

ABSTRACT
Gammaherpesviruses, such as human Epstein-Barr virus (EBV) and murine gammaherpesvirus 68 (MHV68), are species-specific, ubiquitous pathogens that are associated with multiple cancers, including B cell lymphomas. These viruses have a natural tropism for B cells and usurp B cell differentiation to drive a unique and robust polyclonal germinal center response to establish a long-term latent reservoir in memory B cells. The robust polyclonal germinal center response driven by gammaherpesvirus infection increases the risk for B cell transformation. Unsurprisingly, many gammaherpesvirus cancers are derived from germinal center or post-germinal center B cells. The viral and host factors that influence the gammaherpesvirus-driven germinal center response are not clearly defined. We previously showed that host interleukin 17 receptor A (IL-17RA) signaling promotes the establishment of chronic MHV68 infection and the MHV68-driven germinal center response. In this study, we found that T cell-intrinsic IL-17RA signaling recapitulates some proviral aspects of global IL-17RA signaling during MHV68 infection. Specifically, we found that T cell-intrinsic IL-17RA signaling supports the MHV68-driven germinal center response, the establishment of latency in the spleen, and viral reactivation in the spleen and peritoneal cavity. Our study unveils an unexpected finding where the T cell-specific IL-17RA signaling supports the establishment of a latent reservoir of a B cell-tropic gammaherpesvirus.
IMPORTANCE Gammaherpesviruses, such as human EBV, establish lifelong infection in >95% of adults and are associated with B cell lymphomas. Gammaherpesviruses usurp the germinal center response to establish latent infection, and the germinal center B cells are thought to be the target of viral transformation. We previously found that global expression of IL-17RA promotes the establishment of chronic MHV68 infection and the MHV68-driven germinal center response. In this study, we showed that T cell-intrinsic IL-17RA signaling is necessary to promote the MHV68-driven germinal center response by supporting CD4+ T follicular helper cell expansion. We also found that T cell-intrinsic IL-17RA signaling contributes to but is not solely responsible for the systemic proviral role of IL-17RA signaling, highlighting the multifaceted function of IL-17RA signaling during MHV68 infection.
KEYWORDS: IL-17RA, T cells, chronic infection, gammaherpesvirus, germinal center response
INTRODUCTION
Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) are human gammaherpesviruses that infect >95% of all adults, establish lifelong infections (1), and are associated with multiple cancers, including B cell lymphomas (2, 3). While not clearly demonstrated for KSHV, EBV usurps B cell differentiation to establish a latent viral reservoir in memory B cells (4–6). EBV achieves this by infecting naive B cells and inducing a robust polyclonal germinal center response which includes both virus-infected and uninfected B cells (4, 7, 8). In contrast, virus reactivation, the switch from latency to lytic replication, occurs when infected B cells differentiate into plasma cells (4, 9, 10). Importantly, in the context of murine gammaherpesvirus 68 (MHV68) infection, the differentiation of infected germinal center B cells into either memory B cells or plasma cells requires the presence of T follicular helper cells (11, 12).
Unlike that observed in most viral infections, the germinal center response induced by gammaherpesvirus infection is unique in that germinal center B cells actually support the majority of the latent viral reservoir, particularly during the early stages of chronic infection (7, 13). Interestingly, the germinal center stage of B cell differentiation is susceptible to cellular transformation, as germinal center B cells rapidly divide while downregulating tumor suppressors (14) and increasing expression of mutagenic enzymes (15, 16). Consequently, many gammaherpesvirus-driven B cell lymphomas are of germinal center or post-germinal center origin (17). Importantly, increased viral reactivation often precedes tumorigenesis (18–21). The mechanisms by which gammaherpesviruses induce the robust polyclonal germinal center response that seeds viral lymphomagenesis and the factors that promote viral reactivation that contributes to lymphomagenesis are poorly understood.
In vivo study of chronic EBV or KSHV infection is challenging given that these viruses have coevolved with their host and are thus species specific. Thus, to overcome the limitations of species specificity, the current study utilized MHV68, a natural rodent pathogen that is genetically and biologically related to EBV and KSHV (22). MHV68 offers a tractable animal model of chronic gammaherpesvirus infection and pathogenesis that benefits from powerful tools of host and virus genetics along with mouse immunology (23–25). This model allows for the study of host mechanisms that facilitate or attenuate the germinal center response as well as viral reactivation and latency during gammaherpesvirus infection.
Interleukin 17A (IL-17A) is the founding member of the IL-17 family, which consists of IL-17A through IL-17F (26). IL-17A signals through a heterodimeric receptor complex of IL-17 receptor A (IL-17RA) and IL-17RC (27–29). IL-17A is a functionally diverse cytokine that is associated with multiple autoimmune diseases and is critical for the immune response to bacterial and fungal pathogens in part via the recruitment of microbicidal neutrophils (30–34). We previously found that global expression of IL-17RA supports the establishment of chronic MHV68 infection and the virus-driven germinal center response (35). Prior to our published study (35), the role of IL-17RA signaling in gammaherpesvirus infection was unknown, with the exception of an observation that herpesvirus saimiri (HVS), a simian gammaherpesvirus, encodes a viral IL-17, which signals similarly to host IL-17A in cultured cells (36–38). Other gammaherpesviruses, including EBV and MHV68, do not encode a discernible IL-17A homologue, which suggests that these gammaherpesviruses usurp host IL-17A. In support of this, we found that MHV68 infection induces multiple cell types to produce IL-17A (35). Our findings in the animal model were corroborated by clinical observation that EBV-infected infectious mononucleosis patients display significantly elevated numbers of CD4+ T cells producing IL-17A, with these IL-17A-positive T cells persisting for at least 1 month following the resolution of clinical symptoms (39). The clinical data along with our discovery that global IL-17RA signaling promotes the establishment of chronic gammaherpesvirus infection indicate that IL-17A production and subsequent IL-17RA signaling engages multiple cell types to support gammaherpesvirus infection.
In this study, we defined the role of T cell-intrinsic IL-17RA signaling by generating a mouse model of T cell-specific IL-17RA deficiency. T cell-specific IL-17RA deficiency had no impact on the number of CD3+ T cells in naive animals. Interestingly, mice with T cell-specific IL-17RA deficiency had significantly lower MHV68 latent reservoir and reactivation in the spleen along with reduced reactivation in the peritoneal cavity; however, this attenuation of MHV68 parameters was to a lesser extent compared to that observed in global IL-17RA deficiency. Further, there was a significant attenuation of the MHV68-driven germinal center response in mice with T cell-specific IL-17RA deficiency. Interestingly, this attenuation of the germinal center response selectively correlated with a significant reduction in the titers of irrelevant and self-directed, but not total or virus specific immunoglobulins induced by MHV68 infection. In summary, our findings reveal a novel proviral role of T cell-intrinsic IL-17RA signaling that promotes the MHV68-driven germinal center response and production of irrelevant and self-directed antibodies. This study also indicates that IL-17RA expression by other cellular populations, to be defined in the future, is required to fully support the establishment of chronic gammaherpesvirus infection.
