/dL) |
15 mg/kg IV | −50.4 | −51.7 | — | −49.0 | −36.9% | — | — | −90.0% | |
|
| |||||||||||||
| Vupanorsen | 1 (62) | Single dose | — | — | — | — | — | — | — | — | — | ||
| TG>150 mg/dL, T2DM and hepatic steatosis | 2 (63) | 105 | Double-blind, placebo-controlled. Vupanorsen 80 mg vs. placebo Q4W | — | −53 | −18 | −38 | −7 (NS) | — | −9 (NS) | —— | −58 | |
ANGPTL3 = angiopoietin-like 3; HeFH = heterozygous familial hypercholesterolemia; HoFH = homozygous familial hypercholesterolemia; HTG = hypertriglyceridemia; IV = intravenous; NS = not significant; T2DM = type 2 diabetes mellitus; Q2W = every 2 weeks; other abbreviations as in Table 3.
Antisense oligonucleotide.
In a phase 1 study, use of a N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, vupanorsen, was associated with a dose-dependent reduction in LDL-C that ranged from −1.3% to −32.9%, but a larger reduction in TGs of 33.2% to 63.1% (62). A phase 2 trial was conducted in 105 patients with type 2 diabetes, hepatic steatosis, and hypertriglyceridemia treated with vupanorsen for 6 months (63). Treatment with vupanorsen 40 mg every 4 weeks, 80 mg every 4 weeks, and 20 mg every week reduced TGs, RC, non-HDL-C, and apoC-III by −24% to −44%, −24% to −38%, −10% to −19%, and −36% to −58%. No significant changes were observed in LDL-C. ApoB100 was reduced by −9% in the group treated with vupanorsen 80 mg every 4 weeks only.
CLINICAL PATHWAYS.
Evidence supporting treatment of high concentrations of TRLs for ASCVD prevention is limited. Clinical trials with TG-lowering therapies in statin-treated patients have failed to demonstrate clinical benefit with the exception of icosopent ethyl (1). Our approach to patients with mild to moderate hypertriglyceridemia (fasting TGs ≥150 mg/dL to <880 mg/dL) involves a low fat and low glycemic index diet with caloric restriction in overweight patients, and regular exercise, including aerobic and muscle strengthening, and guideline-directed management of major cardiovascular risk factors including intensive control of dysglycemia (Central Illustration). Statins have a dose-dependent effect on TG lowering with efficacy of 40% to 44% in patients with TG levels as high as 800 mg/dL to 850 mg/dL. When TGs remain elevated, particularly in the context of an elevated apolipoprotein B, we add ezetimibe. After lowering LDL-C, we consider icosopent ethyl, while recognizing the higher risk of atrial fibrillation with omega-3 fatty acid medications (28), and recognizing the limitations of the REDUCE-IT trial that used an active comparator (mineral oil) that may have accounted for a part of the differences in clinical outcomes between the 2 treatment arms (35). The role for pemafibrate will be elucidated on completion of the PROMINENT (Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients With Diabetes) study (64). This clinical approach is consistent with the recently published “2021 ACC Expert Consensus Decision Pathway on the Management of ASCVD Risk Reduction in Patients with Persistent Hypertriglyceridemia” (2).
CENTRAL ILLUSTRATION. Clinical Algorithm for Treatment of High Triglyceride-Rich Lipoprotein Remnants.