RESULTS
Mouse model of T cell-specific IL-17RA deficiency.
Having discovered a proviral role of global IL-17RA signaling during gammaherpesvirus infection (35), we next investigated the effect of T cell-intrinsic IL-17RA signaling. T cell-specific IL-17RA deficiency was generated by crossing IL-17RAfl/fL mice (40) with a mouse strain in which expression of the Cre recombinase is driven by a distal promoter of the lymphocyte protein tyrosine kinase gene (dLck Cre) (41, 42). Because the dLck promoter becomes active after positive selection of T cells in the thymus, dLck-driven Cre expression allows for the recombination of the conditional IL-17RA allele at the later stages of T cell development. Successful generation of the T cell-specific IL-17RA-deficient mice was confirmed by genotyping for the IL-17RA flox and dLck Cre alleles (Fig. 1A) and examining cellular expression on CD3+ T cells from the spleen via flow cytometry (Fig. 1B) (40). Loss of IL-17RA in T cells had no impact on the absolute numbers of CD3+ T cells in the spleens (Fig. 1C) and peritoneal cavities (Fig. 1D) of naive dLck Cre-positive compared to Cre-negative littermates.
FIG 1.

Mouse model of T cell-specific IL-17RA deficiency. (A) Representative PCR products of genotyping reactions designed to detect the presence of the LoxP sites for the conditional IL-17RA allele and presence of the dLck Cre allele. For the floxed IL-17RA allele, the wild type (WT) is at ~304 bp and the mutant is at ~350 bp. The internal positive control is at 200 bp in the dLck Cre reaction, while the transgene is at 300 bp. C57BL/6 (BL6) mice were genotyped as a negative control (B). Cellular expression of IL-17RA in CD3+ T cells and CD3− cells from the spleen was determined by flow cytometry. (C and D) Absolute numbers of CD3+ T cells in the spleen (C) and peritoneal cavity (D) isolated from naive dLck Cre-positive and dLck Cre-negative littermates. Data were pooled from 4 to 6 independent experiments, with each symbol representing an individual mouse.
T cell-specific IL-17RA deficiency leads to attenuated establishment of chronic MHV68 infection.
After infection of a naive mouse, acute MHV68 replication occurs at several anatomical locations and is controlled by 12 days postinfection. Concurrent with lytic virus clearance, MHV68 switches to latency, with peak splenic latent reservoir established by 16 days postinfection. We previously found that global IL-17RA signaling supports the establishment of chronic MHV68 infection while having no effect on acute MHV68 replication (35). The T cell-intrinsic role of IL-17RA during the establishment of chronic MHV68 infection was defined by measuring parameters of MHV68 latency in the spleens and peritoneal cavities of dLck Cre-positive and Cre-negative littermates at 16 days postinfection. Consistent with that observed in mice with global IL-17RA deficiency (35), the frequency of MHV68 DNA-positive splenocytes (Fig. 2A) and the frequencies of MHV68 reactivation from splenocytes (Fig. 2B) and peritoneal cells (Fig. 2D) were reduced in dLck Cre-positive mice compared to dLck Cre-negative littermates. In contrast, the frequency of MHV68 DNA-positive peritoneal cells was minimally affected in dLck Cre-positive mice (Fig. 2C). Thus, T cell-intrinsic IL-17RA signaling supports the establishment of chronic MHV68 infection.
FIG 2.

T cell-specific IL-17RA deficiency leads to attenuated establishment of chronic MHV68 infection. dLck Cre-positive and dLck Cre-negative mice were intranasally infected with 1,000 PFU of MHV68. Splenocytes and peritoneal cells were collected at 16 and 42 days postinfection and subjected to limiting-dilution assays to determine the frequency of MHV68 DNA-positive cells (A, C, E, and F) or the frequency of ex vivo viral reactivation (B and D). Each experimental group consisted of 3 to 5 animals; data were pooled from 2 or 3 independent experiments. SEM is displayed. For the limiting-dilution assays, the dotted line is drawn at 63.2% and the x coordinate of intersection of this line with the sigmoid graph represents an inverse of frequency of positive events.
Between 16 and 42 days postinfection, the MHV68 latent viral reservoir subsides and stabilizes, with concomitant contraction of the MHV68-driven germinal center response and decrease in MHV68 reactivation to undetectable levels. Similar to observations in mice with global IL-17RA deficiency (35), there was no longer a difference in the frequencies of MHV68-positive splenocytes (Fig. 2E) and peritoneal cells (Fig. 2F) in long-term (42 days)-infected dLck Cre-positive and dLck Cre-negative littermates. Thus, T cell-intrinsic IL-17RA signaling promotes peak levels of MHV68 latency at 16 days postinfection but does not contribute to the long-term maintenance of the latent viral reservoir.
T cell-specific IL-17RA signaling supports gammaherpesvirus-driven germinal center response during the establishment of latency.
Given the significance of the germinal center response for the establishment of chronic MHV68 infection (4–6) and the decrease in viral latency at 16 days postinfection in the dLck Cre-positive mice (Fig. 2), the germinal center response was examined next. Loss of IL-17RA in T cells resulted in a significant reduction in the frequency of germinal center B cells (Fig. 3A and B) and T follicular helper cells (Fig. 3C and D) at 16 days postinfection. The decrease in germinal center B cells of dLck Cre-positive mice occurred despite the expression of IL-17RA by B cells in this model, indicating that T cell-intrinsic IL-17RA signaling functions in trans to support expansion of germinal center B cells. Following transition to long-term infection (42 days), there was no longer a difference in the frequency of germinal center B cells (Fig. 3E) between dLck Cre-positive and dLck Cre-negative littermates. The frequency of T follicular helper cells, however, remained significantly decreased in long-term-infected dLck Cre-positive mice (Fig. 3F). Thus, T cell-intrinsic IL-17RA signaling supports the expansion of T follicular helper cells throughout chronic MHV68 infection.
FIG 3.
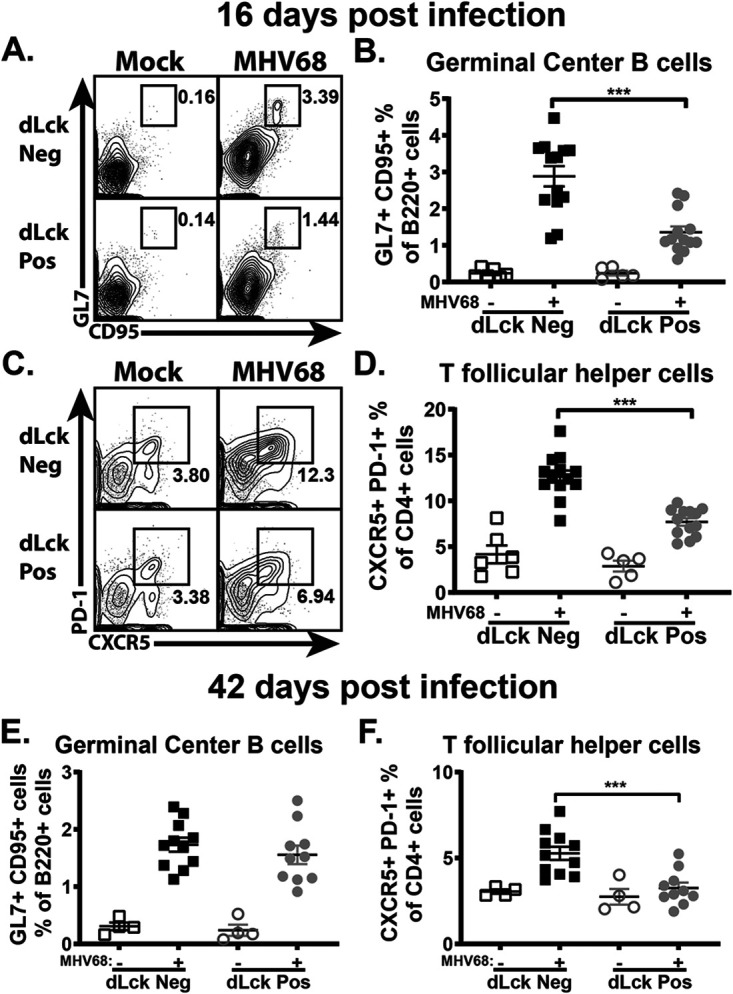
T cell-specific IL-17RA signaling supports MHV68-driven germinal center response during the establishment of latency. dLck Cre-positive and dLck Cre-negative mice were infected as for Fig. 2. The germinal center response was measured at 16 and 42 days postinfection, with germinal center B cells (A, B, and E) defined as B220+ GL7+ CD95+ cells and T follicular helper cells (C, D, and F) defined as CD3+ CD4+ CXCR5+ PD-1+cells. Each experimental group consisted of 3 or 4 animals; data were pooled from 2 or 3 independent experiments. Means and standard errors of the means are shown. ***, P < 0.001.
T cell expression of IL-17RA selectively supports irrelevant and self-directed class-switched antibody titers stimulated by MHV68 infection.
Having observed the decreased germinal center response in MHV68-infected mice with T cell-specific IL-17RA deficiency (Fig. 3), we next measured the humoral response, an outcome of B cell differentiation. Similar to observations by several groups in mice with global IL-17RA deficiency (35, 43), dLck Cre-positive mice had a baseline increase in IgG levels (Fig. 4A) (P = 0.0034), indicating that T cell-intrinsic IL-17RA signaling suppresses elevated baseline production of class-switched IgG. Further, there was no significant difference in total and MHV68-specific IgG (Fig. 4A and C) and IgM (Fig. 4B and D) levels between infected dLck Cre-positive and dLck Cre-negative littermates.
FIG 4.

T cell expression of IL-17RA selectively supports irrelevant and self-directed class-switched antibodies stimulated by MHV68 infection. dLck Cre-positive and dLck Cre-negative mice were infected as for Fig. 2 and serum was collected at 16 days postinfection. Sera were used to determine total IgG (A) and IgM (B), MHV68-specific IgG (C) and IgM (D), and dsDNA IgG (G) antibody titers. (E) Reactivity of mouse sera with HEp-2 monolayers (ANA) using anti-mouse IgG fluorescein isothiocyanate (FITC)-conjugated antibody for detection, with corrected total cell fluorescence (CTCF) quantified in panel F. Data were pooled from 2 or 3 independent experiments, with each symbol representing an individual mouse. Means and standard errors of the means are shown. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Gammaherpesviruses, including EBV and MHV68, drive a uniquely robust non-virus-specific B cell differentiation leading to a rapid (albeit transient) increase in antibody titers directed against self-antigens and foreign-species (irrelevant) antigens (44, 45). In the case of recent EBV infection in humans, this induction of antibodies against irrelevant antigens forms the basis of a diagnostic assay, which detects antibodies against horse red blood cells (46). To fully capture the range of self-antigen and irrelevant-antigen-directed antibodies produced in response to MHV68 infection, a clinical assay used in the diagnosis of autoimmune diseases (antinuclear antibody [ANA]) was repurposed to examine sera from mock- and MHV68-infected mice (45). As expected, MHV68 infection of control Cre-negative mice induced robust levels of antibodies directed against multiple antigens expressed by human HEp-2 cells (Fig. 4E). In contrast, T cell-specific deficiency of IL-17RA resulted in minimal MHV68-driven induction of antibodies directed against self-antigens and irrelevant antigens (Fig. 4E and F). Similar to observations in mice with global IL-17RA deficiency, the titers of MHV68-induced anti-double-stranded DNA (anti-dsDNA) antibodies were significantly lower in dLck Cre-positive than in dLck Cre-negative littermates (Fig. 4G). Taken together, our findings show that T cell-intrinsic IL-17RA signaling selectively supports the induction of irrelevant and self-directed, but not virus-specific, antibodies during MHV68 infection.
Impact of T cell-specific IL-17RA expression on the CD8+ T cell response during MHV68 infection.
IL-17RA signaling promotes CD8+ T cell recruitment, survival, and cytotoxicity in other experimental systems (47–50). Further, CD8+ T cells are needed to control latency and reactivation during MHV68 infection, particularly in the peritoneal cavity (51–53). Having observed decreased MHV68 reactivation from the peritoneal cells (Fig. 2D), we next quantified the CD8+ T cell response. At 16 days postinfection, loss of IL-17RA signaling in T cells had no impact on the frequency of CD8+ T cells in the spleen (Fig. 5A). Interestingly, infected dLck Cre-positive mice had significantly fewer CD8+ T cells in the peritoneal cavity (Fig. 5B). In contrast, T cell-intrinsic IL-17RA signaling did not impact the abundance of MHV68-specific CD8+ T cells in the spleen (Fig. 5C) or peritoneal cavity (Fig. 5D). The functional ability of CD8+ T cells to produce gamma interferon (IFN-γ) in the absence of IL-17RA signaling was examined next. Splenocytes and peritoneal cells from dLck Cre-positive and dLck Cre-negative littermates were restimulated ex vivo with MHV68-derived peptides (ORF6487-495 and ORF61524-531) and the frequency of IFN-γ-producing CD8+ T cells was determined. There was a significant decrease in the frequency of IFN-γ-producing CD8+ T cells in the spleens, but not the peritoneal cavities, of infected dLck Cre-positive compared to dLck Cre-negative littermates (Fig. 5E and F). Thus, despite attenuated MHV68 reactivation from the peritoneal cells (Fig. 2D), the abundance of MHV68-specific CD8 T cells and their ability to express IFN-γ were not altered in the peritoneal cavities of dLck Cre-positive mice. Conversely, decreased ability of MHV68 specific CD8+ T cells to express IFN-γ was observed in the spleen, without loss of control of splenic latency (Fig. 2A).
FIG 5.