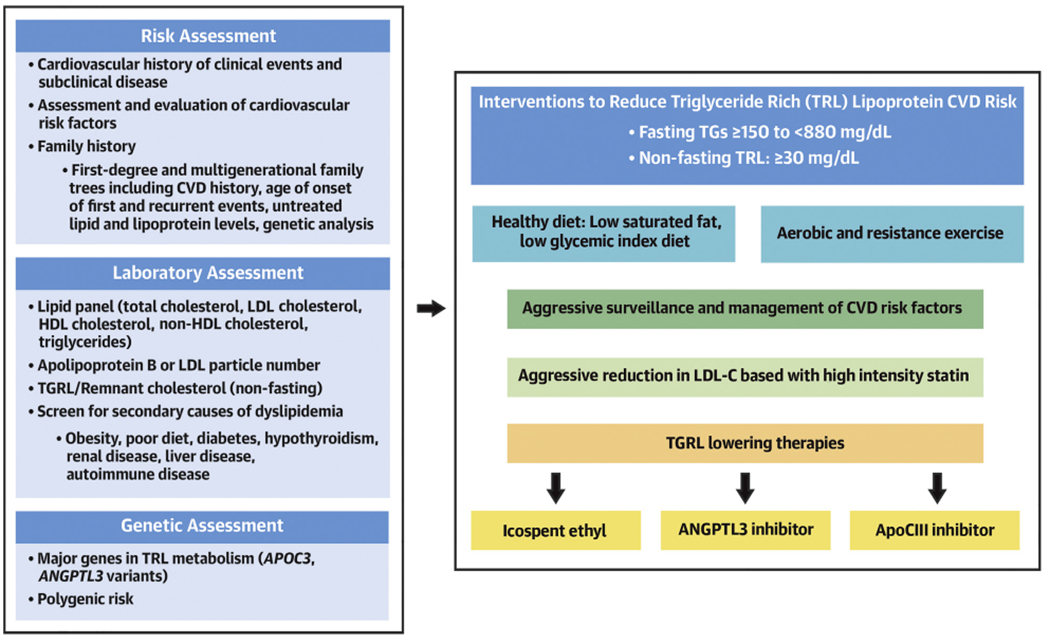
Evaluation of TRL-related ASCVD disease risk begins with evaluation of clinical and subclinical atherosclerosis, and ensuring optimal management of LDL-C and other major modifiable risk factors. We measure remnant cholesterol on nonfasting specimens and consider genetic testing of common traits contributing to impaired hydrolysis of TRLs. Lifestyle changes and weight loss in overweight patients is an essential first step for lowering TRLs. We consider icosopent ethyl in patients with fasting triglycerides ≥135 mg/dL and recognize the importance of ANGPLT3 inhibitors that lower TRLs and LDL-C. For patients with severe hypertriglyceridemia (>880 mg/dL), the use of an apolipoprotein C-III inhibitor is an option, particularly in patients with biallelic LOFV in genes regulating lipoprotein lipase or LPL-related pathways. ANGPTL3 = angiopoietin-like 3; ASCVD = atherosclerotic cardiovascular disease; LDL-C = low-density lipoprotein cholesterol; LOFV = loss-of-function variant; LPL = lipoprotein lipase; TGRL = triglyceride rich remnant lipoprotein; TRL = triglyceride rich lipoprotein.
The 2 major classes of TRL-lowering therapy (ANGPTL3 and apoC-III) inhibitors differ with respect to efficacy in lowering apoB-containing lipoproteins. Large reductions in apoB-100 have been consistently seen with immunotherapy for ANGPTL3 in patients with mild to moderate hypertriglyceridemia, which is a potential advantage for this approach.
SUMMARY
Evidence from observational epidemiological and genomic studies supports the contribution of TGLs in ASCVD development. New therapies for TG lowering have the potential to reduce cardiovascular risk based on natural selection studies. Clinical trials are warranted to establish the efficacy of inhibiting apoC-III and ANGPTL3.
HIGHLIGHTS.
Elevated TRL concentrations are associated with an increased risk of atherosclerotic cardiovascular events.
Most conventional TG-lowering therapies do not reduce the risk of cardiovascular events in statin-treated patients.
Targeting pathways identified by natural selection studies may identify novel therapies that lower TRL levels as well as cardiovascular risk.
ACKNOWLEDGMENTS
The authors acknowledge Priscilla Mejia and Shaunte Baboumian for assisting with the manuscript preparation.
FUNDING SUPPORT AND AUTHOR DISCLOSURES
Prime Medica Global (London, UK) provided assistance with the development of computerized graphics used for Figures 1, 2, and 4 and the Central Illustration. Support for the efforts of Prime Medical Global were provided by Regeneron Pharmaceuticals Inc. (Tarrytown, New York). Dr Rosenson has received research funding through his employer from Amgen, Arrowhead, National Institutes of Health, Novartis, and Regeneron; has received consulting fees and honoraria from Amgen, Amyrt, C5, CVS Caremark, 89 Bio, Kowa, Novartis, and Regeneron; has received royalties from Wolters Kluwer (UpToDate); and has stock holdings in MediMergent, LLC. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- ANGPTL3
angiopoietin-like protein 3
- ASCVD
atherosclerotic cardiovascular disease
- EPA
eicosapentaenoic acid
- HDL-C
high-density lipoprotein cholesterol
- LDL-C
low-density lipoprotein cholesterol
- LOFV
loss-of-function variants
- LPL
lipoprotein lipase
- TGL
triglyceride-rich lipoprotein
- TRL
triglyceride-rich lipoprotein remnants
- VLDL
very low-density lipoproteins
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
REFERENCES
- 1.Shaik A, Rosenson RS. Genetics of triglyceride-rich lipoproteins guide identification of pharmacotherapy for cardiovascular risk reduction. Cardiovasc Drugs Ther. 2021;35(3):677–690. [DOI] [PubMed] [Google Scholar]
- 2.Virani SS, Morris PB, Agarwala A, et al. 2021 ACC expert consensus decision pathway on the management of ASCVD risk reduction in patients with persistent hypertriglyceridemia: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2021;78(9):960–993. [DOI] [PubMed] [Google Scholar]
- 3.Chait A, Ginsberg HN, Vaisar T, et al. Remnants of the triglyceride-rich lipoproteins, diabetes, and cardiovascular disease. Diabetes. 2020;69:508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duran EK, Pradhan AD. Triglyceride-rich lipoprotein remnants and cardiovascular disease. Clin Chem. 2021;67:183–196. [DOI] [PubMed] [Google Scholar]
- 5.Langlois MR, Nordestgaard BG, Langsted A, et al. Quantifying atherogenic lipoproteins for lipid-lowering strategies: consensus-based recommendations from EAS and EFLM. Clin Chem Lab Med. 2020;58:496–517. [DOI] [PubMed] [Google Scholar]
- 6.Varbo A, Benn M, Tybjaerg-Hansen A, et al. Elevated remnant cholesterol causes both low-grade inflammation and ischemic heart disease, whereas elevated low-density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation. 2013;128:1298–1309. [DOI] [PubMed] [Google Scholar]
- 7.Raposeiras-Roubin S, Rosselló X, Oliva B, et al. Triglycerides and residual atherosclerotic risk. J Am Coll Cardiol. 2021;77:3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L, Gill R, Pedersen TL, et al. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res. 2009;50:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin HK, Kim YK, Kim KY, et al. Remnant lipoprotein particles induce apoptosis in endothelial cells by NAD(P)H oxidase-mediated production of superoxide and cytokines via lectin-like oxidized low-density lipoprotein receptor-1 activation: prevention by cilostazol. Circulation. 2004;109:1022–1028. [DOI] [PubMed] [Google Scholar]
- 10.Johansen M, Nielsen SF, Afzal S, et al. Very low-density lipoprotein cholesterol may mediate a substantial component of the effect of obesity on myocardial infarction risk: the Copenhagen general population study. Clin Chem. 2021;67:276–287. [DOI] [PubMed] [Google Scholar]
- 11.Varbo A, Nordestgaard BG. Remnant cholesterol and risk of ischemic stroke in 112,512 individuals from the general population. Ann Neurol. 2019;85:550–559. [DOI] [PubMed] [Google Scholar]
- 12.Castañer O, Pintó X, Subirana I, et al. Remnant cholesterol, not LDL cholesterol, is associated with incident cardiovascular disease. J Am Coll Cardiol. 2020;76:2712–2724. [DOI] [PubMed] [Google Scholar]
- 13.Varbo A, Benn M, Tybjærg-Hansen A, et al. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–436. [DOI] [PubMed] [Google Scholar]
- 14.Quispe R, Martin SS, Michos ED, et al. Remnant cholesterol predicts cardiovascular disease beyond LDL and ApoB: a primary prevention study. Eur Heart J. Published online July 19, 2021. 10.1093/eurheartj/ehab432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saeed A, Feofanova EV, Yu B, et al. Remnant-like particle cholesterol, low-density lipoprotein triglycerides, and incident cardiovascular disease. J Am Coll Cardiol. 2018;72:156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duran EK, Aday AW, Cook NR, et al. Triglyceride-rich lipoprotein cholesterol, small dense ldl cholesterol, and incident cardiovascular disease. J Am Coll Cardiol. 2020;75:2122–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balling M, Afzal S, Varbo A, et al. VLDL cholesterol accounts for one-half of the risk of myocardial infarction associated with apoB-containing lipoproteins. J Am Coll Cardiol. 2020;76:2725–2735. [DOI] [PubMed] [Google Scholar]