Impact of T cell-specific IL-17RA expression on the CD8+ T cell response during MHV68 infection. dLck Cre-positive and dLck Cre-negative mice were infected as for Fig. 2 with splenocytes, and peritoneal cells were analyzed at 16 days postinfection. (A and B) Frequency of CD8+ T cells, defined as CD3+ CD8+ cells, in the spleen and peritoneal cavity. (C and D) Frequency of MHV68-specific CD8+ T cells, defined as CD3+ CD8+ CD44+ Orf6 (MHC class I tetramer)+ cells, in the spleen and peritoneal cavity. (E and F) Frequency of IFN-γ-producing CD8+ T cells, defined as CD3+ CD8+ IFN-γ+ cells, following ex vivo restimulation with MHV68 immunodominant peptides in the spleen and peritoneal cavity. Data were pooled from 2 or 3 independent experiments, with each symbol representing an individual mouse. Means and standard errors of the means are shown. *, P < 0.05; **, P < 0.01.
Impact of T cell-specific IL-17RA expression on the CD4+ T cell response during MHV68 infection.
In the context of autoimmune mouse models, IL-17RA signaling negatively regulates expression of IL-17A by CD4+ T cells (54–56). Thus, we next determined the extent to which T cell-specific loss of IL-17RA led to increased expression of IL-17A and/or affected expression of IFN-γ, a critical antiviral cytokine, by CD4+ T cells. T cell-intrinsic IL-17RA signaling had no impact on the overall frequency of CD4+ T cells in either the spleen (Fig. 6A) or peritoneal cavity (Fig. 6B) in naive or infected mice. Next, cells were ex vivo restimulated with MHV68 immunodominant major histocompatibility complex (MHC) class II epitope (gp15067-83 [57]). Similar to observations for the splenic CD8+ T cells (Fig. 5E), IL-17RA signaling in T cells supported the production of IFN-γ by CD4+ T cells in the spleen following MHV68 infection (Fig. 6C). Interestingly, despite what was observed in autoimmune mouse models (54–56), a lack of T cell-intrinsic IL-17RA signaling during MHV68 infection decreased IL-17A production by CD4+ T cells in the spleen (Fig. 6E). Loss of T cell-specific IL-17RA signaling in the peritoneal cavity, however, had no impact on the ability of CD4+ T cells to produce IFN-γ (Fig. 6D) or IL-17A (Fig. 6F).
FIG 6.

Impact of T cell-specific IL-17RA expression on the CD4+ T cell response during MHV68 infection. dLck Cre-positive and dLck Cre-negative mice were infected as for Fig. 2 with splenocytes, and peritoneal cells were analyzed at 16 days postinfection. (A and B) Frequency of CD4+ T cells, defined as CD3+ CD4+ cells, in the spleen and peritoneal cavity. (C to F) Splenocytes and peritoneal were ex vivo restimulated with MHV68 gp150 peptide. (C and D) IFN-γ-producing CD4+ T cells, defined as CD3+ CD4+ IFN-γ+ cells, in the spleen and peritoneal cavity. (E and F) IL-17A-producing CD4+ T cells, defined as CD3+ CD4+ IL-17A+ cells, in the spleen and peritoneal cavity. Data were pooled from 2 or 3 independent experiments, with each symbol representing an individual mouse. Means and standard errors of the means are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
This study uncovered the proviral T cell-intrinsic role of IL-17RA signaling during MHV68 infection by generating a mouse model of T cell-specific IL-17RA deficiency. Despite B cell tropism of MHV68, T cell-intrinsic IL-17RA signaling supported the establishment of chronic MHV68 infection as well as the MHV68-driven germinal center response and subsequent increase in irrelevant and self-directed class-switched antibodies, unveiling an intriguing connection between T cells responding to IL-17A and viral and B cell parameters of chronic gammaherpesvirus infection.
T cell-intrinsic function of IL-17RA during MHV68 infection.
Our previous study was the first to demonstrate a proviral role for global IL-17RA signaling during the establishment of chronic MHV68 infection (35). Global loss of IL-17RA significantly attenuated MHV68 latent reservoir and reactivation in the spleen and peritoneal cavity along with the MHV68-driven germinal center response. Further, global IL-17RA signaling facilitated the expansion of MHV68-specific and self-directed class-switched antibodies driven by MHV68 infection (35). To better understand the mechanisms underlying the proviral role of IL-17RA signaling during MHV68 infection, this study examined the T cell-intrinsic role of IL-17RA signaling. Loss of IL-17RA signaling in T cells was sufficient to attenuate MHV68 latency and reactivation in the spleen (Fig. 2A and B). In contrast to the case with global IL-17RA deficiency, T cell-intrinsic IL-17RA signaling had little to no impact on viral latency (Fig. 2C) in the peritoneal cavity during MHV68 infection.
Interestingly, despite the intact IL-17RA gene in the B cells, T cell-intrinsic loss of IL-17RA attenuated the MHV68-driven germinal center response (Fig. 3) and production of irrelevant and self-directed class-switched antibodies (Fig. 4E to G). T cell-intrinsic IL-17RA signaling, however, had no impact on MHV68-specific class-switched antibodies (Fig. 4C and D), Thus, given that both MHV68-specific and irrelevant antibody titers were decreased in the context of global IL-17RA deficiency (35), IL-17RA signaling in B cells may facilitate the generation of MHV68-specific class-switched antibodies during infection, a hypothesis to be tested in the future studies. Intriguingly, results of the current study indicate that T cell-intrinsic IL-17RA signaling supports the production of irrelevant and self-directed antibodies driven by MHV68 infection.
The reduction of viral latency and reactivation observed in the spleen (Fig. 2A and B) of dLck Cre-positive mice is likely due to the attenuated MHV68-driven germinal center response (Fig. 3A to D), which is critical for the establishment of chronic infection (4–6). T follicular helper cells are necessary to support the expansion of germinal center B cells during MHV68 infection (12), and this study found that T cell-intrinsic IL-17RA signaling supports expansion of T follicular helper cells (Fig. 3D and F) and expression of IL-17A by CD4+ T cells in the spleen (Fig. 6E), offering a plausible explanation for the proviral role of IL-17RA signaling in the MHV68-driven germinal center response.
Interestingly, despite the attenuation of viral reactivation in the peritoneal cavities of infected dLck Cre-positive mice (Fig. 2D), T cell-intrinsic IL-17RA signaling had no impact on the antiviral CD8+ or CD4+ T cell function in the peritoneal cavity, as measured by the IFN-γ production (Fig. 5 and 6). We have previously shown that IL-17A can promote MHV68 reactivation ex vivo; however, expression of IL-17A by peritoneal CD4+ T cells was not affected by the deficiency of IL-17RA (Fig. 6F) (35). In contrast, decreased IL-17A production by splenic CD4 T cells (Fig. 6E) could contribute to the attenuated MHV68 reactivation. In a nonexclusive alternative, decreased splenic reactivation could also result from the attenuated latent reservoir and MHV68-driven B cell differentiation in the absence of T cell-specific IL-17RA expression.
This study revealed that many of the proviral effects of global IL-17RA signaling (35) during MHV68 infection can be attributed to T cell-intrinsic IL-17RA expression, which was necessary to support the establishment of viral latency and viral reactivation in the spleen, the MHV68-driven germinal center response, the generation of irrelevant and self-directed class-switched antibodies driven by MHV68 infection, and viral reactivation in the peritoneal cavity. Though T cell-intrinsic IL-17RA signaling contributed to these proviral functions, it is clear when comparing the extents of MHV68-dependent phenotypes between the global (35) and T cell-specific IL-17RA deficiency mouse models that T cells are not the only cell type that is responsible for the proviral effects of IL-17A signaling. Nowhere is this highlighted better than in the peritoneal cavity, where loss of T cell-intrinsic IL-17RA signaling has no impact on viral latency (Fig. 2C), in contrast to the attenuation observed in mice with global loss of IL-17RA signaling (35). The additional cell type(s) relying on IL-17RA signaling to promote the peritoneal MHV68 latent reservoir will be identified in future studies.
T cell-intrinsic function of IL-17RA in the germinal center response and autoantibody production.
The production of IL-17A and subsequent IL-17RA signaling promote the germinal center response in autoimmune models, with both germinal center B cells and T follicular helper populations capable of IL-17RA expression (58–62). In an autoimmune mouse model, global loss of IL-17RA did not diminish T follicular helper cell numbers but did disrupt T follicular helper cell localization in and migration to the germinal center (59). We found that both global (35) and T cell-specific deficiency of IL-17RA during MHV68 infection significantly reduced the frequency (Fig. 3D and F) and number (data not shown) of T follicular helper cells in addition to the reduction in germinal center B cells (Fig. 3B), highlighting the significance of T cell-intrinsic IL-17RA signaling for the MHV68-driven germinal center response. Additionally, it is not yet clear whether the contribution of T cell-intrinsic IL-17RA signaling to the germinal center response is selective for MHV68 infection or if the observed germinal center phenotype is a general function of T cell-intrinsic IL-17RA signaling.
IL-17A is associated with multiple autoimmune diseases (33), and there is evidence that in the context of autoimmune mouse models, IL-17RA signaling contributes to the generation of autoantibodies against self-directed antigens (59, 62, 63). A hallmark and a unique feature of gammaherpesvirus infection is a significant, though transient, increase in irrelevant and self-directed antibody titers (44, 45). This study is the first to identify a T cell-intrinsic role for IL-17RA signaling in the generation of irrelevant and self-directed, but not antiviral, antibodies during MHV68 infection (Fig. 4). Importantly, IL-17RA signaling does not contribute to autoantibody production in every mouse model, as IL-17RA-deficient mice in a model of autoimmune lymphoproliferative syndrome (ALPS) and systemic lupus erythematosus (SLE) do not display any difference in antinuclear antibody (ANA) titers (64).
Importantly, this study is, to our knowledge, the first to examine the significance of T cell-intrinsic IL-17RA signaling for the germinal center response and production of irrelevant and self-directed class-switched antibodies, as all published studies to date have relied on mouse models of global IL-17RA deficiency or used IL-17RA neutralizing antibodies (58–64). The mice generated in this study provide an excellent tool to uncover the significance of T cell-intrinsic IL-17RA signaling in an abundance of autoimmune and infection models.
MATERIALS AND METHODS
Animals used.
All experimental manipulations of mice were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (MCW) (protocol number: AUA971). C57BL/6J mice, dLck-hcre3779 [B6/Cg-Tg(Lck-icre)3779Nik/J stock 012837] mice (41, 42), and IL-17RAfl/fL (B6.Cg-IL-17ratm2.1Koll/J stock 031000) mice (40) were obtained from The Jackson Laboratories (Bar Harbor, ME). The dLck-hcre3779 mice were bred to the IL-17RAfl/fL mice in order to generate T cell-specific IL-17RA-deficient mice (Fig. 1A). The presence of the conditional IL-17RAfl/fL allele was detected using the following primer pair: GCCTCCAAGTCTAGCTTTGCTTGGC (forward) and AGGCCCCTGAGAGCGGTTCA (reverse) (40). The dLck-hcre3779 allele was detected using ATGGTGCCCAAGAAGAAGAG (forward), CAGGTGCTGTTGGATGGTCT (reverse), CAAATGTTGCTTGTCTGGTG (internal positive control, forward), and GTCAGTCGAGTGCACAGTTT (internal positive control, reverse) (41, 42). All mice were housed and bred in a specific-pathogen-free facility at MCW. Both male and female mice were used, with no gender-specific phenotypes noted.
Infections.
Between 6 and 10 weeks of age, mice were intranasally inoculated with 1,000 PFU of MHV68 (WUMS) diluted in sterile serum-free Dulbecco’s modified Eagle’s medium (DMEM) (15 μL/mouse), under light anesthesia. MHV68 viral stock was prepared and titers were determined on NIH 3T12 cells. The spleens and peritoneal cells were harvested from euthanized mock-treated and MHV68-infected animals at the indicated times postinfection. Mice were euthanized by CO2 inhalation from a compressed gas source in a nonovercrowded chamber. Mice were bled prior to euthanasia via the submandibular route, and serum was isolated using BD Microtainer blood collection tubes (Becton, Dickinson and Company, Franklin Lakes, NJ).
Limiting-dilution assays.
The frequency of cells harboring viral DNA was determined by limiting-dilution PCR analysis, while the frequency of ex vivo reactivation was determined by limiting-dilution assay as previously described (65). Briefly, to determine the frequency of cells reactivating virus ex vivo, serial 2-fold dilutions of splenocytes or peritoneal cell suspensions harvested from infected mice were plated onto monolayers of mouse embryonic fibroblasts (MEF) immediately following harvest, at 24 replicates per dilution. To control for preformed infectious virus, 2-fold serial dilutions of mechanically disrupted splenocytes or peritoneal cells were plated as described above. MHV68 was allowed to reactivate from explanted cells, and virus was further amplified within the same well via subsequent replication in MEF. At 21 days postplating, all replicates were scored in a binary fashion for the presence of live fibroblasts (no viral reactivation/replication) or absence of such (cytopathic effect driven by lytic replication). Because primary MEF were used to amplify the virus, the sensitivity of limiting-dilution reactivation assay was below a single PFU of MHV68 defined using a 3T12 cell-based plaque assay. Because the endpoint of viral amplification in MEF was measured, the limiting-dilution reactivation assay was not susceptible to variability of titers released from primary cells upon viral reactivation ex vivo (66).
Flow cytometry.