- 18.Hussain A, Sun C, Selvin E, et al. Triglyceride-rich lipoproteins, apolipoprotein C-III, angiopoietin-like protein 3, and cardiovascular events in older adults: Atherosclerosis Risk in Communities (ARIC) study. Eur J Prev Cardiol. Published online January 14, 2021. 10.1093/eurjpc/zwaa152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liou L, Kaptoge S. Association of small, dense LDL-cholesterol concentration and lipoprotein particle characteristics with coronary heart disease: a systematic review and meta-analysis. PLoS One. 2020;15:e0241993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikezaki H, Lim E, Cupples LA, et al. Small dense low-density lipoprotein cholesterol is the most atherogenic lipoprotein parameter in the prospective framingham offspring study. J Am Heart Assoc. 2021;10:e019140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dron JS, Wang J, Cao H, et al. Severe hypertriglyceridemia is primarily polygenic. J Clin Lipidol. 2019;13:80–88. [DOI] [PubMed] [Google Scholar]
- 22.Swanson D, Block R, Mousa SA. Omega-3 fatty acids EPA and DHA: health benefits throughout life. Adv Nutr. 2012;3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shearer GC, Savinova OV, Harris WS. Fish oil-how does it reduce plasma triglycerides? Biochim Biophys Acta. 2012;1821:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aung T, Halsey J, Kromhout D, et al. Associations of omega-3 fatty acid supplement use with cardiovascular disease risks: meta-analysis of 10 trials involving 77 917 individuals. JAMA Cardiol. 2018;3:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369:1090–1098. [DOI] [PubMed] [Google Scholar]
- 26.Manson JE, Cook NR, Lee IM, et al. Marine n-3 fatty acids and prevention of cardiovascular disease and cancer. N Engl J Med. 2019;380:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowman L, Mafham M, Wallendszus K, et al. Effects of n-3 fatty acid supplements in diabetes mellitus. N Engl J Med. 2018;379:1540–1550. [DOI] [PubMed] [Google Scholar]
- 28.Bhatt DL, Steg PG, Miller M, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2019;380:11–22. [DOI] [PubMed] [Google Scholar]
- 29.Nicholls SJ, Lincoff AM, Garcia M, et al. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the STRENGTH randomized clinical trial. JAMA. 2020;324:2268–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marston NA, Giugliano RP, Im K, et al. Association between triglyceride lowering and reduction of cardiovascular risk across multiple lipidlowering therapeutic Classes: a systematic review and meta-regression analysis of randomized controlled trials. Circulation. 2019;140:1308–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherratt SCR, Mason RP. Eicosapentaenoic acid and docosahexaenoic acid have distinct membrane locations and lipid interactions as determined by X-ray diffraction. Chem Phys Lipids. 2018;212:73–79. [DOI] [PubMed] [Google Scholar]
- 32.Mason RP, Jacob RF, Shrivastava S, et al. Eicosapentaenoic acid reduces membrane fluidity, inhibits cholesterol domain formation, and normalizes bilayer width in atherosclerotic-like model membranes. Biochim Biophys Acta. 2016;1858:3131–3140. [DOI] [PubMed] [Google Scholar]
- 33.Williams JA, Batten SE, Harris M, et al. Docosahexaenoic and eicosapentaenoic acids segregate differently between raft and nonraft domains. Biophys J. 2012;103:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le VT, Knight S, Knowlton K, et al. Higher docosahexaenoic acid levels lower the protective impact of eicosapentaenoic acid on long-term MACE in those with and without angiographic CAD. J Am Coll Cardiol. 2021;77(18 Suppl 1):1453. [Google Scholar]
- 35.Doi T, Langsted A, Nordestgaard BG. A possible explanation for the contrasting results of REDUCE-IT vs. STRENGTH: cohort study mimicking trial designs. Eur Heart J. 2021. Aug 29: ehab555. 10.1093/eurheartj/ehab555. [DOI] [PubMed] [Google Scholar]
- 36.Adiels M, Taskinen MR, Björnson E, et al. Role of apolipoprotein C-III overproduction in diabetic dysLipidaemia. Diabetes Obes Metab. 2019;21:1861–1870. [DOI] [PubMed] [Google Scholar]
- 37.Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, et al. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41. [DOI] [PubMed] [Google Scholar]
- 38.Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reyes-Soffer G, Sztalryd C, Horenstein RB, et al. Effects of APOC3 heterozygous deficiency on plasma lipid and lipoprotein metabolism.Arterioscler Thromb Vasc Biol. 2019;39:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larsson M, Vorrsjö E, Talmud P, et al. Apolipoproteins C-I and C-III inhibit lipoprotein lipase activity by displacement of the enzyme from lipid droplets. J Biol Chem. 2013;288:33997–34008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaudet D, Brisson D, Tremblay K, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–2206. [DOI] [PubMed] [Google Scholar]