Single cell suspensions of splenocytes and peritoneal cells from individual mice were prepared in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline [PBS] plus 2% fetal calf serum [FCS] plus 0.05% sodium azide) at 1 × 107 nucleated cells/mL. A total of 1 × 106 cells were treated with Fc block (24G2) prior to extracellular staining for 30 min on ice. For T cell phenotyping, single cell suspensions of splenocytes and peritoneal cells were plated at 6 × 106 nucleated cells/mL in a 96-well plate and stimulated either nonspecifically with 10 ng/mL of phorbol 12-myristate 13-acetate (PMA; P8139-5MG; Sigma-Aldrich, St. Louis, MO), 1 μg/mL of ionomycin (I0634-1MG; Sigma-Aldrich), and 10 μg/mL of brefeldin A (420601; BioLegend, San Diego, CA) in DMEM with 10% FBS for 4 h at 37°C or with relevant virus-specific peptides and 10 μg/mL of brefeldin A (420601; BioLegend) in DMEM with 10% FBS for 6 h at 37°C. For virus-specific CD4+ T cells, 2.5 μg/mL of MHV68-specific viral peptide GP150 (57) (GenScript, Piscataway, NJ) was utilized, while 10 μg/mL of ORF6 and ORF61 viral peptides (Thermo Fisher Scientific, Waltham, MA) were used to stimulate virus-specific CD8+ T cells. Following restimulation, cells were washed in FACS buffer before being treated with Fc block (24G2) and subjected to extracellular staining with an optimal antibody concentration for 30 min on ice. After extracellular staining, the cells were fixed and permeabilized using the BD Cytofix/Cytoperm kit (554714; Fisher Scientific, Hampton, NH). The cells were then intracellularly stained with an optimal antibody concentration for 30 min on ice. Data acquisition was performed on an LSR II flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star, Ashland, OR). The following antibodies were purchased from BioLegend (San Diego, CA) for use in this study: CD3 (17A2), CD4 (RM4-5), CD8a (53-7.3), CD95 (Jo2), PD-1 (29f.1A12), B220 (RA3-6B2), GL7 (GL-7), IL-17A (TC11-18H10.1), and IFN-γ (XMG1.2). CXCR5 (2G8) was purchased from BD Pharmingen (San Jose, CA). IL-17Ra (PAJ-17R; catalog number 12-7182-82) was purchased from Thermo Fisher Scientific (Waltham, MA). Allophycocyanin-conjugated major histocompatibility complex (MHC) class I tetramers specific for MHV68 epitope Db/ORF6487-495 (AGPHNDMEI) were obtained from the NIH Tetramer Core Facility (Emory University, Atlanta, GA). Compensation controls were done using OneComp eBeads (Thermo Fisher Scientific). Briefly, a negative control (beads alone) was used to establish a baseline photomultiplier tube (PMT) voltage and fluorescent background. Positive controls for each fluorochrome (beads with a single fluorochrome) were used to establish spillover of the individual fluorochrome into the other channels being used. PMT values are adjusted for each fluorochrome to minimize spillover. Gating strategies followed what has been previously described (35).
ELISA.
Total, MHV68-specific, and dsDNA immunoglobulin levels were determined as previously described (67). Briefly, Nunc Maxisorp plates (Fisher Scientific, Pittsburgh, PA) were coated with anti-IgG (heavy plus light) or anti-IgM antibodies (Jackson ImmunoResearch, West Grove, PA), UV-irradiated MHV68 virus stock in PBS (740,000 μJ/cm2 × 2) (Stratalinker UV cross-linker 1800; Agilent Technologies, Santa Clara, CA) or dsDNA from Escherichia coli (12.5 μg/mL; Sigma-Aldrich, St. Louis, MO) overnight at 4°C. Plates were washed with PBS-Tween (0.05%), blocked for 1 h with PBS-Tween (0.05%)-bovine serum albumin (BSA; 3%), incubated with 5-fold serial dilutions of serum in PBS-Tween (0.05%)-BSA (1.5%) for 2 h, and washed with PBS-Tween (0.05%). Bound antibody was detected with horseradish peroxidase (HRP)-conjugated goat anti-mouse total IgG (heavy + light chain [H+L]) or IgM (Jackson ImmunoResearch, West Grove, PA) using 3,3′,5,5′-tetramethylbenzidine substrate (Life Technologies, Gaithersburg, MD). HRP enzymatic activity was stopped by the addition of 1 N HCl (Sigma-Aldrich, St. Louis, MO), and the absorbance read at 450 nm on a model 1420 Victor3V multilabel plate reader (PerkinElmer, Waltham, MA).
ANA panels.
Antinuclear antibodies (ANAs) were assessed with an ANA test kit (Antibodies Inc., Davis, CA). Following the manufacturer’s protocol, serum was diluted (1:40 in PBS) and incubated over slides coated with fixed HEp-2 cells. Following serum incubation, the slides were rinsed and stained with anti-mouse IgG Alexa Fluor 488 (H+L) (Thermo Fisher Scientific, Waltham, MA). Fluorescent images were captured using NIS Elements software. Corrected fluorescence was quantified using ImageJ software from a randomly chosen field of ~20 cells in each sample.
Statistical analyses.
Statistical analyses were performed using Student’s t test (Prism; GraphPad Software, Inc.).
ACKNOWLEDGMENTS
This study was supported by grant CA203923 (V.L.T.) and an American Cancer Society Postdoctoral Award (134165-PF-19-176-01-MPC to C.N.J.).
C.N.J. and V.L.T. led the conceptual design of the study and wrote the manuscript. C.N.J. designed, performed, and analyzed experiments.
We declare no competing interests.
Contributor Information
C. N. Jondle, Email: cjondle@mcw.edu.
V. L. Tarakanova, Email: vera@mcw.edu.
Jae U. Jung, Lerner Research Institute, Cleveland Clinic
REFERENCES
- 1.Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, Gale CV, Du MQ, Whitehouse A, Kellam P. 2007. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J Virol 81:13578–13586. 10.1128/JVI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cesarman E. 2014. Gammaherpesviruses and lymphoproliferative disorders. Annu Rev Pathol 9:349–372. 10.1146/annurev-pathol-012513-104656. [DOI] [PubMed] [Google Scholar]
- 3.Jha HC, Banerjee S, Robertson ES. 2016. The role of gammaherpesviruses in cancer pathogenesis. Pathogens 5:18. 10.3390/pathogens5010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flano E, Kim IJ, Woodland DL, Blackman MA. 2002. Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J Exp Med 196:1363–1372. 10.1084/jem.20020890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willer DO, Speck SH. 2005. Establishment and maintenance of long-term murine gammaherpesvirus 68 latency in B cells in the absence of CD40. J Virol 79:2891–2899. 10.1128/JVI.79.5.2891-2899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babcock GJ, Decker LL, Freeman RB, Thorley-Lawson DA. 1999. Epstein-Barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J Exp Med 190:567–576. 10.1084/jem.190.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roughan JE, Thorley-Lawson DA. 2009. The intersection of Epstein-Barr virus with the germinal center. J Virol 83:3968–3976. 10.1128/JVI.02609-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L. 2018. Regulation of the germinal center response. Front Immunol 10.3389/fimmu.2018.02469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. 2009. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog 5:e1000677. 10.1371/journal.ppat.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol 79:1296–1307. 