- 42.Zheng C, Khoo C, Furtado J, et al. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation. 2010;121:1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramms B, Gordts P. Apolipoprotein C-III in triglyceride-rich lipoprotein metabolism. Curr Opin lipidol. 2018;29:171–179. [DOI] [PubMed] [Google Scholar]
- 44.Cohn JS, Patterson BW, Uffelman KD, et al. Rate of production of plasma and very-low-density lipoprotein (VLDL) apolipoprotein C-III is strongly related to the concentration and level of production of VLDL triglyceride in male subjects with different body weights and levels of insulin sensitivity. J Clin Endocrinol Metab. 2004;89:3949–3955. [DOI] [PubMed] [Google Scholar]
- 45.Esan O, Wierzbicki AS. Volanesorsen in the treatment of familial chylomicronemia syndrome or hypertriglyceridaemia: design, development and place in therapy. Drug Des Devel Ther. 2020;14:2623–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaudet D, Alexander VJ, Baker BF, et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. 2015;373:438–447. [DOI] [PubMed] [Google Scholar]
- 47.Witztum JL, Gaudet D, Freedman SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med. 2019;381:531–542. [DOI] [PubMed] [Google Scholar]
- 48.Gouni-Berthold I, Alexander VJ, Yang Q, et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021;9:264–275. [DOI] [PubMed] [Google Scholar]
- 49.Alexander VJ, Xia S, Hurh E, et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J. 2019;40:2785–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watts GF, Schwabe C, Scott R, et al. RNA interference targeting hepatic angiopoietin-like protein 3 results in prolonged reductions in plasma triglycerides and LDL-C in human subjects. Circulation. 2019;140:E987–E988. [Google Scholar]
- 51.Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017;377:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.StitzieL NO, Khera AV, Wang X, et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69:2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chi X, Britt EC, Shows HW, et al. ANGPTL8 promotes the ability of ANGPTL3 to bind and inhibit lipoprotein lipase. Mol Metab. 2017;6:1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haller JF, Mintah IJ, Shihanian LM, et al. ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. J Lipid Res. 2017;58:1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen YQ, Pottanat TG, Siegel RW, et al. Angiopoietin-like protein 8 differentially regulates ANGPTL3 and ANGPTL4 during postprandial partitioning of fatty acids. J Lipid Res. 2020;61:1203–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimizugawa T, Ono M, Shimamura M, et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. 2002;277:33742–33748. [DOI] [PubMed] [Google Scholar]
- 57.Adam RC, Mintah IJ, Alexa-Braun CA, et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J Lipid Res. 2020;61:1271–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reeskamp LF, Millar JS, Wu L, et al. ANGPTL3 inhibition with evinacumab results in faster clearance of IDL and LDL apoB in patients with homozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2021;41(5):1753–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahmad Z, Banerjee P, Hamon S, et al. Inhibition of angiopoietin-like protein 3 with a monoclonal antibody reduces triglycerides in hypertriglyceridemia. Circulation. 2019;140:470–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahmad Z, Pordy R, Rader DJ, et al. Inhibition of angiopoietin-like protein 3 with evinacumab in patients with high and severe hypertriglyceridemia. J Am Coll Cardiol. 2021;78:193–195. [DOI] [PubMed] [Google Scholar]
- 61.Rosenson RS, Gaudet D, Ballantyne C, et al. A phase 2 trial of the efficacy and safety of evinacumab in patients with severe hypertriglyceridemia. American College of Cardiology, Annual Meeting. 2021. Abstract 406-19. [Google Scholar]
- 62.Graham MJ, Lee RG, Brandt TA, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. 2017;377:222–232. [DOI] [PubMed] [Google Scholar]
- 63.Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur Heart J. 2020;41:3936–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pradhan AD, Paynter NP, Everett BM, et al. Rationale and design of the pemafibrate to reduce cardiovascular outcomes by reducing triglycerides in patients with diabetes (PROMINENT) study. Am Heart J. 2018;206:80–93. [DOI] [PubMed] [Google Scholar]
- 65.Rosenson RS, Burgess LJ, Ebenbichler CF, et al. Evinacumab in patients with refractory hypercholesterolemia. N Engl J Med. 2020;383:2307–2319. [DOI] [PubMed] [Google Scholar]
- 66.Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383:711–720. [DOI] [PubMed] [Google Scholar]