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins CM, Speck SH. 2015. Interleukin 21 signaling in B cells is required for efficient establishment of murine gammaherpesvirus latency. PLoS Pathog 11:e1004831. 10.1371/journal.ppat.1004831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins CM, Speck SH. 2014. Expansion of murine gammaherpesvirus latently infected B cells requires T follicular help. PLoS Pathog 10:e1004106. 10.1371/journal.ppat.1004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins CM, Boss JM, Speck SH. 2009. Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J Virol 83:6484–6493. 10.1128/JVI.00297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phan RT, Dalla-Favera R. 2004. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432:635–639. 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 15.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem 274:18470–18476. 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 16.Martin A, Bardwell PD, Woo CJ, Fan M, Shulman MJ, Scharff MD. 2002. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature 415:802–806. 10.1038/nature714. [DOI] [PubMed] [Google Scholar]
- 17.Kuppers R. 2003. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol 3:801–812. 10.1038/nri1201. [DOI] [PubMed] [Google Scholar]
- 18.Campbell TB, Borok M, Gwanzura L, MaWhinney S, White IE, Ndemera B, Gudza I, Fitzpatrick L, Schooley RT. 2000. Relationship of human herpesvirus 8 peripheral blood virus load and Kaposi’s sarcoma clinical stage. AIDS 14:2109–2116. 10.1097/00002030-200009290-00006. [DOI] [PubMed] [Google Scholar]
- 19.Meerbach A, Wutzler P, Hafer R, Zintl F, Gruhn B. 2008. Monitoring of Epstein-Barr virus load after hematopoietic stem cell transplantation for early intervention in post-transplant lymphoproliferative disease. J Med Virol 80:441–454. 10.1002/jmv.21096. [DOI] [PubMed] [Google Scholar]
- 20.Feng WH, Cohen JI, Fischer S, Li L, Sneller M, Goldbach-Mansky R, Raab-Traub N, Delecluse HJ, Kenney SC. 2004. Reactivation of latent Epstein-Barr virus by methotrexate: a potential contributor to methotrexate-associated lymphomas. J Natl Cancer Inst 96:1691–1702. 10.1093/jnci/djh313. [DOI] [PubMed] [Google Scholar]
- 21.Orlandi E, Paulli M, Viglio A, Pagnucco G, Riboni R, Baldanti F, Lazzarino M. 2001. Epstein-Barr virus-positive aggressive lymphoma as a consequence of immunosuppression after multiple salvage treatments for follicular lymphoma. Br J Haematol 112:373–376. 10.1046/j.1365-2141.2001.02579.x. [DOI] [PubMed] [Google Scholar]
- 22.Barton E, Mandal P, Speck SH. 2011. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol 29:351–397. 10.1146/annurev-immunol-072710-081639. [DOI] [PubMed] [Google Scholar]
- 23.Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD, Gompels UA. 1990. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J Gen Virol 71:1365–1372. 10.1099/0022-1317-71-6-1365. [DOI] [PubMed] [Google Scholar]
- 24.Tarakanova VL, Suarez F, Tibbetts SA, Jacoby MA, Weck KE, Hess JL, Speck SH, Virgin HW, IV.. 2005. Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB beta2 microglobulin-deficient mice. J Virol 79:14668–14679. 10.1128/JVI.79.23.14668-14679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Virgin HW, IV, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71:5894–5904. 10.1128/JVI.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolls JK, Lindén A. 2004. Interleukin-17 family members and inflammation. Immunity 21:467–476. 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, Qiu Y, Whitters MJ, Tomkinson KN, Dunussi-Joannopoulos K, Carreno BM, Collins M, Wolfman NM. 2007. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem 282:13447–13455. 10.1074/jbc.M700499200. [DOI] [PubMed] [Google Scholar]
- 28.Chang SH, Dong C. 2007. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res 17:435–440. 10.1038/cr.2007.35. [DOI] [PubMed] [Google Scholar]
- 29.Toy D, Kugler D, Wolfson M, Bos TV, Gurgel J, Derry J, Tocker J, Peschon J. 2006. Cutting edge: Interleukin 17 signals through a heteromeric receptor complex. J Immunol 177:36–39. 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 30.Gu C, Wu L, Li X. 2013. IL-17 family: cytokines, receptors and signaling. Cytokine 64:477–485. 10.1016/j.cyto.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veldhoen M. 2017. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol 18:612–621. 10.1038/ni.3742. [DOI] [PubMed] [Google Scholar]
- 32.Iwakura Y, Ishigame H, Saijo S, Nakae S. 2011. Functional specialization of interleukin-17 family members. Immunity 34:149–162. 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 33.Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. 2017. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflamm 2017:3908061. 10.1155/2017/3908061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mills KH. 2008. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol 38:2636–2649. 10.1002/eji.200838535. [DOI] [PubMed] [Google Scholar]
- 35.Jondle CN, Johnson KE, Aurubin C, Sylvester P, Xin G, Cui W, Huppler AR, Tarakanova VL. 2021. Gammaherpesvirus usurps host IL-17 signaling to support the establishment of chronic infection. mBio 12:e00566-21. 10.1128/mBio.00566-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knappe A, Hiller C, Niphuis H, Fossiez F, Thurau M, Wittmann S, Kuhn EM, Lebecque S, Banchereau J, Rosenwirth B, Fleckenstein B, Heeney J, Fickenscher H. 1998. The interleukin-17 gene of herpesvirus saimiri. J Virol 72:5797–5801. 10.1128/JVI.72.7.5797-5801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. 1995. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 3:811–821. 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 38.Yao Z, Spriggs MK, Derry JM, Strockbine L, Park LS, VandenBos T, Zappone JD, Painter SL, Armitage RJ. 1997. Molecular characterization of the human interleukin (IL)-17 receptor. Cytokine 9:794–800. 10.1006/cyto.1997.0240. [DOI] [PubMed] [Google Scholar]
- 39.Gao A, Shao Y. 2017. Investigation for the roles of TLR2, TLR9 and Th17/Treg in the pathogenesis of infectious mononucleosis. Int J Clin Exp Med 10:14946–14953. [Google Scholar]
- 40.Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, Vikram A, Good M, Schoenborn AA, Bibby K, Montelaro RC, Metzger DW, Gulati AS, Kolls JK. 2016. Intestinal interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity 44:659–671. 10.1016/j.immuni.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wildin RS, Wang HU, Forbush KA, Perlmutter RM. 1995. Functional dissection of the murine lck distal promoter. J Immunol 155:1286–1295. [PubMed] [Google Scholar]
- 42.Wildin RS, Garvin AM, Pawar S, Lewis DB, Abraham KM, Forbush KA, Ziegler SF, Allen JM, Perlmutter RM. 1991. Developmental regulation of lck gene expression in T lymphocytes. J Exp Med 173:383–393. 10.1084/jem.173.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, Evans RT, Gaffen SL. 2007. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood 109:3794–3802. 10.1182/blood-2005-09-010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sangster MY, Topham DJ, D'Costa S, Cardin RD, Marion TN, Myers LK, Doherty PC. 2000. Analysis of the virus-specific and nonspecific B cell response to a persistent B-lymphotropic gammaherpesvirus. J Immunol 164:1820–1828. 10.4049/jimmunol.164.4.1820. [DOI] [PubMed] [Google Scholar]
- 45.Gauld SB, De Santis JL, Kulinski JM, McGraw JA, Leonardo SM, Ruder EA, Maier W, Tarakanova VL. 2013. Modulation of B-cell tolerance by murine gammaherpesvirus 68 infection: requirement for Orf73 viral gene expression and follicular helper T cells. Immunology 139:197–204. 10.1111/imm.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fleisher GR, Collins M, Fager S. 1983. Limitations of available tests for diagnosis of infectious mononucleosis. J Clin Microbiol 17:619–624. 10.1128/jcm.17.4.619-624.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dadaglio G, Fayolle C, Oberkampf M, Tang A, Rudilla F, Couillin I, Torheim EA, Rosenbaum P, Leclerc C. 2020. IL-17 suppresses the therapeutic activity of cancer vaccines through the inhibition of CD8+ T-cell responses. Oncoimmunology 9:1758606. 10.1080/2162402X.2020.1758606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J, Ye X, Pitmon E, Lu M, Wan J, Jellison ER, Adler AJ, Vella AT, Wang K. 2019. IL-17 inhibits CXCL9/10-mediated recruitment of CD8+ cytotoxic T cells and regulatory T cells to colorectal tumors. J Immunother Cancer 7:324. 10.1186/s40425-019-0757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tosello Boari J, Araujo Furlan CL, Fiocca Vernengo F, Rodriguez C, Ramello MC, Amezcua Vesely MC, Gorosito Serrán M, Nuñez NG, Richer W, Piaggio E, Montes CL, Gruppi A, Acosta Rodríguez EV. 2018. IL-17RA-signaling modulates CD8+ T cell survival and exhaustion during Trypanosoma cruzi infection. Front Immunol 9:2347–2347. 10.3389/fimmu.2018.02347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Acharya D, Wang P, Paul AM, Dai J, Gate D, Lowery JE, Stokic DS, Leis AA, Flavell RA, Town T, Fikrig E, Bai F. 2017. Interleukin-17A promotes CD8+ T cell cytotoxicity to facilitate West Nile virus clearance. J Virol 91:e01529-16. 10.1128/JVI.01529-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tibbetts SA, van Dyk LF, Speck SH, Virgin HW, IV.. 2002. Immune control of the number and reactivation phenotype of cells latently infected with a gammaherpesvirus. J Virol 76:7125–7132. 10.1128/jvi.76.14.7125-7132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Braaten DC, Sparks-Thissen RL, Kreher S, Speck SH, Virgin HW, IV.. 2005. An optimized CD8+ T-cell response controls productive and latent gammaherpesvirus infection. J Virol 79:2573–2583. 10.1128/JVI.79.4.2573-2583.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evans AG, Moser JM, Krug LT, Pozharskaya V, Mora AL, Speck SH. 2008. A gammaherpesvirus-secreted activator of Vβ4+ CD8+ T cells regulates chronic infection and immunopathology. J Exp Med 205:669–684. 10.1084/jem.20071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagata T, McKinley L, Peschon JJ, Alcorn JF, Aujla SJ, Kolls JK. 2008. Requirement of IL-17RA in Con A induced hepatitis and negative regulation of IL-17 production in mouse T cells. J Immunol 181:7473–7479. 10.4049/jimmunol.181.11.7473. [DOI] [PubMed] [Google Scholar]
- 55.Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, Wang W, Chen Z, Silver PB, Jittayasothorn Y, Chan CC, Chen J, Horai R, Caspi RR. 2020. The cytokine IL-17A Limits Th17 pathogenicity via a negative feedback loop driven by autocrine induction of IL-24. Immunity 53:384–397.e5. 10.1016/j.immuni.2020.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt T, Luebbe J, Kilian C, Riedel J-H, Hiekmann S, Asada N, Ginsberg P, Robben L, Song N, Kaffke A, Peters A, Borchers A, Flavell RA, Gagliani N, Pelzcar P, Huber S, Huber TB, Turner J-E, Paust H-J, Krebs CF, Panzer U. 2021. IL-17 receptor C signaling controls CD4+ Th17 immune responses and tissue injury in immune-mediated kidney diseases. J Am Soc Nephrol 32:3081–3098. 10.1681/ASN.2021030426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu Z, Blackman MA, Kaye KM, Usherwood EJ. 2015. Functional heterogeneity in the CD4+ T cell response to murine gamma-herpesvirus 68. J Immunol 194:2746–2756. 10.4049/jimmunol.1401928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsu H-C, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le T-VL, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. 2008. Interleukin 17–producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 9:166–175. 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 59.Ding Y, Li J, Wu Q, Yang P, Luo B, Xie S, Druey KM, Zajac AJ, Hsu H-C, Mountz JD. 2013. IL-17RA is essential for optimal localization of follicular Th cells in the germinal center light zone to promote autoantibody-producing B cells. J Immunol 191:1614–1624. 10.4049/jimmunol.1300479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferretti E, Ponzoni M, Doglioni C, Pistoia V. 2016. IL-17 superfamily cytokines modulate normal germinal center B cell migration. J Leukoc Biol 100:913–918. 10.1189/jlb.1VMR0216-096RR. [DOI] [PubMed] [Google Scholar]
- 61.Ferretti E, Di Carlo E, Ognio E, Guarnotta C, Bertoni F, Corcione A, Prigione I, Fraternali-Orcioni G, Ribatti D, Ravetti JL, Ponzoni M, Tripodo C, Pistoia V. 2015. Interleukin-17A promotes the growth of human germinal center derived non-Hodgkin B cell lymphoma. Oncoimmunology 4:e1030560. 10.1080/2162402X.2015.1030560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghali JR, O’Sullivan KM, Eggenhuizen PJ, Holdsworth SR, Kitching AR. 2017. Interleukin-17RA promotes humoral responses and glomerular injury in experimental rapidly progressive glomerulonephritis. Nephron 135:207–223. 10.1159/000453059. [DOI] [PubMed] [Google Scholar]
- 63.Smith C, Buhlmann JE, Wang X, Bartlett A, Lim B, Barrington RA. 2016. CD275-independent IL-17–producing T follicular helper–like cells in lymphopenic autoimmune-prone mice. J Immunol 196:4935–4946. 10.4049/jimmunol.1402193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corneth OBJ, Schaper F, Luk F, Asmawidjaja PS, Mus AMC, Horst G, Heeringa P, Hendriks RW, Westra J, Lubberts E. 2019. Lack of IL-17 receptor A signaling aggravates lymphoproliferation in C57BL/6 lpr mice. Sci Rep 9:4032. 10.1038/s41598-019-39483-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tarakanova VL, Stanitsa E, Leonardo SM, Bigley TM, Gauld SB. 2010. Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in vivo. Virology 405:50–61. 10.1016/j.virol.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 66.Mboko WP, Olteanu H, Ray A, Xin G, Darrah EJ, Kumar SN, Kulinski JM, Cui W, Dittel BN, Gauld SB, Tarakanova VL. 2015. Tumor suppressor interferon-regulatory factor 1 counteracts the germinal center reaction driven by a cancer-associated gammaherpesvirus. J Virol 90:2818–2829. 10.1128/JVI.02774-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Darrah EJ, Jondle CN, Johnson KE, Xin G, Lange PT, Cui W, Olteanu H, Tarakanova VL. 2019. Conserved gammaherpesvirus protein kinase selectively promotes irrelevant B cell responses. J Virol 93:e01760-18. 10.1128/JVI.01760-18. [DOI] [PMC free article] [PubMed] [Google Scholar]